
Introduction
β-N-acetylglucosaminylation (O-GlcNAcylation) is a unique form of glycosylation that occurs on serine and threonine residues of proteins throughout the cytoplasmic and nuclear compartment of cells. It is a monosaccharide that is dynamically cycled akin to that of O-phosphorylation. The enzyme O-GlcNAc transferase (OGT) adds O-GlcNAc to proteins via the substrate UDP-GlcNAc, while O-GlcNAcase (OGA) is responsible for its removal. O-GlcNAc influences the extent of O-phosphorylation of proteins through shared binding sites and steric hindrance [1]. As the end-product of the Hexosamine Biosynthesis pathway (HBP), UDP-GlcNAc and subsequent O-GlcNAcylation is considered a nutrient sensor to the overall metabolic status of the cell. Two to five percent of the glucose that enters the cell is channeled into the HBP. As such, it is not surprising that increased O-GlcNAcylation is implicated in the development of insulin resistance [2]. Aberrant O-GlcNAcylation is a characteristic of heart disease, neurodegenerative disorders such as Alzheimer’s disease, and is a hallmark of many cancers, including endometrial cancer [3]. A relationship between glycosylation and metastasis is also evident in lung cancers, wherein epithelial-mesenchymal transition (EMT) acts through the HBP as an inducer of aberrant glycosylation [4]. Despite these observations, very little is known about the mechanistic actions of O-GlcNAcylation in cancer, specifically, in the EMT process.
Many illnesses associated with aberrant O-GlcNAcylation are also co-morbidities of Type 2 Diabetes (T2D). For instance, women with T2D have a 2-fold greater risk of developing endometrial cancer than their healthy cohorts [5]. While the incidence of many forms of cancer is declining, endometrial cancer remains among those increasing for all women [6]. Endometrial cancer results from the abnormal growth, migration, and invasion of cells that line the uterus. Common genetic alterations in endometrial tumors include PTEN, PIK3CA, CTNNB1 (beta-catenin), and KRAS, all of which are related to metabolism [7]. Moreover, all of these genes influence epithelial-mesenchymal transition (EMT) signaling pathways, and recent studies indicate that the E-cadherin repressors Slug, ZEB1, and HMGA2 are preferentially expressed along the myometrial invasive edge of tumors [8]. Snail1, a key regulator of EMT, is stabilized by O-GlcNAcylation in several cell types [9], and the mRNA of O-GlcNAc cycling enzymes (OGT and OGA) is up-regulated in endometrial tumors [3], suggesting that O-GlcNAcylation influences metastasis.
The gold standard for treatment of endometrial cancer is radiation therapy and surgery; however, 5–30% of women with endometrial cancer are premenopausal and under the age of 50 at the time of diagnosis. For these women, fertility-sparing treatments, such as progestin therapy, are an option [10]. A recent meta-analysis determined that women treated with hormonal therapy methods had a pooled regression rate of 76.2%, with 28% live births reported; however, a 40.6% relapse rate was also noted [11]. These findings underscore the importance of identifying basic mechanisms by which metabolism and O-GlcNAcylation influence the progression of endometrial cancer, with the goal of improving fertility-sparing treatments. The objective of the current study was to determine some of these mechanisms, specifically focusing on the manipulation of O-GlcNAc cycling enzymes (OGT and OGA) and their impact on molecular and cellular aspects of Epithelial-Mesenchymal Transition (EMT).
Results
The O-GlcNAc cycling enzymes, OGT and OGA, are altered in endometrial cancer
Analysis of gene alterations of OGT and OGA (OGT and MGEA5, respectively) using data from the RNAseq and Microarray databases available in cBioPortal (http://cbioportal.org) revealed that of the 18 female reproductive cancer databases available, Uterine/Endometrial Cancer ranks highest for gene alterations to OGT and MGEA5, including both mutational and amplification modifications (Figure 1A). Additionally, both genes are upregulated in screened participants with Diabetes Mellitus compared to non-Diabetic controls (Figure 1B).
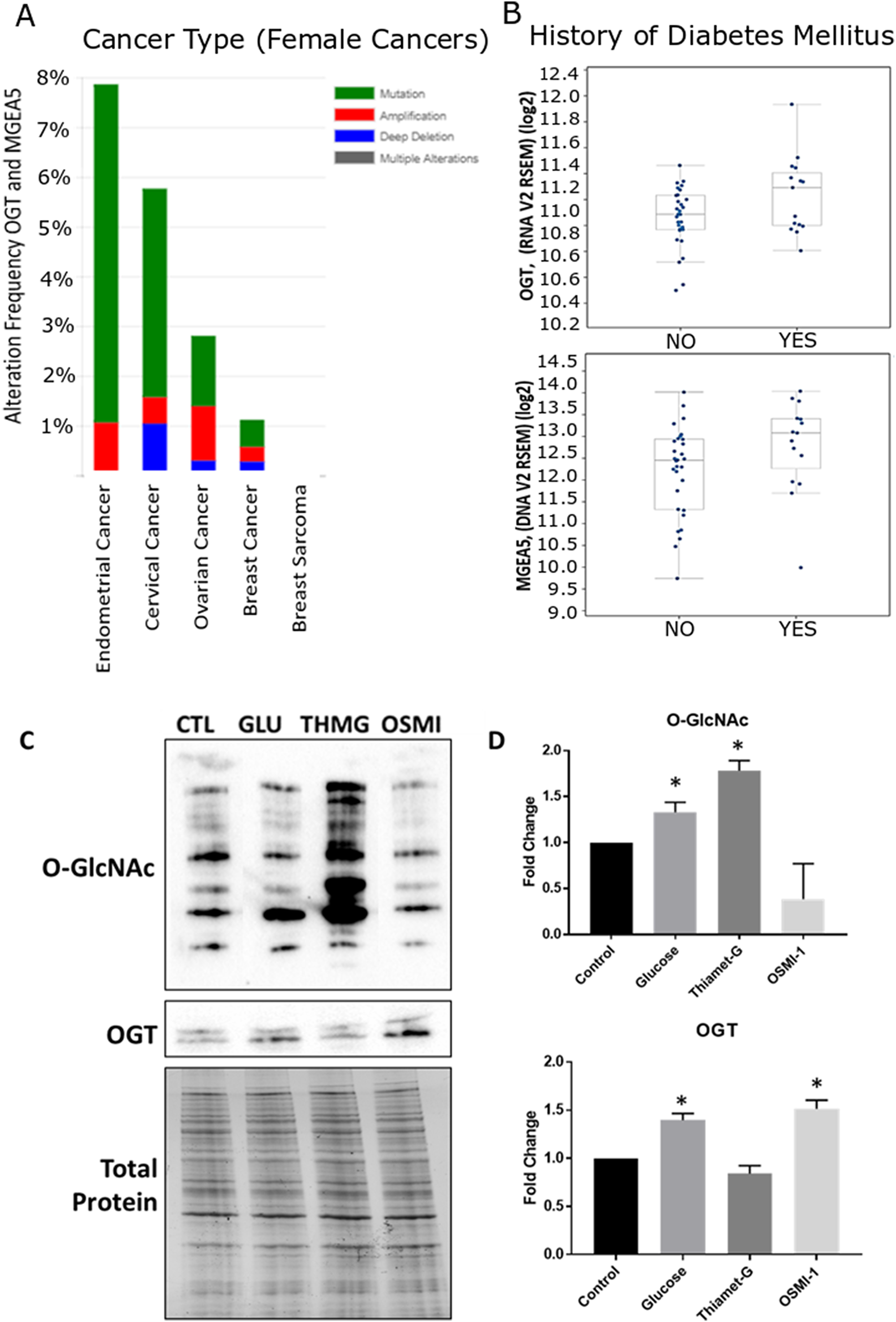
Figure 1: Meta-analysis of O-GlcNAc alteration in female cancers and validation of global O-GlcNAc modification in Ishikawa cells. Cancer genomics data analysis depicting a cross-cancer O-GlcNAc enzyme (OGT, MGEA5) alteration summary of female cancers. (A) Histograms depicting the level of gene amplification, mutation, or deletion in each data set. Data was mined from the TCGA database and was analyzed with the cBioPortal web analysis tool. Of these 18 female cancer datasets, uterine/endometrial cancer ranked the highest for gene alterations of OGT and MGEA5. (B) Box plots comparing the relative mRNA expression of OGT and MGEA5 in patients with or without a diagnosis of diabetes mellitus (DM). Additional analysis of the data set revealed that OGT and MGEA5 mRNA were more highly expressed in endometrial cancer patients with DM. (C) Western Blot analysis of global O-GlcNAc modification and OGT expression, relative to total protein, in whole cell Ishikawa lysates treated for 24 hours with the OGT inhibitor, OSMI-1; the OGA inhibitor, Thiamet-G; or supplemented with 25 mM Glucose. (D) Densitometry analysis of Western Blots, bars represent the mean +/– SEM (n = 4), (*) denotes statistically significant differences in density compared to control (p < 0.05).
Detection and manipulation of O-GlcNAcylation in the endometrial cancer line, Ishikawa
Immunodetection of global O-GlcNAcylation in Ishikawa cells revealed this form of protein modification was upregulated (Hyper-O-GlcNAcylation) in cells by supplementing complete media with 25 mM Glucose or by inhibiting OGA with Thiamet-G (1 μM; ThmG; p < 0.05; Figure 1C and 1D). While a qualitative decrease in O-GlcNAc expression was noted by inhibiting OGT with OSMI-1 (50 μM; OSMI-1), relative expression did not differ from controls (p > 0.05; Figure 1C and 1D). Interestingly, however, high glucose and OGT inhibition each enhanced relative OGT expression (p < 0.05; Figure 1C and 1D). In all subsequent experiments, these same manipulations of O-GlcNAcylation were utilized to determine effects of aberrant O-GlcNAcylation on phenotypic changes in Ishikawa cells (i.e., cell proliferation/migration and invasion), as well as morphological and molecular parameters associated with EMT.
Hyper-O-GlcNAcylation supports endometrial cancer cell proliferation/migration, and promotes invasion
Cell proliferation in response to altered O-GlcNAcylation was assessed via growth curve and MTS assay in serum free conditions (Figure 2A and 2B). Ishikawa cells proliferated in serum free conditions throughout 96 hours of culture, however, inhibition of OGT (OSMI-1) impaired proliferation beginning at 72 hours compared to control and OGA-inhibited (Thiamet-G), hyper-O-GlcNAcylated cells. Similar results were observed in MTS assays. Inhibition of proliferation occurred in OGT-inhibited (OSMI-1) cells compared to all other treatment groups between 72 and 96 hours of culture (p < 0.05, Figure 2B), but cell viability was unchanged in this group during the entire 96 hour culture period (Figure 2A and 2B).
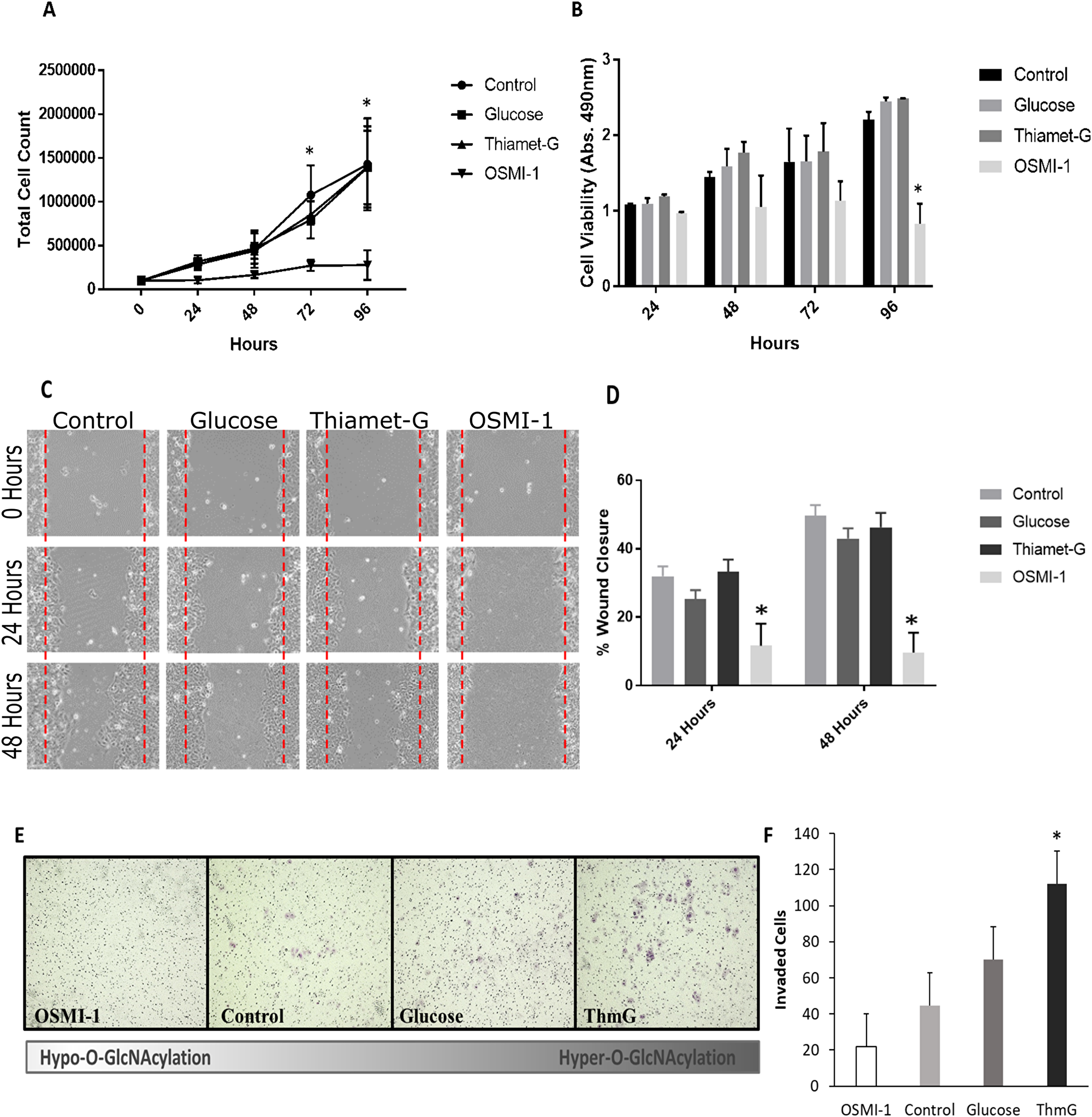
Figure 2: O-GlcNAcylation is necessary for Ishikawa cell proliferation and migration. (A) Cell growth curve depicting cell proliferation over 96 hours under serum free conditions in cells exposed to 25 mM Glucose, Thiamet-G, OSMI-1, or vehicle (media refreshed every 24 hours). Each point in the graph represents the mean +/– SEM of 3 biological replicates. An asterisk (*) indicates a difference between OSMI-1 treated cells and all other treatment groups (p < 0.05). (B) Bar graph representing the mean absorbance (n = 3) +/– SEM of MTS cell viability/proliferation assays. OSMI-1 treated cells did not proliferate, but cell viability was maintained throughout the culture. An asterisk (*) indicates a difference between OSMI-1 treated cells and all other treatment groups (p < 0.05). (C) Representative images of a wound healing assay evaluating the effects of Thiamet-G, Glucose, OSMI-1, or vehicle on migration of Ishikawa cells in serum free conditions. “Wounds” were imaged every 24 hours for 48 hours (100X). (D) Bar graphs of the wound healing assay. Mean +/– SEM (n = 3) of the percent of wound closure depicted. An asterisk (*) indicates a difference between OSMI-1 treated cells and all other treatment groups (p < 0.05). (E) Representative images of invasive cells following a Biocoat Matrigel Transwell Invasion assay (100X). Purple foci depict invasive cells. (F) Bar graph of the invasion assay depictin the mean +/– SEM (n = 4) of invaded cells after 48 hours of culture. An asterisk (*) indicates a difference between ThmG treated cells compared to Control (p < 0.05).
Wound healing assays demonstrated that Hyper-O-GlcNAcylation supported Ishikawa cell migration, with no difference in wound closure observed among Control, Glucose and ThmG-treated cultures (p > 0.05; Figure 2C and 2D). Hence, Hyper-O-GlcNAcylation was conducive to wound closure. Conversely, Hypo-O-GlcNAcylation (via OSMI-1) impaired cell migration (p < 0.05), resulting in ~10% wound closure after 48 hours of culture, compared to ~45% in Control and Hyper-O-GlcNAcylated cells (Figure 2C and 2D).
Although Ishikawa cells are considered relatively-low metastatic cells [12], Hyper-O-GlcNAcylation (i.e., ThmG treatment) augmented invasiveness compared to Control and Hypo-O-GlcNAcylated (OSMI-1-treated) cells (p < 0.05; Figure 2D and 2E). Glucose supplementation provided an intermediate response, that did not differ from either Control or Hyper-O-GlcNAcylated cells (p > 0.05; Figure 2E). Hypo-O-GlcNAcylated cells (OSMI-1-treated) had the least invasion potential, comparable to Controls (p > 0.05; Figure 2E and 2F).
Hyper-O-GlcNAcylation promotes the EMT phenotype
Aspects of the EMT process were examined in Ishikawa cells at both the mRNA and protein levels by manipulating O-GlcNAcylation (as described above). Phalloidin staining of stress filaments in the cells (F-actin) revealed diffuse and epithelial-like staining in Hypo-O-GlcNAcylated (OSMI-1- treated) cells (Figure 3A). Conversely, Hyper-O-GlcNAcylated (Glucose and ThmG-treated) cells had conspicuous actin staining throughout the cytoplasm with stress fiber-like bundles along the marginal borders of the cells (white arrows, Figure 3A), as well as focal adhesions (yellow arrows, Figure 3A), indicative of mesenchymal cell morphology.
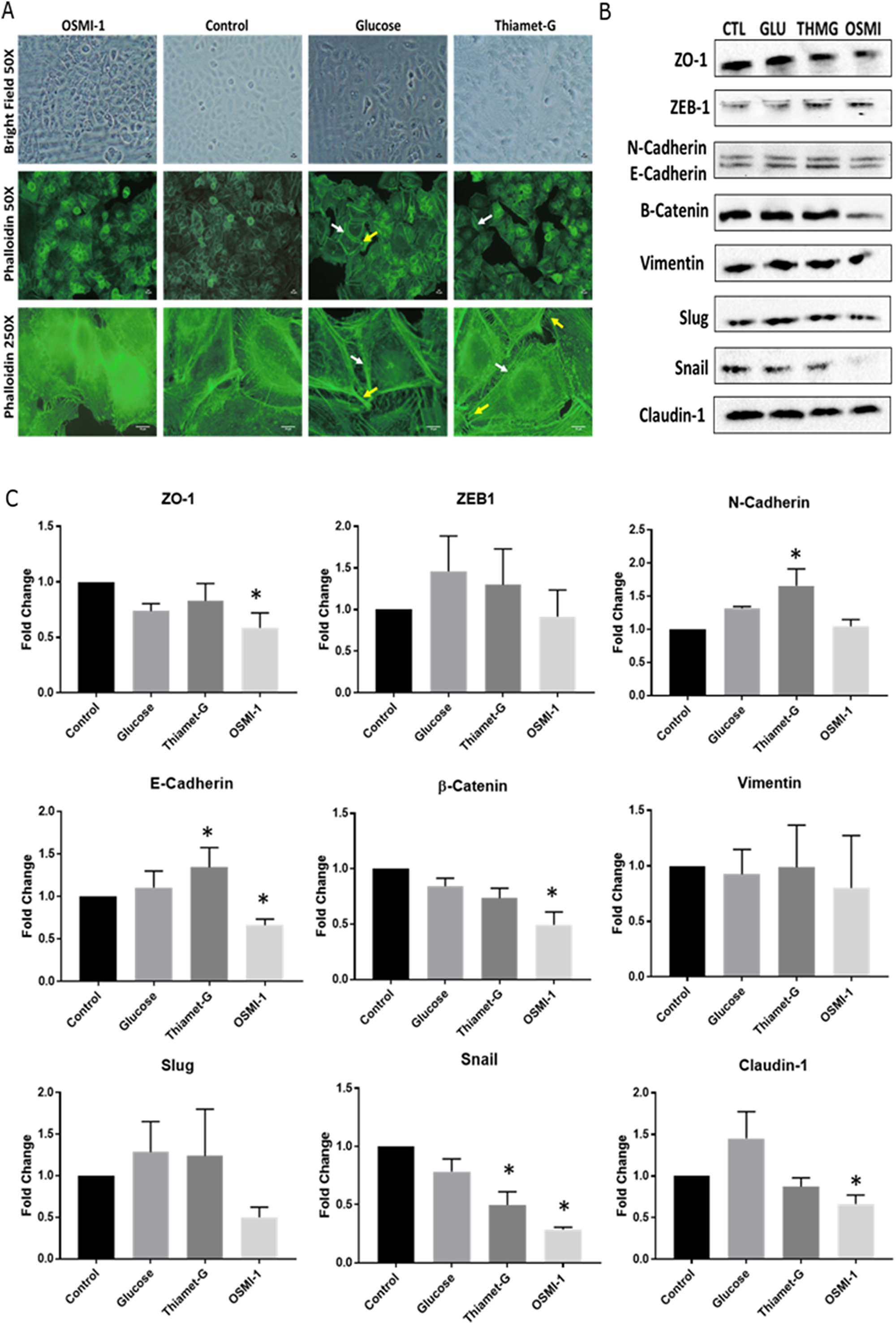
Figure 3: Disruption to O-GlcNAc signaling alters cell morphology and EMT protein expression. (A) Representative images of cultured Ishikawa cells treated with OSMI-1, Thiamet-G, 25 mM glucose, or vehicle for 48 hours. Immunofluorescent staining of actin with phalloidin was assessed for stress fibers (white arrows), and focal adhesions (yellow arrows) as indicators of EMT. (B) Representative Western Blots (n = 6 biological replicates) of whole cell lysates probed for epithelial and mesenchymal protein markers of EMT in Ishikawa cells treated as above for 48 hours. (C) Bar graphs depicting the densitometry analysis of the Western Blots. Bars depict the mean +/– SEM (n = 6 biological replicates for each protein. An asterisk (*) denotes differences compared to control (p < 0.05).
Despite the above-described morphological changes in Hypo- and Hyper-O-GlcNAcylated cells, there was no evidence that transcript abundance for key EMT markers (ZEB1, CDH1, CDH2, CTNNB1, VIM, SNAI1, SNAI2, and CLDN1) changed among the treatment groups as assessed by qPCR analysis (Supplementary Table 1). Conversely, immunoblots indicated an increase in E-Cadherin (CDH1) expression in Hyper-O-GlcNAcylated (ThmG-treated) cells and a decrease in Hypo-O-GlcNAcylated (OMSI-1 treated) cells compared to Controls (P < 0.05; Figure 3B and 3C). Consistently, expression of the E-Cadherin suppressor, Snail, was reduced by these same treatments (p < 0.05; Figure 3C). The expression of β-Catenin was sustained by Hyper-O-GlcNAcylation (ThmG), but down-regulated by Hypo-O-GlcNAcylation (OSMI-1; (p < 0.05; Figure 3C). Claudin-1, a major constituent of tight junction complexes, was markedly decreased in Hypo-O-GlcNAcylatied (OSMI-1-treated) cells compared to Controls (p < 0.05; Figure 3C). Relative expression of ZO-1 was also reduced by OSMI-1 treatment (p < 0.05; Figure 3C). None of the other EMT markers detected (i.e., Vimentin, Slug, ZEB1) were affected by O-GlcNAcylation status (p > 0.05; Figure 3C).
Microarray analysis corroborates dysregulation of O-GlcNAcylation as a mechanism of EMT in endometrial cancer
The above phenotypic and molecular changes in response to altered O-GlcNAcylation were corroborated by mRNA analysis of 86 genes important to the process of EMT. The EMT RT2 Profiler PCR Array and GeneGlobe Data Analysis Center analysis tool (Qiagen), was used to evaluate changes in gene expression (2.5 fold or greater) compared to Control cells (Supplementary Table 2). Three independent experiments yielded an identical mRNA expression profile for both Hyper-O-GlcNAcylation treatments (i.e., Thiamet-G and Glucose), with increased expression of FOXC2 and WNT5B, both promoters of EMT, and KRT14, a cytoskeletal intermediate filament monomer (Figure 4A, 4B and 4D). Hypo-O-GlcNAcylated (OSMI-1-treated) cells also had increased WNT5B and KRT14; however, expression of AHNAK, CALD1, FGFBP1, TGFB2, and TFPI2 genes were decreased (Figure 4C and 4D).
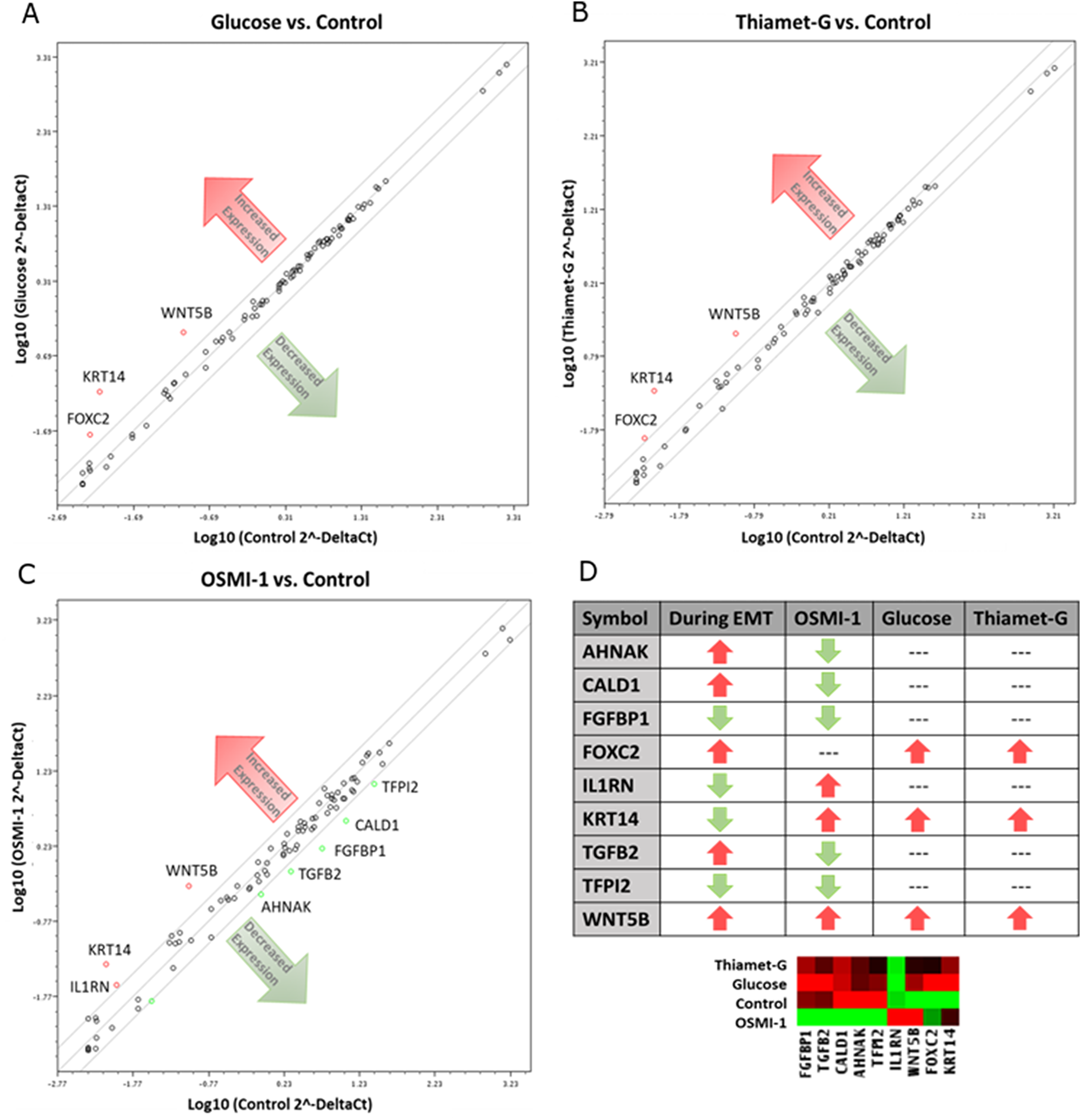
Figure 4: Disruption of O-GlcNAc alters gene expression in EMT related genes of Ishikawa cells. Cells were cultured with OSMI-1, Thiamet-G, 25 mM glucose, or vehicle for 48 hours before harvesting for RNA isolation. The mRNA was the quantified by RT-PCR (n = 3 biological replicates) and the relative expression is depicted for cells grown in (A) 25 mM glucose, (B) Thiamet-G, and (C) OSMI-1 compared to vehicle (control). The lines encompass 2.5-fold changes. Genes up-regulated are denoted by red circles, whereas genes down-regulated appear as green circles. Black circles constitute genes relatively unchanged. (D) Chart and heatmap summarizing the gene expression results compared to anticipated outcomes for EMT based upon literature review.
DISCUSSION
O-GlcNAcylation is a recently discovered post-translational modification of cellular proteins that is highly abundant and ubiquitous throughout the nuclear and cytoplasmic compartments of the cell, yet its influence on cellular processes is poorly understood. First-discovered about 30 years ago [13], O-GlcNAc is an end-product of the hexosamine biosynthesis pathway (HBP), where its relative concentration serves as nutrient sensor indicative of metabolic status [14]. Indeed, elevated O-GlcNAcylation (i.e., Hyper- O-GlcNAcylation), for example, is a hallmark of high metabolism seen in many types of cancers [15], including cancers of the breast [16], liver [17], bladder [18], prostate [19], lung [20], colon [20, 21], and endometrium [3]. Thus, O-GlcNAc, and by extension O-GlcNAcylation, may constitute an important regulator of cancer development and progression, while simultaneously providing a potential, novel connection between the onset of T2D and the increased risk of certain cancers [22]. Endometrial cancers fall into this category of increased risk attributable to T2D [5, 23, 24], and the current study reveals that aberrant O-GlcNAcylation can mechanistically-augment the tumorigenicity of endometrial cancer cells. These findings are consistent with the previous assertion that O-GlcNAc enzyme gene expression is increased along the myometrial invasive edge of endometrial tumors [3]. Similarly, elevated expression of OGT and MGEA5 mRNA is consistent with the nutrient-sensing nature of O-GlcNAcylation. High nutrient intake, hyperglycemia, and other metabolic abnormalities all promote the flow of glucose into the HBP, resulting in elevated O-GlcNAcylation. In this way, the current findings of the meta-analysis add support to the concept that O-GlcNAc modification has a role in the development of diabetic complications and cancer [2, 25, 26].
One mechanism responsible for increased tumorigenicity in many cancers is the phenomenon of epithelial-mesenchymal transition (EMT). Although it is acknowledged that EMT is a critical process of embryogenesis, this same process enhances tumor development and metastasis [27–29]. Some of the key characteristics of EMT include the loss of cell polarity and cell-cell adhesions, and an increase in invasive properties [30, 31]. In the current study, Hyper-O-GlcNAcylation supported cell proliferation, induced reorganization of the cytoskeleton, and promoted migration and invasion of endometrial cancer (Ishikawa) cells. This was evidenced by a significant reduction in cell proliferation at 72 hours of growth compared to hyper and normally O-GlcNAcylated cells when O-GlcNAcylation was reduced by inhibition of OGT, (Figure 2A and 2B). Cytoskeletal reorganization was noted by F-actin staining in hyper-O-GlcNAcylated cells (Glucose and Thiamet-G) by an observable increase in stress fibers and focal adhesions compared to control (DMSO) and hypo-O-GlcNAcylated (OSMI-1) cells (Figure 3A). Results of a matrigel invasion assay provided evidence that increased O-GlcNAcylation by Thiamet-G treatment increased the invasiveness of Ishikawa cells in culture, (Figure 2E and 2F). Similarly, a wound healing assay suggested that reduced O-GlcNAcylation (OSMI-1) impaired cell migration, even when controlling for proliferation (serum free conditions, 48 hours) (Figure 2C and 2D).
Intercellular adhesion, mediated largely by cadherin and catenin proteins, is a critical obstacle for epithelial cells to overcome as they de-differentiate and become more tumorigenic. A decrease in E-Cadherin expression, for instance, is among the initiating steps of EMT [32, 33], and is a notable consequence of Hyper-O-GlcNAcylation in many cell types [34]. Snail1, an E-Cadherin suppressor, is stabilized by O-GlcNAc at Ser112 under hyperglycemic culture conditions [9], and O-GlcNAcylation of E-Cadherin can occur directly via the cytoplasmic domain, which consequently inhibits its transport to the cell surface and prevents intercellular adhesion [35]. In the current study, Hyper-O-GlcNAcylation unexpectedly decreased the expression of Snail1, in turn increasing E-Cadherin expression. However, its mesenchymal counterpart N-Cadherin was also increased by hyper-O-GlcNAcylation. This suggests that the alteration of E-Cadherin expression is perhaps less critical for EMT than that of N-Cadherin in Ishikawa cells, or that the period of time required for this type of transition is much longer than that evaluated in the current experiments (48 hrs).
A major group of cellular signaling pathways associated with EMT are the Wnt signaling pathways. Of the Wnt pathways, 3 have been characterized; the canonical pathway, the noncanonical pathway, and the noncanonical Wnt/Calcium pathway [29]. In the canonical pathway, Wnt induces the expression of the mesenchymal-like protein, Beta-catenin. Beta-catenin accumulates in the cytoplasm and is translocated to the nucleus, where it acts as a transcriptional activator of TCF/LEF family transcription factors to induce a cellular response [36]. A recent study investigating the role of high glucose concentrations in endometrial cancer found that high glucose increased the flux of glucose into the HBP, in turn increasing O-GlcNAcylation of proteins [37]. Hyper-O-GlcNAcylation also increased the expression of B-catenin [37]. In the current study, however, an upregulation of the non-canonical Wnt ligand, WNT5B, by either Hyper- or Hypo-O-GlcNAcylation did not affect the canonical Wnt ligands (i.e. Wnts –1, –2, –3, –8a, –8b, –10a, –10b) [38] or β-catenin expression (Figure 4 and 3B, respectively). The non-canonical Wnt pathways are not as well understood as the canonical, partly due to the diversity and sheer number of pathways. However, one such pathway activated by Wnt5b is the Planar Cell Polarity (PCP) pathway, which controls tissue polarity and cell migration [39]. WNT5B is up-regulated in MCF-7 cells [40] and is associated with cell migration and proliferation in lung cancer cells [41]. In preadipocytes, WNT5B overexpression partially inhibits canonical Wnt/β-catenin signaling, thereby promoting adipogenesis [42]. In addition to its role in cancer and metastasis, non-canonical Wnt signaling also promotes metabolic dysfunction and adipose tissue inflammation, which is critical to the development of insulin resistance [43]. Collectively, these observations lead us to suggest that, in Ishikawa cells, dysregulation of O-GlcNAc signaling could promote EMT through activation of non-canonical Wnt signaling via WNT5B overexpression, which may in turn result in inhibited Wnt/β-catenin signaling. Such a mechanism could also help explain the connection between insulin resistance and increased risk of endometrial cancer.
In addition to increased WNT5B expression, the current study revealed that the transcription factor, Forkhead box protein C2 (FOXC2) is also highly-expressed in Hyper-O-GlcNAcylated Ishikawa cells (Figure 4A, 4B and 4D). Often, increased expression of FOXC2 during EMT is associated with highly-metastatic cancers [44], some of which can be reduced by shRNA therapy [45]. In podocytes, for instance, increased FOXC2 is associated with increased Vimentin expression, cytoskeletal reorganization, disruption to ZO-1 localization, and increased cell motility [46]. In the current study, cytoskeletal re-organization was noted in hyper-O-GlcNAcylated cells (Figure 3A), and although Vimentin expression was consistent among treatments, ZO-1 expression was reduced by inhibition of OGT (hypo-O-GlcNAcylation, OSMI-1) (Figure 3C). Thus, there is support for the concept that Hyper-O-GlcNAcylation of Ishikawa cells induces EMT and metastasis via a FOXC2-mediated mechanism.
Cytoskeletal re-organization is key feature of EMT. Cytokeratin intermediate filaments are often considered biomarkers of cancers, but are more recently gaining attention as mediators of disease [47, 48]. A good example of this is Keratin 18, which is a component of cytokeratin 8/18 intermediate filaments and has long been used as a marker for apoptosis in cancers [49], but the loss of keratin 18 expression in cells is associated with EMT [50]. Previously we have determined that Hypo-O-GlcNAcylation in cervical cancer cells reduces and re-organizes cytokeratin 8/18 filaments, suggesting that O-GlcNAcylation influences filament expression and stability [51]. Similar restructuring of filaments in Ishikawa cells occurred in the current study, with both Hyper- and Hypo-O-GlcNAcylation increasing the overall expression of Keratin 14 mRNA. Co-expression of Vimentin and keratin in MCF-7 breast cancer cells increases cell invasiveness [52]. In the current study, Hyper-O-GlcNAcylation had no effect on Vimentin expression, increased Keratin 14 expression, and yet still increased metastatic potential. These findings warrant further investigation into the regulation of keratin filaments by O-GlcNAcylation, and their possible role in disease progression.
In summary, dysregulation of O-GlcNAcylation supports EMT and cytoskeletal re-organization in endometrial cancer cells, potentially through activation of the noncanonical Wnt signaling pathway via WNT5B. Additionally, Hyper-O-GlcNAcylation induces the expression of FOXC2 and augments the invasion potential of the cells. These findings indicate Hyper-O-GlcNAcylation, such as that observed in diabetic individuals [53], could enhance the aggressiveness of endometrial cancer. Conversely, Hypo-O-GlcNAcylation impaired endometrial cancer cell proliferation and wound healing, and down-regulated expression of pro-EMT genes (AHNAK, CALD1, and TGFB2). These observations suggest that metabolic status, particularly as it relates to O-GlcNAcylation within cells, might be an important consideration in therapeutic approaches to endometrial cancer. Future studies should focus on aberrant O-GlcNAcylation as a potential marker for more aggressive disease in human tissues, and the inhibition of Hyper-O-GlcNAcylation as a potential treatment in diabetic patients.
Materials and Methods
Cell culture/Reagents
Endometrial Cancer cells (Ishikawa) were obtained from Sigma Aldrich (cat. #99040201). Cell Line authentication was performed before, during, and after experimentation in the University of Vermont Cancer Center Advanced Genome Technologies Core and was supported by the University of Vermont Cancer Center, Lake Champlain Cancer Research Organization, and the University of Vermont College of Medicine. O-GlcNAcylation in cells was manipulated by the OGA inhibitor, ThmG (1 μM, Fisher Scientific), the OGT inhibitor, OSMI-1 (50 μM, Aobious), and supplementation of excess glucose (25 mM). ThmG and OSMI-1 were dissolved in DMSO (Fisher Scientific) at a concentration of 1000X, thus control and glucose treated cells received 0.1% DMSO (vehicle). Cell culture media (EMEM, 10% FBS and 10 μl/mL antibiotic-anti-mycotic (Fisher Scientific) was exchanged every 24 hours unless otherwise specified.
Immunoblotting
Cell Lysates were harvested via trypsin digestion and dissolved in RIPA buffer, and then passaged 5 times through a 26G needle. Protein concentration was measured by BCA Assay (BioRad) following the manufacturer’s protocol. Pre-stained SDS-PAGE gels were loaded with 20 μg of proteins per lane. Protein was then transferred to PVDF membrane (Millipore). Membranes were then probed with antibodies associated with EMT (EMT antibody kit), O-GlcNAc (CTD 110.6) (Supplementary Figure 1), and OGT. Goat-anti-rabbit and goat-anti-mouse HRP conjugated secondary antibodies were used in combination with Clarity Western ECL Blotting Substrate (BioRad) for imaging on the BioRad ChemiDoc Imager. All antibodies were purchased from Cell Signaling unless otherwise specified.
Immunohistochemistry
Cells were seeded on glass coverslips and incubated in treatments described above for 72 hours. Media was exchanged daily. Cells were then fixed with 4% paraformaldehyde in PBS (Fisher Scientific) and stained with Alexaflour-488 Phalloidin (Molecular Probes, Eugene, OR) and DAPI (Molecular Probes). Cells were imaged with the Olympus CKX53 at 200–1000X.
Proliferation
In a set of 3 independent experiments, cells were seeded in quadruplicate in flat-sided Thermo Fisher Scientific™ Nunc™ Cell Culture Tubes at an initial seeding density of 50 k cells/ml of serum free culture medium. Cells were treated as described above. The conditioned culture medium was exchanged daily, and the cells for each group were harvested for counting at 24-hour intervals over a four-day period. An MTS assay was also used to measure proliferation and viability. Clear 96 well plates were seeded with 5,000 cells/well in serum free conditions. Cells were treated as described above, and conditioned medium was refreshed daily. Cell proliferation was measured with the CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) kit following manufacturer instructions (Promega, Madison, WI) at 24 and 48 hours as measured by the Synergy HT Plate Reader (Biotek, Winooski, VT).
Migration
A wound-healing assay was used to measure migration of cells treated as described. Confluent monolayers of treated cells grown in 24 well plates were serum starved for 24 hours before “wounds” were created by running a 10 μL pipette tip across the monolayer. Cells were maintained in serum free conditions to inhibit proliferation and were incubated at 37°C and 5% CO2 for 48 hours. Images of the “wound” area were taken every 24 hours (100X) and the “wound” area was measured with ImageJ software to calculate percent wound closure.
Invasion
Corning Matrigel Biocoat Invasion Chambers (24 well, 0.8 μ) (Corning, Bedford, MA) were used to assess the invasion potential of treated cells compared to control. The upper chamber was seeded with 100,000 serum-starved cells. Cells were exposed to O-GlcNAc-modifying treatments at the time of seeding. The lower chamber contained 5% FBS as a chemoattractant. Cells were incubated for 48 hours at 37°C and 5% CO2. Cells were then removed from the upper chamber and the Transwell membranes were fixed with 100% methanol and stained with Crystal Violet. Invaded cells were then counted by two independent researchers and the results were averaged.
RT PCR array
RNA was extracted from treated cell pellets with the RNeasy Mini-Kit (Qiagen) with DNase digestion (RNAse free DNase Kit, QIAGEN) following the manufacturer protocols. The cDNA synthesis and RT2 Profiler PCR Arrays (QIAGEN, PAHS-090Z) (Supplementary Table 2) were performed in the University of Vermont Cancer Center Advanced Genome Technologies Core and was supported by the University of Vermont Cancer Center, Lake Champlain Cancer Research Organization, and the University of Vermont College of Medicine. Results were analyzed via the GeneGlobe Data Analysis Center (QIAGEN) with 2.5 fold change serving as the cut-off.
Oncogenomic data analysis
Multidimensional cancer genomic data analysis was performed using online data-mining tool from the cBioPortal for Cancer Genomics (http://cbioportal.org) and the data sets from the cBioPortal for Cancer Genomics and the TCGA research Network (http://cancergenome.nih.gov) [54, 55]. Tumor types and data sets are chosen in accordance with the publication guidelines from TCGA (last update:30 September 2014). Genomic alterations were identified when the following occurred: (1) gene mutations; (2) putative copy number alteration (amplification or deletion); (3) RNA expression Z-scores (RNA Seq Version 2 RSEM) with Z-score thresholds ± 2.0; and (4) protein/phospho-protein level (RPPA) with Z-score thresholds ± 2.0.
Data analysis
A minimum of 3 independent replicates were completed for each experiment, reported as mean +/– SEMs. Statistical analysis was conducted using 1 or 2-Way ANOVA and Student’s t-tests as appropriate with Graph Pad Prism Statistical Analysis Software. Fold-change differences in mRNA expression were calculated with the GeneGlobe Data Analysis Center (QIAGEN) for RT2 Profiler PCR Arrays.
Abbreviations
O-GlcNAc: β-N-acetylglucosamine; OGT: O-GlcNAc Transferase; OGA: O-GlcNAcase; HBP: Hexosamine Biosynthesis Pathway; T2D: Type 2 Diabetes; ThmG: Thiamet-G; FBS: Fetal Bovine Serum.
CONFLICTS OF INTEREST
The authors declare they have no competing interests.
FUNDING
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE 1450271. (NMJ).
References
1. Bond MR, Hanover JA. A little sugar goes a long way: The cell biology of O-GlcNAc. J Cell Biol. 2015; 208:869–80. https://doi.org/10.1083/jcb.201501101. [PubMed].
2. Issad T, Masson E, Pagesy P. O-GlcNAc modification, insulin signaling and diabetic complications. Diabetes Metab. 2010; 36:423–35. https://doi.org/10.1016/j.diabet.2010.09.001. [PubMed].
3. Krześlak A, Wójcik-Krowiranda K, Forma E, Bieńkiewicz A, Bryś M. Expression of genes encoding for enzymes associated with O-GlcNAcylation in endometrial carcinomas: clinicopathologic correlations. Ginekol Pol. 2012; 83:22–6. [PubMed].
4. Lucena MC, Carvalho-Cruz P, Donadio JL, Oliveira IA, de Queiroz RM, Marinho-Carvalho MM, de Paula IF, Gondim KC, McComb ME, Costello CE, Whelan SA, Todeschini AR, Dias WB. Epithelial mesenchymal transition induces aberrant glycosylation through hexosamine biosynthetic pathway activation. J Biol Chem. 2016; 291:12917–29. https://doi.org/10.1074/jbc.M116.729236. [PubMed].
5. Friberg E, Orsini N, Mantzoros CS, Wolk A. Diabetes mellitus and risk of endometrial cancer: a meta-analysis. Diabetologia. 2007; 50:1365–74. https://doi.org/10.1007/s00125-007-0681-5. [PubMed].
6. Cote ML, Ruterbusch JJ, Olson SH, Lu K, Ali-Fehmi R. The growing burden of endometrial cancer: A major racial disparity affecting black women. Cancer Epidemiol Biomarkers Prev. 2015; 24:1407–15. https://doi.org/10.1158/1055-9965.EPI-15-0316. [PubMed].
7. Catasus L, Gallardo A, Prat J. Molecular genetics of endometrial carcinoma. Diagn Histopathol. 2009; 15:554–63. https://doi.org/10.1016/J.MPDHP.2009.09.002.
8. Stewart CJR, Mccluggage WG. Epithelial-mesenchymal transition in carcinomas of the female genital tract. Histopathology. 2013; 62:31–43. https://doi.org/10.1111/his.12057. [PubMed].
9. Park SY, Kim HS, Kim NH, Ji S, Cha SY, Kang JG, Ota I, Shimada K, Konishi N, Nam HW, Hong SW, Yang WH, Roth J, et al. Snail1 is stabilized by O-GlcNAc modification in hyperglycaemic condition. EMBO J. 2010; 29:3787–96. https://doi.org/10.1038/emboj.2010.254. [PubMed].
10. Park JY, Nam JH. Progestins in the Fertility-Sparing Treatment and Retreatment of Patients With Primary and Recurrent Endometrial Cancer. Gynecol Oncol. 2015; 20:270–8. https://doi.org/10.1634/theoncologist.2013-0445. [PubMed].
11. Gallos ID, Yap J, Rajkhowa M, Luesley DM, Coomarasamy A, Gupta JK. Regression, relapse, and live birth rates with fertility-sparing therapy for endometrial cancer and atypical complex endometrial hyperplasia: a systematic review and metaanalysis. Am J Obstet Gynecol. 2012; 207:266.e1–266.e12. https://doi.org/10.1016/j.ajog.2012.08.011. [PubMed].
12. Ohtani K, Sakamoto H, Rutherford T, Chen Z, Satoh K, Naftolin F. Ezrin, a membrane-cytoskeletal linking protein, is involved in the process of invasion of endometrial cancer cells. Cancer Lett. 1999; 147:31–8. https://doi.org/10.1016/S0304-3835(99)00272-4. [PubMed].
13. Torres CR, Hart GW. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem. 1984; 259:3308–17. http://www.jbc.org/content/259/5/3308.full.pdf. [PubMed].
14. Palin V, O’Neil I, Warwicker J, Paijens S, Aplin J, Westwood M. The hexosamine biosynthetic pathway: a role in nutrient regulation of growth signalling in the human placenta. Placenta. 2014; 35:A92. https://doi.org/10.1016/j.placenta.2014.06.297.
15. Singh JP, Zhang K, Wu J, Yang X. O-GlcNAc signaling in cancer metabolism and epigenetics. Cancer Lett. 2015; 356:244–50. https://doi.org/10.1016/j.canlet.2014.04.014. [PubMed].
16. Caldwell SA, Jackson SR, Shahriari KS, Lynch TP, Sethi G, Walker S, Vosseller K, Reginato MJ. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene. 2010; 29:2831–42. https://doi.org/10.1038/onc.2010.41. [PubMed].
17. Guo K, Gan L, Zhang S, Cui FJ, Cun W, Li Y, Kang NX, Gao MD, Liu KY. Translocation of HSP27 into liver cancer cell nucleus may be associated with phosphorylation and O-GlcNAc glycosylation. Oncol Rep. 2012; 28:494–500. https://doi.org/10.3892/or.2012.1844. [PubMed].
18. Rozanski W, Krzeslak A, Forma E, Brys M, Blewniewski M, Wozniak P, Lipinski M. Prediction of bladder cancer based on urinary content of MGEA5 and OGT mRNA level. Clin Lab. 2012; 58:579–83. [PubMed].
19. Itkonen HM, Minner S, Guldvik IJ, Sandmann MJ, Tsourlakis MC, Berge V, Svindland A, Schlomm T, Mills IG. O-GlcNAc Transferase Integrates Metabolic Pathways to Regulate the Stability of c-MYC in Human Prostate Cancer Cells. Cancer Res. 2013; 73:5277–87. https://doi.org/10.1158/0008-5472.CAN-13-0549. [PubMed].
20. Mi W, Gu Y, Han C, Liu H, Fan Q, Zhang X, Cong Q, Yu W. O-GlcNAcylation is a novel regulator of lung and colon cancer malignancy. Biochim Biophys Acta. 2011; 1812:514–9. https://doi.org/10.1016/j.bbadis.2011.01.009. [PubMed].
21. Kamigaito T, Okaneya T, Kawakubo M, Shimojo H, Nishizawa O, Nakayama J. Overexpression of O-GlcNAc by prostate cancer cells is significantly associated with poor prognosis of patients. Prostate Cancer Prostatic Dis. 2013; 17:18–22. https://doi.org/10.1038/pcan.2013.56. [PubMed].
22. Gallagher EJ, LeRoith D. Obesity and Diabetes: The Increased Risk of Cancer and Cancer-Related Mortality. Physiol Rev. 2015; 95:727–48. https://doi.org/10.1152/physrev.00030.2014. [PubMed].
23. Zelenko Z, Gallagher EJ. Diabetes and cancer. Endocrinol Metab Clin North Am. 2014; 43:167–85. https://doi.org/10.1016/j.ecl.2013.09.008. [PubMed].
24. Byrne FL, Poon IK, Modesitt SC, Tomsig JL, Chow JD, Healy ME, Baker WD, Atkins KA, Lancaster JM, Marchion DC, Moley KH, Ravichandran KS, Slack-Davis JK, Hoehn KL. Metabolic vulnerabilities in endometrial cancer. Cancer Res. 2014; 74:5832–45. https://doi.org/10.1158/0008-5472.CAN-14-0254. [PubMed].
25. Ruan HB, Singh JP, Li MD, Wu J, Yang X. Cracking the O-GlcNAc code in metabolism. Trends Endocrinol Metab. 2013; 24:301–9. https://doi.org/10.1016/j.tem.2013.02.002. [PubMed].
26. Copeland RJ, Han G, Hart GW. O-GlcNAcomics--Revealing roles of O-GlcNAcylation in disease mechanisms and development of potential diagnostics. Proteomics Clin Appl. 2013; 7:597–606. https://doi.org/10.1002/prca.201300001. [PubMed].
27. Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013; 19:1438–49. https://doi.org/10.1038/nm.3336. [PubMed].
28. Kondaveeti Y, Guttilla Reed IK, White BA. Epithelial–mesenchymal transition induces similar metabolic alterations in two independent breast cancer cell lines. Cancer Lett. 2015; 364:44–58. https://doi.org/10.1016/j.canlet.2015.04.025. [PubMed].
29. Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009; 28:15–33. https://doi.org/10.1007/s10555-008-9169-0. [PubMed].
30. Larue L, Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3’ kinase/AKT pathways. Oncogene. 2005; 24:7443–54. https://doi.org/10.1038/sj.onc.1209091. [PubMed].
31. Rojas-Puentes L, Cardona AF, Carranza H, Vargas C, Jaramillo LF, Zea D, Cetina L, Wills B, Ruiz-Garcia E, Arrieta O. Epithelial-mesenchymal transition, proliferation, and angiogenesis in locally advanced cervical cancer treated with chemoradiotherapy. Cancer Med. 2016; 5:1989–99. https://doi.org/10.1002/cam4.751. [PubMed].
32. Yao D, Dai C, Peng S. Mechanism of the Mesenchymal-Epithelial Transition and Its Relationship with Metastatic Tumor Formation. Mol Cancer Res. 2011; 9:1608–20. https://doi.org/10.1158/1541-7786.mcr-10-0568. [PubMed].
33. Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008; 68:3645–54. https://doi.org/10.1158/0008-5472.CAN-07-2938. [PubMed].
34. Fardini Y, Dehennaut V, Lefebvre T, Issad T. O-GlcNAcylation: A New Cancer Hallmark? Front Endocrinol (Lausanne). Frontiers. 2013; 4:99. https://doi.org/10.3389/fendo.2013.00099. [PubMed].
35. Zhu Q, Zhou L, Yang Z, Lai M, Xie H, Wu L, Xing C, Zhang F, Zheng S. O-GlcNAcylation plays a role in tumor recurrence of hepatocellular carcinoma following liver transplantation. Med Oncol. 2012; 29:985–93. https://doi.org/10.1007/s12032-011-9912-1. [PubMed].
36. Polakis P. Wnt signaling and cancer. Genes Dev. 2000; 14:1837–51. [PubMed].
37. Zhou F, Huo J, Liu Y, Liu H, Liu G, Chen Y, Chen B. Elevated glucose levels impair the WNT/Î2-catenin pathway via the activation of the hexosamine biosynthesis pathway in endometrial cancer. J Steroid Biochem Mol Biol. 2016; 159:19–25. https://doi.org/10.1016/j.jsbmb.2016.02.015. [PubMed].
38. Siar CH, Nagatsuka H, Han PP, Buery RR, Tsujigiwa H, Nakano K, Ng KH, Kawakami T. Differential expression of canonical and non-canonical Wnt ligands in ameloblastoma. J Oral Pathol Med. 2012; 41:332–9. https://doi.org/10.1111/j.1600-0714.2011.01104.x. [PubMed].
39. Katoh M. WNT/PCP signaling pathway and human cancer. Oncology Reports. 1994; 14:1583–1588. https://www.spandidos-publications.com/or/14/6/1583.
40. Saitoh T, Katoh M. Expression and regulation of WNT5A and WNT5B in human cancer: Up-regulation of WNT5A by TNFα in MKN45 cells and up-regulation of WNT5B by β-estradiol in MCF-7 cells. Int J Mol Med. 2002; 10:345–9. https://www.spandidos-publications.com/ijmm/10/3/345. [PubMed].
41. Harada T, Yamamoto H, Kishida S, Kishida M, Awada C, Takao T, Kikuchi A. Wnt5b-associated exosomes promote cancer cell migration and proliferation. Cancer Sci. 2017; 108:42–52.https://doi.org/10.1111/cas.13109. [PubMed].
42. Kanazawa A, Tsukada S, Kamiyama M, Yanagimoto T, Nakajima M, Maeda S. Wnt5b partially inhibits canonical Wnt/β-catenin signaling pathway and promotes adipogenesis in 3T3-L1 preadipocytes. Biochem Biophys Res Commun. 2005; 330:505–10. https://doi.org/10.1016/J.BBRC.2005.03.007. [PubMed].
43. Fuster JJ, Zuriaga MA, Ngo DT, Farb MG, Aprahamian T, Yamaguchi TP, Gokce N, Walsh K. Noncanonical Wnt signaling promotes obesity-induced adipose tissue inflammation and metabolic dysfunction independent of adipose tissue expansion. Diabetes. 2015; 64:1235–48. https://doi.org/10.2337/db14-1164. [PubMed].
44. Hollier BG, Tinnirello AA, Werden SJ, Evans KW, Taube JH, Sarkar TR, Sphyris N, Shariati M, Kumar SV, Battula VL, Herschkowitz JI, Guerra R, Chang JT, et al. FOXC2 Expression Links Epithelial–Mesenchymal Transition and Stem Cell Properties in Breast Cancer. Cancer Res. 2013; 73:1981–92. https://doi.org/10.1158/0008-5472.CAN-12-2962. [PubMed].
45. Paranjape AN, Soundararajan R, Werden SJ, Joseph R, Taube JH, Liu H, Rodriguez-Canales J, Sphyris N, Wistuba I, Miura N, Dhillon J, Mahajan N, Mahajan K, et al. Inhibition of FOXC2 restores epithelial phenotype and drug sensitivity in prostate cancer cells with stem-cell properties. Oncogene. 2016; 35:5963–76. https://doi.org/10.1038/onc.2015.498. [PubMed].
46. Datta N, Lindfors S, Miura N, Saleem MA, Lehtonen S. Overexpression of transcription factor FOXC2 in cultured human podocytes upregulates injury markers and increases motility. Exp Cell Res. 2016; 340:32–42. https://doi.org/10.1016/J.YEXCR.2015.10.035. [PubMed].
47. Karantza V. Keratins in health and cancer: more than mere epithelial cell markers. Oncogene. 2011; 30:127–38. https://doi.org/10.1038/onc.2010.456. [PubMed].
48. Pan X, Hobbs RP, Coulombe PA. The expanding significance of keratin intermediate filaments in normal and diseased epithelia. Curr Opin Cell Biol. 2013; 25:1–10. https://doi.org/10.1016/j.ceb.2012.10.018. [PubMed].
49. Lomnytska M, Souchelnytsky S. Markers of breast and gynecological malignancies: The clinical approach of proteomics-based studies. Proteomics Clin Appl. 2007; 1:1090–101. https://doi.org/10.1002/prca.200700179. [PubMed].
50. Fortier AM, Asselin E, Cadrin M. Keratin 8 and 18 loss in epithelial cancer cells increases collective cell migration and cisplatin sensitivity through claudin1 up-regulation. J Biol Chem. 2013; 288:11555–71. https://doi.org/10.1074/jbc.M112.428920. [PubMed].
51. Jaskiewicz NM, Hermawan C, Parisi S, Townson DH, Morin Jaskiewicz N. O-GlcNAcylation enhances the tumorigenic properties of cervical cancer cells in vitro. Clin Obstet Gynecol Reprod Med. 2017; 3:1–6. https://doi.org/10.15761/COGRM.1000183.
52. Hendrix MJC, Seftor EA, Seftor REB, Trevort KT. Experimental Co-Expression of Vimentin and Keratin Intermediate Filaments in Human Breast Cancer Cells Results in Phenotypic Interconversion and Increased Invasive Behavior. Am J Pathol. 1997; 150:483–95. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1858294/pdf/amjpathol00026-0099.pdf. [PubMed].
53. Medford HM, Chatham JC, Marsh SA. Chronic ingestion of a Western diet increases O-linked-β-N-acetylglucosamine (O-GlcNAc) protein modification in the rat heart. Life Sci. 2012; 90:883–8. https://doi.org/10.1016/j.lfs.2012.04.030. [PubMed].
54. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012; 2:401–4. https://doi.org/10.1158/2159-8290.CD-12-0095. [PubMed].
55. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci Signal. 2013; 6:pl1. https://doi.org/10.1126/scisignal.2004088. [PubMed].