
INTRODUCTION
Pancreatic cancer is among the most intractable of all human cancers and is also a leading cause of cancer-related death in the world [1, 2]. Besides the difficulty of early detection, which leads to dismally low surgical resection rate, pancreatic cancer is characterized by pervasive therapy resistance, being refractory to both chemotherapy and radiotherapy [3–5]. In the past decades, the survival of patients with pancreatic cancer has been improved with the development of fluorouracil (FU)- and gemcitabine (GEM)-based chemotherapeutic regimens, however, the prognosis of pancreatic cancer still remains miserably poor with only less than 10% of patients surviving 5 years [5–7].
Cancer stem cells (CSCs), a subpopulation of cancer cells specifically endowed with tumor-initiating capacity, have often been associated with higher therapy resistance compared with non-stem cancer cells and are therefore increasingly deemed a major culprit of post-treatment progression and/or recurrence of a variety of, albeit not all, human cancers [8–12]. Since the identification of pancreatic CSCs, studies have consistently shown that pancreatic CSCs do have higher chemoresistance than non-stem pancreatic cancer cells and have also implicated pancreatic CSCs in the poor prognosis of pancreatic cancer, suggesting that overcoming the chemoresistance inherently associated with pancreatic CSCs is apparently an indispensible step to improve overall management of pancreatic cancer [13, 14]. However, to date, the molecular basis on which pancreatic CSCs acquire resistance to conventional chemotherapeutic agents against pancreatic cancer has been poorly understood.
Here in this study, while investigating the molecular mechanism underlying the differential chemoresistance to 5-FU and GEM between pancreatic CSCs and their non-stem counterparts, we discovered that the JNK pathway is critically involved in pancreatic CSCs’ resistance to these chemotherapeutic agents through suppression of the intracellular level of reactive oxygen species (ROS). Our findings suggest that targeting this JNK – “ROS defense” axis in combination with current chemotherapeutic regimens may be a rational approach to overcome the therapy resistance of pancreatic cancer.
RESULTS
Self-renewing pancreatic cancer stem cells are highly resistant to 5-fluorouracil and gemcitabine compared with their differentiated counterparts
CSCs are in general considered to be highly resistant to chemotherapeutic drugs [9, 10]. To examine whether this applies to the pancreatic CSCs we have established, we used PANC-1 CSLCs, CSCs derived from the pancreatic cancer cell line PANC-1 [15], and determined the sensitivity of self-renewing (undifferentiated) PANC-1 CSLCs and their serum-differentiated counterparts to 5-FU and GEM, which are current chemotherapeutic agents of choice for the treatment of pancreatic cancer. Treatment of self-renewing and differentiated PANC-1 CSLCs with 5-FU for 3 days reduced the number of viable cells in a concentration-dependent manner, with the IC50 values being 16.8 μM and 1.7 μM, respectively (Figure 1A). Essentially similar results were obtained when GEM was used instead of 5-FU, with the IC50 values being 1.5 μM and 0.12 μM for self-renewing and differentiated PANC-1 CSLCs, respectively (Figure 1B). Thus, self-renewing PANC-1 CSLCs were by far more resistant to 5-FU and GEM than their differentiated counterparts. We then conducted similar analyses using PSN-1 CSLCs, CSCs derived from another pancreatic cancer cell line PSN-1 [15]. The results indicated that the IC50 values of 5-FU and GEM for self-renewing PSN-1 CSLCs were again much higher than those for their differentiated counterparts [5-FU: approximately 1 μM and 0.3 μM for self-renewing and differentiated PSN-1 CSLCs, respectively; GEM: 0.11 μM and < 0.01 μM for self-renewing and differentiated PSN-1 CSLCs, respectively] ((Supplementary Figure S1). Thus, the self-renewing stem cell status was indeed associated with a highly chemoresistant phenotype in pancreatic cancer cells in this study.
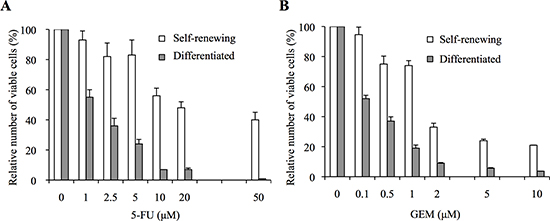
Figure 1: Pancreatic cancer stem cells show higher resistance to 5-fluorouracil and gemcitabine than their non-stem cell counterparts. (A, B) PANC-1 CSLCs, either maintained under the stem cell culture condition (Self-renewing, white bars) or induced to undergo differentiation in the presence of serum (Differentiated, gray bars), were treated with the indicated concentrations of 5-fluorouracil (A, 5-FU) or gemcitabine (B, GEM) for 3 days. Then, the number of viable cells was determined using trypan blue as a vital dye. Values in the graphs are expressed as relative to controls (i.e., drug concentration = 0 μM) and represent means + SD from triplicate samples of a representative experiment repeated with similar results.
Pivotal role of JNK in the maintenance of the chemoresistant phenotype of pancreatic cancer stem cells
We have recently demonstrated that the JNK signaling pathway is activated in self-renewing pancreatic CSCs compared with their differentiated counterparts [15]. Because our present data indicated that self-renewing pancreatic CSCs were more resistant to 5-FU and GEM (Figure 1; (Supplementary Figure 1), we questioned whether JNK signaling is involved in the chemoresistance of pancreatic CSCs to these drugs. To address this question, we determined whether the effects of 5-FU/GEM on pancreatic CSCs were altered by treatment with the JNK inhibitor SP600125, which effectively inhibited JNK activity in both PANC-1 CSLCs and PSN-1 CSLCs (Figure 2A, 2B; (Supplementary Figure S2A, S2B). When the pancreatic CSCs were exposed to 5-FU or GEM for 3 days after first treating them with SP600125, the number of viable cells decreased and the proportion of dead cells increased significantly compared with 5-FU or GEM treatment alone (Figure 2C, 2D; (Supplementary Figure S2C, S2D), suggesting that SP600125 sensitized pancreatic CSCs to these drugs through promotion of 5-FU/GEM-induced cell death. For confirmation, cell death was also analyzed by treating cell cultures in situ with a fluorescent vital dye propidium iodide (PI) instead of using trypan blue, which enabled us to count the number of dead cells without losing dead cells during the cell harvesting procedure. The results were essentially similar to those obtained by using trypan blue as a vital dye, with SP600125 pretreatment combined with 5-FU/GEM treatment causing remarkable cell death in pancreatic CSCs. In contrast, treatment with 5-FU/GEM alone again had little effect on cytotoxicity (Figure 2E, 2F; (Supplementary Figure S2E, S2F).
We also asked here whether the timing of treatment affects the effect of SP600125. PANC-1 CSLCs were either treated with SP600125 before 5-FU treatment (pre-treatment), treated simultaneously with SP600125 and 5-FU (co-treatment), or treated with SP600125 before and simultaneously with 5-FU (pre- and co-treatment). The results indicated that “co-treatment” was significantly less effective than “pre-treatment” and “pre- and co-treatment” ((Supplementary Figure S3), suggesting that inhibition of JNK prior to 5-FU treatment may be critical for efficient sensitization of pancreatic CSCs to 5-FU.
To ascertain whether the effect of SP600125 on pancreatic CSCs was mediated by inhibiting JNK activity, we next examined the effect of JNK knockdown on resistance to 5-FU/GEM. Transfection of pancreatic CSCs with combinations of siRNAs directed against JNK1 and JNK2 (siJNK1/2) mRNAs decreased the expression of JNK1 and JNK2 (Figure 3A; (Supplementary Figure S4A), and the proportion of dead cells was substantially increased when cells were exposed to 5-FU (Figure 3B, 3D; (Supplementary Figure S4B, S4D) or GEM (Figure 3C, 3E; (Supplementary Figure S4C, S4E) in combination with JNK knockdown. Thus, these results strongly suggest that JNK signaling is critically involved in the chemoresistance of pancreatic CSCs to 5-FU/GEM.
JNK contributes to the chemoresistance of pancreatic cancer stem cells through suppression of 5-fluorouracil/gemcitabine-induced increase in intracellular reactive oxygen species
We next investigated the mechanism by which JNK contributes to the chemoresistance of pancreatic CSCs. Since reactive oxygen species (ROS) have been implicated in the cytotoxicity of chemotherapeutic agents such as 5-FU and GEM [16–21], we examined the intracellular ROS levels of pancreatic CSCs after drug treatment. We noted that treatment of PANC-1 CSLCs with 5-FU (Figure 4A) or GEM (Figure 4B) alone modestly increased the proportion of cells with increased intracellular ROS compared with controls. We also noted at the same time that SP600125 treatment of PANC-1 CSLCs increased the intracellular ROS levels (Figure 4A, 4B). Strikingly, pretreating of PANC-1 CSLCs with SP600125 combined with 5-FU/GEM treatment remarkably increased the proportion of ROS-positive cells compared with 5-FU/GEM treatment alone (Figure 4A, 4B).
To determine whether the increase in intracellular ROS caused by SP600125 pretreatment has a role in SP600125-mediated sensitization of PANC-1 CSLCs to 5-FU and GEM, we next examined the effect of the antioxidant N-acetylcysteine (NAC), which scavenges free radicals by increasing intracellular glutathione levels (GSH) [22]. NAC, which effectively suppressed the increase in intracellular ROS, inhibited sensitization of pancreatic CSCs to 5-FU and GEM by SP600125; NAC treatment significantly decreased the proportion of dead cells after treatment of PANC-1 CSLCs with 5-FU (Figure 4C; (Supplementary Figure S5) and GEM (Figure 4D) in combination with SP600125. Essentially similar results were obtained when PSN-1 CSLCs ((Supplementary Figure S6) were used instead of PANC-1 CSLCs. Together, the results suggested that SP600125 sensitized pancreatic CSCs to 5-FU and GEM through promotion of 5-FU/GEM-induced increase in intracellular ROS.
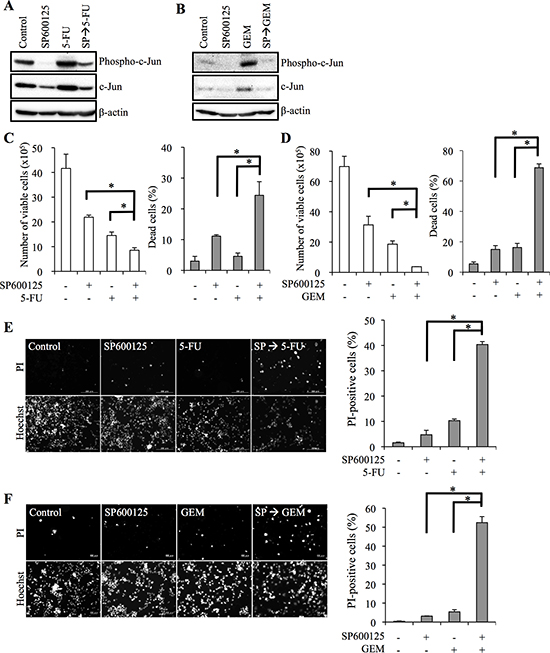
Figure 2: JNK inhibitor pretreatment sensitizes pancreatic cancer stem cells to 5-fluorouracil and gemcitabine. (A-F) PANC-1 CSLCs pretreated with or without SP600125 (SP, 20 μM) for 3 days and subsequently treated with or without 5-fluorouracil (5-FU, 20 μM) or gemcitabine (GEM, 2 μM) as indicated for 3 days in the absence of SP600125 were analyzed as follows. (A, B) Cells were subjected to immunoblot analyses for the expression of phospho- and total c-Jun. (C, D) The number of viable cells (left panels) and the percentage of dead cells (right panels) were determined using trypan blue as a vital dye. Values represent means + SD from triplicate samples of a representative experiment repeated with similar results. *P < 0.05. (E, F) Cells were subjected to cell death analysis using propidium iodide (PI) as a vital dye. Left, representative fluorescence images of PI- (upper rows) and Hoechst- (lower rows) positive cells are shown. Right, the percentage of PI-positive cells (dead cells) relative to Hoechst-positive cells (total cells) was determined. Values in the graphs represent means + SD from triplicate samples of a representative experiment repeated with similar results. *P < 0.05.
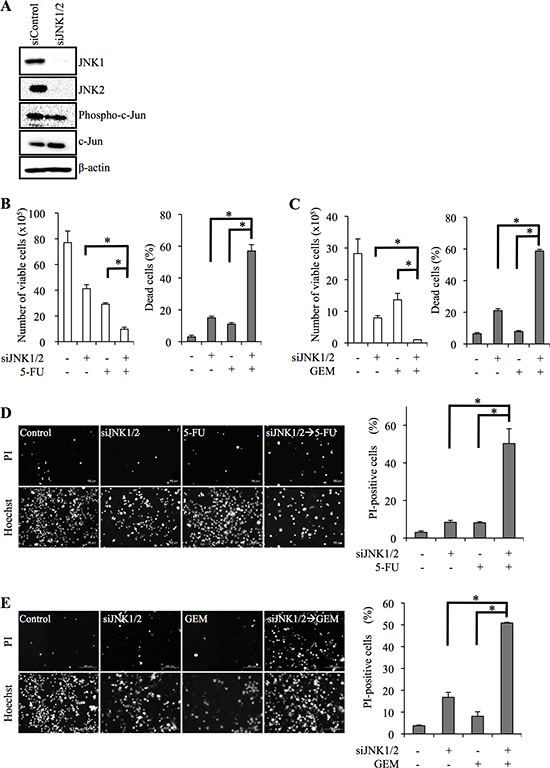
Figure 3: siRNA-mediated JNK knockdown sensitizes pancreatic cancer stem cells to 5-fluorouracil and gemcitabine. (A) PANC-1 CSLCs were transiently transfected with siRNAs against JNK1 and JNK2 (siJNK1/2) or with a control siRNA (siControl), as detailed in Materials and methods. After 8 days, the transfected cells were subjected to immunoblot analyses for the expression of the indicated proteins. (B, C) PANC-1 CSLCs were transfected as in (A) followed by treatment with 5-fluorouracil (5-FU, 20 μM) or gemcitabine (GEM, 2 μM) as indicated for 3 days. Then, the number of viable cells (left panels) and the percentage of dead cells (right panels) were determined using trypan blue. Values represent means + SD from triplicate samples of a representative experiment repeated with similar results. *P < 0.05. (D, E) PANC-1 CSLCs were transfected as in (A) followed by treatment with 5-fluorouracil (5-FU, 20 μM) or gemcitabine (GEM, 2 μM) for 3 days. Then the cells treated as indicated were subjected to cell death analysis using propidium iodide (PI). Left, representative fluorescence images of PI- (upper rows) and Hoechst- (lower rows) positive cells are shown. Right, the percentage of PI-positive cells (dead cells) relative to Hoechst-positive cells (total cells) was determined. Values in the graphs represent means + SD from triplicate samples of a representative experiment repeated with similar results. *P < 0.05.
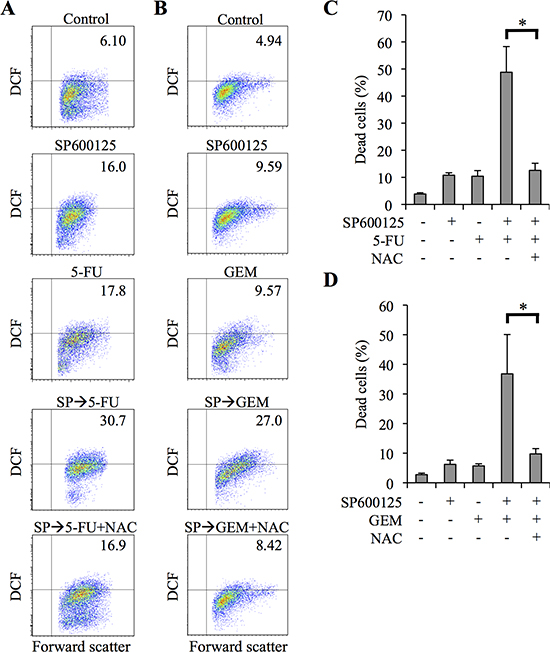
Figure 4: JNK inhibitor pretreatment sensitizes pancreatic cancer stem cells to 5-fluorouracil and gemcitabine in ROS - dependent manner. (A-D) PANC-1 CSLCs pretreated with or without SP600125 (20 μM) for 3 days were, after being cultured in the presence or absence of N-acetylcysteine (NAC, 10 mM) for 30 min, further treated with or without 5-fluorouracil (5-FU, 20 μM) or gemcitabine (GEM, 2 μM) as indicated for 3 days in the absence of SP600125. Then, the cells were analyzed as follows. (A, B) Cells were stained with 2′,7′-dichlorofluorescein diacetate (DCF-DA) and subjected to flow cytometric analysis to detect intracellular ROS. Representative flow cytometric plots with the percentages of ROS-positive cells are shown. (C, D) The percentage of dead cells was determined using trypan blue. Values represent means + SD from triplicate samples of a representative experiment repeated with similar results. *P < 0.05.
To ascertain that the effect of SP600125 on ROS accumulation induced by 5-FU/GEM, as well as on ROS-mediated sensitization of pancreatic CSCs to 5-FU/GEM, was dependent on the inhibition of JNK, we next examined the effect of JNK knockdown on the intracellular ROS level of PANC-1 CSLCs treated with 5-FU or GEM. Transient transfection of PANC-1 CSLCs with siJNK1/2 increased the proportion of ROS-positive cells following treatment with 5-FU/GEM compared with 5-FU/GEM treatment combined with control siRNA transfection (Figure 5A, 5B). Importantly, we confirmed that NAC treatment inhibited the augmentation of 5-FU/GEM-induced increase in intracellular ROS and cytotoxicity by JNK knockdown (Figure 5C, 5D). Together, these results suggested that JNK may contribute to the chemoresistance of pancreatic CSCs by preventing drug-induced increase in intracellular ROS.
JNK inhibition followed by treatment with 5-fluorouracil or gemcitabine in the absence of simultaneous JNK inhibition does not cause toxicity to normal human fibroblasts
We next evaluated the effects of combination treatment with SP600125 and 5-FU/GEM on IMR90 normal human lung fibroblasts. When IMR90 cells were treated with the drugs at their respective highest concentrations used in combination treatments in this study (20 μM for SP600125, 20 μM for 5-FU, and 2 μM for GEM), the viability of IMR90 cells was not appreciably affected either by single or combination treatments, so long as SP600125 treatment was applied prior to 5-FU or GEM in case of combination treatments (Figure 6A, 6B). Of note, the intracellular ROS level of IMR90 cells remained unchanged irrespective of whether the drugs were applied alone or in combination, which may explain the lack of toxicity of these drugs to IMR90 cells (Figure 6C, 6D). We also asked here whether the timing of SP600125 treatment affects its toxicity to IMR90 cells. Whereas “pre-treatment” and “pre- and co-treatment” of SP600125 were similarly effective at sensitizing pancreatic CSCs to 5-FU ((Supplementary Figure S3), “pre- and co-treatment” but not “pre-treatment” turned out to have a growth inhibitory effect on IMR90 cells though neither treatment induced cell death ((Supplementary Figure S7). Thus, the results suggest that the “pre-treatment” protocol might have the widest therapeutic window when JNK inhibition is to be combined with 5-FU/GEM to target pancreatic CSCs.
DISCUSSION
Pancreatic cancer is one of the most malignant human cancers, which is so often unresectable already at the time of diagnosis and, above all, responds poorly to either chemotherapy or radiotherapy [3–5]. A growing body of evidence has come to suggest that pancreatic CSCs, endowed with both tumor-initiating capacity as well as with high therapy resistance, is among the key factors underlying the extreme refractoriness of pancreatic cancer to conventional cancer therapies. Identification of the molecules and/or signaling pathways that confer chemoresistance on pancreatic CSCs, therefore, is expected to contribute to better management of pancreatic cancer. However, currently, the molecular basis of the increased chemoresistance of pancreatic CSCs remains poorly understood.
Here in this study, we first found that pancreatic CSCs, which we have shown to have activated JNK [15], were highly resistant to 5-FU and GEM compared with their non-stem counterparts, essentially consistent with earlier reports [13, 14]. We then demonstrated that inhibition of the activated JNK in pancreatic CSCs sensitized them to 5-FU and GEM and that JNK inhibition did so through elevation of the intracellular ROS level in combination with these chemotherapeutic agents. These results suggest that a JNK-regulated ROS defense mechanism plays a key role in the chemoresistance of pancreatic CSCs and that targeting this mechanism may be a useful measure to overcome the chemoresistance of pancreatic CSCs.
Although JNK was originally identified as a positive regulator of cell death and overwhelming evidence does support the idea, JNK has also been shown to have a pro-survival function depending on the context [23–25]. Indeed, there are studies suggesting that the JNK pathway may play a positive role in chemoresistance of some cancer types [26]. However, the role of JNK in chemoresistance/sensitivity of pancreatic cancer cells, let alone pancreatic CSCs, to clinically relevant chemotherapeutic agents has remained largely unknown except for a report showing that GEM-induced apoptosis of pancreatic cancer cells was dependent on JNK-mediated DUSP1 expression [27]. In this regard, the present study is the very first to demonstrate that JNK has a protective role against 5-FU or GEM in pancreatic cancer cells, more specifically, in pancreatic CSCs. On the other hand, although the mechanisms involved in pancreatic CSC chemoresistance are only beginning to be understood, recent studies have suggested that uPA and c-Met may also be involved in the chemoresistance of pancreatic CSCs, which is quite intriguing in light of the previous observations that JNK could function upstream and downstream of uPA and c-Met, respectively [28–30]. Although not pursued in the present study, investigation on the possible functional links among uPA, c-Met, and JNK may help better understand the whole picture of the molecular mechanism underlying the chemoresistance of pancreatic CSCs.
In this study, we have provided evidence that JNK contributes to the chemoresistance of pancreatic CSCs through suppression of the intracellular ROS level. It has been demonstrated that CSCs have lower intracellular ROS levels and that the low ROS levels are associated with the radioresistance of CSCs [31, 32]. In contrast to radioresistance, however, the role of enhanced ROS defense of CSCs in chemoresistance has yet to be established. Significantly, lines of evidence suggest that ROS may participate in the cytotoxic mechanisms of chemotherapeutic agents such as 5-FU and GEM [17, 33–37], lending support to the idea that cellular antioxidant capacity may determine cells’ sensitivity/resistance to such chemotherapeutic agents. The results of the present study, indicating that the increase in intracellular ROS caused by JNK inhibition sensitizes pancreatic CSCs to 5-FU and GEM, are quite in line with the idea. Although only pancreatic CSCs were examined in the present study, future studies may reveal that this paradigm can be extended to CSCs of other cancer types. It would also be worth mentioning here that our results clearly demonstrated the key role of JNK in maintaining the intracellular ROS level low in pancreatic CSCs. In sharp contrast to the well-documented role of ROS in JNK activation [38, 39], the role of JNK in the control of the intracellular ROS level remains largely uninvestigated. Yet there have been two studies reporting the ROS-inhibitory role of JNK. Interestingly, these two studies showed that activated JNK was localized to mitochondria and that JNK inhibition led to accumulation of mitochondrial superoxide [40, 41], pointing to the intriguing possibility that JNK may have a role in keeping the mitochondrial ROS level in check.
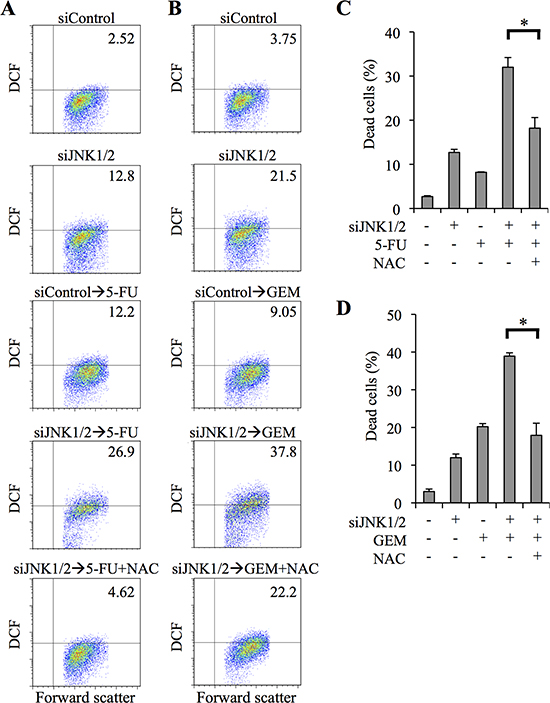
Figure 5: siRNA-mediated JNK knockdown sensitizes pancreatic cancer stem cells to 5-fluorouracil and gemcitabine in ROS - dependent manner. (A-D) PANC-1 CSLCs were transiently transfected with siRNAs against JNK1 and JNK2 (siJNK1/2) or with a control siRNA (siControl), as detailed in Materials and methods. After 8 days, the transfected cells were cultured in the presence or absence of N-acetylcysteine (NAC, 10 mM) for 30 min and further treated with or without 5-fluorouracil (5-FU, 20 μM) or gemcitabine (GEM, 2 μM) as indicated for 3 days. Then, the cells were analyzed as follows. (A, B) Cells were stained with 2′,7′-dichlorofluorescein diacetate (DCF-DA) and subjected to flow cytometric analysis to detect intracellular ROS. Representative flow cytometric plots with the percentages of ROS-positive cells are shown. (C, D) The percentage of dead cells was determined using trypan blue. Values represent means + SD from triplicate samples of a representative experiment repeated with similar results. *P < 0.05.
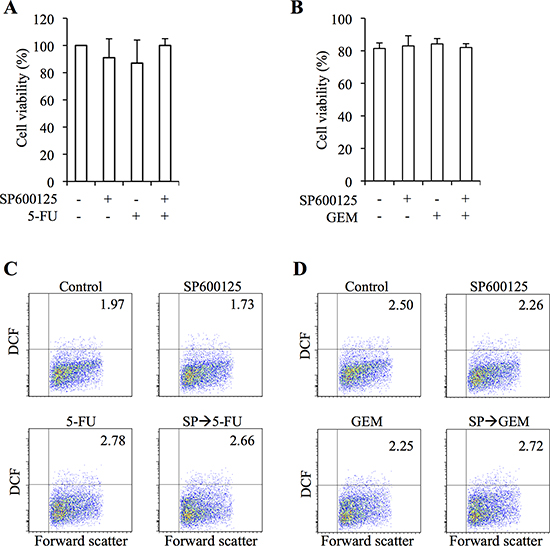
Figure 6: JNK inhibitor pretreatment does not increase the toxic effects of 5-fluorouracil and gemcitabine on normal human fibroblasts. (A-D) IMR90 normal human fibroblasts were pretreated with or without SP600125 (SP, 20 μM) for 3 days and subsequently treated with or without 5-fluorouracil (5-FU, 20 μM) or gemcitabine (GEM, 2 μM) as indicated in the absence of SP600125 for 3 days. The cells were then analyzed as follows. (A, B) Cell viability was determined using trypan blue. Values represent means + SD from triplicate samples of a representative experiment repeated with similar results. (C, D) Cells were stained with 2′,7′-dichlorofluorescein diacetate (DCF-DA) and subjected to flow cytometric analysis to detect intracellular ROS. Representative flow cytometric plots with the percentages of ROS-positive cells are shown.
Unlike in normal pancreas, JNK is activated in the majority of pancreatic cancers [42]. The significance of JNK activation in pancreatic cancer biology remained unclear, but we have only recently demonstrated that JNK activation by KRas, which is mutationally activated in the majority of pancreatic cancers, plays a key role in the maintenance of self-renewal and tumor-initiating capacity of pancreatic CSCs [15]. The findings of the present study now add a new role of JNK in pancreatic CSCs, namely, the maintenance of chemoresistance. The role of KRas in the chemoresistance of pancreatic CSCs still remains to be explored, but a previous study showed that Ras inhibition using farnesylthiosalicylic acid sensitized pancreatic cancer cells to GEM [43]. It might therefore be tempting to speculate that KRas contributes to the chemoresistance of pancreatic CSCs as well through the activation of the JNK-ROS defense axis.
Of importance from a therapeutic perspective, we found in this study that JNK inhibitor pre-treatment as well as pre- and co-treatment was superior to co-treatment in augmenting the cytotoxicity of 5-FU to pancreatic CSCs, whereas pre- and co-treatment was apparently more toxic to normal human fibroblasts than the others. The results, though being in vitro findings, suggest that a time-staggered treatment protocol might be beneficial in maximizing the therapeutic effect while minimizing adverse effects if JNK inhibition is to be combined with FU-based regimens (and possibly GEM-based regimens) in future pancreatic cancer treatment. As an explanation for the superior effect of JNK inhibitor pre-treatment, we found that the increase in the intracellular ROS level caused by JNK inhibition persisted at least for days after cessation of JNK inhibition (S.S, and M.O., unpublished observation), suggesting that “rewiring” of signaling pathways [44] may have taken place with some stability during the pre-treatment period, i.e., by the time cells were treated with 5-FU. On the other hand, GEM for instance reportedly exerts its cytotoxic effect on lung cancer cells through activation of JNK [45], suggesting the possibility that co-treatment could render the cells even more resistant to chemotherapeutic agents in case their cytotoxic effect is JNK-dependent. If we take this possibility into consideration, the pre-treatment protocol may be superior to the others in sensitizing pancreatic CSCs to chemotherapeutic agents, which is to be investigated using GEM in combination with JNK inhibitor treatment.
In conclusion, we have elucidated in this study the mechanism underlying the increased chemoresistance of pancreatic CSCs, namely, JNK-mediated suppression of the intracellular ROS level. Although preclinical studies are warranted, our findings also suggest that targeting JNK in combination with FU- or GEM-based regimens may be a promising approach to elimination of pancreatic CSCs and therefore successful treatment of pancreatic cancer.
MATERIALS AND METHODS
Antibodies and reagents
Anti-c-Jun (#9165) and anti-phospho-c-Jun (#9261) antibodies were purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA). Anti-β-actin (A1978) was from Sigma (St. Luis, MO, USA). Anti-JNK1 (sc-474) and anti-JNK2 (sc-7345) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). SP600125 was purchased from Calbiochem (La Jolla, CA, USA) and was dissolved in dimethylsulfoxide (DMSO) to prepare a 50 mM stock solution. 5-FU, GEM, 2’,7’-dichlorofluorescein diacetate (DCF-DA), and N-acetyl-L-cysteine (NAC) were from Sigma and were dissolved in DMSO to prepare 200 mM, 1 mM, 20 mM, and 5 M stock solutions, respectively.
Cell culture
The establishment of PANC-1 CSLCs and PSN-1 CSLCs, CSCs derived from human pancreatic cancer cell lines PANC-1 and PSN-1, respectively, was previously reported [15]. The authenticity of PANC-1 CSLCs and PSN-1 CSLCs as cells derived from PANC-1 and PSN-1, respectively, was verified by genotyping of short tandem repeat (STR) loci (Bio-Synthesis, Inc., Lewisville, TX, USA) followed by comparison to the ATCC STR database for Human Cell Lines. Unless otherwise indicated, these pancreatic CSCs were stably maintained and used for experiments under the monolayer stem cell culture condition, as previously described [15]. Briefly, cells were cultured on collagen-I-coated dishes (IWAKI, Tokyo, Japan) in the stem cell culture medium (DMEM/F12 medium supplemented with 1% B27 [Gibco-BRL, Carlsbad, CA, USA], 20 ng/mL EGF and FGF2 [Peprotech, Inc.,Rocky Hill, NJ, USA], D-(+)-glucose [final concentration, 26.2 mM], L-glutamine [final concentration, 4.5 mM], 100 units/mL penicillin and 100 μg/mL streptomycin). In principle, the stem cell culture medium was changed every 3 days, and EGF and FGF2 were added to the culture medium every day. Differentiation of pancreatic CSCs was induced by culturing the cells under the differentiation-inducing condition (DMEM/F12 containing 10% FBS) for 2 weeks, as previously described [15]. Normal human IMR90 fetal lung fibroblasts [46, 47] were obtained from American Type Culture Collection and maintained in DMEM/F12 supplemented with 10% fetal bovine serum (FBS) and 100 units/mL penicillin and 100 μg/mL streptomycin. All IMR90 experiments were performed using low passage number (less than 9) cells.
Gene silencing by siRNA
siRNAs against human JNK1 (VHS40722) and JNK2 (VHS40726) as well as Medium GC Duplex #2 of Stealth RNAi™ siRNA Negative Control Duplexes (as a control for siRNA experiments) were purchased from Invitrogen Life Technologies (Carlsbad, CA, USA). Transfection of siRNAs was performed using Lipofectamine RNAiMAX™ (Life Technologies) according to the manufacturer's instructions. To achieve sustained knockdown of the target genes, siRNA transfection was repeated 4 days after the initial transfection.
5-Fluorouracil and gemcitabine treatment and cell viability assays
Unless otherwise indicated, throughout this study, PANC-1 CSLCs were treated with 20 μM of 5-FU or 2 μM of GEM for 3 days. PSN-1 CSLCs were treated with 2.5 μM of 5-FU or 0.25 μM of GEM for 3 days. In each set of treatment, cells were examined for their viability at the end of pretreatment (i.e., SP600125 treatment [20 μM for PANC-1 CSLCs, 10 μM for PSN-1 CSLCs] and/or knockdown of JNK1/2), and equal numbers of viable cells were used for subsequent 5-FU, GEM, or control (DMSO) treatment. Viable and dead cells were identified by their ability and inability to exclude vital dyes, respectively [48]. In brief, cells were stained with 0.2% trypan blue, and the numbers of viable and dead cells were determined using a hemocytometer. Cell viability (%) was defined as 100 × ‘the number of viable cells’/ (‘the number of viable cells’ + ‘the number of dead cells’), whereas the percentage of dead cells was defined as 100 × ‘the number of dead cells’/ (‘the number of viable cells’ + ‘the number of dead cells’). Alternatively, cells were incubated in situ with propidium iodide (PI, 1 μg/ml) and Hoechst 33342 (10 μg/ml) for 10 min at 37 °C in the CO2 incubator, to stain dead cells and the cell nuclei, respectively. Then the numbers of PI- and Hoechst-positive cells were scored under a fluorescence microscope (CKX41; Olympus, Tokyo, Japan), and the percentage of PI-positive cells (dead cells) against Hoechst-positive cells (total cells) was determined.
Detection and measurement of intracellular ROS
Cells were incubated in the stem cell culture medium containing 10 μM DCF-DA for 10 min at 37 °C, washed twice with phosphate-buffered saline (PBS), and re-suspended in PBS. The cells were then subjected to flow cytometric analysis, during which care was taken not to expose the samples to light. For flow cytometric analysis, at least 1 × 104 cells were evaluated and gated using side and forward scatters to identify viable cell populations. All flow cytometric experiments were run on FACSCanto™ II Flow Cytometer (BD Biosciences, Franklin Lakes, NJ, USA) and the data were analyzed using FlowJo software, version 7.6.5 (Treestar Inc., Ashland, OR, USA).
Immunoblot analysis
Cells were washed with ice-cold PBS and lysed in RIPA buffer (10 mM Tris-HCl [pH 7.4], 0.1% SDS, 0.1% sodium deoxycholate, 1% NP-40, 150 mM NaCl, 1 mM EDTA, 1.5 mM Na3VO4, 10 mM NaF, 10 mM sodium pyrophosphate, 10 mM sodium β-glycerophosphate and 1% protease inhibitor cocktail set III [Calbiochem]). After centrifugation for 10 min at 14,000 × g at 4 °C, the supernatants were recovered as the cell lysates, and the protein concentration of the cell lysates was determined by the BCA protein assay kit (Pierce Biotechnology, Inc., Rockford, IL, USA). Cell lysates containing equal amounts of protein were separated by SDS-PAGE and transferred to a polyvinylidene difluoride membrane. The membrane was probed with a primary antibody and then with an appropriate HRP-conjugated secondary antibody according to the protocol recommended by the manufacturer of each antibody. Immunoreactive bands were visualized using Immobilon Western Chemiluminescent HRP Substrate (Millipore, Billerica, MA, USA).
Statistical analysis
Results are expressed as means and standard deviation (SD), and differences were compared using the 2-tailed Student's t-test. P-values < 0.05 were considered statistically significant and indicated with asterisks in the figures.
ACKNOWLEDGEMENTS
We thank Ms. Eriko Watanabe and Ms. Asuka Sugai for their technical and secretarial contributions to this study, respectively. This work was supported by Grants-in-Aid for Scientific Research, for Challenging Exploratory Research, and for Young Scientists from the Ministry of Education, Culture, Sports, Science and Technology of Japan, by a Grant-in-Aid from the Global COE Program of the Japan Society for the Promotion of Science, by the National Cancer Center Research and Development Fund (23-A-20), and by a grant from the Japan Brain Foundation.
Conflict of interest statement
The authors declare no conflict of interest.
REFERENCES
1. Ma J, Jemal A. The rise and fall of cancer mortality in the USA: why does pancreatic cancer not follow the trend? Future Oncol. 2013; 9:917–919.
2. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013; 63:11–30.
3. Hidalgo M. Pancreatic cancer. N Engl J Med. 2010; 362:1605–1617.
4. Partensky C. Toward a better understanding of pancreatic ductal adenocarcinoma: glimmers of hope? Pancreas. 2013; 42:729–739.
5. Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011; 378:607–620.
6. Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardiere C, Bennouna J, Bachet JB, Khemissa-Akouz F, Pere-Verge D, Delbaldo C, Assenat E, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011; 364:1817–1825.
7. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013; 369:1691–1703.
8. Bao B, Ahmad A, Azmi AS, Ali S, Sarkar FH. Overview of cancer stem cells (CSCs) and mechanisms of their regulation: implications for cancer therapy. Curr Protoc Pharmacol. 2013; Chapter 14:Unit 14.25.
9. Malik B, Nie D. Cancer stem cells and resistance to chemo and radio therapy. Front Biosci (Elite Ed). 2012; 4:2142–2149.
10. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001; 414:105–111.
11. Yu Y, Ramena G, Elble RC. The role of cancer stem cells in relapse of solid tumors. Front Biosci (Elite Ed). 2012; 4:1528–1541.
12. Yu Z, Pestell TG, Lisanti MP, Pestell RG. Cancer stem cells. Int J Biochem Cell Biol. 2012; 44:2144–2151.
13. Li Y, Kong D, Ahmad A, Bao B, Sarkar FH. Pancreatic cancer stem cells: emerging target for designing novel therapy. Cancer Lett. 2013; 338:94–100.
14. Xia J, Chen C, Chen Z, Miele L, Sarkar FH, Wang Z. Targeting pancreatic cancer stem cells for cancer therapy. Biochim Biophys Acta. 2012; 1826:385–399.
15. Okada M, Shibuya K, Sato A, Seino S, Suzuki S, Seino M, Kitanaka C. Targeting the K-Ras - JNK axis eliminates cancer stem-like cells and prevents pancreatic tumor formation. Oncotarget. 2014; 5:5100–5112.
16. Aresvik DM, Pettersen RD, Abrahamsen TG, Wright MS. 5-fluorouracil-induced death of Jurkat T-cells—a role for caspases and MCL-1. Anticancer Res. 2010; 30:3879–3887.
17. Donadelli M, Costanzo C, Beghelli S, Scupoli MT, Dandrea M, Bonora A, Piacentini P, Budillon A, Caraglia M, Scarpa A, Palmieri M. Synergistic inhibition of pancreatic adenocarcinoma cell growth by trichostatin A and gemcitabine. Biochim Biophys Acta. 2007; 1773:1095–1106.
18. Hwang IT, Chung YM, Kim JJ, Chung JS, Kim BS, Kim HJ, Kim JS, Yoo YD. Drug resistance to 5-FU linked to reactive oxygen species modulator 1. Biochem Biophys Res Commun. 2007; 359:304–310.
19. Hwang PM, Bunz F, Yu J, Rago C, Chan TA, Murphy MP, Kelso GF, Smith RA, Kinzler KW, Vogelstein B. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat Med. 2001; 7:1111–1117.
20. Maehara S, Tanaka S, Shimada M, Shirabe K, Saito Y, Takahashi K, Maehara Y. Selenoprotein P, as a predictor for evaluating gemcitabine resistance in human pancreatic cancer cells. Int J Cancer. 2004; 112:184–189.
21. Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat. 2004; 7:97–110.
22. Kelly GS. Clinical applications of N-acetylcysteine. Altern Med Rev. 1998; 3:114–127.
23. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000; 103:239–252.
24. Sui X, Kong N, Ye L, Han W, Zhou J, Zhang Q, He C, Pan H. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014; 344:174–179.
25. Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995; 270:1326–1331.
26. Vasilevskaya I, O'Dwyer PJ. Role of Jun and Jun kinase in resistance of cancer cells to therapy. Drug Resist Updat. 2003; 6:147–156.
27. Liu F, Gore AJ, Wilson JL, Korc M. DUSP1 is a novel target for enhancing pancreatic cancer cell sensitivity to gemcitabine. PLoS One. 2014; 9:e84982.
28. Aguirre Ghiso JA, Alonso DF, Farias EF, Gomez DE, de Kier Joffe EB. Deregulation of the signaling pathways controlling urokinase production. Its relationship with the invasive phenotype. Eur J Biochem. 1999; 263:295–304.
29. Garcia-Guzman M, Dolfi F, Zeh K, Vuori K. Met-induced JNK activation is mediated by the adapter protein Crk and correlates with the Gab1 - Crk signaling complex formation. Oncogene. 1999; 18:7775–7786.
30. Rodrigues GA, Park M, Schlessinger J. Activation of the JNK pathway is essential for transformation by the Met oncogene. Embo j. 1997; 16:2634–2645.
31. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, Joshua B, Kaplan MJ, Wapnir I, Dirbas FM, Somlo G, Garberoglio C, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009; 458:780–783.
32. Kim YS, Kang MJ, Cho YM. Low production of reactive oxygen species and high DNA repair: mechanism of radioresistance of prostate cancer stem cells. Anticancer Res. 2013; 33:4469–4474.
33. Afzal S, Jensen SA, Sorensen JB, Henriksen T, Weimann A, Poulsen HE. Oxidative damage to guanine nucleosides following combination chemotherapy with 5-fluorouracil and oxaliplatin. Cancer Chemother Pharmacol. 2012; 69:301–307.
34. Chen SH, Li DL, Yang F, Wu Z, Zhao YY, Jiang Y. Gemcitabine-induced pancreatic cancer cell death is associated with MST1/cyclophilin D mitochondrial complexation. Biochimie. 2014; 103:71–79.
35. Dalla Pozza E, Fiorini C, Dando I, Menegazzi M, Sgarbossa A, Costanzo C, Palmieri M, Donadelli M. Role of mitochondrial uncoupling protein 2 in cancer cell resistance to gemcitabine. Biochim Biophys Acta. 2012; 1823:1856–1863.
36. Das R, Bhattacharya K, Sarkar S, Samanta SK, Pal BC, Mandal C. Mahanine synergistically enhances cytotoxicity of 5-fluorouracil through ROS-mediated activation of PTEN and p53/p73 in colon carcinoma. Apoptosis. 2014; 19:149–164.
37. Pan X, Zhang X, Sun H, Zhang J, Yan M, Zhang H. Autophagy inhibition promotes 5-fluorouraci-induced apoptosis by stimulating ROS formation in human non-small cell lung cancer A549 cells. PLoS One. 2013; 8:e56679.
38. Shen HM, Liu ZG. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med. 2006; 40:928–939.
39. Son Y, Kim S, Chung HT, Pae HO. Reactive oxygen species in the activation of MAP kinases. Methods Enzymol. 2013; 528:27–48.
40. Lopez-Sanchez N, Rodriguez JR, Frade JM. Mitochondrial c-Jun NH2-terminal kinase prevents the accumulation of reactive oxygen species and reduces necrotic damage in neural tumor cells that lack trophic support. Mol Cancer Res. 2007; 5:47–60.
41. Yue WY, Clark JJ, Fernando A, Domann F, Hansen MR. Contribution of persistent C-Jun N-terminal kinase activity to the survival of human vestibular schwannoma cells by suppression of accumulation of mitochondrial superoxides. Neuro Oncol. 2011; 13:961–973.
42. Takahashi R, Hirata Y, Sakitani K, Nakata W, Kinoshita H, Hayakawa Y, Nakagawa H, Sakamoto K, Hikiba Y, Ijichi H, Moses HL, Maeda S, Koike K. Therapeutic effect of c-Jun N-terminal kinase inhibition on pancreatic cancer. Cancer Sci. 2013; 104:337–344.
43. Gana-Weisz M, Halaschek-Wiener J, Jansen B, Elad G, Haklai R, Kloog Y. The Ras inhibitor S-trans,trans-farnesylthiosalicylic acid chemosensitizes human tumor cells without causing resistance. Clin Cancer Res. 2002; 8:555–565.
44. Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, Yaffe MB. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012; 149:780–794.
45. Teraishi F, Zhang L, Guo W, Dong F, Davis JJ, Lin A, Fang B. Activation of c-Jun NH2-terminal kinase is required for gemcitabine's cytotoxic effect in human lung cancer H1299 cells. FEBS Lett. 2005; 579:6681–6687.
46. Nichols WW, Murphy DG, Cristofalo VJ, Toji LH, Greene AE, Dwight SA. Characterization of a new human diploid cell strain, IMR-90. Science. 1977; 196:60–63.
47. Zahnreich S, Krunic D, Melnikova L, Szejka A, Drossel B, Sabatier L, Durante M, Ritter S, Fournier C. Duplicated chromosomal fragments stabilize shortened telomeres in normal human IMR-90 cells before transition to senescence. J Cell Physiol. 2012; 227:1932–1940.
48. Okada M, Sato A, Shibuya K, Watanabe E, Seino S, Suzuki S, Seino M, Narita Y, Shibui S, Kayama T, Kitanaka C. JNK contributes to temozolomide resistance of stem-like glioblastoma cells via regulation of MGMT expression. Int J Oncol. 2014; 44:591–599.