
The question about molecular risk in anaplastic lymphoma kinase (ALK)-positive non-small cell lung cancer (NSCLC) is mainly posed by the recent therapeutic advances: prior to the availability of ALK tyrosine kinase inhibitors (TKI) and other targeted therapies, metastatic NSCLC was a rapidly lethal disease with a median overall patient survival (OS) below one and a half years [1]. In contrast, under sequential treatment with ALK TKI the median life expectancy of ALK+ lung cancer patients currently exceeds 5 years [2]. This impressive extension of patient life-span creates both the opportunity and the necessity to characterize early events, as their mechanistic understanding, prediction and tailored management will be crucial for further therapeutic progress.
Based on several retrospective analyses, it is well known that clinical parameters, e.g., advanced age [3], male sex [3], current smoking [2] and worse performance status [2], can predict worse survival of ALK+ NSCLC patients. These associations are plausible considering the naturally limited life expectancy of older individuals, the longer survival for women of any age [4], and the experience with other NSCLC, including EGFR+ lung adenocarcinoma, in which a positive smoking history and worse clinical condition at baseline are also associated with inferior outcome [5-7]. However, unfortunately, at the same time, predictive and prognostic implications of clinical variables are of limited utility, since clinical factors are neither a good source of mechanistic insights, nor informative for causal therapies that would improve the course of disease in individual patients.
Therefore, the translation of clinical profiles associated with higher risk into molecular features is an important, but challenging task in ALK+ NSCLC. Special obstacles include the rarity and genetic heterogeneity of the disease due to multiple ALK fusion variants [8], which are further potentiated by its complex management, including highly variable sequences of TKI and local ablative treatments [9], long patient survival of several years [2], limited availability of tissue from small biopsies, and variable ability of next-generation sequencing (NGS) assays to detect gene fusions in tissue or circulating tumor DNA (ctDNA) [10].
Recently, however, several studies combining state-of-the-art molecular profiling with detailed clinical annotation of large patient cohorts with long clinical follow-up identified two key molecular risk factors in ALK+ NSCLC: EML4-ALK fusion variant 3 (E6;A20) [11-16] and the presence of TP53 mutations [17-19]. Among newly diagnosed patients, these genetic events occur independent from each other in about 30–40% and 20–25% of cases, respectively, have synergistic effects and are both associated with shorter progression-free survival (PFS) after treatment with first- and second-generation ALK inhibitors and with worse OS (Table 1) [11, 16, 18]. In addition, detection of TP53 mutations in tissue or liquid rebiopsies at the time of disease progression in previously TP53 negative patients, identifies another approximately 20% of cases with a poor outcome, comparable to that with primarily TP53 mutated tumors (TP53 status conversion in approximately 25% of cases x initially wildtype TP53 result in approximately 75-80% of cases) [20].
Table 1: Baseline molecular risk in ALK+ NSCLC
(% at diangosis) |
Metastatic spread |
PFS under TKI |
OS |
|
V3+ |
30-40% |
↑13,14,16,18 |
↓11,14,18 (12,16) |
↓14,18 (16) |
TP53mut |
20-25% |
(depends on the oncogene) |
↓18,19 (31) |
↓18,19 (17) |
V3+TP53mut |
6-10% |
↑↑18 |
↓↓18 |
↓↓18 |
Frequency of EML4-ALK V3 and TP53 mutations in newly-diagnosed ALK+ NSCLC patients and their effect on main clinical characteristics of the disease, based on the collective insight from several studies [11-14,16-19,31].
Abbreviations: PFS: progression free survival, OS: overall survival; TKI: tyrosine kinase inhibitors.
Thus, the biology of ALK+ NSCLC displays some basic similarities with that of the other major oncogene-driven lung cancer subtype, namely EGFR+ NSCLC, in which the oncogene variant (e.g. exon 19 indels vs. other alterations [6]) and the presence of TP53 mutations [21] influence benefit from TKI and patient survival, as well [22]. However, there are important differences. First, while in EGFR+ NSCLC the oncogene variants, such as exon 19 indels, L858R, “rare” point mutations and exon 20 insertions, cause a largely similar oncogenic drive [23-25], which nevertheless translates into a different clinical course only after institution of EGFR-directed therapies due to differential TKI sensitivity [26], the unfavourable EML4-ALK V3 variant in ALK+ NSCLC has a different biology per se. There is evidence that the shorter V3 oncoprotein is more stable [11, 27, 28], causes stronger ALK phosphorylation [11] and promotes cell motility and metastasis more efficiently [13, 16], resulting in a higher a priori clinical risk [29]. These data are supported by clinical observations: a higher incidence of metastatic disease [13] and as a higher number of metastatic sites in stage IV patients [14] at diagnosis, i.e., a more adverse course before and independent of the type of treatment (Figure 1A and Table 1) [16, 29]. Interestingly, in keeping with the finding of earlier and broader metastatic dissemination, the presence of V3 is also associated with a worse performance status of newly diagnosed ALK+ NSCLC patients (Figure 1B), which has already been recognized as a clinical risk factor in ALK+ disease [2], but is itself neither biologically insightful nor druggable. This observation further underlines the importance of defining disease risk at the molecular level in order to facilitate therapeutic advances. Of note, preclinical and limited clinical data suggest that besides EML4-ALK V3, other “short” EML4-ALK variants, such as V5 (E2;A20) [11], and non-EML4-ALK fusions [30] are also associated with worse outcome, while the longer EML4-ALK V2 (E20;A20) appears to be favourable [31]. The stronger kinase effects and weaker suppression of the V3 oncoprotein by first- and second-generation ALK inhibitors [11] may also facilitate earlier TKI escape through the development of ALK resistance mutations, which are another salient and clinically relevant characteristic of V3-driven disease [12]. Their occurrence depends not only on the type of ALK fusion, but also on TKI sequencing and is important for the choice of next-line therapy [9, 32, 33]. A recently published analysis in a subset (for example 37% or 112/303 cases with detection of V1, V2, V3 in tissue) of patients from the ALEX trial suggested a superior outcome with alectinib compared to crizotinib for patients with all three main EML4-ALK fusion variants V1, V2, V3, but also a trend that the benefit from alectinib, i.e. the response rate (p = 0.10) and the PFS (p = 0.11), might be lower in case of non-V1 EML4-ALK variants, as detected in tissue samples [34]. Since these data are still immature (based on the data cut-off of December 2017), and the patient subgroups are small (n = 8-25, i.e. smaller than in previous retrospective analyses), these results have to be interpreted with some caution.
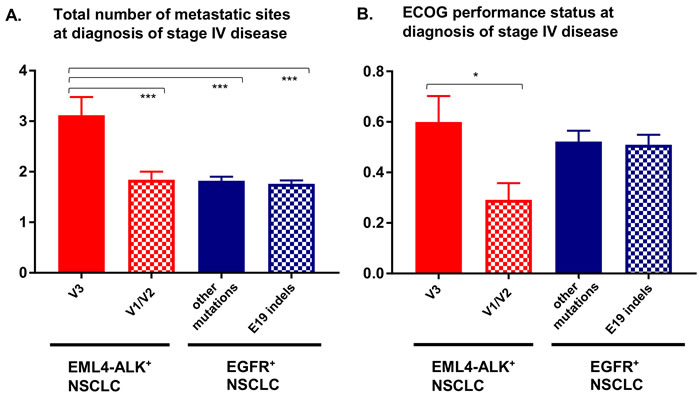
Figure 1: Number of metastatic sites and performance status in newly diagnosed stage IV EML4-ALK+ and EGFR+ NSCLC patients. A. The total number of metastatic sites (intrathoracic, brain, liver, bone, adrenal, other) for newly diagnosed stage IV EML4-ALK+ (n = 34 V3 cases, n = 44 V1/V2 cases) and stage IV EGFR+ (n = 221 EGFR exon 19 [E19] indel cases, n = 197 cases with other EGFR mutations) NSCLC patients typed at our institution with available data [18,22]. Statistical comparison was performed with ANOVA (p < 0.001) followed by the Dunnett’s post-hoc test. Columns and error bars show mean values and their standard errors: 3.12 and 0.36 for EML4-ALK V3, 1.84 and 0.16 for EML4-ALK V1/V2, 1.76 and 0.07 for EGFR exon 19 indels, 0.82 and 0.08 for other EGFR alterations. Statistically significant results are shown in the graph; ***: p < 0.001. B. The Eastern Cooperative Oncology Group (ECOG) performance status for newly diagnosed stage IV EML4-ALK+ (n = 35 V3 cases, n = 48 V1/V2 cases) and EGFR+ (n = 210 EGFR exon 19 [E19] indel cases, n = 178 with other EGFR mutations) NSCLC patients from our institution with available data [18,22]. Statistical comparison was performed with ANOVA (p < 0.05) followed by the Dunnett’s post-hoc test. Columns and error bars show mean values and their standard errors: 0.60 and 0.10 for EML4-ALK V3, 0.29 and 0.07 for EML4-ALK V1/V2, 0.52 and 0.04 for EGFR exon 19 indels, 0.51 and 0.04 for other EGFR alterations); *:p = 0.037.
A second major difference that distinguishes ALK-driven lung cancers from their EGFR+ counterparts is an apparently even lower genetic complexity based on a very low tumor mutational burden (TMB, mean 2.0 vs. 5.0 mutations/Mbp in the MSKCC cohort [35-37], p < 0.001, Figure 2A), a lower frequency of TP53 mutations (25% vs. 42%, p < 0.01, Figure 2B, 2C) [18, 19, 35], and few other co-mutations (Figure 2D) [19, 22]. Of note, both the very low TMB (mean < 3, Figure 2A) and the very low TP53 mutation rate (about 20-25%, Figure 2B) are unique features of ALK+ tumors distinguishing them from all other NSCLC [18, 38]. Since TMB of oncogene-driven lung cancer is higher in the presence of TP53 mutations (Figure 2C), the two parameters appear to be linked, possibly through DNA damage facilitated by TP53 loss due to genetic instability [39]. Such an accumulation of genetic abnormalities is crucial for the development of TKI failure [40-42], which is associated not only with the presence of TP53 mutations [21], but also with a higher TMB in EGFR+ NSCLC [43]. For example, specific co-occuring genetic alterations acquired with disease progression (e.g. in CTNNB1 and PIK3CA), were shown to cooperatively promote tumor metastasis, while other evolutionary paths impair the apoptotic response and cause resistance to EGFR inhibitors [44, 45]. According to very recent data on HER2-amplified gastroesophageal adenocarcinoma, increased genomic complexity can reduce efficacy of targeted therapies even when more than one target is hit at the same time [46, 47]. Future studies are warranted to analyze whether this observation can be conceptualized: is the degree of intratumoral heterogeneity a predictor of response to targeted drugs across cancer types independent of the druggable target itself [48]? Conversely, the lower baseline TMB and TP53 mutation rate of ALK+ compared to EGFR+ NSCLC (Figure 2A, 2B) suggest a more “benign” biology. Consistent with this notion, ALK+ NSCLC patients treated with even just one TKI have a longer OS than TKI-treated EGFR+ NSCLC patients (Figure 3, Table 2), and survival has generally been longer in trials of ALK inhibitors compared to trials of EGFR inhibitors (for example median OS was > 45 months in the Profile 1014 trial of the first-generation ALK inhibitor crizotinib [49] vs. 28 months in the LUX-Lung-3 study of the second-generation EGFR inhibitor afatinib [50]). This lower genetic complexity of ALK-driven NSCLC compared to other lung tumors may explain why therapeutic progress has been much faster and survival gains much larger for ALK+ compared to other lung cancer patients. Interestingly, the worse outcome of smoker ALK+ and EGFR+ NSCLC patients [2, 6] appears to correlate with a higher TMB [51], which illustrates again how “traditional” clinical risk factors can be redefined at the molecular level in order to promote deeper understanding of basic pathogenetic processes.
Table 2: Characteristics of the ALK+ and EGFR+ NSCLC patients included in Figure 3
ALK+ patients |
ALK+ NSCLC |
EGFR+ NSCLC TKI-treated, (n = 344)3 |
|||
>1 TKI, (n = 74) |
1 TKI, (n = 109) |
||||
Age (median; IQR) |
51; 14 |
59; 11 |
65; 12 |
||
Never/light-smokers (<10 py), n (%) 1 |
44/50 (88%) |
53/78 (68%) |
154/341 (45%) |
||
ECOG PS (median; IQR) 1 |
0; 1 |
0; 1 |
1;1 |
||
TKI treatment, n (%) |
crizotinib |
72 (97%) |
92 (85%) |
erlotinib |
183 (53%) |
ceritinib |
50 (68%) |
7 (6%) |
gefitinib |
79 (23%) |
|
alectinib |
46 (62%) |
8 (7%) |
afatinib |
117 (34%) |
|
brigatinib |
14 (19%) |
2 (2%) |
osimertinib |
61 (18%) |
|
lorlatinib |
6 (8%) |
- |
|||
Summary of the complete treatment |
|||||
no. of TKI treatment lines (mean; SD) |
2.6; 0.8 |
1.0; 0.0 |
1.3; 0.6 |
||
no. of treatment lines for St. IV (mean; SD) |
4.2; 1.6 |
2.3; 1.6 |
2.2; 1.3 |
||
patients with additional radiotherapy (%) |
43 (58%) |
48 (44%) |
176 (51%) |
||
patients with additional surgical treatment 2 |
9 (12%) |
10 (9%) |
25 (7%) |
||
1 Data not available for all patients.
2 Excluding video-assisted thoracoscopy for pleural effusion.
3 Most patients were treated before availability of osimertinib, and some received more than one EGFR inhibitors, due to poor tolerability of the first compound or in different treatment lines alternating with chemotherapy.
Abbreviations: IQR: interquartile range; py: pack-years; PS: performance status; TKI: tyrosine kinase inhibitors; SD: standard deviation; no.: number.
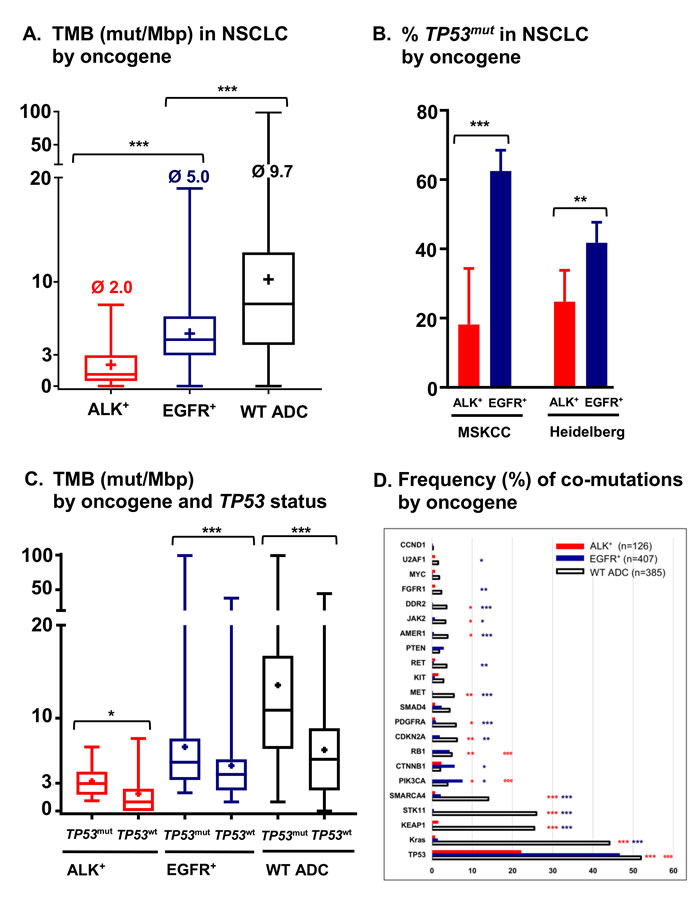
Figure 2: Tumor mutational burden, frequency of TP53 mutations and frequency of co-mutations in metastatic ALK+ and EGFR+ NSCLC. A. Tumor mutational burden (TMB) of metastatic ALK+ (n = 33, mean 2.0 mutations[mut]/Mbp), EGFR+ (n = 232, mean 5.0 mut/Mbp) and wildtype (WT, i.e. ALK/EGFR/RET/ROS-negative, n = 557, mean 9.7 mut/Mbp) cases from the publicly available MSKCC lung adenocarcinoma (ADC) cohort (http://www.cbioportal.org) as estimated by targeted sequencing with the IMPACT341 and IMPACT411 panels [35-37]. For cases with multiple sampling time-points, only the earliest one in the disease course was analyzed, and among multiple samples at the earliest time-point, that with the highest number of mutations was chosen. Boxplots show medians, means (“+”) and range; ***p < 0.001 with the Kruskal-Wallis test followed by the Dunn’s post-hoc test. B. Frequency of TP53 mutations in metastatic ALK+ (18%, n = 33) and EGFR+ (63%, n = 232) NSCLC cases of the MSKCC cohort [35-37], as well as in untreated metastatic ALK+ (25%, n = 105) and EGFR+ (42%, n = 273) tumors sequenced for exons 4-10 of TP53 at our institution [18, 22]. Columns and error bars show percentages and 95% confidence intervals, respectively; ***p < 0.001, and **p = 0.0022 with a chi-square test. C. Tumor mutational burden (TMB) according to TP53 status for ALK+ (mean 3.2 mut/Mbp for TP53 mutated, n = 6, vs. 1.8 mut/Mbp for TP53 wildtype cases, n = 27, p = 0.039 with a Mann-Whitney test), EGFR+ (mean 5.6 mut/Mbp, n = 145, vs. 4.0 mut/Mbp, n = 87, p < 0.001) and WT cases (mean 13.6 mut/Mbp, n = 291, vs. 6.6 mut/Mbp, n = 266, p < 0.001) from the publicly available MSKCC lung adenocarcinoma cohort (http://www.cbioportal.org) [35-37]. In a bivariable linear regression analysis among ALK+ and EGFR+ patients, type of oncogene (EGFR vs. ALK, beta = 0.248, p < 0.001) and TP53 status (mutated vs. wildtype, beta = 0.256, p < 0.001) were similarly strong determinants of TMB. Boxplots show medians, means (“+”) and range. D. Frequency of co-mutations in untreated ALK+, EGFR+ and WT NSCLC patients. Analyzed were untreated ALK+ (n = 105) and EGFR+ (n = 273) patients sequenced with PCR-based DNA NGS using our custom panel of 38 genes as previously described [22], as well as the untreated ALK+ (n = 21), EGFR+ (n = 134) and WT (n = 385) patients of the MSKCC lung adenocarcinoma cohort sequenced with the MSK-IMPACT341 and MSK-IMPACT411 panels (http://www.cbioportal.org) [35-37]. Visualized are all common genes among the three panels with at least one detectable mutation. Statistical comparisons for ALK+ vs. WT, and EGFR+ vs. WT were performed with a chi-square test, and results with p < 0.05 and Benjamini-Hochberg q < 0.05 are shown in the graph in red and dark blue color, respectively: *:p < 0.05, **:p < 0.01, ***:p < 0.001. Statistical comparison for ALK+ vs. EGFR+ was performed similarly, and significant results are shown in red color: e:p < 0.05, ee:p < 0.01, eee:p < 0.001.
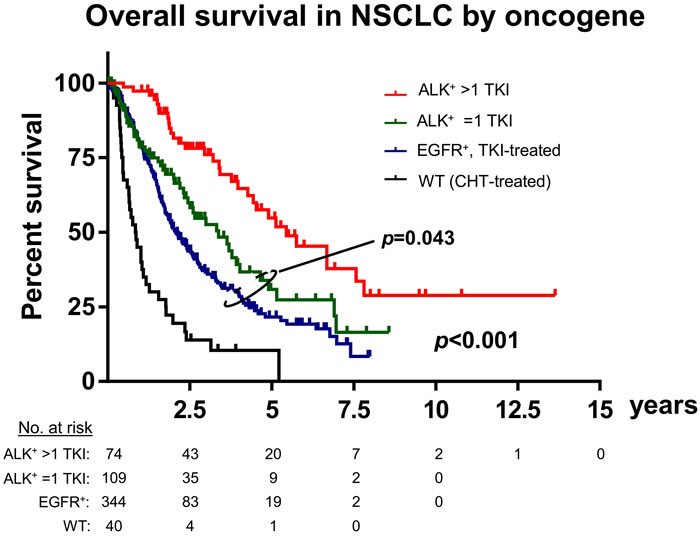
Figure 3: Differential outcome of ALK+, EGFR+ and chemotherapy-treated wildtype NSCLC. Retrospective analysis of tyrosine kinase inhibitor (TKI)-treated ALK+ (n = 74 with >1 TKI, n = 109 with just 1 TKI) and EGFR+ (n = 344) NSCLC patients, along with a random sample of n = 40 EGFR/ALK-wildtype NSCLC patients that received chemotherapy in the Thoraxklinik at Heidelberg University Hospital [22]. Basic clinical and treatment characteristics of ALK+ and EGFR+ patients are shown in Table 2. Median overall survival was 65 months for ALK+ patients that received >1 TKI, 40 months for ALK+ patients that received just 1 TKI, 25 months for TKI-treated EGFR+ patients and 10 months for wildtype patients; p < 0.001 across groups and p = 0.043 for the comparison between the EGFR+ and ALK/1-TKI subgroups with a log-rank test; WT: wildtype; CHT: chemotherapy.
The scarcity of additional genetic alterations is presumably an important reason why TP53 mutations have a major effect on the clinical phenotype of ALK+ NSCLC: they are associated with increased disease aggressiveness and metastatic dissemination synergistically with EML4-ALK V3, and they are linked with shorter PFS under TKI and shorter OS independently from EML4-ALK V3, so that double positive V3+TP53+ patients have a very high risk of death with a median OS of around 2 years in our series (Table 1) [18]. In contrast, a predictive and prognostic impact of TP53 mutations has been hard to discern in case of EGFR/ALK-negative NSCLC [52], in which the much higher number of genetic alterations at baseline [53] presumably obscures the effect of TP53 status and dilutes the consequences of genetic instability [39]. A analogous difference becomes apparent if solid cancers [54, 55] are collectively considered against hematologic malignancies, for example acute myeloid leukemia, multiple myeloma, chronic lymphocytic leukemia and mantle-cell lymphoma: in the latter TMB is generally lower (median <3 mutations/Mbp) [56] and TP53 abnormalities are less frequent (generally <10-15%), but more important for clinical course and crucial for patient management [57-60]. In the model disease of precision medicine, chronic myeloid leukemia, TP53 abnormalities and other cytogenetic aberrations or co-mutations are also associated with clonal evolution, TKI failure and poor outcome [61-63]. Transgenic mouse models of oncogene-, for example Kras- or Egfr-driven NSCLC, demonstrate this principle nicely through a paucity of concomitant genetic alterations [64], but a dramatic phenotypic change upon TP53 loss with metastases and earlier death [65]. Presumably along the same lines, TP53 alterations impair outcome of ALK+ NSCLC patients more in case of TKI treatment compared to chemotherapy [18], which is itself genotoxic [66].
Thus, although scarce, concomitant genetic alterations appear to be another important determinant of tumor biology and patient outcome in ALK-driven NSCLC, beside the oncogenic driver, i.e. the ALK fusion itself. For example, KRAS amplifications promote resistance to ALK inhibitors by activating RAS-MAPK signaling [67], which is amenable to SHP2 inhibition [68]. A similar picture emerges for EGFR+ NSCLC, in which however the spectrum of oncogene alterations and co-mutations is much broader [43, 69]. The independent and possibly synergistic effects of both the ALK fusion variant and TP53 mutations (as well as other, yet to be identified molecular features) on the clinical course of ALK+ NSCLC patients [18] mean that considerable biological and clinical variability is to be expected, if a study would take only one of these molecular factors into account. For effective guidance of patient management that is based on the molecular properties of the tumor, broad profiling approaches will be required, which could for example utilize combined targeted RNA and DNA NGS [22].
From a clinical standpoint, there is currently little that can be done for higher-risk, i.e. V3+, TP53mut and particularly V3+TP53mut ALK+ NSCLC patients. When discussing life-expectancy, some reservation is warranted, especially for “double-positive” V3+TP53mut cases, for which the 5-year landmark does not apply [18]. At the time of disease progression, a more aggressive strategy regarding local ablative therapies should be considered, otherwise some of these patients can rush through all available ALK TKI lines, for example with a change of therapy every 4-6 months, and end up with palliative chemotherapy within the first 2 years [18]. More frequent radiologic surveillance, additional ctDNA monitoring [20], upfront administration of more potent ALK inhibitors and combination with experimental compounds, such as TP53-directed drugs [70] may also be beneficial, but their clinical utility needs to be tested in prospective trials. Moreover, in vitro and animal studies of the tumor-promoting effects from different ALK fusion variants and TP53 mutations in ALK+ disease will be instrumental for deeper mechanistic insights towards uncovering of therapeutic susceptibilities and new drug development.
In summary, ALK+ NSCLC is currently spearheading the advent of “precision medicine” in thoracic oncology [11-14, 16, 18, 19, 27, 28, 30]. Distinguished by the lowest genetic complexity and the longest patient survival among NSCLC, ALK+ disease is serving as a model for the illustration of basic biological principles and for the development of novel therapeutic strategies, which will probably prove useful in other lung cancer subtypes and tumor entities as well. As our understanding of its pathogenesis deepens, additional molecular features critical for patient outcome will be identified and used to further refine our concepts. Elaboration of a baseline molecular risk stratification—complemented by profiling and targeting of treatment resistance—is a crucial step towards tailored, more effective patient management and better preclinical modeling that will foster therapeutic progress.
Acknowledgments
We would like to thank Petra Kettenring of our Clinical Trial Unit for assistance with the collection of patient data and samples, and we thank all technical assistants of the Center for Molecular Pathology (CMP) for expert handling of tissue and technical support.
Conflicts of interest
PC reports lecture fees from Roche, Chugai and Novartis; SD reports lecture fees from Roche; HS reports advisory board and speaker’s honoraria from Roche; MT reports advisory board honoraria from Novartis, Lilly, BMS, MSD, Roche, Celgene, Takeda, AbbVie, Boehringer, speaker’s honoraria from Lilly, MSD, Takeda, research funding from Ast raZeneca, BMS, Celgene, Novartis, Roche and travel grants from BMS, MSD, Novartis, Boehringer; AS reports advisory board honoraria from Bayer, Novartis, AstraZeneca, ThermoFisher, BMS, Illumina, and lecture fees from Bayer, Roche, BMS, Illumina, AstraZeneca, Novartis, ThermoFisher; MK and JB declare no potential conflicts of interest.
Funding
German Center for Lung Research (DZL) and the German Cancer Consortium (DKTK).
References
1. Pilkington G, Boland A, Brown T, Oyee J, Bagust A, Dickson R. A systematic review of the clinical effectiveness of first-line chemotherapy for adult patients with locally advanced or metastatic non-small cell lung cancer. Thorax. 2015; 70:359-67. https://doi.org/10.1136/thoraxjnl-2014-205914.
2. Duruisseaux M, Besse B, Cadranel J, Perol M, Mennecier B, Bigay-Game L, Descourt R, Dansin E, Audigier-Valette C, Moreau L, Hureaux J, Veillon R, Otto J, et al. Overall survival with crizotinib and next-generation ALK inhibitors in ALK-positive non-small-cell lung cancer (IFCT-1302 CLINALK): a French nationwide cohort retrospective study. Oncotarget. 2017; 8:21903-17. https://doi.org/10.18632/oncotarget.15746.
3. Kayaniyil S, Hurry M, Wilson J, Wheatley-Price P, Melosky B, Rothenstein J, Cohen V, Koch C, Zhang J, Osenenko K, Liu G. Treatment patterns and survival in patients with ALK-positive non-small-cell lung cancer: a Canadian retrospective study. Curr Oncol. 2016; 23:e589-e97. https://doi.org/10.3747/co.23.3273.
4. Rochelle TL, Yeung DK, Bond MH, Li LM. Predictors of the gender gap in life expectancy across 54 nations. Psychol Health Med. 2015; 20:129-38. https://doi.org/10.1080/13548506.2014.936884.
5. Tseng CH, Chiang CJ, Tseng JS, Yang TY, Hsu KH, Chen KC, Wang CL, Chen CY, Yen SH, Tsai CM, Huang MS, Ho CC, Yu CJ, et al. EGFR mutation, smoking, and gender in advanced lung adenocarcinoma. Oncotarget. 2017; 8:98384-93. https://doi.org/10.18632/oncotarget.21842.
6. Lin JJ, Cardarella S, Lydon CA, Dahlberg SE, Jackman DM, Janne PA, Johnson BE. Five-Year Survival in EGFR-Mutant Metastatic Lung Adenocarcinoma Treated with EGFR-TKIs. J Thorac Oncol. 2016; 11:556-65. https://doi.org/10.1016/j.jtho.2015.12.103.
7. Liu Y, Zhang Y, Zhang L, Liu B, Wang Y, Zhou X, Li Y, Zhao Q, Gong Y, Zhou L, Zhu J, Ding Z, Wang J, et al. Efficacy of epidermal growth factor receptor-tyrosine kinase inhibitors for lung squamous carcinomas harboring EGFR mutation: A multicenter study and pooled analysis of published reports. Oncotarget. 2017; 8:49680-8. https://doi.org/10.18632/oncotarget.17915.
8. Bayliss R, Choi J, Fennell DA, Fry AM, Richards MW. Molecular mechanisms that underpin EML4-ALK driven cancers and their response to targeted drugs. Cell Mol Life Sci. 2016; 73:1209-24. https://doi.org/10.1007/s00018-015-2117-6.
9. Lin JJ, Riely GJ, Shaw AT. Targeting ALK: Precision Medicine Takes on Drug Resistance. Cancer Discov. 2017; 7: 137-55. https://doi.org/10.1158/2159-8290.CD-16-1123.
10. Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, Liu CL, Neal JW, Wakelee HA, Merritt RE, Shrager JB, Loo BW Jr, Alizadeh AA, Diehn M. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014; 20:548-54. https://doi.org/10.1038/nm.3519.
11. Woo CG, Seo S, Kim SW, Jang SJ, Park KS, Song JY, Lee B, Richards MW, Bayliss R, Lee DH, Choi J. Differential protein stability and clinical responses of EML4-ALK fusion variants to various ALK inhibitors in advanced ALK-rearranged non-small cell lung cancer. Ann Oncol. 2017; 28:791-7. https://doi.org/10.1093/annonc/mdw693.
12. Lin JJ, Zhu VW, Yoda S, Yeap BY, Schrock AB, Dagogo-Jack I, Jessop NA, Jiang GY, Le LP, Gowen K, Stephens PJ, Ross JS, Ali SM, et al. Impact of EML4-ALK Variant on Resistance Mechanisms and Clinical Outcomes in ALK-Positive Lung Cancer. J Clin Oncol. 2018; 36:1199–206.
13. Noh KW, Lee MS, Lee SE, Song JY, Shin HT, Kim YJ, Oh DY, Jung K, Sung M, Kim M, An S, Han J, Shim YM, et al. Molecular breakdown: a comprehensive view of anaplastic lymphoma kinase (ALK)-rearranged non-small cell lung cancer. J Pathol. 2017; 243:307-19. https://doi.org/10.1002/path.4950.
14. Christopoulos P, Endris V, Bozorgmehr F, Elsayed M, Kirchner M, Ristau J, Buchhalter I, Penzel R, Herth FJ, Heussel CP, Eichhorn M, Muley T, Meister M, et al. EML4-ALK fusion variant V3 is a high-risk feature conferring accelerated metastatic spread, early treatment failure and worse overall survival in ALK+ NSCLC. Int J Cancer. 2018; 142:2589-2598. https://doi.org/10.1002/ijc.31275.
15. Sabir SR, Yeoh S, Jackson G, Bayliss R. EML4-ALK Variants: Biological and Molecular Properties, and the Implications for Patients. Cancers (Basel). 2017; 9:118. https://doi.org/10.3390/cancers9090118.
16. O’Regan L, Barone G, Adib R, Woo CG, Jeong HJ, Richardson EL, Richards MW, Muller PAJ, Collis SJ, Fennell DA, Choi J, Bayliss R, Fry AM. EML4-ALK V3 drives cell migration through NEK9 and NEK7 kinases in non-small-cell lung cancer. bioRxiv. 2019: 567305. https://doi.org/10.1101/567305.
17. Aisner DL, Sholl LM, Berry LD, Rossi MR, Chen H, Fujimoto J, Moreira AL, Ramalingam SS, Villaruz LC, Otterson GA, Haura E, Politi K, Glisson B, et al, and LCMC2 investigators. The Impact of Smoking and TP53 Mutations in Lung Adenocarcinoma Patients with Targetable Mutations-The Lung Cancer Mutation Consortium (LCMC2). Clin Cancer Res. 2018; 24:1038-47. https://doi.org/10.1158/1078-0432.CCR-17-2289.
18. Christopoulos P, Kirchner M, Bozorgmehr F, Endris V, Elsayed M, Budczies J, Ristau J, Penzel R, Herth FJ, Heussel CP, Eichhorn M, Muley T, Meister M, et al. Identification of a highly lethal V3+TP53+ subset in ALK+ l ung adenocarcinoma. Int J Cancer. 2019; 144:190-199. https://doi.org/10.1002/ijc.31893.
19. Kron A, Alidousty C, Scheffler M, Merkelbach-Bruse S, Seidel D, Riedel R, Ihle MA, Michels S, Nogova L, Fassunke J, Heydt C, Kron F, Ueckeroth F, et al. Impact of TP53 mutation status on systemic treatment outcome in ALK-rearranged non-small-cell lung cancer. Ann Oncol. 2018; 29:2068–75. https://doi.org/10.1093/annonc/mdy333.
20. Christopoulos P, Dietz S, Kirchner M, Volckmar AL, Endris V, Neumann O, Ogrodnik S, Heussel CP, Herth FJ, Eichhorn M, Meister M, Budczies J, Allgauer M, et al. Detection of TP53 Mutations in Tissue or Liquid Rebiopsies at Progression Identifies ALK+ Lung Cancer Patients with Poor Survival. Cancers (Basel). 2019; 11:E124. https://doi.org/10.3390/cancers11010124.
21. Canale M, Petracci E, Delmonte A, Chiadini E, Dazzi C, Papi M, Capelli L, Casanova C, De Luigi N, Mariotti M, Gamboni A, Chiari R, Bennati C, et al. Impact of TP53 Mutations on Outcome in EGFR-Mutated Patients Treated with First-Line Tyrosine Kinase Inhibitors. Clin Cancer Res. 2017; 23:2195-202. https://doi.org/10.1158/1078-0432.CCR-16-0966.
22. Volckmar AL, Leichsenring J, Kirchner M, Christopoulos P, Neumann O, Budczies J, Morais de Oliveira CM, Rempel E, Buchhalter I, Brandt R, Allgäuer M, Talla SB, von Winterfeld M, et al. Combined targeted DNA and RNA sequencing of advanced NSCLC in routine molecular diagnostics: analysis of the first 3,000 Heidelberg cases. Int J Cancer. 2019; Epub Ahead of Print. https://doi.org/10.1002/ijc.32133.
23. Cho J, Kim S, Du J, Meyerson M. Autophosphorylation of the carboxyl-terminal domain is not required for oncogenic transformation by lung-cancer derived EGFR mutants. Int J Cancer. 2018; 143:679-85. https://doi.org/10.1002/ijc.31332.
24. Furuyama K, Harada T, Iwama E, Shiraishi Y, Okamura K, Ijichi K, Fujii A, Ota K, Wang S, Li H, Takayama K, Giaccone G, Nakanishi Y. Sensitivity and kinase activity of epidermal growth factor receptor (EGFR) exon 19 and others to EGFR-tyrosine kinase inhibitors. Cancer Sci. 2013; 104:584-9. https://doi.org/10.1111/cas.12125.
25. Greulich H, Chen TH, Feng W, Janne PA, Alvarez JV, Zappaterra M, Bulmer SE, Frank DA, Hahn WC, Sellers WR, Meyerson M. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005; 2:e313. https://doi.org/10.1371/journal.pmed.0020313.
26. Kobayashi Y, Mitsudomi T. Not all epidermal growth factor receptor mutations in lung cancer are created equal: Perspectives for individualized treatment strategy. Cancer Sci. 2016; 107:1179-86. https://doi.org/10.1111/cas.12996.
27. Richards MW, Law EW, Rennalls LP, Busacca S, O’Regan L, Fry AM, Fennell DA, Bayliss R. Crystal structure of EML1 reveals the basis for Hsp90 dependence of oncogenic EML4-ALK by disruption of an atypical beta-propeller domain. Proc Natl Acad Sci U S A. 2014; 111:5195-200. https://doi.org/10.1073/pnas.1322892111.
28. Heuckmann JM, Balke-Want H, Malchers F, Peifer M, Sos ML, Koker M, Meder L, Lovly CM, Heukamp LC, Pao W, Kuppers R, Thomas RK. Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clin Cancer Res. 2012; 18:4682-90. https://doi.org/10.1158/1078-0432.CCR-11-3260.
29. Christopoulos P, Kirchner M, Endris V, Stenzinger A, Thomas M. EML4-ALK V3, treatment resistance, and survival: refining the diagnosis of ALK+ NSCLC. J Thorac Dis. 2018; 10:S1989–91. https://doi.org/10.21037/jtd.2018.05.61.
30. Rosenbaum JN, Bloom R, Forys JT, Hiken J, Armstrong JR, Branson J, McNulty S, Velu PD, Pepin K, Abel H, Cottrell CE, Pfeifer JD, Kulkarni S, et al. Genomic heterogeneity of ALK fusion breakpoints in non-small-cell lung cancer. Mod Pathol. 2018; 31:791-808. https://doi.org/10.1038/modpathol.2017.181.
31. Li Y, Zhang T, Zhang J, Li W, Yuan P, Xing P, Zhang Z, Chuai S, Li J, Ying J. Response to crizotinib in advanced ALK-rearranged non-small cell lung cancers with different ALK-fusion variants. Lung Cancer. 2018; 118:128-33. https://doi.org/10.1016/j.lungcan.2018.01.026.
32. Yoda S, Lin JJ, Lawrence MS, Burke BJ, Friboulet L, Langenbucher A, Dardaei L, Prutisto-Chang K, Dagogo-Jack I, Timofeevski S, Hubbeling H, Gainor JF, Ferris LA, et al. Sequential ALK Inhibitors Can Select for Lorlatinib-Resistant Compound ALK Mutations in ALK-Positive Lung Cancer. Cancer Discov. 2018; 8:714-29. https://doi.org/10.1158/2159-8290.CD-17-1256.
33. Shaw AT, Solomon BJ, Besse B, Bauer TM, Lin CC, Soo RA, Riely GJ, Ou SI, Clancy JS, Li S, Abbattista A, Thurm H, Satouchi M, et al. ALK Resistance Mutations and Efficacy of Lorlatinib in Advanced Anaplastic Lymphoma Kinase-Positive Non-Small-Cell Lung Cancer. J Clin Oncol. 2019: JCO1802236. https://doi.org/10.1200/JCO.18.02236.
34. Camidge DR, Dziadziuszko R, Peters S, Mok T, Noe J, Nowicka M, Gadgeel SM, Cheema P, Pavlakis N, de Marinis F, Cho BC, Zhang L, Moro-Sibilot D, et al. Updated Efficacy and Safety Data and Impact of the EML4-ALK Fusion Variant on the Efficacy of Alectinib in Untreated ALK-positive Advanced Non-small-cell Lung Cancer in the Global Phase III ALEX Study. J Thorac Oncol. 2019; S1556-0864: 30210-2. https://doi.org/10.1016/j.jtho.2019.03.007.
35. Jordan EJ, Kim HR, Arcila ME, Barron D, Chakravarty D, Gao J, Chang MT, Ni A, Kundra R, Jonsson P, Jayakumaran G, Gao SP, Johnsen HC, et al. Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov. 2017; 7:596-609. https://doi.org/10.1158/2159-8290.CD-16-1337.
36. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401-4. https://doi.org/10.1158/2159-8290.CD-12-0095.
37. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1. https://doi.org/10.1126/scisignal.2004088.
38. Spigel DR, Schrock AB, Fabrizio D, Frampton GM, Sun J, He J, Gowen K, Johnson ML, Bauer TM, Kalemkerian GP, Raez LE, Ou SH, Ross JS, et al. Total mutation burden (TMB) in lung cancer (LC) and relationship with response to PD-1/PD-L1 targeted therapies. J Clin Oncol. 2016; 34:9017. https://doi.org/10.1200/JCO.2016.34.15_suppl.9017.
39. Hanel W, Moll UM. Links between mutant p53 and genomic instability. J Cell Biochem. 2012; 113:433-9. https://doi.org/10.1002/jcb.23400.
40. Sotillo R, Schvartzman JM, Socci ND, Benezra R. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature. 2010; 464:436-40. https://doi.org/10.1038/nature08803.
41. Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, Dagogo-Jack I, Gadgeel S, Schultz K, Singh M, Chin E, Parks M, Lee D, et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016; 6:1118-33. https://doi.org/10.1158/2159-8290.CD-16-0596.
42. Westover D, Zugazagoitia J, Cho BC, Lovly CM, Paz-Ares L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol. 2018; 29:i10-i9. https://doi.org/10.1093/annonc/mdx703.
43. Offin M, Rizvi H, Tenet M, Ni A, Sanchez-Vega F, Li BT, Drilon A, Kris MG, Rudin CM, Schultz N, Arcila ME, Ladanyi M, Riely GJ, et al. Tumor Mutation Burden and Efficacy of EGFR-Tyrosine Kinase Inhibitors in Patients with EGFR-Mutant Lung Cancers. Clin Cancer Res. 2019; 25:1063-1069. https://doi.org/10.1158/1078-0432.CCR-18-1102.
44. Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, Maruvka YE, Ji F, Bhang HE, Krishnamurthy Radhakrishna V, Siravegna G, Hu H, Raoof S, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016; 22:262-9. https://doi.org/10.1038/nm.4040.
45. Blakely CM, Watkins TBK, Wu W, Gini B, Chabon JJ, McCoach CE, McGranahan N, Wilson GA, Birkbak NJ, Olivas VR, Rotow J, Maynard A, Wang V, et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet. 2017; 49:1693-704. https://doi.org/10.1038/ng.3990.
46. Klempner SJ, Catenacci DVT. Variety Is the Spice of Life, but Maybe Not in Gastroesophageal Adenocarcinomas. Cancer Discov. 2019; 9:166-8. https://doi.org/10.1158/2159-8290.CD-18-1447.
47. Sanchez-Vega F, Hechtman JF, Castel P, Ku GY, Tuvy Y, Won H, Fong CJ, Bouvier N, Nanjangud GJ, Soong J, Vakiani E, Schattner M, Kelsen DP, et al. EGFR and MET Amplifications Determine Response to HER2 Inhibition in ERBB2-Amplified Esophagogastric Cancer. Cancer Discov. 2019; 9:199-209. https://doi.org/10.1158/2159-8290.CD-18-0598.
48. Turajlic S, Sottoriva A, Graham T, Swanton C. Resolving genetic heterogeneity in cancer. Nat Rev Genet. 2019; Epub Ahead of Print. https://doi.org/10.1038/s41576-019-0114-6.
49. Solomon BJ, Kim DW, Wu YL, Nakagawa K, Mekhail T, Felip E, Cappuzzo F, Paolini J, Usari T, Tang Y, Wilner KD, Blackhall F, Mok TS. Final Overall Survival Analysis From a Study Comparing First-Line Crizotinib Versus Chemotherapy in ALK-Mutation-Positive Non-Small-Cell Lung Cancer. J Clin Oncol. 2018; 36:2251-8. https://doi.org/10.1200/JCO.2017.77.4794.
50. Yang JC, Wu YL, Schuler M, Sebastian M, Popat S, Yamamoto N, Zhou C, Hu CP, O’Byrne K, Feng J, Lu S, Huang Y, Geater SL, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015; 16:141-51. https://doi.org/10.1016/S1470-2045(14)71173-8.
51. Chae YK, Davis AA, Raparia K, Agte S, Pan A, Mohindra N, Villaflor V, Giles F. Association of Tumor Mutational Burden With DNA Repair Mutations and Response to Anti-PD-1/PD-L1 Therapy in Non-Small-Cell Lung Cancer. Clin Lung Cancer. 2019; 20:88-96.e6. https://doi.org/10.1016/j.cllc.2018.09.008.
52. Viktorsson K, De Petris L, Lewensohn R. The role of p53 in treatment responses of lung cancer. Biochem Biophys Res Commun. 2005; 331:868-80. https://doi.org/10.1016/j.bbrc.2005.03.192.
53. Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL, Maher CA, Fulton R, Fulton L, Wallis J, Chen K, Walker J, McDonald S, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012; 150:1121-34. https://doi.org/10.1016/j.cell.2012.08.024.
54. Robles AI, Harris CC. Clinical outcomes and correlates of TP53 mutations and cancer. Cold Spring Harb Perspect Biol. 2010; 2:a001016. https://doi.org/10.1101/cshperspect.a001016.
55. Li VD, Li KH, Li JT. TP53 mutations as potential prognostic markers for specific cancers: analysis of data from The Cancer Genome Atlas and the International Agency for Research on Cancer TP53 Database. J Cancer Res Clin Oncol. 2019; 145:625-636. https://doi.org/10.1007/s00432-018-2817-z.
56. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, et al. Signatures of mutational processes in human cancer. Nature. 2013; 500:415-21. https://doi.org/10.1038/nature12477.
57. Rajkumar SV. Multiple myeloma: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016; 91:719-34. https://doi.org/10.1002/ajh.24402.
58. Hallek M. Chronic lymphocytic leukemia: 2017 update on diagnosis, risk stratification, and treatment. Am J Hematol. 2017; 92:946-65. https://doi.org/10.1002/ajh.24826.
59. Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, Dombret H, Ebert BL, Fenaux P, Larson RA, Levine RL, Lo-Coco F, Naoe T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017; 129:424-47. https://doi.org/10.1182/blood-2016-08-733196.
60. Eskelund CW, Dahl C, Hansen JW, Westman M, Kolstad A, Pedersen LB, Montano-Almendras CP, Husby S, Freiburghaus C, Ek S, Pedersen A, Niemann C, Raty R, et al. TP53 mutations identify younger mantle cell lymphoma patients who do not benefit from intensive chemoimmunotherapy. Blood. 2017; 130:1903-10. https://doi.org/10.1182/blood-2017-04-779736.
61. Wendel HG, de Stanchina E, Cepero E, Ray S, Emig M, Fridman JS, Veach DR, Bornmann WG, Clarkson B, McCombie WR, Kogan SC, Hochhaus A, Lowe SW. Loss of p53 impedes the antileukemic response to BCR-ABL inhibition. Proc Natl Acad Sci U S A. 2006; 103:7444-9. https://doi.org/10.1073/pnas.0602402103.
62. Abraham SA, Hopcroft LE, Carrick E, Drotar ME, Dunn K, Williamson AJ, Korfi K, Baquero P, Park LE, Scott MT, Pellicano F, Pierce A, Copland M, et al. Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature. 2016; 534:341-6. https://doi.org/10.1038/nature18288.
63. Kim T, Tyndel MS, Kim HJ, Ahn JS, Choi SH, Park HJ, Kim YK, Kim SY, Lipton JH, Zhang Z, Kim DD. Spectrum of somatic mutation dynamics in chronic myeloid leukemia following tyrosine kinase inhibitor therapy. Blood. 2017; 129:38-47. https://doi.org/10.1182/blood-2016-04-708560.
64. McFadden DG, Politi K, Bhutkar A, Chen FK, Song X, Pirun M, Santiago PM, Kim-Kiselak C, Platt JT, Lee E, Hodges E, Rosebrock AP, Bronson RT, et al. Mutational landscape of EGFR-, MYC-, and Kras-driven genetically engineered mouse models of lung adenocarcinoma. Proc Natl Acad Sci U S A. 2016; 113:E6409-E17. https://doi.org/10.1073/pnas.1613601113.
65. DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc. 2009; 4:1064-72. https://doi.org/10.1038/nprot.2009.95.
66. Cheung-Ong K, Giaever G, Nislow C. DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol. 2013; 20:648-59. https://doi.org/10.1016/j.chembiol.2013.04.007.
67. Hrustanovic G, Olivas V, Pazarentzos E, Tulpule A, Asthana S, Blakely CM, Okimoto RA, Lin L, Neel DS, Sabnis A, Flanagan J, Chan E, Varella-Garcia M, et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat Med. 2015; 21:1038-47. https://doi.org/10.1038/nm.3930.
68. Dardaei L, Wang HQ, Singh M, Fordjour P, Shaw KX, Yoda S, Kerr G, Yu K, Liang J, Cao Y, Chen Y, Lawrence MS, Langenbucher A, et al. SHP2 inhibition restores sensitivity in ALK-rearranged non-small-cell lung cancer resistant to ALK inhibitors. Nat Med. 2018; 24:512-7. https://doi.org/10.1038/nm.4497.
69. Yu HA, Suzawa K, Jordan E, Zehir A, Ni A, Kim R, Kris MG, Hellmann MD, Li BT, Somwar R, Solit DB, Berger MF, Arcila M, et al. Concurrent Alterations in EGFR-Mutant Lung Cancers Associated with Resistance to EGFR Kinase Inhibitors and Characterization of MTOR as a Mediator of Resistance. Clin Cancer Res. 2018; 24:3108-18. https://doi.org/10.1158/1078-0432.CCR-17-2961.
70. Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. 2018; 18:89-102. https://doi.org/10.1038/nrc.2017.109.