
Introduction
Metformin (1,1-dimethylbiguanide hydrochloride) is a biguanide used worldwide to treat type II diabetes and pre-diabetic syndromes [1]. The physiological mechanism of its action in diabetes is to enhance glucose utilization, increase insulin sensitivity and reduce hepatic glucose production and free fatty acid utilization [2]. Its primary effect is based on interfering with respiratory complex I, reducing ATP production [3], and leading to the activation of adenosine monophosphate-activated kinase (AMPK) [4, 5]. Diabetic patients treated with metformin, but not other anti-diabetic drugs, have reduced incidence and better survival from cancer of many organs, including colorectal, liver, pancreatic, rectal, breast, prostate cancer and upper-tract urothelial carcinoma [6-12]. However, discordance exists in prostate cancer as some retrospective studies have reported that metformin use was not associated with risk reduction in systemic progression and all-cause mortality in patients treated with radical prostatectomy [13-16].
AMPK, a serine/threonine kinase, functions as an energy sensor and metabolic master switch, and is activated under conditions of increasing cellular AMP:ATP ratios, such as hypoxia, heat shock and ischemia [17]. AMPK is a heterotrimeric protein composed of a catalytic α subunit (63 kDa) and regulatory β (40 kDa) and γ (38 kDa) subunits. Each is expressed as functionally redundant isoforms including α1, α2, β1, β2, γ1, γ2 and γ3, giving 12 different possible combinations of holoenzyme. Upon activation, the α subunit is phosphorylated at the Thr172 residue (reviewed in [18]). Activated AMPK elevates cellular energy levels by stimulating energy producing catabolic pathways and inhibiting anabolic energy consuming pathways. AMPK can be activated by up-stream proteins including liver kinaseB1 (LKB1) [19] and ataxia teleangiectasia mutated (ATM) kinase [20]. The activation of AMPK inactivates mammalian target of rapamycin (mTOR), a stimulator of cancer cell growth and proliferation frequently hyper-activated by genetic alterations in cancer [21]. The anti-tumour effects of metformin have been shown to be dependent on such AMPK activation [22-25], however antineoplastic effects may also be independent of AMPK activation with, for example, altered NF-kB signalling being implicated [26].
Metformin and AMPK have recently been shown to be involved in regulating the radioresponse of cancer cells. Metformin radiosensitised FSaII mouse fibrosarcoma cells and human breast cancer MCF7 [27], lung cancer cells A549 and H1299 [28, 29] and preferentially killed cancer stem cells, by activating AMPK and suppressing mTOR [27]. AMPK inhibition induced radioresistance of lung cancer cells A549 and H1299 in normal culture conditions [28], with glucose starvation reversing this [30]. AMPK is phosphorylated by irradiation in an ATM dependent manner [30], but independent of LKB1 [28]. Metformin is reported to be involved in redox regulation; reducing intracellular reactive oxygen species (ROS) levels in primary human aortic endothelial cells by upregulating expression of thioredoxin (Trx) via the AMPK-FOXO3 pathway [31]. It has also been shown to inhibit thioredoxin-interacting protein (Txnip) mRNA as well as protein expression in HeLa cells [32].
The Trx system is a central enzyme family that regulates intracellular redox homeostasis and plays an important role in regulating the effects of irradiation on cancer cells [33]. Trx is a central part of the Trx system that also includes thioredoxin reductase (TrxR) and Txnip [34]. Trx is reduced, into its biologically active form, by TrxR in a NADPH-dependent manner and in turn reduces oxidized cysteine groups on down-stream proteins [35]. Txnip is the negative regulator of Trx, which directly interacts with the catalytic active centre to block the reducing activity of Trx as well as the interaction between Trx and its down-stream factors [36].
The aims of this study were to determine the expression, and clinical importance, of total- and phospho(Thr172)- AMPKα in early-stage invasive breast cancer from patients treated with radiotherapy and to investigate the effect of metformin on the radiosensitivity of different phenotypes of breast cancer cells, assessing if changes in redox homeostasis, due to alterations in Trx system proteins, played a role in any altered radiosensitivity.
Results
AMPKα and pAMPKα(Thr172) staining location and frequency – in the discovery cohort
Both pAMPKα(Thr172) and AMPKα demonstrated a mixture of diffuse and granular cytoplasmic staining. Heterogeneous staining was shown between, as well as within, certain tumour cores for both markers, varying from weak to intense staining. Cytoplasmic staining of both markers was scored: pAMPKα(Thr172) had a median H-score of 98, ranging between 0 and 200; and AMPKα had a median H-score of 93, ranging between 0 and 228. Figure 1A and B illustrates the staining pattern for both markers. There was a marginal positive correlation between both markers (r=0.305, P=0.000084). X-tile bioinformatics software was used to obtain an unbiased optimal H-score cut-point for each protein based on patient outcome, and dichotomised H-scores into low and high scores. The cut-point for pAMPKα(Thr172) was 65 with 30% (48 of 162) of cases having a low score; and the cut-point for AMPKα was 90 with 49% (80 of 163) of cases having a low score. A small number of tissue microarray (TMA) cores were not assessed due to missing cores or insufficient representative tumour within a core.
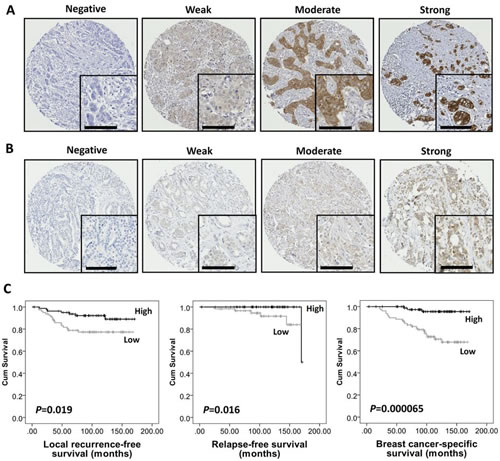
Figure 1: High AMPKα expression was associated with good prognosis in breast cancer in the discovery cohort. Representative photomicrographs of negative, weak, moderate and strong staining of (A) AMPKα and (B) pAMPKα(Thr172). Photomicrographs are at ×10 magnification with ×20 magnification inset box where scale bar shows 100 μm. (C) Kaplan-Meier analysis of local recurrence-free, relapse-free and breast cancer-specific survival showing the impact of AMPKα expression in the discovery cohort of 166 patients with significance determined using the log-rank test. High AMPKα expression is associated with lower local recurrence risk (P=0.019), better relapse-free survival (P=0.016) and breast cancer-specific survival (P=0.000065).
Relationship of AMPKα and pAMPKα(Thr172) expression with clinicopathological variables and clinical outcome – in the discovery cohort
The expression of AMPKα and pAMPKα(Thr172) was assessed for association with clinicopathological variables in the discovery cohort (Table 1). High AMPKα expression was significantly associated with smaller tumour size (χ2=3.97, d.f.=1, P=0.046), low grade (χ2=28.338, d.f.=2, P=0.000001), low Nottingham Prognostic Index (NPI) score (χ2=18.84, d.f.=2, P=0.000081) and estrogen receptor (ER) positive status (χ2=38.69, d.f.=1, P<0.000001). The expression of pAMPKα(Thr172) was not significantly associated with any of the clinicopathological variables.
Table 1: Association between AMPKα protein expression and clinicopathological variables.
Variable |
Discovery cohort AMPKα (n=163) |
Validation cohort AMPKα (n=479) |
||||
|
Low |
High |
P-value |
Low |
High |
P-value |
Age (years) |
|
|
|
|
|
|
≤40 |
3 |
4 |
1 |
22 |
22 |
0.017 |
>40 |
77 |
79 |
|
140 |
295 |
|
Size (cm) |
|
|
|
|
|
|
≤2 |
63 |
73 |
0.046 |
121 |
257 |
0.105 |
>2 |
17 |
8 |
|
41 |
60 |
|
Stage |
|
|
|
|
|
|
I |
65 |
63 |
0.763 |
117 |
235 |
0.904 |
II |
13 |
17 |
|
38 |
69 |
|
III |
2 |
2 |
|
7 |
13 |
|
Grade |
|
|
|
|
|
|
I |
6 |
25 |
0.000001 |
9 |
59 |
<0.000001 |
II |
24 |
37 |
|
42 |
139 |
|
III |
50 |
19 |
|
111 |
119 |
|
Node status |
|
|
|
|
|
|
Negative |
64 |
62 |
0.595 |
112 |
220 |
0.982 |
Positive |
16 |
19 |
|
39 |
77 |
|
NPI |
|
|
|
|
|
|
Good (<3.4) |
22 |
47 |
0.000081 |
34 |
143 |
0.000001 |
Intermediate (3.4-5.4) |
55 |
28 |
|
109 |
153 |
|
Poor (>5.4) |
3 |
6 |
|
19 |
21 |
|
Vascular invasion |
|
|
|
|
|
|
Negative |
60 |
65 |
0.518 |
113 |
200 |
0.109 |
Positive |
20 |
17 |
|
46 |
114 |
|
ER |
|
|
|
|
|
|
Negative |
33 |
1 |
<0.000001 |
70 |
45 |
<0.000001 |
Positive |
47 |
80 |
|
90 |
261 |
|
PgR |
|
|
|
|
|
|
Negative |
ND |
|
|
90 |
87 |
<0.000001 |
Positive |
|
|
|
62 |
207 |
|
HER2 |
|
|
|
|
|
|
Negative |
ND |
|
|
135 |
277 |
0.481 |
Positive |
|
|
|
21 |
35 |
|
Basal phenotype |
|
|
|
|
|
|
Non-basal |
ND |
|
|
101 |
254 |
0.000003 |
Basal |
|
|
|
52 |
45 |
|
Classification |
|
|
|
|
|
|
Luminal |
ND |
|
|
91 |
265 |
<0.000001 |
Triple negative |
|
|
|
55 |
28 |
|
HER2+ |
|
|
|
12 |
11 |
|
The P-values are resultant from the Pearson χ2 test of association. Significant P-values are indicated in bold. The discovery and validation cohorts were comprised of 166 and 609 patients respectively; however, scores were not available for every patient (163 of 166 in the discovery cohort and 479 of 609 in the validation cohort). The number of observations for each cohort is shown for each clinicopathological variable; the table does not include the number of observations where clinicopathological data were not available. Abbreviations: ND, not determined.
Kaplan-Meier analysis showed that high AMPKα expression was associated with better outcome in terms of lower local recurrence risk (P=0.019), longer relapse-free survival (P=0.016) and breast cancer-specific survival (P=0.000065) (Figure 1C), while pAMPKα(Thr172) was not associated with breast cancer outcome in this cohort. Multivariate Cox regression analysis including tumour size, stage (i.e. TNM stage), grade, node status, NPI, vascular invasion and ER status (with individual Kaplan-Meier statistics of P=0.048, P=0.000079, P=0.000025, P=0.019, P=0.000482, P=0.003 and P=0.007, respectively), showed that AMPKα expression was independently associated with breast cancer-specific survival (Hazard Ratio (HR) = 0.16; 95% confidence interval (CI) = 0.04-0.63; P=0.009); and such analysis, including tumour stage (with individual Kaplan-Meier statistics of P=0.001), showed that AMPKα expression was also independently associated with relapse-free survival (HR = 0.36; 95% CI = 0.15-0.87; P=0.023) (Table 2). AMPKα expression was not, however, independently associated with local recurrence in multivariate analysis that included ER status and tumour size (HR = 0.52; 95% CI = 0.19-1.43; P=0.206).
Table 2: Multivariate Cox Regression analysis of factors associated with breast cancer-specific survival and relapse-free survival for the discovery cohort.
|
Breast cancer-specific survival |
Relapse-free survival |
||||
Variable |
HR |
95% CI |
P-value |
HR |
95% CI |
P-value |
AMPKα expression |
0.16 |
0.04 to 0.63 |
0.009 |
0.36 |
0.15 to 0.87 |
0.023 |
Stage |
2.24 |
0.54 to 9.37 |
0.269 |
1.17 |
0.51 to 2.69 |
0.708 |
Grade |
2.61 |
0.68 to 10.08 |
0.164 |
N/A |
|
|
Size (≤ / > 2 cm) |
1.48 |
0.49 to 4.45 |
0.488 |
N/A |
|
|
Node status (-/+) |
0.84 |
0.09 to 7.72 |
0.880 |
N/A |
|
|
NPI |
1.21 |
0.24 to 6.17 |
0.821 |
N/A |
|
|
Vascular invasion |
2.19 |
0.91 to 5.29 |
0.081 |
N/A |
|
|
ER (-/+) |
0.93 |
0.37 to 2.37 |
0.882 |
N/A |
|
|
Abbreviation: HR, Hazard Ratio; CI, confidence interval; N/A, not applicable. Significant P-values are indicated by bold.
Relationship of AMPKα expression with clinicopathological variables and clinical outcome – in the validation cohort
To verify the finding that AMPKα expression was associated with prognosis in early-stage invasive breast cancer patients treated by breast-conserving surgery plus radiotherapy, an independent patient validation cohort was investigated. The staining pattern of AMPKα was similar to that of the discovery cohort. The median H-score was 145 and ranged between 0 and 300. The X-tile determined cut-point was 115 with 34% (162 of 479) of cases having a low score. Similar to the discovery cohort, high AMPKα expression was significantly associated with features of good prognosis including low grade (χ2=43.42, d.f.=2, P<0.000001), low NPI score (χ2=27.32, d.f.=2, P=0.000001) and ER positive status (χ2=47.68, d.f.=1, P<0.000001). High AMPKα expression was also associated with some of the additional biomarkers available for this cohort, including progesterone receptor (PgR) positive status (χ2=36.72, d.f.=1, P<0.000001), and basal-phenotype negative status (χ2=21.53, d.f.=1, P=0.000003). High AMPKα expression was significantly associated with older patients (χ2=5.67, d.f.=1, P=0.017), but not with tumour size (Table 1).
Kaplan-Meier survival analysis showed that high AMPKα expression was associated with longer breast cancer-specific survival (P=0.005, Figure 3A), but not with local recurrence-free (P=0.328) or relapse-free survival (P=0.057, Figure 2A). In multivariate Cox regression analysis, including tumour size, stage, grade, node status, NPI, vascular invasion, ER, PgR, human epidermal growth factor receptor 2 (HER2) and basal-phenotype status (with individual Kaplan-Meier statistics of P=0.041, P<0.000001, P=0.00002, P=0.000005, P<0.000001, P=0.002, P=0.004, P=0.000428, P=0.002 and P=0.000073, respectively), AMPKα expression was not independently associated with breast cancer-specific survival in all breast cancer phenotypes (HR = 0.66; 95% CI = 0.41-1.07; P=0.089) but significant results were obtained with the luminal-phenotype group separately and are described below.
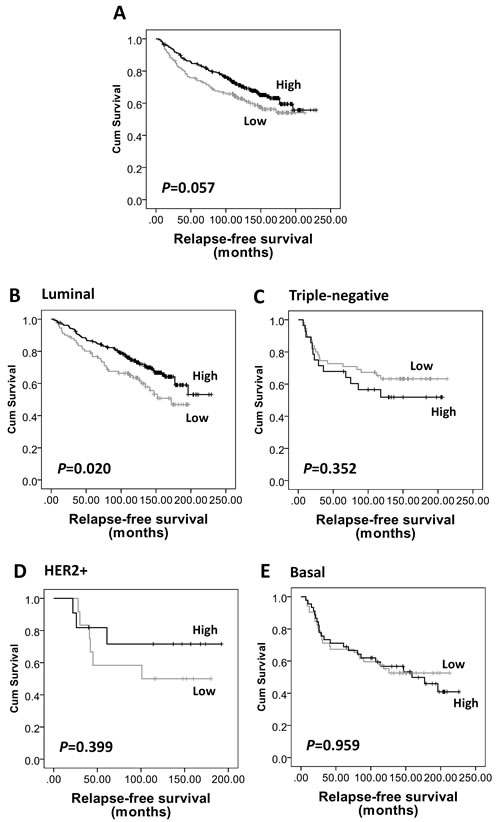
Figure 2: Kaplan-Meier analysis of relapse-free survival showing the impact of AMPKα expression in the validation cohort of 609 patients with significance determined using the log-rank test. (A) AMPKα expression is not associated with relapse-free survival in the whole cohort (P=0.057). (B) High AMPKα expression is associated with a low risk of disease relapse in luminal breast cancer subgroup (n=453, P=0.020), but not in (C) triple-negative (n=108, P=0.352), (D) HER2+ (n=26, P=0.399), and (E) basal-like (n=117, P=0.959) breast cancer subgroups.
Relationship of AMPKα expression with clinical outcome of luminal-phenotype breast cancer – in the validation cohort
As AMPKα expression was significantly associated with ER, PgR and basal-phenotype status, we further investigated the prognostic significance of AMPKα expression in different subtypes of breast cancer in the validation cohort. As shown in Supplementary Table S1, of the 609 patients, 77% (n=453) tumours were of luminal-phenotype, 18% were triple-negative (n=108), 4% (n=26) were HER2+ and 21% (n=117) were basal-like. High AMPKα expression was associated with longer relapse-free survival (P=0.02, Figure 2B) and breast cancer-specific survival (P=0.004, Figure 3B) in luminal-phenotype tumours. No similar associations were identified in the HER2+, basal-like or triple-negative classes. Multivariate Cox regression analysis demonstrated that AMPKα expression was independently associated with relapse-free survival (HR = 0.57; 95% CI = 0.38-0.86; P=0.008; potential confounding factors: stage, node status, NPI, vascular invasion and HER2 with individual Kaplan-Meier statistics of P=0.001, P=0.015, P=0.000282, P=0.004 and P=0.001, respectively) and breast cancer-specific survival (HR = 0.48; 95% CI = 0.28-0.82; P=0.007; potential confounding factors: stage, grade, node status, NPI, vascular invasion, PgR and HER2 with individual Kaplan-Meier statistics of P<0.000001, P=0.000331, P=0.000001, P<0.000001, P=0.002, P=0.01 and P=0.000005, respectively) in luminal-phenotype breast cancer cases when potential confounding factors were included (Table 3).
Table 3: Multivariate Cox Regression analysis of factors associated with breast cancer-specific survival and relapse-free survival for luminal phenotype disease in the validation cohort.
|
Breast cancer-specific survival |
Relapse-free survival |
||||
Variable |
HR |
95% CI |
P-value |
HR |
95% CI |
P-value |
AMPKα expression |
0.48 |
0.28 to 0.82 |
0.007 |
0.57 |
0.38 to 0.86 |
0.008 |
Stage |
3.60 |
1.75 to 7.42 |
0.001 |
2.62 |
1.38 to 4.97 |
0.003 |
Grade |
1.71 |
0.88 to 3.33 |
0.114 |
N/A |
|
|
Node status (-/+) |
1.07 |
0.48 to 2.42 |
0.863 |
0.66 |
0.29 to 1.48 |
0.310 |
NPI |
0.82 |
0.36 to 1.86 |
0.633 |
0.85 |
0.57 to 1.28 |
0.435 |
Vascular invasion |
1.53 |
0.90 to 2.63 |
0.119 |
1.68 |
1.12 to 2.51 |
0.011 |
PgR (-/+) |
0.51 |
0.29 to 0.90 |
0.020 |
N/A |
|
|
HER2 (-/+) |
2.59 |
1.28 to 5.25 |
0.008 |
2.12 |
1.22 to 3.70 |
0.008 |
Abbreviation: HR, Hazard Ratio; CI, confidence interval; N/A, not applicable. Significant P-values are indicated by bold.
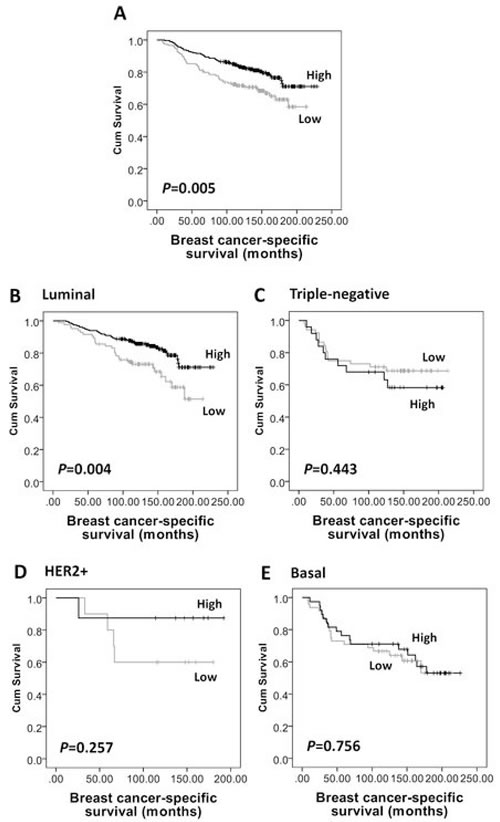
Figure 3: Kaplan-Meier analysis of breast cancer-specific survival showing the impact of AMPKα expression in the validation cohort of 609 patients with significance determined using the log-rank test. (A) High AMPKα expression is associated with a low risk of breast specific death in the whole cohort (P=0.005) and in (B) luminal breast cancer subgroup (n=453, P=0.004), but not in (C) triple-negative (n=108, P=0.443), (D) HER2+ (n=26, P=0.257), or (E) basal-like (n=117, P=0.756) breast cancer subgroups.
Metformin sensitises luminal breast cancer cells to irradiation but not basal phenotype
As radiotherapy is a crucial component of breast-conserving therapy, and to take such IHC based expression studies forward, we assessed whether metformin, a known modulator of AMPK activity could affect the radiosensitivity of different breast cancer phenotypes. Both MCF7 and MDA-MB-231 cells exhibited similar radiosensitivities under control conditions (Figure 4A). The IC50 for metformin in both cell lines, determined from cell growth curve experiments (data not shown), was 10 mM. This drug concentration was used for radiosensitivity studies and was initially assessed for drug alone cytotoxicity using clonogenic survival. As shown in Figure 4B, metformin exhibited significant cytotoxicity to both cell lines; with an increased level of cytotoxicity observed with the basal-like breast cancer MDA-MB-231 cells (surviving fraction (SF)=21.75% of control, P<0.01) compared to the luminal MCF7 cells (SF=59.14% of control, P<0.01). Interestingly, the radiosensitisation effect of metformin did not parallel its cytotoxic effect. Metformin caused little change in the radiosensitivity of MDA-MB-231 cells but a substantial increase in radiosensitivity of MCF7 cells (sensitiser enhancement ratio (SER) =1.5 at 1% SF) (Figure 4C). The SF at 6 Gray (Gy) for MCF7 cells treated with metformin in combination was 0.03% but 0.55% for radiation alone (P<0.01, Figure 4C).
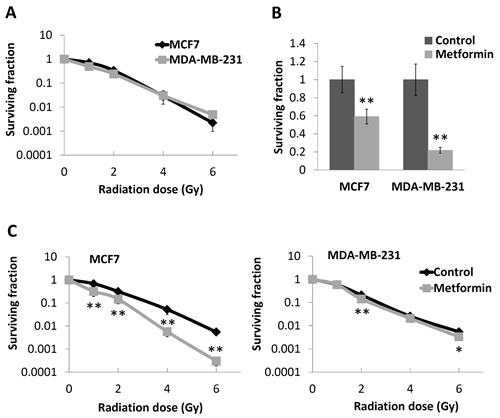
Figure 4: Effects of metformin on survival and radioresponse of breast cancer cell lines. (A) Luminal MCF7 and basal-like MDA-MB-231 breast cancer cells were subjected to single dose irradiation and compared with sham irradiated cells (0 Gy) as control. Clonogenic assay was performed to assess the surviving fraction of cells. (B) Both cell lines were treated with 10 mM of metformin for 48 hours (cells without metformin treatment as control), and clonogenic assays were performed to assess the surviving cells. Values were plotted as percentage of control. (C) MCF7 and MDA-MB-231 cells after metformin treatment were subjected to single dose irradiation, and clonogenic assay was used to assess the surviving cells. PEs for MDA-MB-231 and MCF7 cells in clonogenic assay were 52.83% (± 7.43%) and 30.33% (± 6.30%), respectively. Data represent the mean ± SD of three independent experiments, with each experiment containing six parallel data sets. *P<0.05, **P<0.01 vs. control.
Metformin elevated intracellular ROS production in luminal breast cancer cells but not basal phenotype
To explore the reason for the differential radiosensitising effects of metformin on breast cancer cells, intracellular ROS levels were assessed by flow cytometry. As shown in Figure 5A H2O2 induced ROS to a similar level in both lines but after metformin treatment, intracellular ROS levels were elevated to 4- fold of control in MCF7 cells (P<0.05), with no significant alterations in MDA-MB-231 cells (P>0.05).
As radiosensitivity can be influenced by the mode of cell death and by perturbations in cell cycle distribution, flow cytometry assessments of apoptosis and the cell cycle were conducted. As shown in Figure 5B and 5C, metformin had no effect on either cell cycle or apoptosis of MCF7 cells. In MDA-MB-231 cells, metformin induced a slight increase in the percentage of necrotic cells (1.94 -fold of control, P<0.05, Figure 5C), however it did not affect the apoptotic or cell cycle response of this line.
Metformin differentially regulates expression of Trx family proteins in breast cancer cells
Western blot analysis of Trx, TrxR and Txnip was conducted to assess whether any altered ROS homeostasis by metformin might involve the Trx system. The endogenous expression levels of Trx and Txnip were lower in MCF7 than in MDA-MB-231, whilst TrxR expression was similar. Metformin decreased Trx expression levels in MCF7 but had no effect on MDA-MB-231 cells. In both cell lines, expression of Txnip was dramatically attenuated by metformin treatment. TrxR expression was not affected by metformin treatment in either cell line (Figure 5D).
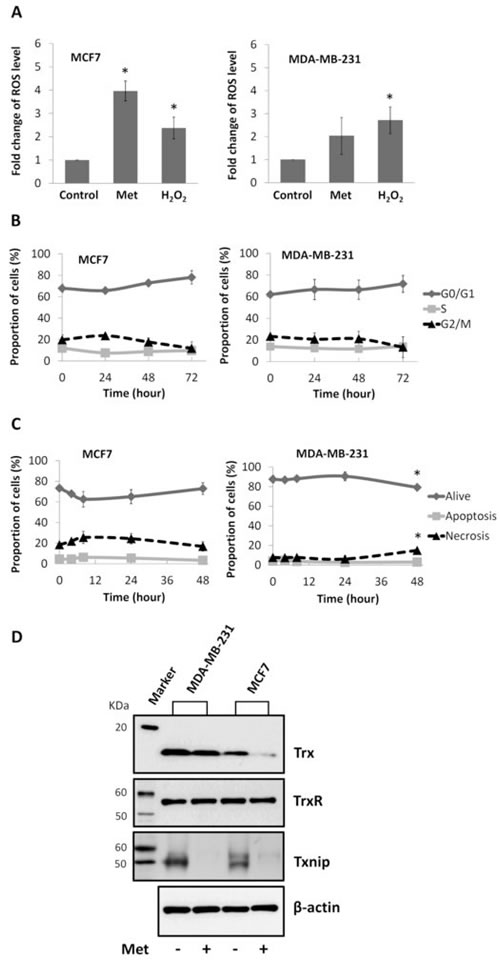
Figure 5: Effects of metformin on ROS level, cell cycle, apoptosis, and expression of Trx system proteins in breast cancer cell lines. (A) Flow cytometry analysis of intracellular ROS level of MCF7 and MDA-MB-231 breast cancer cells treated with 10 mM metformin (Met) for 48 hours, positive control (H2O2) and negative control cells. MFIs were calculated and plotted as fold change of control. Cells treated with 10 mM metformin for different period of time as indicated (72 hours means that cells were treated with metformin for 48 hours and spared for extra 24 hours) were subject to flow cytometry analysis of (B) cell cycle and (C) apoptosis. Percentages of cells were calculated and presented as mean ± SD of three independent experiments. *P<0.05 vs. control. (D) Cells were treated with 10 mM metformin for 48 hours (cells without treatment as control). Western blot was performed to assess the expression of Trx (MW=12KDa), TrxR (MW=55KDa) and Txnip (MW=50KDa) in cells, with β-actin (MW=42KDa) as internal control. Experiments were repeated three times and the representative blots are presented.
Discussion
Retrospective studies have found that use of the antidiabetic drug metformin in diabetic patients’ results in a reduced incidence of, and better survival from, breast cancer [12, 37-39]. However, in a randomized pre-surgical trial including 200 non-diabetic breast cancer patients, no alteration to tumour proliferation, as assessed by Ki67, was observed [40]. Metformin has been reported to be involved in regulating the radioresponse of a number of different cancer cell types, including breast cancer, in an AMPK-dependent manner [27-29, 41]. In order to assess the possible value of metformin as a sensitiser for radiotherapy of breast cancer and AMPK as a potential prognostic factor, the present study first sought to evaluate the expression of AMPK, the target of metformin, in radiotherapy treated early-stage invasive breast cancer patients. Similar to others [42], the current study did not observe any association between pAMPKα(Thr172) expression and prognosis. However, when total-AMPKα expression was assessed, in two independent patient cohorts, expression was associated with patient prognosis. The reason why total-AMPKα but not pAMPKα(Thr172) associated with prognosis is uncertain, but may be due to the differential phosphorylation patterns that can occur with AMPK: in addition to Thr172 AMPKα is also phosphorylated at Thr258 and Ser485 for AMPKα1 and Ser491 for AMPKα2 [43], expression of pAMPKα at other phosphorylation sites, rather than Thr172, may be important for breast cancer prognosis, and should perhaps be addressed in future work.
Although pAMPKα(Thr172) expression has been previously studied using immunohistochemical approaches, total-AMPKα has rarely been assessed. Small studies in thyroid and breast cancer measured levels between normal and cancerous tissue [44, 45] but the current study is the first of its type to determine associations with clinical outcomes. In both the discovery and validation cohorts, high AMPKα expression was significantly associated with low grade, low NPI score and ER positive status, all of which are indicative of better prognosis. High AMPKα expression was associated with patient age in the validation cohort but not in the discovery cohort. The age distribution was, however, different in these two cohorts: the age of patients in the discovery cohort ranged from 31 to 70 vs. 18 to 72 in the validation cohort; and the number of patients aged 40 or less occupied 8% of the whole population in the validation cohort, which is nearly twice of that in the discovery cohort (4.2%). AMPKα expression was associated with two additional clinicopathological variables in the validation cohort: PgR and basal-phenotype status; these clinicopathological variables were not available for the discovery cohort. The association of high AMPKα expression with ER, PgR positive and non basal-like tumours may indicate differential expression of AMPKα in different breast cancer phenotypes and requires further verification.
High AMPKα expression was associated with lower local recurrence risk, better relapse-free and breast cancer-specific survival. In multivariate Cox regression analysis AMPKα significantly associated with relapse-free and breast cancer-specific survival independent of possible confounding factors in the discovery cohort. AMPKα expression was significantly associated with breast cancer-specific survival in the validation cohort. As AMPKα expression was related to breast cancer phenotype, the importance of AMPKα expression in prognosis of different subtypes of breast cancer was assessed in the validation cohort. Interestingly, high AMPKα expression associated with better relapse-free and breast cancer-specific survival and in multivariate Cox regression analysis AMPKα expression was also independently associated with relapse-free and breast cancer-specific survival in luminal phenotype breast cancer. Our study is the first to examine the expression of total-AMPKα in breast cancer tissue and to report on its prognostic significance in radiotherapy treated breast cancer, especially in luminal phenotype disease.
Interestingly, such phenotype preference is also observed in the tumour inhibitory effects of metformin, the activator of AMPK, in breast cancer patients. Retrospective studies have shown that metformin use is associated with improved breast cancer-specific survival of diabetic women with luminal [38] and HER2+ breast cancers [39], but did not significantly impact survival outcomes in diabetic patients with triple-negative breast cancer [46]. As a result, we further assessed the effect of metformin on the radioresponse of different phenotypes of breast cancer in-vitro. Luminal breast cancer MCF7 and basal-like breast cancer MDA-MB-231 cells were used in this study, and when treated by single-dose irradiation both lines exhibited a similar response. However, when metformin treatment was combined with irradiation, a substantial increase in radiosensitivity of MCF7 cells was observed, with an SER of 1.5, but with little effect on the radiosensitivity of MDA-MB-231 cells. Such results suggest a possible phenotype related mechanism of action for metformin, which can apparently radiosensitise luminal phenotype cells but has limited effect on basal phenotype breast cancer.
To explore the mechanisms of this interesting phenomenon, alterations to redox homeostasis were assessed by examining the change in ROS levels following metformin treatment. Intracellular ROS production is the major mediator for low-LET radiation-induced cell killing in radiotherapy [47]. Metformin induced a significant increase of ROS levels in luminal breast cancer MCF7 cells but had no effect in basal-like breast cancer MDA-MB-231 cells. The differential effects of metformin on ROS production in luminal vs. basal-like breast cancer cells may provide a potential explanation for its phenotype preference in radiosensitisation. Others have reported upon metformin regulating intracellular ROS levels but with contradictory findings: metformin reduced the intracellular ROS level in human aortic endothelial cells [31]; but elevated it in primary human fibroblasts [48], which, together with our results, suggest that the effect of metformin in regulating intracellular ROS production may be largely dependent upon the intrinsic characteristics of the different cell types, with no uniform pattern. Current work suggests that the radiosensitisation effects of metformin were not attributable to it altering cell cycle progression or the level of apoptosis. Others have shown, however, that AMPK deficient cells are radioresistant and that such resistance is linked to cell cycle progression and proliferation [49].
Cells have a number of systems available to regulate intracellular ROS production and maintain redox homeostasis including the Trx system [33], which has been shown to be associated with clinical outcome in radiotherapy treated early stage breast cancer [50]. We performed Western blots to analyse the expression changes of Trx family proteins in metformin treated cells: metformin decreased the Trx expression level in MCF7 cells but had no effect on MDA-MB-231 cells. Interestingly, Hou et al. demonstrated that metformin inhibited intracellular ROS by upregulation of Trx expression via the AMPK-FOXO3 pathway in human aortic endothelial cells [31], whilst the current study shows the opposite in breast cancer MCF7 cells, with metformin increasing ROS production by decreasing Trx expression. There are probably many factors responsible for such conflicting results, including the concentration and duration of metformin exposure (i.e. 250 μM for 24 hours in the endothelial study vs. 10 mM for 48 hours in the current work), and the particular cell line used; however, both studies show the importance of Trx in metformin regulated ROS production. It is also interesting to note the effect of metformin on Txnip, with metformin totally abrogating its expression in both cell lines, which is consistent with previous reports suggesting that metformin inhibits Txnip expression in human hepatocellular carcinoma HepG2 and cervical cancer HeLa cells [32], as well as rat insulinoma INS-1E beta-cells [51].
In summary, the present study shows that high AMPKα expression is associated with a better prognosis of early-stage invasive breast cancer patients treated by breast-conserving surgery and radiotherapy in two independent cohorts. This result was more apparent in luminal phenotype disease with further in-vitro characterisations showing that metformin, the activator of AMPK, induced ROS production in luminal breast cancer cells possibly through altered Trx system expression; with little effect in basal phenotype cells. Although further confirmation is required, both in-vitro and in-vivo, current results suggest that metformin may be a clinically effective radiosensitiser and that its use should potentially be focused on patients with luminal phenotype breast cancer.
Materials and Methods
Clinical samples
This study is reported according to REMARK (Reporting Recommendations for Tumour Marker Prognostic Studies) criteria [52]. Ethical approval for the study was granted by Nottingham Research Ethics committee 2 under the title “Development of a molecular genetic classification of breast cancer” (C202313). Two independent cohorts were used; a discovery cohort of 166 cases and a validation cohort of 609 cases. Both cohorts were comprised of primary operable early-stage (stage I-III) invasive breast cancers from patients treated by breast-conserving surgery (wide local excision) and radiotherapy at Nottingham University Hospitals. Information on clinical history and outcome is prospectively maintained and patients were assessed in a standardised manner for clinical history and tumour characteristics (see Supplementary Table S1). Breast cancer-specific survival was defined as the time interval (in months) between the date of primary surgery and death resultant from breast cancer; local recurrence-free survival was defined as the time interval (in months) between the start of primary treatment and date of first histological confirmation of recurrent cancer (invasive or in-situ) at any site within the treated breast; relapse-free survival was defined as the time interval (in months) between the start of primary treatment and date of disease relapse.
The 166 cases in the discovery cohort were selected from all of the early-stage primary operable invasive breast cancer patients treated by wide local excision and radiotherapy at Nottingham University Hospitals between 1998 and 2006. The selection scheme was based on the local recurrence cases in a chronologic order, where every local recurrence case was included in the study: when a case with local recurrence was chosen, the immediate following 5 suitable cases without local recurrence were included. The median age at diagnosis of this cohort was 56 years (ranging from 31 to 70) and 78% (130 of 166) of patients had stage I disease. The follow-up time was 171 months (median follow-up time 108 months). Supplementary Table S1 shows the full clinicopathological characteristics of this cohort.
The 609 samples in the validation cohort were chosen from a well-characterised consecutive series of early-stage invasive breast cancer patients treated at Nottingham University Hospitals, between 1986 and 1998. From the initial whole series (n=1802), cases treated by wide local excision plus radiotherapy were selected (n=609). The median age of the validation cohort was 54 years (ranging from 18 to 72) and 74% (449 of 609) of patients had stage I disease. The follow-up time was 247 months (median follow-up time 134 months). Data on a wide range of biomarkers was available (Supplementary Table S1); estrogen receptor (ER), progesterone receptor (PgR), human epidermal growth factor receptor 2 (HER2) and basal phenotype status were available for this cohort and have been described previously [53, 54], with basal phenotype being defined as cytokeratin (CK)-5/6 and/or CK-14 positivity [55]. Current cases were classified into three molecular subgroups: ER and/or PgR positive (regardless of HER2 status) was defined as luminal; ER, PgR and HER2 negative was defined as triple-negative; and ER and PgR negative plus HER2 positive was defined as HER2+ [56].
Patient treatment, tissue microarray (TMA) construction and immunohistochemistry (IHC) are described in Supplementary Materials and Methods.
Cell culture
Two human breast cancer cell lines: MCF7 (luminal subtype), MDA-MB-231 (basal subtype) were used in this study (both were from American Type Culture Collection). All cell lines were cultured at 37˚C in a humidified incubator with 5% CO2. MCF7 (passage window 15) were maintained in RPMI1640 (Sigma, Dorset, UK) supplemented with 10% iron supplemented donor calf serum (PAA laboratories, Austria) and 1% penicillin/ streptomycin (Sigma). MDA-MB-231 (passage window 15) were maintained in minimal essential medium EAGLE (Sigma) supplemented with 0.1 mM non-essential amino acids solution (Sigma), 2 mM L-glutamine(Sigma), 1% penicillin/ streptomycin and 10% iron supplemented donor calf serum.
Cell irradiation and clonogenic survival assay
Cells were sub-confluent at irradiation. X-ray-irradiation was conducted using an RS225 Xstrahl X-ray cabinet irradiation system (Xstrahl Limited, UK) with a single dose of 1, 2, 4 or 6 Gray (Gy). X-rays were delivered at 195 kV, 10 mA, with a dose rate of 0.87 Gy/min. The cabinet was fitted with a 0.5 mm copper filter and used at a 48.4 cm focus-to-skin distance. Sham irradiated cells were used as controls. Cells were exposed to 10 mM of metformin (BioVision, Milpitas, CA) for 48 hours prior to irradiation. Cells were then trypsinised and plated for clonogenic survival assay with six parallel sets. After 18 days colonies were fixed (50% methanol in 0.9% saline solution for 15 min followed by methanol for another 15 min), stained (0.5% crystal violet) and counted as a survivor if containing more than 50 cells. Surviving fraction (SF) was calculated as: number of colonies / (number of cells plated × plating efficiency), where plating efficiency (PE) was defined as: number of control colonies obtained / number of control cells plated. The sensitizer enhancement ratio (SER) was calculated as the radiation dose yielding 1% SF divided by the radiation dose giving the same survival where cells were treated with metformin.
Measurement of intracellular ROS levels
Sub-confluent cells were treated with 10 mM metformin for 48 hours or, as a ROS positive control, 1 mM of H2O2 for 1 hour then further incubated with the membrane permeable dye 2’,7’-dichlorodihydrofluorescein diacetate (H2DCF-DA, Sigma) in fresh medium at a final concentration of 1 μM for 30 min, at 37˚C. Cells were then collected and intracellular ROS levels assessed by measurement of the fluorescence (excitation 500 and emission 520 nm respectively) using a Beckman Coulter FC500 MCL flow cytometer system (Beckman, USA). Data exported from the flow cytometer were analysed using FlowJo7.6.1 software (Tree Star) to obtain the median fluorescence intensity (MFI) of each group.
Protein extraction and Western blot
Sub-confluent cells, following incubation with 10 mM metformin for 48 hours, were harvested and re-suspended in RIPA buffer (Sigma) supplemented with protease inhibitor cocktail (Sigma) . A Bio-Rad protein assay kit (Bio-Rad Laboratories, USA) was used to determine the protein concentration of each sample. Lysates were separated by SDS-PAGE and transferred onto a nitrocellulose membrane. After blocking with 5% (w/v) milk powder in 0.1% PBS/Tween, the nitrocellulose membrane was incubated with primary antibody at 4°C overnight. The primary antibodies used in this study were: goat anti- human Trx antibody (1:1000 dilution; American Diagnostica, Stamford, CT), mouse anti- human TrxR antibody [19A1] (1:2000 dilution; Abcam, Cambridge, UK) and mouse anti- human Txnip antibody (1:1000 dilution; MBL International Corporation, Woburn, USA). HRP-conjugated β-actin antibody (Invitrogen) was used as internal control. Membranes were developed with Amersham ECL reagent (GE Healthcare, UK).
Cell cycle analysis
Sub-confluent cells were treated with 10 mM metformin for 24 or 48 hours, or treated with metformin for 48 hours and spared for an extra 24 hours (72 hours), then collected and fixed in 70% ethanol in PBS overnight. Fixed cells were then stained using a PBS solution containing 2.5 μg/ml of propidium iodide and 200 μg/ml of RNase for 30 min at 37°C and analyzed using a Beckman Coulter FC500 MCL flow cytometer system (Beckman, USA).
Annexin V-FITC apoptosis assay
Sub-confluent cells were treated with 10 mM metformin for 4, 8, 24 or 48 hours. Apoptosis of cells was then assessed by using an annexin V-FITC apoptosis detection kit (Invitrogen) according to the manufacturer’s instructions and analyzed using a Beckman Coulter FC500 MCL flow cytometer system (Beckman, USA).
Statistical Analysis
The relationship between categorised protein expression and clinicopathological variables was assessed using the Pearson Chi Square (χ2) test of association or Fishers Exact test if a cell count was less than five. Spearman rank order correlations were performed to test the association between expression of AMPKα and pAMPKα(Thr172). Survival curves were plotted according to the Kaplan-Meier method and significance determined using the log-rank test. Multivariate survival analysis was performed by the Cox proportional hazards regression model and included only those parameters that were significant in univariate analysis. Data from in-vitro experiments were expressed as the mean ± standard deviation (SD) and analysed using the Student t-test and ANOVA one-way tests. All differences were deemed statistically significant at the level of P<0.05. Statistical analysis was performed using SPSS 21.0 software (IBM, Armonk, NY).
Acknowledgments
The authors acknowledge Christopher Nolan for his excellent technical assistance. The authors also gratefully acknowledge the Chinese Scholarship Council for funding Yimin Zhang.
Author Contributions
Y. Zhang and S.G. Martin conceived the study, developed the methodology, analysed and interpreted data; Y. Zhang carried out experiments and wrote the manuscript; S.J. Storr developed the methodology, constructed databases and analysed data; S.G. Martin, S.J. Storr reviewed and revised the manuscript; K. Johnson helped carry out histopathology experiments; A.R. Green, E.A. Rakha, I.O. Ellis assisted with histopathology and, along with D.A. Morgan, constructed and maintained clinical databases; S.G. Martin supervised the study. All authors had final approval of the submitted version.
References
1. Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996; 334(9):574-579.
2. Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005; 310(5754):1642-1646.
3. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000; 348 Pt 3:607-614.
4. Hardie DG. (2011). AMP-activated protein kinase: a cellular energy sensor with a key role in metabolic disorders and in cancer. Biochem Soc Trans. (England, pp. 1-13.
5. Thompson AM. Molecular pathways: preclinical models and clinical trials with metformin in breast cancer. Clin Cancer Res. 2014; 20(10):2508-2515.
6. Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009; 32(9):1620-1625.
7. Bo S, Ciccone G, Rosato R, Villois P, Appendino G, Ghigo E, Grassi G. Cancer mortality reduction and metformin: a retrospective cohort study in type 2 diabetic patients. Diabetes Obes Metab. 2012; 14(1):23-29.
8. Lee MS, Hsu CC, Wahlqvist ML, Tsai HN, Chang YH, Huang YC. Type 2 diabetes increases and metformin reduces total, colorectal, liver and pancreatic cancer incidences in Taiwanese: a representative population prospective cohort study of 800,000 individuals. BMC Cancer. 2011; 11:20.
9. Skinner HD, Crane CH, Garrett CR, Eng C, Chang GJ, Skibber JM, Rodriguez-Bigas MA, Kelly P, Sandulache VC, Delclos ME, Krishnan S, Das P. Metformin use and improved response to therapy in rectal cancer. Cancer Med. 2013; 2(1):99-107.
10. Margel D, Urbach DR, Lipscombe LL, Bell CM, Kulkarni G, Austin PC, Fleshner N. Metformin use and all-cause and prostate cancer-specific mortality among men with diabetes. J Clin Oncol. 2013; 31(25):3069-3075.
11. Sadeghi N, Abbruzzese JL, Yeung SC, Hassan M, Li D. Metformin use is associated with better survival of diabetic patients with pancreatic cancer. Clin Cancer Res. 2012; 18(10):2905-2912.
12. Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, Hsu L, Hung MC, Hortobagyi GN, Gonzalez-Angulo AM. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. 2009; 27(20):3297-3302.
13. Rieken M, Kluth LA, Xylinas E, Fajkovic H, Becker A, Karakiewicz PI, Herman M, Lotan Y, Seitz C, Schramek P, Remzi M, Loidl W, Pummer K, et al. Association of diabetes mellitus and metformin use with biochemical recurrence in patients treated with radical prostatectomy for prostate cancer. World J Urol. 2013.
14. Kaushik D, Karnes RJ, Eisenberg MS, Rangel LJ, Carlson RE, Bergstralh EJ. Effect of metformin on prostate cancer outcomes after radical prostatectomy. Urol Oncol. 2013.
15. Zhang P, Li H, Tan X, Chen L, Wang S. Association of metformin use with cancer incidence and mortality: a meta-analysis. Cancer Epidemiol. 2013; 37(3):207-218.
16. Allott EH, Abern MR, Gerber L, Keto CJ, Aronson WJ, Terris MK, Kane CJ, Amling CL, Cooperberg MR, Moorman PG, Freedland SJ. Metformin does not affect risk of biochemical recurrence following radical prostatectomy: results from the SEARCH database. Prostate Cancer Prostatic Dis. 2013.
17. Carling D. The AMP-activated protein kinase cascade--a unifying system for energy control. Trends Biochem Sci. 2004; 29(1):18-24.
18. Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev. 2009; 89(3):1025-1078.
19. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009; 9(8):563-575.
20. Zhou K, Bellenguez C, Spencer CC, Bennett AJ, Coleman RL, Tavendale R, Hawley SA, Donnelly LA, Schofield C, Groves CJ, Burch L, Carr F, Strange A, et al. Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat Genet. 2011; 43(2):117-120.
21. Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Research. 2006; 66(21):10269-10273.
22. Liu BL, Fan ZY, Edgerton SM, Deng XS, Alimova IN, Lind SE, Thor AD. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle. 2009; 8(13):2031-2040.
23. Rattan R, Graham RP, Maguire JL, Giri S, Shridhar V. Metformin Suppresses Ovarian Cancer Growth and Metastasis with Enhancement of Cisplatin Cytotoxicity In Vivo. Neoplasia. 2011; 13(5):483-U130.
24. Alimova IN, Liu B, Fan Z, Edgerton SM, Dillon T, Lind SE, Thor AD. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle. 2009; 8(6):909-915.
25. Martinez Marignac VL, Smith S, Toban N, Bazile M, Aloyz R. Resistance to Dasatinib in primary chronic lymphocytic leukemia lymphocytes involves AMPK-mediated energetic re-programming. Oncotarget. 2013; 4(12):2550-2566.
26. Moiseeva O, Deschenes-Simard X, St-Germain E, Igelmann S, Huot G, Cadar AE, Bourdeau V, Pollak MN, Ferbeyre G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell. 2013; 12(3):489-498.
27. Song CW, Lee H, Dings RPM, Williams B, Powers J, Dos Santos T, Choi BH, Park HJ. Metformin kills and radiosensitizes cancer cells and preferentially kills cancer stem cells. Scientific Reports. 2012; 2.
28. Sanli T, Rashid A, Liu C, Harding S, Bristow RG, Cutz JC, Singh G, Wright J, Tsakiridis T. Ionizing radiation activates AMP-activated kinase (AMPK): a target for radiosensitization of human cancer cells. Int J Radiat Oncol Biol Phys. 2010; 78(1):221-229.
29. Storozhuk Y, Hopmans SN, Sanli T, Barron C, Tsiani E, Cutz JC, Pond G, Wright J, Singh G, Tsakiridis T. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br J Cancer. 2013; 108(10):2021-2032.
30. Zannella VE, Cojocari D, Hilgendorf S, Vellanki RN, Chung S, Wouters BG, Koritzinsky M. AMPK regulates metabolism and survival in response to ionizing radiation. Radiother Oncol. 2011; 99(3):293-299.
31. Hou XG, Song J, Li XN, Zhang L, Wang XL, Chen L, Shen YH. Metformin reduces intracellular reactive oxygen species levels by upregulating expression of the antioxidant thioredoxin via the AMPK-FOXO3 pathway. Biochemical and Biophysical Research Communications. 2010; 396(2):199-205.
32. Chai TF, Hong SY, He H, Zheng L, Hagen T, Luo Y, Yu FX. A potential mechanism of metformin-mediated regulation of glucose homeostasis: inhibition of Thioredoxin-interacting protein (Txnip) gene expression. Cell Signal. 2012; 24(8):1700-1705.
33. Zhang Y, Martin SG. Redox proteins and radiotherapy. Clin Oncol (R Coll Radiol). 2014; 26(5):289-300.
34. Powis G, Mustacich D, Coon A. The role of the redox protein thioredoxin in cell growth and cancer. Free Radical Biology and Medicine. 2000; 29(3-4):312-322.
35. Powis G, Montfort WR. Properties and biological activities of thioredoxins. Annual Review of Pharmacology and Toxicology. 2001; 41:261-295.
36. Nishiyama A, Matsui M, Iwata S, Hirota K, Masutani H, Nakamura H, Takagi Y, Sono H, Gon Y, Yodoi J. Identification of thioredoxin-binding protein-2/vitamin D-3 up-regulated protein 1 as a negative regulator of thioredoxin function and expression. Journal of Biological Chemistry. 1999; 274(31):21645-21650.
37. Yin M, Zhou J, Gorak EJ, Quddus F. Metformin Is Associated With Survival Benefit in Cancer Patients With Concurrent Type 2 Diabetes: A Systematic Review and Meta-Analysis. Oncologist. 2013.
38. Xiao Y, Zhang S, Hou G, Zhang X, Hao X, Zhang J. Clinical pathological characteristics and prognostic analysis of diabetic women with luminal subtype breast cancer. Tumour Biol. 2013.
39. He X, Esteva FJ, Ensor J, Hortobagyi GN, Lee MH, Yeung SC. Metformin and thiazolidinediones are associated with improved breast cancer-specific survival of diabetic women with HER2+ breast cancer. Ann Oncol. 2012; 23(7):1771-1780.
40. Bonanni B, Puntoni M, Cazzaniga M, Pruneri G, Serrano D, Guerrieri-Gonzaga A, Gennari A, Trabacca MS, Galimberti V, Veronesi P, Johansson H, Aristarco V, Bassi F, et al. Dual effect of metformin on breast cancer proliferation in a randomized presurgical trial. J Clin Oncol. 2012; 30(21):2593-2600.
41. Zhang T, Zhang L, Fan J, Wu K, Guan Z, Wang X, Li L, Hsieh JT, He D, Guo P. Metformin Sensitizes Prostate Cancer Cells to Radiation Through EGFR/p-DNA-PKCS In Vitro and In Vivo. Radiat Res. 2014; 181(6):641-649.
42. Hadad SM, Baker L, Quinlan PR, Robertson KE, Bray SE, Thomson G, Kellock D, Jordan LB, Purdie CA, Hardie DG, Fleming S, Thompson AM. Histological evaluation of AMPK signalling in primary breast cancer. BMC Cancer. 2009; 9:307.
43. Woods A, Vertommen D, Neumann D, Turk R, Bayliss J, Schlattner U, Wallimann T, Carling D, Rider MH. Identification of phosphorylation sites in AMP-activated protein kinase (AMPK) for upstream AMPK kinases and study of their roles by site-directed mutagenesis. J Biol Chem. 2003; 278(31):28434-28442.
44. Vidal AP, Andrade BM, Vaisman F, Cazarin J, Pinto LF, Breitenbach MM, Corbo R, Caroli-Bottino A, Soares F, Vaisman M, Carvalho DP. AMP-activated protein kinase signaling is upregulated in papillary thyroid cancer. Eur J Endocrinol. 2013; 169(4):521-528.
45. Fox MM, Phoenix KN, Kopsiaftis SG, Claffey KP. AMP-Activated Protein Kinase alpha 2 Isoform Suppression in Primary Breast Cancer Alters AMPK Growth Control and Apoptotic Signaling. Genes Cancer. 2013; 4(1-2):3-14.
46. Bayraktar S, Hernadez-Aya LF, Lei X, Meric-Bernstam F, Litton JK, Hsu L, Hortobagyi GN, Gonzalez-Angulo AM. Effect of metformin on survival outcomes in diabetic patients with triple receptor-negative breast cancer. Cancer. 2012; 118(5):1202-1211.
47. Hall EJ, Giaccia AJ. (2012). Radiobiology for the radiologist. (Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins).
48. Algire C, Moiseeva O, Deschenes-Simard X, Amrein L, Petruccelli L, Birman E, Viollet B, Ferbeyre G, Pollak MN. Metformin reduces endogenous reactive oxygen species and associated DNA damage. Cancer Prev Res (Phila). 2012; 5(4):536-543.
49. Sanli T, Storozhuk Y, Linher-Melville K, Bristow RG, Laderout K, Viollet B, Wright J, Singh G, Tsakiridis T. Ionizing radiation regulates the expression of AMP-activated protein kinase (AMPK) in epithelial cancer cells: modulation of cellular signals regulating cell cycle and survival. Radiother Oncol. 2012; 102(3):459-465.
50. Woolston CM, Storr SJ, Ellis IO, Morgan DAL, Martin SG. Expression of thioredoxin system and related peroxiredoxin proteins is associated with clinical outcome in radiotherapy treated early stage breast cancer. Radiotherapy and Oncology. 2011; 100(2):308-313.
51. Shaked M, Ketzinel-Gilad M, Cerasi E, Kaiser N, Leibowitz G. AMP-activated protein kinase (AMPK) mediates nutrient regulation of thioredoxin-interacting protein (TXNIP) in pancreatic beta-cells. PLoS One. 2011; 6(12):e28804.
52. McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM. REporting recommendations for tumor MARKer prognostic studies (REMARK). Nat Clin Pract Urol. 2005; 2(8):416-422.
53. Abdel-Fatah TM, Powe DG, Agboola J, Adamowicz-Brice M, Blamey RW, Lopez-Garcia MA, Green AR, Reis-Filho JS, Ellis IO. The biological, clinical and prognostic implications of p53 transcriptional pathways in breast cancers. J Pathol. 2010; 220(4):419-434.
54. Rakha EA, El-Rehim DA, Paish C, Green AR, Lee AH, Robertson JF, Blamey RW, Macmillan D, Ellis IO. Basal phenotype identifies a poor prognostic subgroup of breast cancer of clinical importance. Eur J Cancer. 2006; 42(18):3149-3156.
55. Rakha EA, El-Sayed ME, Green AR, Paish EC, Lee AH, Ellis IO. Breast carcinoma with basal differentiation: a proposal for pathology definition based on basal cytokeratin expression. Histopathology. 2007; 50(4):434-438.
56. Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98(19):10869-10874.