
Introduction
High grade serous ovarian cancer (HGSOC) is the most lethal gynecologic malignancy in the United States (U.S.), with an overall 5-year survival rate under 40% [1]. More than 80% of HGSOC present at an advanced stage, and recurrence is nearly universal leading to incurable disease [2]. Approximately 40% of HGSOC harbors defects in genes involved in homologous recombination (HR) repair (HRR) for double-stranded DNA breaks, such as BRCA1 and BRCA2 [3, 4]. While approximately 15% of HGSOC patients have a germline mutation in BRCA1 or BRCA2 (gBRCAm) [4, 5], HRR deficiency may be caused by other molecular alterations such as RAD51C, RAD51D, BRIP1, and CHEK2 [6–8]. Deficient HRR leads to activation of alternate DNA repair pathways including base excision repair (BER) and non-homologous end-joining (NHEJ), which require poly (ADP-ribose) polymerase (PARP) activity [9]. Increased PARP-1 activity is demonstrated in several tumor types including HGSOC [10, 11]. In preclinical studies, PARP inhibitors (PARPi) showed enhanced cytotoxicity in both HGSOC with BRCA mutations and HRR-deficient HGSOC with wild-type BRCA (BRCAwt), suggesting a broad applicability of PARPi in clinic [12–14].
There are three PARPi - olaparib, rucaparib and niraparib - now U.S. Food and Drug Administration (FDA)-approved for ovarian cancer treatment or maintenance. Olaparib is the first approved PARPi for use as monotherapy in gBRCAm patients with recurrent HGSOC who had >3 prior treatment regimens [15]. It is also FDA-approved as a maintenance therapy in platinum-sensitive recurrent HGSOC patients regardless of their BRCA status, as well as for frontline maintenance in BRCAm patients [16–19]. Rucaparib, another PARPi, is approved for third-line treatment in patients with a germline or somatic BRCA mutation as well as for maintenance therapy [20, 21]. Like olaparib and rucaparib, niraparib is approved as a maintenance therapy in platinum-sensitive recurrent HGSOC patients who achieved response following chemotherapy [22]. So far, clinical benefits of PARPi in HGSOC appear strongest in BRCAm women in the platinum-sensitive recurrent treatment setting (response rates around 40–60%) [20, 23, 24]. Less activity is observed in heavily pretreated and non-BRCA mutant patients (response rates around 10–30% for platinum-sensitive and <10% for platinum-resistant) [20, 23], establishing the need to test combination strategies for this population.
Cisplatin, and now preferentially carboplatin, are the backbone of ovarian cancer treatment. Platinum agents form DNA-platinum adducts that damage DNA leading to cell death [25]. This is counteracted by the DNA repair mechanisms of BER and nucleotide excision repair [25–27]. Increased levels of poly(ADP-ribose) (PAR) polymers have been shown after cisplatin treatment in O-342 rat ovarian tumor cell lines [28] and PARP upregulation after cisplatin exposure was also demonstrated in normal renal tubular and human colon carcinoma cells [29, 30]. Concomitant use of PARPi with a platinum agent has been tested in several types of cancer, demonstrating increased cytotoxicity [31–35]. PARP inhibition potentiated platinum cytotoxicity in the O-342/DDP and CH1cisR platinum-resistant ovarian cancer cell lines [31, 32], as well as in the BRCAwt and BRCA2-restored OV90 and PEO4 ovarian cancer cell lines, respectively [33]. The PARPi CEP-6800 and olaparib also enhanced platinum-induced cytotoxicity in HRR-proficient non-small cell lung and colorectal carcinomas, respectively [34, 35]. These data support clinical testing of the PARPi and carboplatin combination in non-BRCA mutant ovarian cancer patients to assess for an additive or synergistic benefit of the doublet.
We previously reported the safety data and recommended phase 2 doses (RP2Ds) of olaparib in combination with carboplatin for patients with gBRCAm recurrent HGSOC or breast cancer and BRCAwt recurrent triple negative breast cancer (TNBC), finding different maximum tolerated doses (MTDs) for each population [36, 37]. We expanded these findings to women with HGSOC who do not have gBRCAm and now report safety, RP2D and activity. We also evaluated potential predictive and pharmacodynamic biomarkers for the PARPi and carboplatin combination.
Results
Patients
The study schema is depicted in Figure 1 and the Consort diagram is in Figure 2 (N = 30). All but 6 patients had negative deleterious gBRCAm commercial testing. Six patients enrolled between 2009–2013 had no BRCA testing prior to enrollment, and were eligible per protocol based on the negative family history of breast and ovarian cancer and a BRCAPro score of ≤20% [38]. Approximately two-thirds had platinum-resistant disease (63% [19/30]). Most patients (90% [27/30]) were heavily treated, having received more than three prior regimens. Other patient characteristics are shown in Table 1.
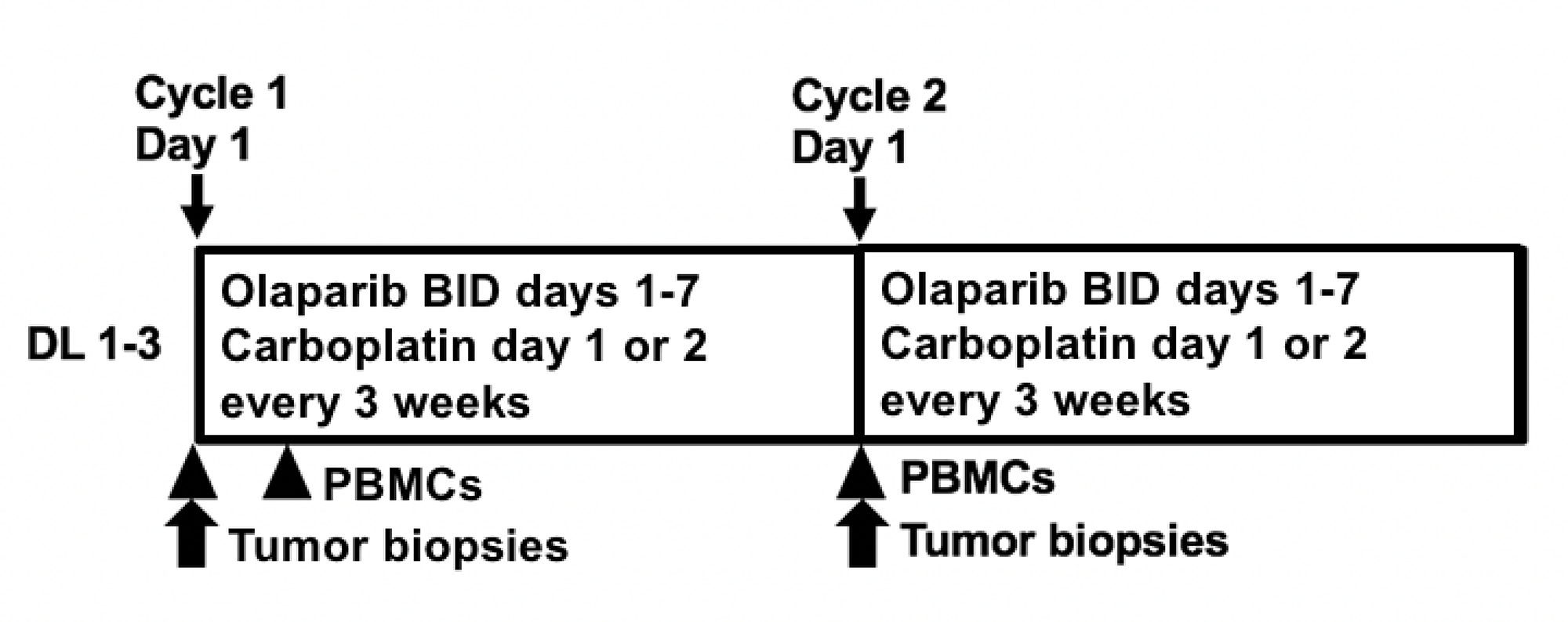
Figure 1: Study Schema. Abbreviations: DL: dose level; bid: twice daily; PBMCs: peripheral blood mononuclear cells.
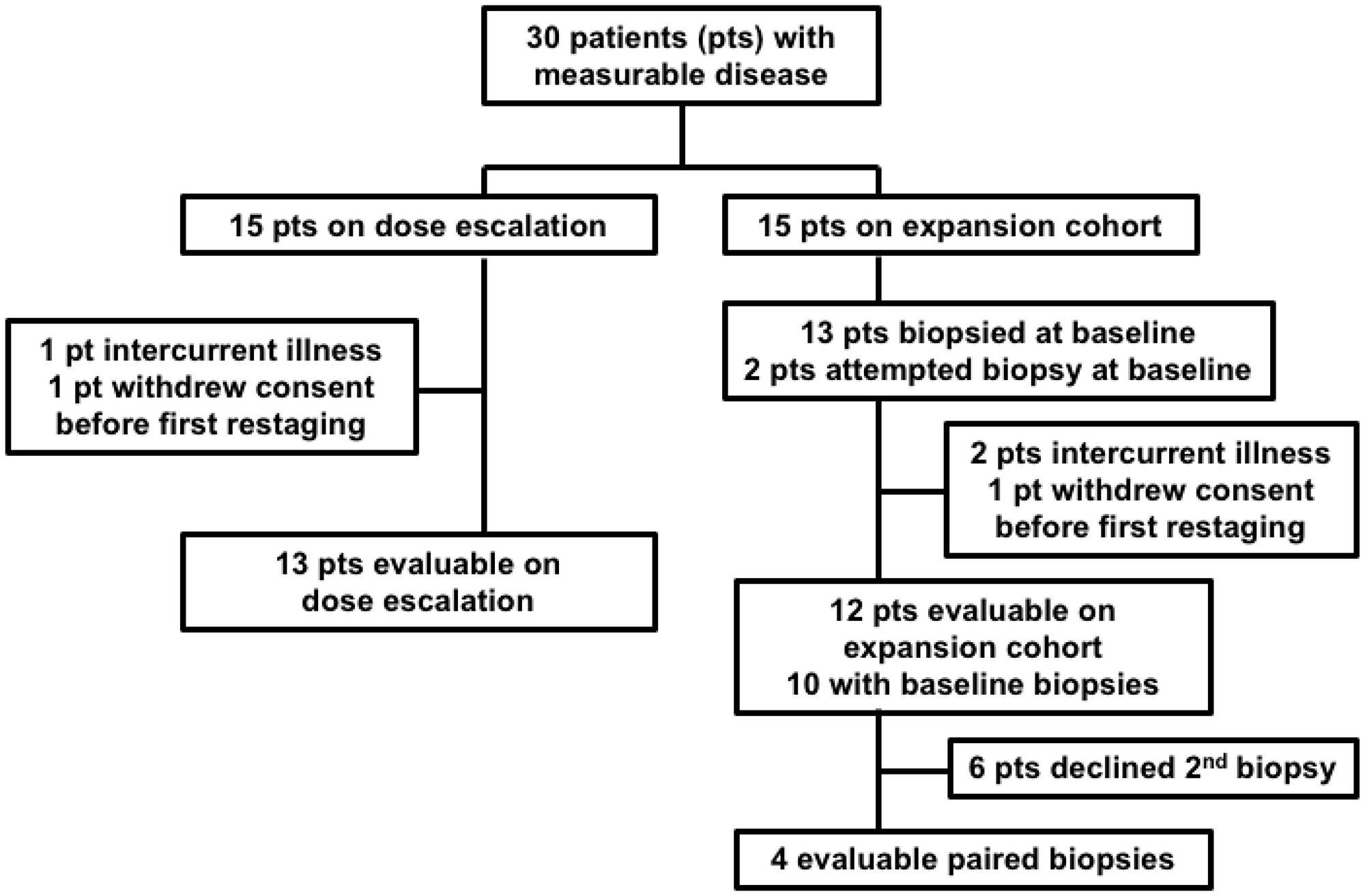
Figure 2: Consort Diagram. Of the 13 expansion cohort patients successfully biopsied at baseline, 10 had evaluable disease, 2 were non-evaluable due to intercurrent illness, and 1 withdrew consent. 6 of the 10 evaluable patients with a baseline biopsy declined a second biopsy, leaving 4 evaluable paired biopsies. Both of the non-evaluable patients with a baseline biopsy had a post-treatment biopsy, resulting in 2 non-evaluable paired biopsies for a total of 6 paired biopsies.
Table 1: Patient characteristics (N = 30)
| Age in years, median (range) | 65 (49–71) |
| ECOG Performance Status, N (%) | |
| 0 | 5 (17%) |
| 1 | 24 (80%) |
| 2 | 1 (3%) |
| Median number of prior regiments (range) | 7 (2–12) |
| Median prior chemotherapeutic agents (range) | 6 (2–10) |
| Median prior biologic agents (range) | 1 (0–3) |
| Prior bevacizumab treatment, N (%)* | 21 (70%) |
| Prior vaccine treatment, N (%) | 3 (10%) |
| Median months since last platinum (range) | 16.5 (7–154) |
| Platinum sensitivity+, N (%) | |
| Platinum resistant recurrent disease | 19 (63%) |
| Platinum sensitive recurrent disease | 11 (37%) |
| Race/Ethnicity, N (%) | |
| White | 27 (90%) |
| Black | 2 (7%) |
| Asian | 1 (3%) |
| Hispanic | 0 (0%) |
Dose optimization
Patients received olaparib 400 mg capsules twice daily on days 1–7 and carboplatin AUC 3–5 on day 1 of each 21-day cycle (Table 2 and Figure 1). Olaparib 400 mg twice a day maintenance therapy was continued after a maximum of 8 carboplatin-containing cycles. No dose-limiting toxicities (DLT) were observed at dose level 2 (DL2) with carboplatin AUC4 during the 2-cycle evaluation period. Increasing to DL3 with carboplatin AUC5 resulted in 2 of 6 patients having DLT (grade 3 thrombocytopenia and neutropenia after one cycle [n = 1] and two concurrent grade 3 infections with an absolute neutrophil count (ANC) within normal range requiring IV antibiotics [n = 1]). One patient in DL3 required carboplatin dose reduction to AUC4 at cycle 4 for persistent neutropenia and treatment delays despite pegfilgrastim supplementation. Another DL3 patient was put on olaparib maintenance therapy after carboplatin discontinuation at cycle 6 due to neutropenic fever. No patients required olaparib dose reduction or (peg)filgrastim supplementation during maintenance therapy. The recommended phase 2 dose is olaparib capsules 400 mg twice daily days 1–7 with carboplatin AUC4 day 1 in 21-day cycles.
Table 2: Dose levels (N = 30)
| Schedule and Dose | ||||
|---|---|---|---|---|
| Dose Level [DL] | Olaparib capsule, BID | Carboplatin IV, q3weeks | DLT | Best response (duration of response) |
| DL 1 (N = 3) | 400 mg, days 1–7 | AUC3, day 1 or 2 | 0 | 1 PR (7.5 mo) 2 SD (3 mo, 3 mo) |
| DL2 (N = 6)* | 400 mg, days 1–7 | AUC4, day 1 or 2 | 0 | 2 PR (3.3 mo, 4.5 mo) 2 SD (5.0 mo, 7.8 mo) 1 PD (2.4 mo) 1 NE (intercurrent illness) |
| DL 3 (N = 6) | 400 mg, days 1–7 | AUC5, day 1 or 2 | 2 | 1 PR (9.5 mo) 4 SD (8.5mo, 9.3mo, 10.8mo, 11.8mo) 1 NE (withdrew consent) |
| Expansion cohort (N = 15) | 400 mg, days 1–7 | AUC4, day 1 or 2 | 1 PR (4 mo) 7 SD (3.0mo, 3.5mo, 4.0 mo, 4.2 mo, 4.8 mo, 5.5mo, 10.6 mo) 4 PD (1.5 mo, 1.8 mo, 1.8 mo, 2.4 mo) 3 NE (1 withdrew consent; 2 intercurrent illness) | |
Adverse Events
Treatment-related adverse events (AEs) are summarized in Table 3. Hematologic toxicity was the most common AE (Tables 3, 4). Neutropenia occurred in 20 out of 30 patients (67%), with grade 3 or 4 neutropenia observed in 7 of 30 (23%) including one episode of febrile neutropenia. Ten of 30 (33%) patients received (peg)filgrastim to prevent treatment delay during the combination treatment. Grade 3 or 4 thrombocytopenia was observed in 6 of 30 patients (20%), and 1 patient (3%) required platelet transfusion after cycle 4 for grade 4 thrombocytopenia without bleeding. Grade 3 or 4 anemia occurred in four patients (13%) during cycles 2, 3, 4 and 5, respectively for each patient. Common (>10%) non-hematologic events included gastrointestinal side effects (nausea 50%, vomiting 27%, dyspepsia 30%), fatigue (33%) and headache (23%), and were predominantly grade 1 or 2, self-limited, and manageable with standard treatments.
Table 3: Drug-related adverse events by maximum grade per patient (N = 30)
| Adverse Event | Grade 1 | Grade 2 | Grade 3 | Grade 4 | Grade 3/4 |
|---|---|---|---|---|---|
| Hematology* | |||||
| Lymphocytopenia | 4 (13%) | 6 (20%) | 11 (37%) | 1 (3%) | 40% |
| White Blood Count | 4 (13%) | 16 (53%) | 6 (20%) | 1 (3%) | 23% |
| Neutropenia | 2 (7%) | 11 (37%) | 3 (10%) | 4 (13%) | 23% |
| Thrombocytopenia | 13 (43%) | 4 (13%) | 4 (13%) | 2 (7%) | 20% |
| Anemia | 6 (20%) | 16 (53%) | 3 (10%) | 1 (3%) | 13% |
| Gastrointestinal disorders | |||||
| Mucositis | 3 (10%) | 1 (3%) | 0 | 0 | 0% |
| Nausea | 13 (43%) | 2 (7%) | 0 | 0 | 0% |
| Vomiting | 5 (17%) | 3 (10%) | 1 (3%) | 0 | 3% |
| Dyspepsia | 8 (27%) | 1 (3%) | 0 | 0 | 0% |
| Gastroesophageal reflux disease | 2 (7%) | 0 | 0 | 0 | 0% |
| Constipation | 6 (20%) | 2 (7%) | 0 | 0 | 0% |
| Diarrhea | 4 (13%) | 0 | 0 | 0 | 0% |
| Chemistry | |||||
| Hyponatremia | 11 (37%) | 0 | 0 | 0 | 0% |
| Hypokalemia | 5 (17%) | 0 | 1 (3%) | 0 | 3% |
| Hypomagnesemia | 11 (37%) | 2 (7%) | 1 (3%) | 0 | 3% |
| Increased AST | 5 (17%) | 2 (7%) | 0 | 0 | 0% |
| Increased ALT | 8 (27%) | 0 | 0 | 0 | 0% |
| Other | |||||
| Fatigue | 7 (23%) | 3 (10%) | 0 | 0 | 0% |
| Skin rash | 3 (10%) | 0 | 0 | 0 | 0% |
| Headache | 7 (23%) | 0 | 0 | 0 | 0% |
| Neuropathy** | 4 (13%) | 0 | 0 | 0 | 0% |
Table 4: Drug-related hematologic adverse events by dose level (N = 30)
| Adverse event | Grade 1 | Grade 2 | Grade 3 | Grade 4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DL1 | DL2 | DL3 | DL1 | DL2 | DL3 | DL1 | DL2 | DL3 | DL1 | DL2 | DL3 | |
| Lymphopenia | 1(33%) | 3(14%) | 0 | 2 (67%) | 3 (14%) | 1 (17%) | 0 | 8(38%) | 3 (50%) | 0 | 1 (5%) | 0 |
| Leukopenia | 1(33%) | 3(14%) | 0 | 2 (67%) | 11 (52%) | 3 (50%) | 0 | 3(14%) | 3 (50%) | 0 | 1 (5%) | 0 |
| Neutropenia | 1(33%) | 1(5%) | 0 | 1 (33%) | 8 (38%) | 2 (33%) | 0 | 2(10%) | 1 (17%) | 0 | 1 (5%) | 3 (50%) |
| Thrombocytopenia | 3(100%) | 8(38%) | 2(33%) | 0 | 4 (19%) | 0 | 0 | 1(5%) | 3 (50%) | 0 | 1 (5%) | 1 (17%) |
| Anemia | 2(67%) | 3(14%) | 1(17%) | 0 | 12 (57%) | 4 (67%) | 0 | 2(10%) | 1 (17%) | 0 | 1 (5%) | 0 |
Clinical activity
Twenty-five patients had disease evaluable for RECIST response determination (Figure 3A and 3B). Five patients attained a PR (median 4.5 months, range 3.3–9.5) yielding an overall response rate (ORR) of 20% (Table 5). 15 patients had stable disease (SD; median 5 months, range 3–11.8) for a clinical benefit rate (CBR, defined as PR+SD≥4 months) of 64% (16/25). Median progression-free survival (PFS) in the 25 evaluable patients was 4.2 months [1.5–11.8 months]. ORR was 7% (1/14) in platinum-resistant and 36% (4/11) in platinum-sensitive disease (Table 6). One patient with platinum-resistant disease who achieved PR (9.5 months) had 9 prior therapies. Nine patients with platinum-resistant disease also had SD ≥ 4 months, for a CBR of 71% (10/14). In platinum-sensitive disease, 4 patients achieved PR and 2 SD ≥ 4 months yielding a CBR of 55% (6/11; Table 6).
Table 5: RECIST response (N = 25 with evaluable disease)
| Best Response | #(%) | Median Duration, mo (range) |
|---|---|---|
| CR | 0 | NA |
| PR | 5 (20%) | 4.5 (3.3–9.5) |
| SD ≥ 4 mo | 11 (44%) | 7.8 (4.0–11.8) |
| Total SD | 15 (60%) | 5 (3.0–11.8) |
| PD | 5 (20%) | 1.8 (1.5–2.4) |
| Clinical Benefit Rate (CR+ PR + SD) | 20/25 (80%; for all SD) 16/25 (64%; for SD ≥ 4 mo) | |
| Overall Response Rate (CR+PR) | 5/25 (20%) | |
Table 6: RECIST response by platinum-sensitivity
| Best Response | Platinum Sensitive (N = 11 evaluable disease) | Platinum Resistant (N = 14 evaluable disease) | ||
|---|---|---|---|---|
| #(%) | Median Duration, mo (range) | #(%) | Median Duration, mo (range) | |
| CR | 0 | NA | 0 | NA |
| PR | 4 (36%) | 4.3 (3.3–7.5) | 1 (7%) | 9.5 |
| SD ≥ 4 mo | 2 (18%) | 10.0 (9.3–10.8) | 9 (64%) | 5.5 (4.0–11.8) |
| Total SD | 4 (36%) | 6.4 (3.0–10.8) | 11 (79%) | 5.0 (3.0–11.8) |
| PD | 3 (27%) | 1.8 (1.5–1.8) | 2 (14%) | 2.4 |
| CBR (CR+ PR + SD) | 8/11 (73%; for all SD) 6/11 (55%; for SD ≥ 4 mo) | 12/14 (86%; for all SD) 10/14 (71%; for SD ≥ 4 mo) | ||
| ORR (CR + PR) | 4/11 (36%) | 1/14 (7%) | ||
![Waterfall plot (A) and duration on the study (B). (A) Twenty-five patients with baseline and subsequent imaging reassessment are shown. Best RECIST response is graphed for each patient. Five patients did not have reassessment scans for comparison (withdrew consent [n = 2], and off-treatment due to intercurrent illness [n = 3]). (B) All 30 patients are shown in a swimmer plot. Color code defines dose level of treatment. Dose level 1: olaparib 400 mg bid days 1–7 with carboplatin AUC3; dose level 2 and expansion cohort: olaparib 400 mg bid days 1–7 with carboplatin AUC4; dose level 3: olaparib 400 mg bid days 1–7 with carboplatin AUC5.](https://oncotarget-cdn.atl1.digitaloceanspaces.com/article/26869/figure/1028004/26869-1028004-1-PB.png)
Figure 3: Waterfall plot (A) and duration on the study (B). (A) Twenty-five patients with baseline and subsequent imaging reassessment are shown. Best RECIST response is graphed for each patient. Five patients did not have reassessment scans for comparison (withdrew consent [n = 2], and off-treatment due to intercurrent illness [n = 3]). (B) All 30 patients are shown in a swimmer plot. Color code defines dose level of treatment. Dose level 1: olaparib 400 mg bid days 1–7 with carboplatin AUC3; dose level 2 and expansion cohort: olaparib 400 mg bid days 1–7 with carboplatin AUC4; dose level 3: olaparib 400 mg bid days 1–7 with carboplatin AUC5.
Correlative studies
PAR levels
We first assessed the target effect induced by PARP inhibition as measured by PAR incorporation in peripheral blood mononuclear cells (PBMCs) and PAR expression levels in tissue biopsies. There was a significant decrease in PBMC PAR levels after treatment with olaparib in 12 evaluable paired samples (pre-treatment median 459.76 pg/mL [45.3–4, 243.5] vs post-treatment median 10.19 pg/mL [0–185.6], p = 0.0005; Figure 4A), demonstrating that olaparib reached pharmacologically effective concentrations. No correlation was seen between fold change from baseline to cycle 1 day 3 PAR incorporation and RECIST response (Figure 4B).
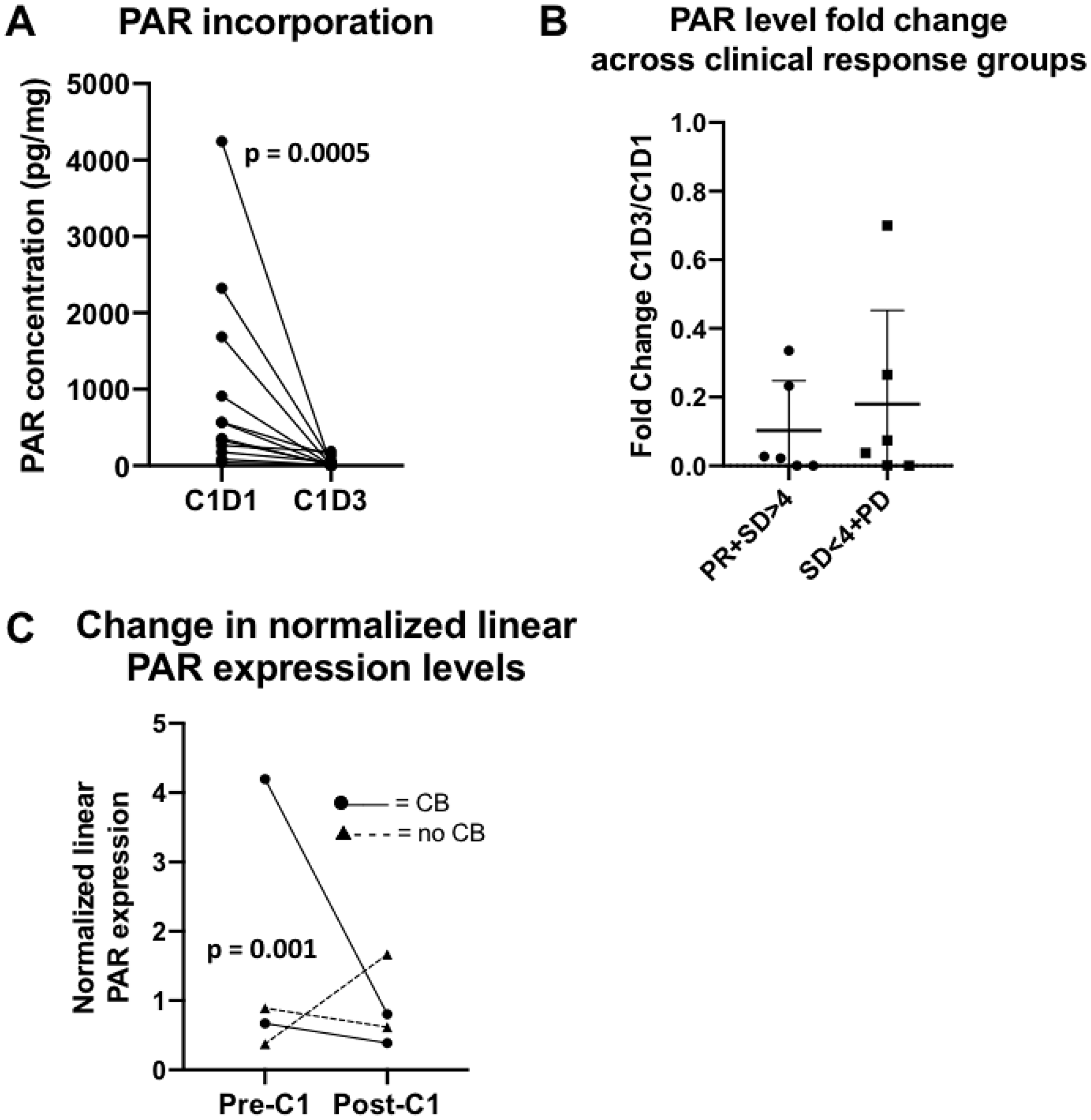
Figure 4: PAR translational studies. (A) PAR incorporation in peripheral blood. There was an expected decrease in PAR levels after treatment with olaparib in 12 evaluable paired samples. (B) Fold change in PAR concentration in peripheral blood by clinical response. No significant differences between fold change of PAR concentrations (C1D3/C1D1) and clinical response, defined as PR+SD>4 months versus SD<4 months+PD were observed. Abbreviations: C1D1 = cycle 1, day 1; C1D3 = cycle 1, day 3; PR = partial response; SD = stable disease; PD = progressive disease. (C) Change in normalized linear PAR expression levels between pre- and post-cycle 1 biopsy. A greater decrease of PAR levels was seen in the clinical benefit group (PR+SD≥4 months) versus no benefit group (SD<4 months+PD), p = 0.001. Abbreviations: C1 = cycle 1; CB = clinical benefit.
Of the 4 evaluable patients with paired biopsies, two had SD (4.2 and 10.6 months, respectively) and two had PD. A greater decrease in the normalized linear PAR expression levels was seen in the clinical benefit group (median change in normalized linear expression -1.84 [−3.39–−0.28]) compared to no benefit (0.51 [−0.27– 1.29]) (Figure 4C).
PARP1/XRCC1 polymorphisms
No PARP1 or XRCC1 polymorphisms were associated with RECIST response or clinical benefit.
Reverse phase protein microarray (RPPA) and a DNA repair score
Protein expression or protein post-translational modifications were assessed by RPPA [39]. Our modified version of the DNA repair score reported by Cardnell et al [40], incorporating baseline expression levels of 11 of the 17 originally used repair proteins that predicted PARPi sensitivity in small cell lung cancer, did not correlate with PFS (Supplementary Table 1; Supplementary Figure 1) [40].
Separately, the relationship between clinical response and pretreatment expression of all 274 proteins or post-translationally modified proteins on the RPPA array at the time was examined. Seven of 10 evaluable patients in the expansion cohort for whom baseline biopsies were obtained achieved clinical benefit and 3 patients had progressive disease. A false discovery rate (FDR) of 10% was used to control for multiple comparisons. Pretreatment biopsies were analyzed for linear correlation to PFS for each patient, without findings of significance in this small cohort.
Tissue apoptotic index
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was done on the available paired biopsies to evaluate DNA damage after treatment. There was no significant difference in mean apoptotic index before (47.9% [24.3–62.2]) and after (50.9% [28.09–75.61]) cycle 1 treatment with combination therapy (Supplementary Figure 2).
DISCUSSION
This phase I/Ib study identified the schedule of olaparib, 400 mg capsules twice daily for days 1–7, administered with carboplatin AUC4 every 3 weeks as tolerable and providing modest activity in heavily pretreated BRCAwt HGSOC patients, including a majority who had platinum-resistant disease. A 400 mg twice daily olaparib capsule dose corresponds to a 250 to 300 mg twice daily olaparib tablet dose [41]. Consistent with our previously reported cohort of BRCAwt triple negative breast cancer patients, the intermittent olaparib schedule resulted in dose-limiting myelotoxicity, requiring a lower carboplatin dose (AUC4) than recommended for gBRCAm patients (AUC5) [36, 37]. This difference may be partly because more heavily pretreated patients were enrolled in the current cohort (median 7 prior regimens [2–12]) compared with the gBRCAm cohort (median 5 prior regimens [2–11]) [36].
The addition of PARPi to cytotoxic chemotherapy has previously been hindered by overlapping marrow toxicity, limiting the dose or treatment duration of PARPi and/or chemotherapy [42–45]. We had to reduce the standard carboplatin dose by over 50% in our phase I study of gBRCAm breast and ovarian cancer patients due to early hematologic toxicity with continuous daily olaparib, which led to successful use of the intermittent olaparib schedule [36]. We found manageable myelotoxicity with carboplatin and intermittently-dosed olaparib in the current cohort. Common (>10%) grade 3 and 4 toxicities were all hematological and managed with appropriate supplementation. Importantly, no grade 5 events were observed. Common non-hematologic adverse events, including gastrointestinal side effects were expected given the known and overlapping toxicities of each drug, and were predominantly grade 1 or 2 and either self-limited or addressed with standard care. These results align with a phase I trial of olaparib with cisplatin in advanced solid tumors that also required intermittent and lower dose olaparib (50–100 mg capsules days 1–5 or 1–10) with cisplatin every 21 days due to marrow toxicity [42]. Similarly, after a phase I trial of olaparib with carboplatin and paclitaxel reported that continuous olaparib intensified hematologic toxicities [45], a phase II trial used intermittent olaparib dosing (200 mg capsules days 1–10) with paclitaxel (175 mg/m2 every 3 weeks) and carboplatin (AUC4 every 3 weeks) in recurrent ovarian cancer patients for better tolerability and no clear loss of activity due to the reduced carboplatin dose [44]. Our results support the safety and tolerability of intermittent olaparib with carboplatin for combination therapy in BRCAwt recurrent HGSOC patients.
Platinum resistance is associated with a poor prognosis for women with ovarian cancer and almost all patients with recurrent disease ultimately develop resistance to platinum-based therapy [46]. Historically, the use of single agent cytotoxic chemotherapy or targeted therapy has resulted in RRs of around 5–15% and a median PFS of 3–4 months in the heavily pretreated, platinum-resistant patient population [47–52]. Our cohort of BRCAwt, heavily pretreated, largely platinum-resistant patients, thus represents a particularly difficult-to-treat population. In our study, 20% of patients achieved PR and two thirds had clinical benefit, although patients were heavily pretreated with a median of 7 prior therapies. We noted comparable clinical benefit in the patients with platinum-resistant (CBR 71%) and platinum-sensitive (CBR 55%) disease. It is possible that this clinical benefit is in part related to the required at least 6-month interval from last platinum exposure to initiation of protocol therapy.
It has been demonstrated preclinically that platinum-resistant clones present in the original tumor persist after platinum-sensitive clones are eradicated by chemotherapy [53, 54]. An in vitro study of the cisplatin-resistant A2780-CR human ovarian cancer cell line showed that following initial cisplatin exposure, cells became significantly more sensitive to cisplatin reintroduction after a 4-week drug-free interval, suggesting that platinum-resistant clones can be sensitized in a time-dependent manner [55]. Clinical studies also demonstrated that extending the interval from last platinum exposure in platinum-resistant or refractory ovarian cancer patients increases the likelihood of response to platinum retreatment [56, 57]. These data suggest re-challenging with platinum-based therapy may yield clinical activity in some platinum-resistant patients, thus the PARPi and carboplatin combination may be a therapeutic opportunity independent of platinum sensitivity. Future preclinical and clinical studies are warranted to explore this possibility.
Identifying and validating effective predictive biomarkers remains a challenge in the BRCAwt population. Among four evaluable paired biopsies, a greater decrease in PAR expression levels was observed in patients who achieved clinical benefit from treatment compared to those who did not, and this should be interpreted with caution due to small numbers. A measure of DNA repair deficiency has been widely investigated as a potential biomarker for the response to PARPi, and proteomic analysis has been employed to study functional DNA repair deficiencies [58, 59]. We applied a modified version of the “DNA repair protein score” developed by Cardnell et al. [40], consisting of expression levels of 17 DNA repair proteins shown to predict response to the PARPi BMN673 (talazoparib) in an in vitro small cell lung cancer model, and found no correlation to PFS. While the RPPA analysis used in the Cardnell study included 193 total and/or phosphorylated proteins, only 11 of the original 17 DNA repair proteins overlapped in the 274-antibody panel used here. Thus, we had to calculate a modified DNA repair score by taking the average expression levels of the overlapping 11 DNA repair proteins which may partly explain the lack of correlation. Also, genomic HR deficiency (HRD) was assessed in the ARIEL2 phase 2 trial of the PARPi rucaparib using next-generation sequencing combining BRCA mutation status with a measure of loss of heterozygosity (LOH) [20]. Both BRCAm and BRCAwt patients with high LOH had improved PFS and objective responses to PARPi treatment compared to BRCAwt/LOH-low patients (hazard ratio 0.27, p < 0.0001 and 0.62, p = 0.011 for BRCAm and LOH high subgroups, respectively, compared to LOH low) [20]. Similarly, the phase 3 NOVA study of niraparib maintenance therapy assessed HRD as a biomarker using the myChoice test from Myriad Genetics and found HRD-positive BRCAwt patients derived greater benefit from therapy versus placebo compared to the overall non-gBRCAm cohort (hazard ratio 0.38, p < 0.001 versus 0.45, p < 0.001, respectively) [22]. Further studies are warranted to identify predictive biomarkers of PARPi-combination treatment response in the BRCA-proficient population.
Our study has several limitations. First, our primary objective was to determine a safe dose and schedule in this patient population. Thus, we had insufficient power, even with an expansion cohort for the purpose of pharmacodynamic correlative exploration, to make conclusions of clinical benefit. We controlled for multiple comparisons to reduce the incidence of type 1 errors, however, due the exploratory nature of the translational endpoints all findings should be examined and validated prospectively before definitive conclusions can be made. We also did not prospectively plan to sequence genes involved in HRR and were unable to do so because of insufficient clinical samples remaining, and lack of proper consent for germline testing. Instead, HRR function was explored by assessing DNA repair protein expression, activation, and an experimental composite score.
There are now a number of PARPi combination studies in phase I to III clinical trials in ovarian cancer, though primarily focusing on the platinum-sensitive and BRCAm population. Our findings present an opportunity to further investigate a PARPi and carboplatin combination in heavily-pretreated BRCAwt patients. The observed modest activity of this combination, regardless of platinum sensitivity, supports the potential for platinum re-sensitization and highlights the need for continued biomarker analysis.
Materials and Methods
Patients
Accrual occurred between 2009 and 2014. Eligible patients had recurrent or refractory HGSOC with known negative gBRCAm testing or a negative family history and a BRCAPro score of ≤20% calculated on the basis of detailed family history [38], an ECOG score of 0–2 and normal organ and bone marrow function. There was no limit on number of prior therapies, although patients had to be at least six months from last platinum exposure. Patients previously treated with a PARPi were excluded from the study although patients with platinum-resistant ovarian cancer were eligible. All patients provided written informed consent. This study was approved by the Institutional Review Board of the Center for Cancer Research, National Cancer Institute. ClinicalTrials.gov identifier: NCT01445418.
Drug administration and determination of MTD
This open label 3+3 dose escalation study examined the combination of olaparib 400 mg capsules every 12 hours on days 1–7 with carboplatin AUC3, 4, or 5 on day 1 (DLs 1–3), in 21-day cycles (Figure 1). Carboplatin infusion was allowed on day 2 for those with schedule conflicts. No more than 8 cycles of combination therapy were given, followed by continuous daily maintenance olaparib monotherapy with 400 mg capsules every 12 hours. Safety and adverse events were graded every cycle (CTCAEv3.0). DLTs were defined as grade 4 hematologic and grade 3 or 4 nonhematologic adverse events related to study medications during the first two cycles of therapy. Grade 3 neutropenia for ≥7 days or with fever, and grade 3 thrombocytopenia lasting ≥7 days or requiring transfusion were also dose-limiting. Exceptions included grade 3 diarrhea, nausea, or vomiting, which had to be unresponsive to optimal medical care, and asymptomatic grade 3 reductions in electrolytes readily reversed with medical management. Complete blood counts and serum chemistries were monitored weekly during the DLT period. MTD was defined as the highest dose level at which ≤33% of patients experienced a DLT.
Granisetron (days 1–7) and dexamethasone (days 1–4) were given prophylactically for nausea during each combination therapy cycle and discontinued during olaparib maintenance. (Peg)filgrastim was not permitted during the first 2 cycles of the dose escalation phase but was indicated for use in subsequent cycles if the day 1 ANC was 1500–1800/mL or if the day 1 ANC was less than 1500/mL, necessitating treatment delay. Once initiated, (peg)filgrastim was continued during all combination treatment cycles, but was not used during olaparib maintenance therapy.
Clinical response was assessed by imaging using RECISTv1.0 criteria every 2 cycles. Study treatment was discontinued for progression of disease, intercurrent illness, adverse events not recovering to grade 1 within 3 weeks, and patient preference.
Correlative studies
PBMCs were collected at baseline and cycle 1 day 3 for PAR incorporation (Figure 1). PBMCs were separated within 4 hours of collection, then stored in aliquots at –80° C until use. PBMC DNA PAR incorporation was measured with a commercial PAR immunoassay (Trevigen, Gaithersburg, MD, USA) as previously described [60]. PBMC DNA was isolated for polymorphism analysis of PARP1 V762A, XRCC1 R194W and XRCC1 Q399R using a commercial DNA purification kit (Qiagen, Germantown, MD), as reported [36].
Paired tumor biopsies were performed in an expansion cohort at MTD pre-and post-cycle 1 for proteomics and apoptosis endpoints (Figure 1). Percutaneous biopsies were obtained by interventional radiologists under CT or ultrasound guidance with independent procedure consent. Samples were processed in real time in optimal cutting temperature compound and stored at −80° C, then cut and stained immediately prior to use as previously described [61]. Optimal tissue quality was defined as paired sequential biopsies with solid tissue areas containing at least 50% tumor cells and less than 25% necrosis [61]. Tissue area was measured as reported [62] and RPPA was performed in 2014 by the MD Anderson RPPA Core facility using their 274-antibody protocol including key proteins for DNA repair [39]. A modified DNA repair score, consisting of expression levels of 11 of 17 DNA repair proteins previously reported to predict PARPi sensitivity in small cell lung cancer in a study by Cardnell et al, was applied (Supplementary Table 1) [40]. Apoptotic cells were counted using the DeadEnd colorimetric TUNEL kit (Promega, Madison, WI, USA) [36]. The apoptotic index was defined as the percentage of TUNEL-positive single cells in five high-power fields.
Statistical analyses
An expansion group was added at the MTD for exploratory biomarker analyses. For any given endpoint comparison, 10 paired biopsies would provide 80% power to detect a difference between pre- and on-treatment values of one standard deviation of the difference (α2 = 0.05). Normalized values for each protein in the RPPA were analyzed for linear correlation to PFS using JMP 9.0 (SAS Institute, Cary, NC, USA). The Hochberg method was used with an alpha of 0.05 to control the FDR [63]. Baseline PAR levels were correlated with response and PFS using the Fisher exact test. PARP1 and XRCC1 polymorphisms were correlated with response and PFS using the Fisher’s exact test and log-rank test, respectively. Pre- and post-cycle 1 treatment TUNEL percent positivity was compared using a two-tailed paired t-test (Prism 6, La Jolla, CA, USA), and TUNEL results were correlated with response by Pearson correlation coefficient (JMP 9.0). A p-value of less than 0.05 was considered statically significant, and all tests were two-sided.
Abbreviations
MTD: maximum tolerated dose; BRCAwt: BRCA wild-type; HGSOC: high grade serous ovarian cancer; HR: homologous recombination; HRR: homologous recombination repair; gBRCAm: germline BRCA mutation; PARPi: poly (ADP-ribose) polymerase inhibitors; ORR: objective response rate; PFS: progression free survival; DL: dose level; DLT: dose-limiting toxicity; ANC: absolute neutrophil count; PR: partial response; SD: stable disease; PD: progressive disease; CBR: clinical benefit rate; PBMCs: peripheral blood mononuclear cells; TNBC: triple negative breast cancer; HRD: homologous recombination deficiency.
ACKNOWLEDGMENTS
Olaparib was supplied to the Center for Cancer Research, NCI under a Cooperative Research and Development Agreement between the CCR/NCI and AstraZeneca.
CONFLICTS OF INTEREST
All authors maintain no conflicts of interest to report.
FUNDING
This clinical trial and translational studies were funded by the Intramural Program of the Center for Cancer Research, NCI, NIH (JML, #ZIA BC011525).
References
1. Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, Gaudet MM, Jemal A, Siegel RL. Ovarian cancer statistics, 2018. CA Cancer J Clin. 2018; 68:284–96. https://doi.org/10.3322/caac.21456. [PubMed].
2. Martin LP, Schilder RJ. Management of recurrent ovarian carcinoma: current status and future directions. Semin Oncol. 2009; 36:112–25. https://doi.org/10.1053/j.seminoncol.2008.12.003. [PubMed].
3. Ledermann JA, Drew Y, Kristeleit RS. Homologous recombination deficiency and ovarian cancer. Eur J Cancer. 2016; 60:49–58. https://doi.org/10.1016/j.ejca.2016.03.005. [PubMed].
4. Bell D, Berchuck A, Birrer M, Chien J, Cramer DW, Dao F, Dhir R, DiSaia P, Gabra H, Glenn P, Godwin AK, Gross J, Hartmann L, et al. Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474:609–15. https://doi.org/10.1038/nature10166. [PubMed].
5. Pal T, Permuth-Wey J, Betts JA, Krischer JP, Fiorica J, Arango H, LaPolla J, Hoffman M, Martino MA, Wakeley K, Wilbanks G, Nicosia S, Cantor A, Sutphen R. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer. 2005; 104:2807–16. https://doi.org/10.1002/cncr.21536. [PubMed].
6. Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015; 5:1137–54. https://doi.org/10.1158/2159-8290.CD-15-0714. [PubMed].
7. Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, Nones K, Cowin P, Alsop K, Bailey PJ, Kassahn KS, Newell F, Quinn MC, et al, and Australian Ovarian Cancer Study Group. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015; 521:489–94. https://doi.org/10.1038/nature14410. [PubMed].
8. Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, Thornton A, Norquist BM, Casadei S, Nord AS, Agnew KJ, Pritchard CC, Scroggins S, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014; 20:764–75. https://doi.org/10.1158/1078-0432.CCR-13-2287. [PubMed].
9. Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017; 355:1152–58. https://doi.org/10.1126/science.aam7344. [PubMed].
10. Ossovskaya V, Koo IC, Kaldjian EP, Alvares C, Sherman BM. Upregulation of Poly (ADP-Ribose) Polymerase-1 (PARP1) in Triple-Negative Breast Cancer and Other Primary Human Tumor Types. Genes Cancer. 2010; 1:812–21. https://doi.org/10.1177/1947601910383418. [PubMed].
11. Barnett JC, Bean SM, Nakayama JM, Kondoh E, Murphy SK, Berchuck A. High poly(adenosine diphosphate-ribose) polymerase expression and poor survival in advanced-stage serous ovarian cancer. Obstet Gynecol. 2010; 115:49–54. https://doi.org/10.1097/AOG.0b013e3181c2d294. [PubMed].
12. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005; 434:917–21. https://doi.org/10.1038/nature03445. [PubMed].
13. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005; 434:913–17. https://doi.org/10.1038/nature03443. [PubMed].
14. McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O’Connor MJ, Tutt AN, Zdzienicka MZ, Smith GC, Ashworth A. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006; 66:8109–15. https://doi.org/10.1158/0008-5472.CAN-06-0140. [PubMed].
15. Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, Mitchell G, Fried G, Stemmer SM, Hubert A, Rosengarten O, Steiner M, Loman N, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015; 33:244–50. https://doi.org/10.1200/JCO.2014.56.2728. [PubMed].
16. Mirza MR, Pignata S, Ledermann JA. Latest clinical evidence and further development of PARP inhibitors in ovarian cancer. Ann Oncol. 2018; 29:1366–76. https://doi.org/10.1093/annonc/mdy174. [PubMed].
17. Pujade-Lauraine E, Ledermann JA, Selle F, Gebski V, Penson RT, Oza AM, Korach J, Huzarski T, Poveda A, Pignata S, Friedlander M, Colombo N, Harter P, et al, and SOLO2/ENGOT-Ov21 investigators. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017; 18:1274–84. https://doi.org/10.1016/S1470-2045(17)30469-2. [PubMed].
18. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott CL, Meier W, Shapira-Frommer R, Safra T, Matei D, Fielding A, Spencer S, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014; 15:852–61. https://doi.org/10.1016/S1470-2045(14)70228-1. [PubMed].
19. Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, Lisyanskaya A, Floquet A, Leary A, Sonke GS, Gourley C, Banerjee S, Oza A, et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med. 2018; 379:2495–505. https://doi.org/10.1056/NEJMoa1810858. [PubMed].
20. Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, Konecny GE, Coleman RL, Tinker AV, O’Malley DM, Kristeleit RS, Ma L, Bell-McGuinn KM, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017; 18:75–87. https://doi.org/10.1016/S1470-2045(16)30559-9. [PubMed].
21. Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, Colombo N, Weberpals JI, Clamp A, Scambia G, Leary A, Holloway RW, Gancedo MA, et al, and ARIEL3 investigators. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017; 390:1949–61. https://doi.org/10.1016/S0140-6736(17)32440-6. [PubMed].
22. Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, Fabbro M, Ledermann JA, Lorusso D, Vergote I, Ben-Baruch NE, Marth C, Mądry R, et al, and ENGOT-OV16/NOVA Investigators. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N Engl J Med. 2016; 375:2154–64. https://doi.org/10.1056/NEJMoa1611310. [PubMed].
23. Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M, Gilks B, Yerushalmi R, Macpherson E, Carmichael J, Oza A. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011; 12:852–61. https://doi.org/10.1016/S1470-2045(11)70214-5. [PubMed].
24. Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, De Greve J, Lubinski J, Shanley S, Messiou C, A’Hern R, Tutt A, Ashworth A, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010; 28:2512–19. https://doi.org/10.1200/JCO.2009.26.9589. [PubMed].
25. Jung Y, Lippard SJ. Direct cellular responses to platinum-induced DNA damage. Chem Rev. 2007; 107:1387–407. https://doi.org/10.1021/cr068207j. [PubMed].
26. Slyskova J, Sabatella M, Ribeiro-Silva C, Stok C, Theil AF, Vermeulen W, Lans H. Base and nucleotide excision repair facilitate resolution of platinum drugs-induced transcription blockage. Nucleic Acids Res. 2018; 46:9537–49. https://doi.org/10.1093/nar/gky764. [PubMed].
27. Martin SA, Lord CJ, Ashworth A. DNA repair deficiency as a therapeutic target in cancer. Curr Opin Genet Dev. 2008; 18:80–86. https://doi.org/10.1016/j.gde.2008.01.016. [PubMed].
28. Bürkle A, Chen G, Küpper JH, Grube K, Zeller WJ. Increased poly(ADP-ribosyl)ation in intact cells by cisplatin treatment. Carcinogenesis. 1993; 14:559–61. https://doi.org/10.1093/carcin/14.4.559. [PubMed].
29. Shino Y, Itoh Y, Kubota T, Yano T, Sendo T, Oishi R. Role of poly(ADP-ribose)polymerase in cisplatin-induced injury in LLC-PK1 cells. Free Radic Biol Med. 2003; 35:966–77. https://doi.org/10.1016/S0891-5849(03)00470-2. [PubMed].
30. Gambi N, Tramontano F, Quesada P. Poly(ADPR)polymerase inhibition and apoptosis induction in cDDP-treated human carcinoma cell lines. Biochem Pharmacol. 2008; 75:2356–63. https://doi.org/10.1016/j.bcp.2008.03.015. [PubMed].
31. Chen G, Zeller WJ. Reversal of acquired cisplatin resistance by nicotinamide in vitro and in vivo. Cancer Chemother Pharmacol. 1993; 33:157–62. https://doi.org/10.1007/BF00685335. [PubMed].
32. Nguewa PA, Fuertes MA, Cepeda V, Alonso C, Quevedo C, Soto M, Pérez JM. Poly(ADP-ribose) polymerase-1 inhibitor 3-aminobenzamide enhances apoptosis induction by platinum complexes in cisplatin-resistant tumor cells. Med Chem. 2006; 2:47–53. https://doi.org/10.2174/157340606775197697. [PubMed].
33. Shen YT, Evans JC, Zafarana G, Allen C, Piquette-Miller M. BRCA Status Does Not Predict Synergism of a Carboplatin and Olaparib Combination in High-Grade Serous Ovarian Cancer Cell Lines. Mol Pharm. 2018; 15:2742–53. https://doi.org/10.1021/acs.molpharmaceut.8b00246. [PubMed].
34. Miknyoczki SJ, Jones-Bolin S, Pritchard S, Hunter K, Zhao H, Wan W, Ator M, Bihovsky R, Hudkins R, Chatterjee S, Klein-Szanto A, Dionne C, Ruggeri B. Chemopotentiation of temozolomide, irinotecan, and cisplatin activity by CEP-6800, a poly(ADP-ribose) polymerase inhibitor. Mol Cancer Ther. 2003; 2:371–82. https://doi.org/10.1158/1535-7163.mct-07-0062. [PubMed].
35. Xu K, Chen Z, Cui Y, Qin C, He Y, Song X. Combined olaparib and oxaliplatin inhibits tumor proliferation and induces G2/M arrest and γ-H2AX foci formation in colorectal cancer. OncoTargets Ther. 2015; 8:3047–54. https://doi.org/10.2147/OTT.S89154. [PubMed].
36. Lee JM, Hays JL, Annunziata CM, Noonan AM, Minasian L, Zujewski JA, Yu M, Gordon N, Ji J, Sissung TM, Figg WD, Azad N, Wood BJ, et al. Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation-associated breast or ovarian cancer with biomarker analyses. J Natl Cancer Inst. 2014; 106:dju089. https://doi.org/10.1093/jnci/dju089. [PubMed].
37. Lee JM, Hays JL, Chiou VL, Annunziata CM, Swisher EM, Harrell MI, Yu M, Gordon N, Sissung TM, Ji J, Figg WD, Minasian L, Lipkowitz S, et al. Phase I/Ib study of olaparib and carboplatin in women with triple negative breast cancer. Oncotarget. 2017; 8:79175–87. https://doi.org/10.18632/oncotarget.16577. [PubMed].
38. Berry DA, Iversen ES
39. MD Anderson Cancer Center. Texas, USA. Retrieved November 16, 2018, from https://www.mdanderson.org/research/research-resources/core-facilities/functional-proteomics-rppa-core.html.
40. Cardnell RJ, Feng Y, Diao L, Fan YH, Masrorpour F, Wang J, Shen Y, Mills GB, Minna JD, Heymach JV, Byers LA. Proteomic markers of DNA repair and PI3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clin Cancer Res. 2013; 19:6322–28. https://doi.org/10.1158/1078-0432.CCR-13-1975. [PubMed].
41. Lee JM, Peer CJ, Yu M, Amable L, Gordon N, Annunziata CM, Houston N, Goey AK, Sissung TM, Parker B, Minasian L, Chiou VL, Murphy RF, et al. Sequence-Specific Pharmacokinetic and Pharmacodynamic Phase I/Ib Study of Olaparib Tablets and Carboplatin in Women’s Cancer. Clin Cancer Res. 2017; 23:1397–406. https://doi.org/10.1158/1078-0432.CCR-16-1546. [PubMed].
42. Balmaña J, Tung NM, Isakoff SJ, Graña B, Ryan PD, Saura C, Lowe ES, Frewer P, Winer E, Baselga J, Garber JE. Phase I trial of olaparib in combination with cisplatin for the treatment of patients with advanced breast, ovarian and other solid tumors. Ann Oncol. 2014; 25:1656–63. https://doi.org/10.1093/annonc/mdu187. [PubMed].
43. Dent RA, Lindeman GJ, Clemons M, Wildiers H, Chan A, McCarthy NJ, Singer CF, Lowe ES, Watkins CL, Carmichael J. Phase I trial of the oral PARP inhibitor olaparib in combination with paclitaxel for first- or second-line treatment of patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2013; 15:R88. https://doi.org/10.1186/bcr3484. [PubMed].
44. Oza AM, Cibula D, Benzaquen AO, Poole C, Mathijssen RH, Sonke GS, Colombo N, Špaček J, Vuylsteke P, Hirte H, Mahner S, Plante M, Schmalfeldt B, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol. 2015; 16:87–97. https://doi.org/10.1016/S1470-2045(14)71135-0. [PubMed].
45. van der Noll RN, Ang JE, Jager A, Marchetti S, Mergui-Roelvink M, De Bono JS, Lolkema M, Brunetto A, Arkenau HT, De Jonge MJ, van der Biessen D, Tchakov I, Bowen K, et al. Phase I study of olaparib in combination with carboplatin and/or paclitaxel in patients with advanced solid tumors. J Clin Oncol. 2013; 31:2579.
46. Herzog TJ, Pothuri B. Ovarian cancer: a focus on management of recurrent disease. Nat Clin Pract Oncol. 2006; 3:604–11. https://doi.org/10.1038/ncponc0637. [PubMed].
47. Vergote I, Finkler NJ, Hall JB, Melnyk O, Edwards RP, Jones M, Keck JG, Meng L, Brown GL, Rankin EM, Burke JJ, Boccia RV, Runowicz CD, Rose PG. Randomized phase III study of canfosfamide in combination with pegylated liposomal doxorubicin compared with pegylated liposomal doxorubicin alone in platinum-resistant ovarian cancer. Int J Gynecol Cancer. 2010; 20:772–80. https://doi.org/10.1111/IGC.0b013e3181daaf59. [PubMed].
48. Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore ME, Lacave AJ. Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol. 2001; 19:3312–22. https://doi.org/10.1200/JCO.2001.19.14.3312. [PubMed].
49. Markman M, Blessing J, Rubin SC, Connor J, Hanjani P, Waggoner S, and Gynecologic Oncology Group. Phase II trial of weekly paclitaxel (80 mg/m2) in platinum and paclitaxel-resistant ovarian and primary peritoneal cancers: a Gynecologic Oncology Group study. Gynecol Oncol. 2006; 101:436–40. https://doi.org/10.1016/j.ygyno.2005.10.036. [PubMed].
50. Cannistra SA, Matulonis UA, Penson RT, Hambleton J, Dupont J, Mackey H, Douglas J, Burger RA, Armstrong D, Wenham R, McGuire W. Phase II study of bevacizumab in patients with platinum-resistant ovarian cancer or peritoneal serous cancer. J Clin Oncol. 2007; 25:5180–86. https://doi.org/10.1200/JCO.2007.12.0782. [PubMed].
51. Mutch DG, Orlando M, Goss T, Teneriello MG, Gordon AN, McMeekin SD, Wang Y, Scribner DR
52. ten Bokkel Huinink W, Gore M, Carmichael J, Gordon A, Malfetano J, Hudson I, Broom C, Scarabelli C, Davidson N, Spanczynski M, Bolis G, Malmström H, Coleman R, et al. Topotecan versus paclitaxel for the treatment of recurrent epithelial ovarian cancer. J Clin Oncol. 1997; 15:2183–93. https://doi.org/10.1200/JCO.1997.15.6.2183. [PubMed].
53. Cooke SL, Ng CK, Melnyk N, Garcia MJ, Hardcastle T, Temple J, Langdon S, Huntsman D, Brenton JD. Genomic analysis of genetic heterogeneity and evolution in high-grade serous ovarian carcinoma. Oncogene. 2010; 29:4905–13. https://doi.org/10.1038/onc.2010.245. [PubMed].
54. Cooke SL, Brenton JD. Evolution of platinum resistance in high-grade serous ovarian cancer. Lancet Oncol. 2011; 12:1169–74. https://doi.org/10.1016/S1470-2045(11)70123-1. [PubMed].
55. Horowitz NS, Hua J, Gibb RK, Mutch DG, Herzog TJ. The role of topotecan for extending the platinum-free interval in recurrent ovarian cancer: an in vitro model. Gynecol Oncol. 2004; 94:67–73. https://doi.org/10.1016/j.ygyno.2004.03.047. [PubMed].
56. Kavanagh J, Tresukosol D, Edwards C, Freedman R, Gonzalez de Leon C, Fishman A, Mante R, Hord M, Kudelka A. Carboplatin reinduction after taxane in patients with platinum-refractory epithelial ovarian cancer. J Clin Oncol. 1995; 13:1584–88. https://doi.org/10.1200/JCO.1995.13.7.1584. [PubMed].
57. Rose PG, Mossbruger K, Fusco N, Smrekar M, Eaton S, Rodriguez M. Gemcitabine reverses cisplatin resistance: demonstration of activity in platinum- and multidrug-resistant ovarian and peritoneal carcinoma. Gynecol Oncol. 2003; 88:17–21. https://doi.org/10.1006/gyno.2002.6850. [PubMed].
58. Nishizuka SS, Mills GB. New era of integrated cancer biomarker discovery using reverse-phase protein arrays. Drug Metab Pharmacokinet. 2016; 31:35–45. https://doi.org/10.1016/j.dmpk.2015.11.009. [PubMed].
59. Kamel D, Brady B, Tabchy A, Mills GB, Hennessy B. Proteomic classification of breast cancer. Curr Drug Targets. 2012; 13:1495–509. https://doi.org/10.2174/138945012803530080. [PubMed].
60. Kinders RJ, Hollingshead M, Khin S, Rubinstein L, Tomaszewski JE, Doroshow JH, Parchment RE, and National Cancer Institute Phase 0 Clinical Trials Team. Preclinical modeling of a phase 0 clinical trial: qualification of a pharmacodynamic assay of poly (ADP-ribose) polymerase in tumor biopsies of mouse xenografts. Clin Cancer Res. 2008; 14:6877–85. https://doi.org/10.1158/1078-0432.CCR-08-0214. [PubMed].
61. Lee JM, Hays JL, Noonan AM, Squires J, Minasian L, Annunziata C, Wood BJ, Yu M, Calvo KR, Houston N, Azad N, Kohn EC. Feasibility and safety of sequential research-related tumor core biopsies in clinical trials. Cancer. 2013; 119:1357–64. https://doi.org/10.1002/cncr.27916. [PubMed].
62. Azad N, Yu M, Davidson B, Choyke P, Chen CC, Wood BJ, Venkatesan A, Henning R, Calvo K, Minasian L, Edelman DC, Meltzer P, Steinberg SM, et al. Translational predictive biomarker analysis of the phase 1b sorafenib and bevacizumab study expansion cohort. Mol Cell Proteomics. 2013; 12:1621–31. https://doi.org/10.1074/mcp.M112.026427. [PubMed].
63. Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J R Stat Soc B. 1995; 57:289–300. https://doi.org/10.1111/j.2517-6161.1995.tb02031.x.