
INTRODUCTION
Three decades’ worth of accumulating data have shown that chromosomal rearrangements, including fusion genes, are frequent events and can act as genetic drivers of hematologic malignancies and mesenchymal tumors [1–3]. However, the occurrence and the biological and clinical effects of gene fusions in epithelial tumors have been less well recognized. The first fusion gene found in epithelial tumors (RET-CCDC6) was discovered in papillary thyroid carcinoma in the early 1990s [4, 5]. Other fusion gene discoveries soon followed in epithelial tumors and other malignancies [6]. Since then, even though the finding of novel fusion genes in epithelial tumors has increased tremendously, they remained poorly described until the last decade. Technical limits once precluded their characterization, but recent rapid advances in genome-sequencing technologies, analytical tools, and application of these technologies have propelled the discovery of fusion genes in epithelial cancers. Such discoveries have revealed thousands of fusion transcripts in hematologic malignancies and mesenchymal tumors as well as in epithelial tumors. However, only a small fraction has been assessed for transforming ability and mechanistic alterations, leaving a large fraction for functional characterization. Therefore, functional characterization of fusion genes needs to be performed to identify those that are ‘driver’ mutations rather than ‘passenger’ mutations and importantly to ensure that the proposed fusion genes are indeed expressed and functional in different tumor lineages. Furthermore, an improved understanding of the mechanisms underlying the formation of fusion transcripts and their effect on cell function will be crucial to translate the rapidly emerging ability to identify fusion transcripts to clinical application. The protein products of fusion genes can serve as therapeutic targets or as biomarkers for diagnosis or disease progression. Here, we focus on mechanisms that lead to the development of fusion genes and alternate transcript formation and the subsequent pathobiology of recurrent gene fusions.
FUSION GENES IN EPITHELIAL TUMORS
Accumulated data in human cancers from recent efforts by The Cancer Genome Atlas (TCGA), International Collaboration for Clinical Genomics (ICCG), International Cancer Genome Consortium (ICGC), and individually generated studies provide evidence that fusion genes/transcripts are much more common in epithelial tumors than previously thought. Indeed, the latest discoveries have demonstrated that fusion genes can be drivers in epithelial tumors in the same way as in hematologic malignancies and mesenchymal tumors. Initially, the genes involved in fusion formation were mainly classified into two classes: 1) transcription factors (Supplementary Figure 1B) or associated cofactors (e.g., RARA, MYC) and 2) receptor and non-receptor tyrosine kinases (e.g., ABL1, ALK, FGFR, NTRK) (Supplementary Figure 1A). In the last decade, genomic analyses dramatically changed this view so that a broader spectrum of genes was recognized as being involved in the formation of fusion genes and consequently in carcinogenic transformation. As described herein, genes involved in fusion formation that are implicated in transformation, progression, or resistance to therapy extend beyond tyrosine kinases and transcription factors or associated cofactors and include genes that mediate nucleo-cytoplasmic transport of protein and RNA (e.g., nucleoporin), a lysine-specific methyltransferase (e.g., MLL), and genes that are involved in metabolic pathways (e.g., PLAG2G4B), signal transduction (e.g., R-spondin in Wnt signaling), DNA repair (e.g., RAD51C), chromosome segregation (e.g., TACC), tumor suppressor genes (e.g. TP53, PTEN, CBFB), and oncogenes (e.g. GNAS, ERBB2). However, the role of most fusion genes in tumorigenesis remains unknown. While most fusion genes are tumor specific (and indeed many are private and found only in single tumors) [7], some fusion genes are involved in the tumorigenesis of multiple epithelial cancers as well as hematologic malignancies [8] and mesenchymal tumors.
FORMATION OF FUSION GENES AND BIOLOGICAL IMPACT OF ONCOGENESIS
The first fusion gene discovered in human cancer was the BCR-ABL gene, described in 1960. The breakpoint cluster region (BCR)-c-abl oncogene, non-receptor tyrosine kinase (ABL) fusion gene is the result of a reciprocal translocation between the q arms of chromosomes 9 and 22 (i.e., an interchromosomal translocation; Figure 1A) and occurs in more than 96% of patients with chronic myelogenous leukemia (CML) [2]. Soon after, the discovery of fusion genes involving MYC and promyelocytic leukemia (PML) followed. Fusions caused by interchromosomal translocation are thought to account for only a fraction of fusion genes. Recent technological advances have revealed that, in addition to being caused by interchromosomal translocations, fusion transcripts also arise by intrachromosomal translocation, insertion, deletion, tandem duplication, inversion, chromothripsis, and aberrant splicing (read-through) (Figure 2).
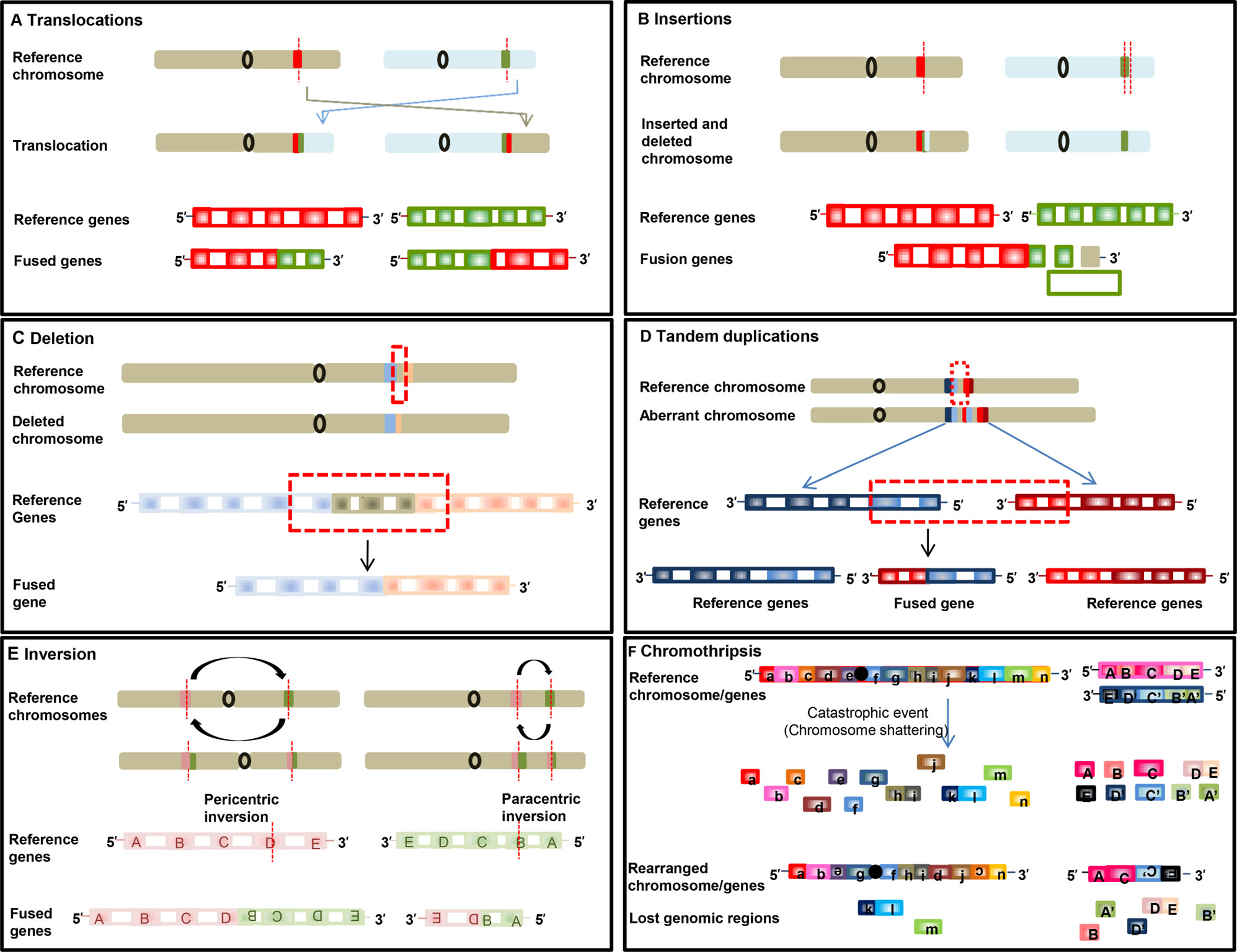
Figure 1: Schematic illustrations of formation of fusion genes. Formation of fusion gene via translocation (A), insertion (B), deletion (C), tandem duplication (D), inversion (E), chromothripsis (F).
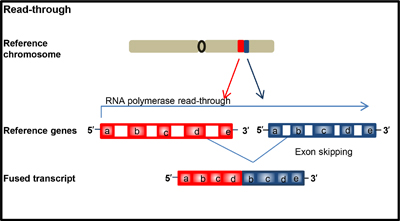
Figure 2: Schematic illustration of formation of chimeric transcript.
Pathogenesis of fusion gene formation
As aforementioned, oncogenic fusion genes or transcripts can arise through multiple different types of rearrangement (Figures 1 and 2), which, with two exceptions, create tail-to-tail and head-to-head fusions. Fusion genes can occur through alterations at the DNA level by six different rearrangements (Figure 1). Furthermore, fusion transcripts can form through read-through during RNA transcription without structural chromosomal changes (Figure 2).
The first type of rearrangement is reciprocal translocation. It can be balanced or unbalanced interchromosomal translocation. Balanced translocation is caused by an exchange of DNA sequences without missing or extra genetic information between two different chromosomes (Figure 1A). Unbalanced translocation, in which the exchange of sequences is unequal, results in missing or extra genetic information. As shown in Supplementary Table 1, fusion of solute carrier family 34, member 2 (SLC34A2) with the gene encoding c-ros oncogene 1 (ROS1) in non-small cell lung cancer (NSCLC) [9, 10] is an example of reciprocal translocation.
The second type of rearrangement is insertion. Insertions are due to movement of a DNA fragment from one region into another in the same chromosome (intrachromosomal) or from one chromosome into other (interchromosomal), the latter also known as nonreciprocal translocation (Figure 1B).
The third type of rearrangement is the formation of fusion genes by juxtaposition of two genes through deletion of regions between two genes that transcribe in the same direction (Figure 1C). Fusion genes such as ATG7-RAF1 in pancreatic cancer (Supplementary Table 2) and EIF3E-RSPO2 in colon cancer are examples of fusion resulting from deletion [7].
The fourth type of rearrangement is tandem duplication. In this scenario, a genomic region is duplicated, resulting in a fusion with a gene in the original region. Fibroblast growth factor receptor 3 (FGFR3)-transforming, acidic coiled-coil containing protein 3 (TACC3) in glioblastoma [11] and chromosome 2 open reading frame 14 (C2orf44)-ALK fusions in colorectal cancer [12] are examples of tandem duplications (Figure 1D).
The fifth type of rearrangement is inversion, in which chromosomal segments flip with (pericentric) or without (paracentric) relationship with the centromere (Figure 1E); examples include kinesin family member 5B (KIF5B)-RET in lung adenocarcinoma [12] and echinoderm microtubule-associated protein-like 4 (EML4)-ALK in non–small cell lung cancer [13].
The sixth type of rearrangement is chromothripsis, in which fusion arises when one chromosome or chromosome region or a few chromosomes shatter into many fragments, and fragments reassemble inaccurately (Figure 1F). Examples of this characteristic event are PVT1-MYC and PVT1-NDRG1 fusions in medulloblastoma [14], and NDUFAF2-MAST4 in prostate cancer cell lines [15].
Aberrant fusion transcript formation can occur due to read-through transcripts, which is different from the other six rearrangements due to occurring at the RNA level. Importantly, this is the only type of fusion transcript that does not involve rearrangement of genomic material. Chimeric transcripts arise when an RNA polymerase does not properly terminate transcription at the end of a gene and continues transcribing until the end of the following gene due to aberrant splicing (Figure 2). Chimeric protein of FGFR3-BAI associated protein 2 like 1 (BAIAP2L1) in bladder carcinoma is example of this rearrangement [16]. This type of rearrangement is not restricted to cancer as genes such as MDS1 and EVI1 can create the read through MECOM in normal or malignant cells.
Interestingly, formation of the same fusion can occur through different rearrangements. An example of this scenario is TMPRSS2-EGR fusion (Supplementary Table 3), which can arise through inversion of chromosome 21q22 in some cases and by interstitial deletion of chromosome 21q22 in other cases [17]. When fusion genes are caused by deletion or tandem duplication, both genes are likely located on the same direction and strand of a chromosome, whereas in fusion genes caused by inversion, the two genes are more likely to be found on opposite chromosome strands. Notably, fusion genes can be complex by involving multiple genes. Fusion genes that express a fusion protein can gain functional properties, or alternatively there can be increased amounts or activity of a single component of the fusion gene. Both mechanisms can lead to cell transformation.
Heterogeneity in fusion genes
Fusion genes can demonstrate marked heterogeneity based on different partners, variable breakpoints, co-mutations, and tissue specific effects. For example ALK, ROS1, and BRAF can have multiple fusion partners and different breakpoints (Supplementary Figure 1) that can alter functional and therapeutic consequences of the fusion genes.
Processes underlying the formation of fusion genes
Repair of double-strand breaks caused by defects in DNA damage repair or bridge-breakage-fusion events [18] by canonical non-homologous end joining (NHEJ) [19], alt-NHEJ [20, 21], break induced replication repair or synthesis induced end joining [21] can alter the structure of the breaks. Further, interphase gene proximity (spatial proximity) is tissue specific and can facilitate generation of fusion genes. For example PML and RARa are found in close spatial proximity in hematopoietic cells resulting in frequent fusion in hematopoietic but not other cells [22]. Similar spatial proximity during interphase has been proposed to mediate RET and H4 fusion in papillary thyroid carcinoma [23], and TMPRSS2 and ERG in prostate cancer [24]. Thus understanding the processes leading to the formation of fusion genes could alter the functional and therapeutic relevance of fusion genes.
Pathobiology of oncogenic fusion genes
Fusion genes can influence biology through six mechanisms. A first mechanism is overexpression of an oncogene through promoter exchange or through linking of an open reading frame to transcriptional control elements (e.g., TMPRSS2 to ETS, IgH to MYC, IgH to BCL2) that are active in the target tissue. As seen in prostate cancer, the forced expression of normal proteins (i.e. 3′ gene, ETS family of transcription factors) through an androgen-responsive promoter (TMPRSS2, a transmembrane serine protease) or by fusion to ubiquitous promoters results in overexpression of a wild-type protein [17, 25–27]. Where the major consequence of a fusion is overexpression of a wild-type protein. TMPRSS2-ERG is the most common fusion in prostate adenocarcinomas (50%) and in precursor high-grade prostatic intraepithelial neoplasia (approximately 20%) [26, 28], and promotes metastasis to bone [29]. TMPRSS2 has been shown to fuse to 20 different partners in prostate adenocarcinomas (Supplementary Table 3) [26, 28]. ETS family members can also be fused with other 5′ partners (e.g., SLC45A3, kallikrein 2 (KLK2), and calcium-activated nucleotidase 1 (CANT1)) [30, 31].
A second mechanism affects biology by causing truncations, which may result in loss of negative regulatory microRNA binding sites. MYB-nuclear factor I/B (NFIB) fusion results in loss of miR-15a/16 and miR-150 binding sites, thereby resulting in overexpression of MYB protein [32]. This fusion occurs in adenoid cystic carcinoma of the breast (67%) and salivary gland (28%) as well as in benign sporadic, dermal cylindromas (60%) [33]. Similar mechanisms occur in other fusion genes such as high mobility group AT-Hook2 (HMGA2)-NFIB, in which loss of negatively regulating let-7 miRNA–binding sequences in the 3′ UTR of HMGA2 enhances oncogenic transformation [34]. This fusion is recurrent in benign pleomorphic salivary gland adenomas [35]. The FGFR3-TACC3 fusion also loses a negatively regulatory binding site of miR-99a, which causes overexpression of the protein and contributes to aneuploidy in glioblastoma cells [11]. As a further example, TFEB–CADM2 fusions, which occur in type 2 papillary renal cell carcinoma, also lose several miRNA-binding sites, [36].
The third mechanism influences cell function by destroying the intrinsic control mechanism through introducing dimerization or oligomerization domain(s) of fusion partner genes. A dimerization or oligomerization domain of partner proteins leads to constitutive activation of the chimeric protein itself as well as downstream signaling pathways. The best example of this is the first identified fusions of BCR-ABL1 in CML. The ABL1 kinase is constitutively activated by BCR-ABL1 oligomerization by the coiled-coil domain in BCR and is necessary for the transforming activity of ABL [37]. Transmembrane TK are activated by ligand binding to their extracellular domains, which leads to receptor dimerization and transphosphorylation of tyrosine residues located in the kinase activation loop or in binding sites for linker molecules, thus activating downstream signaling pathways. Fusion proteins frequently co-opt the normal activation mechanism of tyrosine kinases through dimerization domains in partner proteins. Notably, most if not all transmembrane tyrosine-kinase fusion gene (e.g., ALK, FGFR, NTRK, RET, and ROS1) partners contain dimerization or oligomerization domains; either coiled-coil or SAM, LisH, BAR, and SPFH [16, 38–41]. The TK frequently fuse with multiple partner genes. For instance, anaplastic lymphoma kinase gene (ALK) fuses with CLTC, TPM3, TFG, ATIC, KIFB, C2orf44, and EML4 (Supplementary Figure 1C), which all contain dimerization domains, rendering ALK constitutively active and resulting in activation of downstream signaling pathways. Fusions that lead to ALK activation can promote cell transformation, proliferation, aberrant development, and clonal expansion. Activation of wild-type ALK induces downstream signaling pathways including phosphatidylinositol 3-kinase–AKT, mitogen-activated protein kinase kinase (MEK)–extracellular signal-regulated kinase (ERK), and STAT [8]. However, the EML4-ALK chimeric protein activates AKT and ERK pathways in EGFR mutation–positive NSCLC [42], while NPM-ALK fusion promotes ERK and STAT3 pathways in anaplastic large cell lymphoma [43]. Thus downstream signaling pathways and roles of chimeric proteins can vary in the context of different cancer subtypes. ALK fusions are frequently observed in glioblastoma, non-small lung cancer, and papillary thyroid cancers (Supplementary Table 1).
FGFR TK family (FGFR1, FGFR2, FGFR3, and FGFR4) members encode transmembrane proteins that contain immunoglobulin-like and kinase domains (Figure 2) that play diverse roles in controlling cell proliferation, cell differentiation, angiogenesis, and tumor development. The most common fusion partners of FGFR1 or FGFR3 are the transforming acidic coiled-coil (TACC)-coding domains of TACC1 or TACC3 (Supplementary Figure S1D). The distinctive feature of TACC proteins is a coiled-coil domain at the C terminus, known as the TACC domain, which exerts ligand-independent activation by dimerization and mediates localization to the mitotic spindle [44–46]. TACC3 plays a crucial role in the stabilization and organization of the mitotic spindle and thus proper chromosome segregation [44]. The FGFR3-TACC3 fusion protein localizes to mitotic spindle poles, leading to mitotic and chromosomal segregation errors that contribute to aneuploidy in glioblastoma [11]. In addition, overexpression of FGFR is a consequence of formation of FGFR3-TACC3 fusion due to loss of a suppressive effect of miR-99a on the 3′ UTR miRNA–binding site [47]. FGFR kinase inhibition by FGFR TKIs (PD173074, AZD4547, or BGJ398) prevents acquisition of aneuploidy defects [11]. Binding of FGF ligands to FGF receptors engages downstream signaling pathways, including PI3K-AKT and RAS-ERK, while the constitutively activated FGFR3-TACC3 protein promotes ERK and STAT3 signaling cascades in glioblastoma, suggesting acquisition of neomorphic functions [11, 47]. A truncated form of FGFR3 protein is not sufficient to cause kinase activation or transforming activity; thus the ability of the fusion protein [46] to dimerize is necessary for transformation. BAI1-associated protein 2–like 1 (also known as insulin receptor kinase substrate) is a fusion partner with FGFR3 in bladder cancer [46]. The FGFR3-BAIAP2L1 fusion promotes ligand-independent phosphorylation of FGFR3-BAIAP2L1 in Rat-2_F3-B cells and tumor formation in nude mice inoculated with Rat-2_F3-B cells [16] by activating ERK and STAT1 signaling [48], similar to FGFR2-CCDC6 [48].
As lesson learned from RARA fusions in APL, with the product generated by the reciprocal translocations of RARA with multiple partners, contributed to the discovery of the biological response of APL to retinoids. Each RARA-containing fusion gene imparts a different treatment response, emphasizing the need to characterize fusion partners.
RET encodes a receptor tyrosine kinase protein containing an N-terminal extracellular domain with four cadherin-like repeats and a cysteine-rich region, a transmembrane domain, and C-terminal cytoplasmic tyrosine kinase domain. Wild-type RET is activated through ligand binding. Fusion proteins are activated through ligand-independent fusion of its intracellular kinase-encoding domain to coiled-coil domain containing 6 (CCDC6) or nuclear receptor coactivator 4 (NCOA4), among other partners. RET fusions mostly occur in irradiation-induced papillary thyroid carcinoma [49] as well as in lung adenocarcinoma. One common fusion partner in lung adenocarcinoma is KIF5B. The RET-KIF5B fusion retains a kinase domain from RET and a coiled-coil domain from KIF5B, which induces homodimerization and activates the oncogenic TK domain by autophosphorylation [50, 51]. Not only do the fusion proteins lead to dimerization but chimeric proteins lack the inhibitory domain of RET (Supplementary Table 1). Other partners of RET (Supplementary Figure 1F) include TRIM24, TRIM27, ELKS, GOLGA5, PRKAR1A, RAB6IP2, MBD1, HOOK3, PCM1, and ERC1 in papillary thyroid carcinoma.
A fourth mechanism affects function through loss of pivotal domains: autoinhibitory segment, membrane, or nuclear localization domains. One example of this mechanism is BRAF fusion (Supplementary Table 2). BRAF encodes a protein called B-raf, a member of the RAF family of cytoplasmic serine/threonine protein kinases that contains an amino terminal RAS-binding domain and a carboxy-terminal kinase domain. BRAF and other RAF family members (A-RAF and C-RAF) are downstream effectors of activated RAS through activating the MEK-ERK signaling pathway and regulating multiple key functions of cells including growth, differentiation, apoptosis, and survival. Recently, BRAF fusions with different partners have been identified in a variety of epithelial tumors, including melanoma, pilocytic astrocytoma, papillary thyroid, rectal and prostate cancers. SLC45A3-BRAF (solute carrier family 45, member 3-v-raf murine sarcoma viral oncogene homolog B1) and ESRP1-RAF1 (epithelial splicing regulatory protein-1–v-raf-1 murine leukemia viral oncogene homolog-1) gene fusions have been identified in prostate cancer, and AGTRAP-BRAF (encoding angiotensin II, type I receptor–associated protein–v-raf murine sarcoma viral oncogene homolog B1) has been found in gastric cancer [52]. SLC45A3 is a transmembrane transporter protein containing a transmembrane domain, and ESRP1 is a splicing factor containing RNA recognition and vascular domains. The expression of SLC45A3-BRAF or ESRP1-RAF1 in prostate cells has been found to induce a neoplastic phenotype that is sensitive to RAF and MEK inhibitors [52]. The fusion proteins all lack the N-terminal RAS-binding domain and N-terminal autoinhibitory region of BRAF while retaining the kinase domain. Thus the fusion protein is constitutively active. In addition to the loss of the autoinhibitory region of BRAF, promoter regulatory elements from SLC45A3, which may be regulated by androgen (SLC45A3-BRAF) or promoter regulatory elements from ESRP1, lead to high expression of the chimeric transcript, which correlates with disease progression in prostate cancer metastases [53].
PSF-TFE3 fusion occurs in papillary renal cell carcinoma [54]. TFE3 is a member of the class III helix-loop-helix (HLH) family, and PSF-A plays a role as a transcriptional repressor [55]. PSF-TFE3 fusion contains DBD and the dimerization domain (LZ region) of TFE3 and the repression domain of PSF, but this fusion lacks the N-terminal activation domain of TFE3 [54]. Parental TFE3 and PSF are nuclear proteins, whereas chimeric proteins localize in the endosomal compartment. Interestingly, the chimeric protein sequesters wild-type TFE3 and TP53 in the cytoplasm, leading to their degradation and to functionally null TP53 and TFE3, with subsequent transformation of cells [54].
EWS-Oct-4 and EWS-Oct-4B fusions are common in sarcoma. OCT-4B encodes a nuclear protein that binds DNA with the same sequence specificity as the Oct-4 protein. Chimeric proteins comprise the N-terminal domain (NTD) of EWS and the POU and C-terminal domains of Oct-4 or Oct-4B. The POU domain is a conserved DNA-binding domain and mediates nuclear localization. EWS-Oct-4 and EWS-Oct-4B chimeric proteins localize in the nucleus, whereas Oct-4B mainly localizes in the cytoplasm. Both fusion proteins have transforming ability in nude mice [56, 57]. Another example is RAD51C-ATXN7 fusion, which has been reported in 36% of colorectal cancers. The chimeric protein contains Rad51c N-terminal domains (ATP binding site and BRC (BRCA1) interacting domains) and the SCA7 (spino-cerebral ataxia 7) domain of ATXN7, and lacking C-terminal nuclear localization signal of Rad51C and CAG (polyglutamine tract) repeat sequence of ATXN7 [58]. This likely decreases the loading of Rad51 onto DNA during homologous recombination resulting in a shift to non-homologous end joining or persistence of double strand breaks resulting in increased genomic instability. TRA2B-DNAH5 fusion has been found in the cytoplasm in lung squamous cell carcinoma (3.1%) and has transformation capacity in Ink4a-/- MEFs. It promotes cell invasion in Beas2B and CRL-5889 cells through activation of the MAPK pathway by decreasing the nuclear localization of SIRT6 [59]. It is important to note that localization of the protein itself is crucial for its role in cells. Even if the gene itself is not inactivated, changing the protein localization through fusion with other partners leads to degradation of the protein or an altered function in cells.
The AKAP9-BRAF fusion has been preferentially reported in radiation-induced papillary carcinomas, whereas the BRAFv600 mutation occurs in sporadic types of cancer [60]. The fusion protein contains the protein kinase domain but lacks the autoinhibitory N-terminal region of BRAF and the C-terminal centrosomal domain of AKAP9. The parental AKAP9 protein is found as a single dot in the centrosome (perinuclear) in normal cells, whereas the chimeric protein has a diffuse distribution in the cytoplasm. Chimeric protein is constitutively activated and has transforming ability in NIH3T3 cells and regulates the MAPK pathway [60]. In parental cells, however, growth factors, hormones, and cytokines regulate BRAF by binding and activating specific receptors on the cell surface [61]. Moreover, it is important to stress that chimeric protein formation may lead to loss of the transmembrane domain of protein and consequently to aberrant localization of chimeric proteins (Supplementary Table 1 and Figure 3).
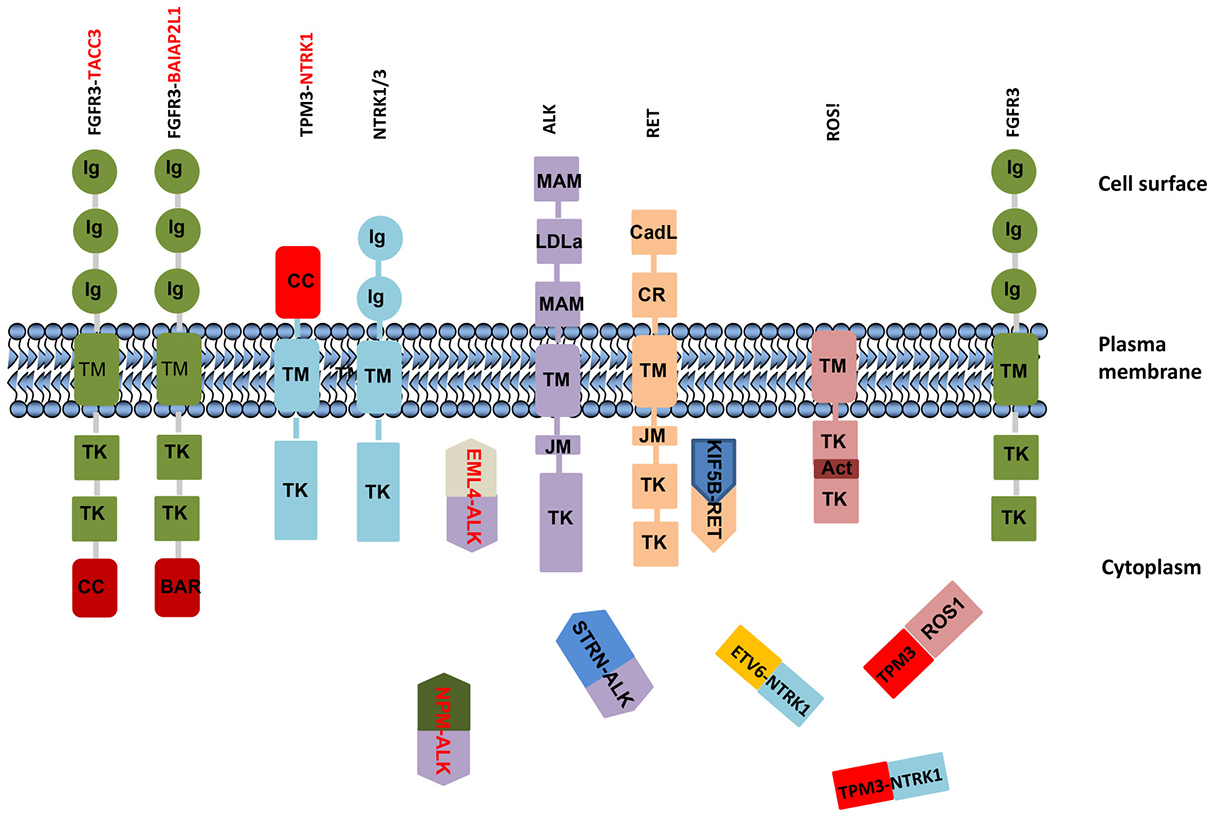
Figure 3: Schematic illustration of localization of parental and chimeric proteins.
A fifth mechanism impairs function by destroying the folding capacity of protein. MLL fusions occur in acute leukemia with a variety of partners (approximately 80). The breakpoints cluster mostly in MLL introns 9-11, with an age difference (in introns 9 and 10 in adults, and mostly in intron 11 in infants), with rare exceptional cases [62, 63]. MLL exons 11 to 16 code the PHD1-3 domains. When fusion breakpoints impair exon 11 in MLL, this results in the loss of two crucial cysteine residues, which prevents correct folding of the PHD domain, and influences its dimerization ability and binding capacity to its targets CYP33 and maybe to ECSASB2 [64, 65].
A sixth mechanism affects function by the destruction of the regulatory role of a protein. Two scenarios are possible for this mechanism. Examples of the first scenario are the AML1-ETO and PML-RARA fusion genes. In this scenario, both fusion genes convert their transcription regulatory role from an ‘activator of gene transcription’ into a ‘repressor of transcription.’ Changing the transcription regulatory role of fusion genes depends on their interacting proteins, recruited corepressors or coactivators, and context-dependent cross talk with other proteins, which can bind to their target genes. An example of the second scenario is the NAB2-STAT6 fusion, which converts the transcriptional repressor into a transcriptional activator in solitary fibrous tumor [66]. EGR1 induces NAB2, and NAB2 represses EGR1 by a negative feedback loop, while STAT is a transcriptional activator. In the context of solitary fibrous tumor, in NAB2-STAT6 fusion, NAB2 inherits an activation domain from the STAT6, thus converting a ‘transcriptional repressor’ role into a ‘transcriptional activator’ of EGR1. This results in increasing cell proliferation by constitutive activation of EGR1, and through activation of EGR1 target genes (e.g., NAB2, NAB1, IGF2, FGF2, PDGFD, FGFR1, and NTRK1) rather than activation of STAT6 [66].
It is important to stress that multiple mechanisms may contribute to the transforming ability of fusion genes in cells. In MYB-QKI fusions, for example, the N-terminal HTH DNA-binding and transactivation domains of MYB and C-terminal region of QKI (QUA2 and Y rich) are retained, whereas the C-terminal negative regulatory domains of MYB and N-terminal KH RNA-binding domain of QKI are lost [67]. The nuclear localization sequence of QKI in C-terminal is retained in splice variant QKI5, with the protein being found in the nucleus, but nuclear localization sequence of QKI is lost in the splice variant QKI6 and it is likely found in the cytoplasm [68] Herein, MYB-QKI5 may also localize in nucleus, whereas MYB-QKI6 may localize in cytoplasm. It has been reported that 1621 and 1947 genes with the common 1029 genes are significantly differentially expressed in mouse neural stem cells that stably over-expressed MYB-QKI5 and MYB-QKI6 fusions compared to eGFP-expressing cells, respectively [67]. The distinct genes that differentially expressed may due to localization of these two splice variants. The oncogenic potential of MYB-QKI is mediated by multiple mechanisms. One possible mechanism can be through the autoregulatory feedback loop by binding the MYB-QKI fusion protein to the MYB promoter, resulting in its activation. The second mechanism is through loss of the N-terminal of QKI, resulting in downregulation of the tumor-suppressor activity of QKI. The third mechanism is through movement of the H3K27ac-bound enhancers in the 3′ region of QKI to the MYB promoter, resulting in activation of MYB [67]. The fourth mechanism is lack of negatively regulatory elements, miR15/miR16 binding sites of MYB are lost due to truncation of C-terminal domains of MYB. MYB belongs to a family of leucine zipper transcription factors and has a role in regulating cell proliferation, apoptosis, and differentiation [69]. Parental MYB lacks transforming ability in vitro, but C-terminus–truncated MYB proteins are oncogenic [70]. Destruction of MYB by fusion formation leads to disruption of target genes as well. QKI encodes the STAR (signal transduction and activation of RNA) RNA-binding protein Quaking, which has a pivotal role in oligodendroglial differentiation [71], and also has a role in regulating alternative splicing, microRNA processing, and circular RNA formation that involves in epithelial-mesenchymal transition [72–74]. MYB-QKI rearrangement occurs in most cases of angiocentric glioma (85.7%), and promote tumorigenesis both in vitro and in vivo [67]. The involvement of multiple mechanisms in transformation may be related to the aggressiveness of cancer and can contribute to patient prognosis.
TARGETING FUSION GENES
It is important to stress that different gene partner domains or different breakpoints in fusion genes can alter the response to targeted therapy. Further, the intrinsic gene expression patterns in different tumors can also alter therapeutic sensitivity. As an example, CCDC6-RET and NCOA4-RET, despite both containing the same RET effector, drive different phenotypes and signaling resulting in differences in response to targeted therapeutics. Indeed, in model systems, different therapeutic approaches are necessary to optimally inhibit CCDC6-RET and NCOA4-RET [75].
Resistance to targeting fusion genes can occur through multiple mechanisms including gate-keeper mutations in the partner-kinase gene, amplification of the fusion gene or alternatively activating signaling pathways that can bypass the effects of the inhibitor (Supplementary Table 5). CCDC6-RET fusion expressing cells can become resistant to multi-targeted inhibitors such as sunitib or lenvatinib through EGFR activation and subsequent increased ERK-AKT signaling. Indeed, sunitib or lenvatinib resistant CCDC6-RET fusion expressing cells are resensitized by inhibition of the EGFR [76]. Similar processes have been observed in ALK, ROS, RET and NTRK1 fusion expressing cells [77, 78]. Importantly, intra-tumoral heterogeneity of fusion genes, similar to mutations, has been identified in tumors that could markedly alter the sensitivity to targeted therapeutics [79].
In addition, different fusion partners can affect the tumorigenicity of the fusion. For example, BAIAP2L1-MET and TFG-MET fusions are transforming, while CAPZA2-MET fusions are not transforming at least as assessed in Ba/F3 cells likely due to differences in subcellular localization [80]. Similarly, while FAM114A2-BRAF and ATG7-BRAF are transforming in Ba/F3 cells, AHCYL2-BRAF is not likely due to retention of an inhibitory domain within the N-terminus of BRAF [80].
It is important to note that in addition to altering responses to targeted therapy, gene fusions found in tumor cells can function as neoantigens, altering responses to immunotherapy [81]. Thus, immunotherapy could provide an alternative approach for therapy of some fusion gene containing tumors. Detailed experimental and modeling studies will be required to fully elucidate the effects of different partner genes, variable breakpoints, co-existence of other gene alterations, and tissue specific effects on sensitivity or resistance to targeted therapies.
A subset of oncogenic fusions can alter p53 activity and deregulate other check point and DNA-damage repair proteins. As noted above PFS-TFE3 fusions can increase degradation of p53. Furthermore, FGFR3-BAIAP2L1 fusions can suppress TP53 and CDKN2A expression as well as RB1 and p27 phosphorylation, associated with E2F2, E2F1, CDK2/Cyclin E and MAPK pathway activation [16]. Similarly, NPM-ALK JNK and MDM2-dependent inactivation of p53 function as well as PI3K-dependent p53 nuclear exclusion in anaplastic large cell lymphoma. Consistent with this concept inhibition of MDM2, JNK and PI3K induces apoptosis in NPM-ALK-expressing cells [82]. In addition, the MLL-ELL fusion inhibits functional activity of p53 in leukemia [83]. PLZF-RARa inhibits p53 and CDKN1A expression while increasing p53 degradation in acute promyelocytic leukemia [84]. Thus, p53 is a frequent target of fusion proteins.
In addition to p53, fusion proteins interact with other components of the DNA damage repair pathways. For example, BCR-ABL interacts with RAD51 resulting in stabilization and phosphorylation of RAD51 with subsequent increases in double strand break repair and drug resistance [85]. TMPRSS2-ERG fusion can block XRCC4-mediated NHEJ by inhibiting DNA-PKcs auto/trans-phosphorylation and thus sensitizes prostate cancer cells to PARP inhibition [86]. TMPRSS2-ERG and ETS family members can downregulate CHK1 expression and elevate DNA damage response in prostate tumor cells again potentially sensitizing cells to PARP inhibition [87].
Moreover, the effects of fusion genes can be both direct and indirect and are conditioned on co-occurring aberrations in other genes. For example, PTEN loss and p53 mutations frequently co-occur with ERG fusion proteins in prostate cancers [88–90]. In the presence of PTEN loss or p53 mutation, the effects of ERG fusion proteins would be modified. Notably expression of TMPRSS2(e1)-ERG(e4) in PC-3 cells inhibits expression a number of cell cycle-related genes including CDK1 and CCND1 thus promoting RB activation and E2F1 repression. This effect of ERG fusion proteins would interact with aberrations in either PTEN or p53. Thus, ERG fusion proteins alter the response of prostate cancer cells to palbociclib which has been demonstrated in human cell lines and mouse xenograft models [91].
TUMOR TYPE–SPECIFIC FUSION TRANSCRIPTS
Fusion genes were at first thought to be specific for tumor types and hence useful as diagnostic and prognostic markers and for monitoring response to therapy. However, recent accumulated data has indicated that only a small portion of fusion genes are specific for certain types of cancer (Supplementary Table 4), and a number have been shown to occur in multiple tumor lineages. It is important to stress that even the same fusion gene can exhibit different oncogenic properties in different types of malignancies, for example, the breakpoints may differ from each other, as in BCR-ABL fusion genes. In this latter case, the fusion gene can be used as a diagnostic marker. This fusion gene is a defining marker of chronic myeloid leukemia (CML) (96%) and breakpoints in the majority of samples occur in the 5.8-kb major breakpoint cluster region (between introns 11 and 16), resulting in a p210 BCR-ABL protein. The same fusion gene with different breakpoints (minor breakpoint cluster region, within the 72-kb BCR intron 1) occurs in adult acute lymphocytic leukemia (30%), resulting in a p190 chimeric protein. It also occurs rarely in childhood ALL (3-5%) and acute myeloid leukemia (1%) [3]. Defining fusion genes have been reported in epithelial tumors as well. ESRRA-C11orf20 fusion gene has been found in a subset of serous ovarian cancers (15%) [92], while EIF3E–RSPO2 and PTPRK–RSPO3 fusion genes (in which chimeric protein activate Wnt signaling) have been found in 10% of colorectal cancers [7]. Other examples of defining fusions are SLC3A2-NRG1 (in which chimeric protein activates AKT and ERK pathways) in invasive mucinous adenocarcinoma of the lung (27%) [93], and EGFR-SEPT14 fusion gene in glioblastoma [94]. JMJD7-PLA2G4B has been reported in HNSCC [95], and chimeric protein promotes cell proliferation and survival by modulating phosphorylation of AKT and regulates cell cycle progression by regulating SKP2 in HNSCC. PLAG2G4B encodes a calcium-dependent phospholipase that hydrolyzes phospholipids to lysophospholipids and fatty acids with lysophospholipids being potent signaling molecules [95]. DNAJB1-PRKACA fusion is a defining biomarker in fibrolamellar hepatocellular carcinoma (100%) and has not been seen in other subtypes of hepatocellular carcinoma [96]. DNAJB1 encodes a heat shock protein, Hsp40, which is involved in protein folding within cells [97]. A chimeric protein contains the N-terminal of DNAJB1 and the C-terminal of PRKACA. Parental PRKACA involves glucose and lipid metabolism and mitochondrial biogenesis [96, 98, 99]. BRD-NUT fusions have been reported in NUT midline carcinoma (66%) [100, 101], and MAML2 fusions—CRTC3-MAML2 (<1%) and CRC1-MAML2 (30–75%)—have been reported in mucoepidermoid carcinoma (MEC) [102, 103], in infantile lung MEC [104], in cervix [105], in thyroid and salivary glands and Warthin’s tumor [106], as well as in hidradenoma of the breast parenchyma [107]. Subsets of breast cancer and head and neck cancer are characterized by MYB-NFIB gene fusions (Supplementary Figure 1I) [32]. TFE3-TFEB fusions are characteristic of renal cell carcinoma [108]. ETV6-NTRK3 has been found in infantile fibrosarcoma, secretory breast carcinoma (>90%) [109], and acute myeloid leukemia [110]. As a result of this fusion, transforming occurs through activation of RAS-MAPK and PI3K-AKT and AP1 transcription complex [111]. MTAP-ANRIL fusion has been reported in melanoma [112] and CLDN18-ARHGAP26 in gastric cancer, the latter promoting loss of the epithelial phenotype in gastric cancer [113].TMPRSS2 fusions may be defining markers for prostate cancer. MYB-QKI fusion genes are defined biomarkers in angiocentric glioma [67]. Of note, ESR1-YAP and ESR1-PCDH11X fusions have crucial role in resistance to endocrine therapy, and trigger metastasis to lung in ER-positive breast cancer [114].
FUSION GENES COMMON IN MULTIPLE TYPES OF CANCER
In contrast, a number of TK fusion genes, such as ALK, ROS1, FGFR, NTRK, and RET, have been identified in multiple cancer lineages. The ALK gene rearrangement was first identified in anaplastic large cell lymphoma (50%) in 1994 [115, 116]. Soon afterwards, this rearrangement was identified in NSCLC [117, 118] papillary thyroid cancer, colorectal cancer [12, 13, 119], renal cell cancer [120], and esophageal [121], breast, and gastric cancers and acute myeloid leukemia [117], as well as in spitzoid tumors (10%) [122] (Supplementary Table 1). ALK’s most common fusion partners are the NPM1 gene in anaplastic large cell lymphoma [115] and EML4 in epithelial tumors. EML4-ALK fusion gene occurs mostly in young NSCLC patients who are non-smokers or light smokers, and it is more common among patients of Asian ancestry (7.4%) than among patients of European ancestry (3.0%) [12, 117, 123]. Similar to ALK fusions, FGFR fusions have been reported in a wide range of tumors (cholangiocarcinoma, breast cancer, prostate cancer, NSCLC, gastric adenocarcinoma, colorectal adenocarcinoma, carcinoma of unknown primary, and glioblastoma) with a variety of partners, including TACC3, PPAPDC1A, AFF3, SLC45A3 and AHCYL1, C10orf68, JAKMIP1, KIAA1598, NCALD, NOL4, NTM, PPAPDCA, TNIP2, and WHSC1 [12, 13, 117, 119, 123]. Indeed, ROS1 fusion was first identified in 1987 in a glioblastoma multiforme cell line [124]. Over the last two decades, discovery of ROS1 fusion has followed in lung adenocarcinoma (1.0-2.5%) [125], gastric cancer (0.61%), cholangiocarcinoma including biliary tract carcinoma (3.9%), colon cancer (0.85%), Spitz nevus (benign) (25.3%), atypical Spitz tumors (6.2%), and spitzoid melanomas (9.1%) [122, 126–130]. These findings raise the possibility that breakpoints may differ among the tumor types or downstream signaling pathways. On the other hand, some of the partner genes are common among fusions: CD74-NRG1 and CD74-ROS1, KIF5B-ALK and KIF5B-RET, TPM3-ALK and TPM3-NTRK1 (Supplementary Figure 1C, 1F–1H) [93].
ARE FUSION GENES THE CAUSE OR CONSEQUENCE OF TUMORIGENESIS?
The causes of structural rearrangements remain poorly understood. As aforementioned DNA rearrangements resulting in fusion genes can be triggered by multiple factors, including cellular stress, inappropriate repair or recombination of DNA (e.g., antigen receptor diversity-generating enzymes, homologous recombination, and non-homologous end joining), DNA sequence and chromatin features (e.g., chromatin modification, repetitive elements, CpG dinucleotides, non-B DNA structure), and spatial proximity (the distance between an oncogene and its translocation partner) [131]. Cellular stress includes genotoxic, oxidative, replication, and transcriptional stresses. Exposure to radiation or chemical compounds such as alkylating agents can lead to genotoxic stress affecting genome stability [131–133]. A high level of reactive oxygen species is also associated with genomic rearrangements [131]. Replication stress is associated with inefficient DNA replication, due to multiple factors. These factors include imbalance of DNA replication enzymes, low level of folate, presence of low-copy number repeats, or fragile sites [131, 134]. Replication stress can also be caused by inhibition of DNA polymerases such as that induced by aphidicolin [131]. The most prevalent examples of fusion genes caused by radiation-induced genotoxic stress are RET fusions found in irradiation-induced papillary thyroid carcinoma [135]. Dietary bioflavonoids, which are a component of natural foods and dietary supplements, cause site-specific DNA cleavage in the mixed-lineage leukemia (MLL) breakpoint cluster region in vivo, representing the most prominent example of fusion genes caused by chemical component–induced genotoxic stress [136]. Bioflavonoid-induced DNA breaks may be due to inhibition of topoisomerase II [136]. Treatment with topoisomerase II inhibitors can lead to double-stand breaks and MLL gene translocation in acute leukemia patients as well as to the development of secondary leukemia [137, 138]. Topoisomerases can both cause and repair double-strand breaks, indicating that the toxicity of current cancer treatments may also cause the formation of fusion genes and the development of secondary malignancies or resistance to therapy. The interaction of androgen receptor (genotoxic stress) and activation-induced cytidine deaminase (AID) (transcriptional stress) is likely to be the cause of the double-strand breaks that drive the specific changes in the genome and/or the chromatin remodeling that bring a TMPRSS2 into proximity with ETS genes to form gene fusions (TMPRSS2-ERG, TMPRSS2-ETV1, and SLC45A3-ETV1) in prostate cancer [24, 139, 140].
FUSION GENES IN NORMAL EPITHELIAL CELLS, DEVELOPMENTAL DISEASES, AND BENIGN TUMORS
Fusion genes occur mostly in tumor cells and have been implicated in tumorigenesis, progression or resistance to therapy. They also occur in benign tumors [122] and developmental disorders [141], as well as in normal cells [142]. One example is the JAZF1-JJAZ1 fusion, which occurs through trans-splicing in normal endometrial stromal cells [143] but not in other cells. The same chimeric gene has been observed in human endometrial stromal tumors [144]. Multiple read-through fusion transcripts, ELAVL1-TIMM44, FAM162B-ZUFSP, IFNAR2-IL10RB, INMT-FAM188B, KIAA1841-C2orf74, NFATC3-PLA2G15, SIRPB1-SIRPD, and SHANK3-ACR, also have been found in normal lung tissue from patients with lung adenocarcinoma [142]. In addition, YPEL5-PPP1CB fusion transcript has been detected in normal samples and in different hematologic malignancies [145]. Notably, fusion genes may be more common in normal epithelial cells than was at first thought. ADAMTS6-ARID1B or ARID1B-ADAMTS6 fusion gene formation occurs by translocation in patients with developmental delay; however, no fusion transcripts were formed due to the opposing transcriptional direction in both genes. However, the formation of fusion led to a small deletion in ARID1B, which may be a cause of developmental delay [141]. ADAMTS6 encodes a zinc metalloprotease, and ARID1B (AT-rich interactive domain 1B (SWI-1-like) encodes the DNA-binding subunit of the SWI/SNF chromatin-remodeling complex, which is involved in DNA replication, repair, and transcriptional regulation [146]. Even if the fusion gene lacks a chimeric transcript or protein, it still may be important. Other changes may occur during the fusion formation, such as deletion or duplication of part of a gene. Therefore, such a gene may still be relevant in development of complex diseases including cancers.
CONCLUSION AND FUTURE DIRECTIONS
It has been known that oncogenic fusion genes influence tumorigenesis through a number of different mechanisms. However, much work remains in elucidating the role of chimeric proteins in tumorigenesis. Fusion genes can compromise transcription factors, tyrosine kinases, metabolic pathways, DNA repair, and signaling pathways such as Wnt. Fusion genes that disrupt transcription factor genes can result in chimeric proteins that have enhanced, repressed, or aberrant transcriptional activity. Fusions that involve transcription factors retain the DNA-binding domain of the transcription factor and have a potent transactivation or suppression motif that induces or suppresses the transcription of target genes. This mediates tumor growth, suppresses target genes needed for normal cell differentiation, and contributes to the accumulation of immature cells. Fusion genes that disrupt tyrosine kinase can result in chimeric proteins with aberrant tyrosine kinase activity promoting downstream signaling pathways. In many kinases, the TK domain locates in the C-terminus, whereas an inhibitory domain that inactivates the kinase activity is found in the N-terminus. In many fusion proteins, the partner gene replaces the N-terminal portion of the protein, while the C-terminal TK domain is retained. Additionally, expression of the fusion product is controlled by the promoter of the partner gene. The activation of chimeric proteins that contain tyrosine kinase is due to dimerization or oligomerization domain of partner genes, which is essential for cell transformation. For example, coiled-coil domains in EML4, C20rf44, KIF5B, CCDC6, NCOA4, TACC3, and TACC1 are fused to kinase domains of ALK, RET, FGFR3, or FGFR1 in EML4-ALK, C2orf44-ALK, KIF5B-RET, CCDC6-RET, NCOA4-RET, FGFR3-TACC3, and FGFR1-TACC1 fusion genes, respectively. As a result, the fusion protein contains a protein kinase domain and a coiled-coil domain with a lack of an inactivation segment. The coiled-coil domain confers ligand-independent dimerization and oligomerization on the fusion kinase, resulting in autophosphorylation-induced constitutive kinase activity. In these cases, inhibiting the dimerization domain with drugs such as stapled peptides may provide an effective strategy for inhibiting the tyrosine kinase activity of the fusion protein. On the other hand, targeting the other distracted genes or pathways such as DNA repair and metabolic pathways may be considered in the treatment of disease. Effective, non-toxic agents that block only the rearranged protein but not normal ones are urgently needed. Identifying the factors that cause gene fusions will improve our understanding of cancer pathogenesis, lead to the development of effective anticancer agents, and help in developing cancer-preventive agents. Defining markers can serve as diagnostic biomarkers in cancer patients.
A number of questions remain to be answered. Do fusion genes activate the same downstream signaling pathways and cause the same clinical outcome in the different types of epithelial tumors? Do all fusion partners across diseases have the same response to therapeutic agents? Current knowledge about fusion genes and their effect in tumorigenesis remains only the tip of the iceberg, and there is still much more to discover.
ACKNOWLEDGMENTS
The authors thank to Michael Worley from Department of Scientific Publication for editing the manuscript.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
FUNDING
This study is supported by MD Anderson’s Cancer Center Support Grant (CCSG) to CA016672.
REFERENCES
1. Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau G, Aurias A, Thomas G. Gene fusion with an ets DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992; 359:162–165.
2. Nowell PC, Hungerford DA. Chromosome studies on normal and leukemic human leukocytes. J Natl Cancer Inst. 1960; 25:85–109.
3. Rowley JD. The critical role of chromosome translocations in human leukemias. Annu Rev Genet. 1998; 32:495–519.
4. Fusco A, Grieco M, Santoro M, Berlingieri MT, Pilotti S, Pierotti MA, Della Porta G, Vecchio G. A new oncogene in human thyroid papillary carcinomas and their lymph-nodal metastases. Nature. 1987; 328:170–172.
5. Grieco M, Santoro M, Berlingieri MT, Melillo RM, Donghi R, Bongarzone I, Pierotti MA, Della Porta G, Fusco A, Vecchio G. Ptc is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell. 1990; 60:557–563.
6. Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007; 7:233–245.
7. Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, Chaudhuri S, Guan Y, Janakiraman V, Jaiswal BS, Guillory J, Ha C, Dijkgraaf GJ, et al. Recurrent r-spondin fusions in colon cancer. Nature. 2012; 488:660–664.
8. Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008; 8:11–23.
9. Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, Massion PP, Siwak-Tapp C, Gonzalez A, Fang R, Mark EJ, Batten JM, Chen H, et al. Ros1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012; 30:863–870.
10. Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, Shefler E, Ramos AH, Stojanov P, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011; 333:1157–1160.
11. Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, Liu EM, Reichel J, Porrati P, Pellegatta S, Qiu K, Gao Z, Ceccarelli M, et al. Transforming fusions of fgfr and tacc genes in human glioblastoma. Science. 2012; 337:1231–1235.
12. Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, Curran JA, Balasubramanian S, Bloom T, Brennan KW, Donahue A, Downing SR, Frampton GM, et al. Identification of new alk and ret gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012; 18:382–384.
13. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Jänne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010; 363:1693–1703.
14. Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, Stütz AM, Korshunov A, Reimand J, Schumacher SE, Beroukhim R, Ellison DW, Marshall CR, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012; 488:49–56.
15. Teles Alves I, Hiltemann S, Hartjes T, van der Spek P, Stubbs A, Trapman J, Jenster G. Gene fusions by chromothripsis of chromosome 5q in the vcap prostate cancer cell line. Human Genet. 2013; 132:709–713.
16. Nakanishi Y, Akiyama N, Tsukaguchi T, Fujii T, Satoh Y, Ishii N, Aoki M. Mechanism of oncogenic signal activation by the novel fusion kinase fgfr3-baiap2l1. Mol Cancer Ther. 2015; 14:704–712.
17. Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, Lee C, Montie JE, Shah RB, et al. Recurrent fusion of tmprss2 and ets transcription factor genes in prostate cancer. Science. 2005; 310:644–648.
18. Gisselsson D, Pettersson L, Hoglund M, Heidenblad M, Gorunova L, Wiegant J, Mertens F, Dal Cin P, Mitelman F, Mandahl N. Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc Natl Acad Sci U S A. 2000; 97:5357–5362.
19. Ally A, Balasundaram M, Carlsen R, Chuah E, Clarke A, Dhalla N, Holt RA, Jones SJ, Lee D, Ma Y, Marra MA, Mayo M, Moore RA, et al, and Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017; 169:1327–1341.e23.
20. Ghezraoui H, Piganeau M, Renouf B, Renaud JB, Sallmyr A, Ruis B, Oh S, Tomkinson AE, Hendrickson EA, Giovannangeli C, Jasin M, Brunet E. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol Cell. 2014; 55:829–842.
21. Seki Y, Mizukami T, Kohno T. Molecular process producing oncogene fusion in lung cancer cells by illegitimate repair of DNA double-strand breaks. Biomolecules. 2015; 5:2464–2476.
22. Neves H, Ramos C, da Silva MG, Parreira A, Parreira L. The nuclear topography of abl, bcr, pml, and raralpha genes: Evidence for gene proximity in specific phases of the cell cycle and stages of hematopoietic differentiation. Blood. 1999; 93:1197–1207.
23. Nikiforova MN, Stringer JR, Blough R, Medvedovic M, Fagin JA, Nikiforov YE. Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science. 2000; 290:138–141.
24. Mani RS, Tomlins SA, Callahan K, Ghosh A, Nyati MK, Varambally S, Palanisamy N, Chinnaiyan AM. Induced chromosomal proximity and gene fusions in prostate cancer. Science. 2009; 326:1230.
25. Cerveira N, Ribeiro FR, Peixoto A, Costa V, Henrique R, Jeronimo C, Teixeira MR. Tmprss2-erg gene fusion causing erg overexpression precedes chromosome copy number changes in prostate carcinomas and paired hgpin lesions. Neoplasia. 2006; 8:826–832.
26. Kumar-Sinha C, Tomlins SA, Chinnaiyan AM. Recurrent gene fusions in prostate cancer. Nat Rev Cancer. 2008; 8:497–511.
27. Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, Menon A, Jing X, Cao Q, Han B, Yu J, Wang L, Montie JE, et al. Distinct classes of chromosomal rearrangements create oncogenic ets gene fusions in prostate cancer. Nature. 2007; 448:595–599.
28. Tomlins SA, Bjartell A, Chinnaiyan AM, Jenster G, Nam RK, Rubin MA, Schalken JA. Ets gene fusions in prostate cancer: From discovery to daily clinical practice. Eur Urol. 2009; 56:275–286.
29. Deplus R, Delliaux C, Marchand N, Flourens A, Vanpouille N, Leroy X, de Launoit Y, Duterque-Coquillaud M. TMPRSS2-ERG fusion promotes prostate cancer metastases in bone. Oncotarget. 2017; 8:11827–40. https://doi.org/10.18632/oncotarget.14399.
30. Paulo P, Barros-Silva JD, Ribeiro FR, Ramalho-Carvalho J, Jeronimo C, Henrique R, Lind GE, Skotheim RI, Lothe RA, Teixeira MR. Fli1 is a novel ets transcription factor involved in gene fusions in prostate cancer. Genes Chromosomes Cancer. 2012; 51:240–249.
31. Hermans KG, Bressers AA, van der Korput HA, Dits NF, Jenster G, Trapman J. Two unique novel prostate-specific and androgen-regulated fusion partners of etv4 in prostate cancer. Cancer Res. 2008; 68:3094–3098.
32. Persson M, Andren Y, Mark J, Horlings HM, Persson F, Stenman G. Recurrent fusion of myb and nfib transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci U S A. 2009; 106:18740–18744.
33. Fehr A, Kovacs A, Loning T, Frierson H, Jr., van den Oord J, Stenman G. The myb-nfib gene fusion-a novel genetic link between adenoid cystic carcinoma and dermal cylindroma. J Pathol. 2011; 224:322–327.
34. Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and hmga2 enhances oncogenic transformation. Science. 2007; 315:1576–1579.
35. Geurts JM, Schoenmakers EF, Roijer E, Astrom AK, Stenman G, van de Ven WJ. Identification of nfib as recurrent translocation partner gene of hmgic in pleomorphic adenomas. Oncogene. 1998; 16:865–872.
36. Linehan WM, Spellman PT, Ricketts CJ, Creighton CJ, Fei SS, Davis C, Wheeler DA, Murray BA, Schmidt L, Vocke CD, Peto M, Al Mamun AA, Shinbrot E, et al, and Cancer Genome Atlas Research Network. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med. 2016; 374:135–145.
37. Nussenzweig A, Nussenzweig MC. Origin of chromosomal translocations in lymphoid cancer. Cell. 2010; 141:27–38.
38. Browman DT, Hoegg MB, Robbins SM. The spfh domain-containing proteins: More than lipid raft markers. Trends Cell Biol. 2007; 17:394–402.
39. Chai J, Wu Q, Shiozaki E, Srinivasula SM, Alnemri ES, Shi Y. Crystal structure of a procaspase-7 zymogen: Mechanisms of activation and substrate binding. Cell. 2001; 107:399–407.
40. Mateja A, Cierpicki T, Paduch M, Derewenda ZS, Otlewski J. The dimerization mechanism of lis1 and its implication for proteins containing the lish motif. J Mol Biol. 2006; 357:621–631.
41. Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJ, Evans PR, McMahon HT. Bar domains as sensors of membrane curvature: The amphiphysin bar structure. Science. 2004; 303:495–499.
42. Ota K, Azuma K, Kawahara A, Hattori S, Iwama E, Tanizaki J, Harada T, Matsumoto K, Takayama K, Takamori S, Kage M, Hoshino T, Nakanishi Y, Okamoto I. Induction of pd-l1 expression by the eml4-alk oncoprotein and downstream signaling pathways in non-small cell lung cancer. Clin Cancer Res. 2015; 21:4014–4021.
43. Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, Wang HY, Wysocka M, Cheng M, Ruggeri BA, Wasik MA. Oncogenic kinase npm/alk induces through stat3 expression of immunosuppressive protein cd274 (pd-l1, b7-h1). Proc Natl Acad Sci U S A. 2008; 105:20852–20857.
44. Hood FE, Royle SJ. Pulling it together: The mitotic function of tacc3. Bioarchitecture. 2011; 1:105–109.
45. Peset I, Vernos I. The tacc proteins: Tacc-ling microtubule dynamics and centrosome function. Trends Cell Biol. 2008; 18:379–388.
46. Williams SV, Hurst CD, Knowles MA. Oncogenic fgfr3 gene fusions in bladder cancer. Hum Mol Genet. 2013; 22:795–803.
47. Parker BC, Annala MJ, Cogdell DE, Granberg KJ, Sun Y, Ji P, Li X, Gumin J, Zheng H, Hu L, Yli-Harja O, Haapasalo H, Visakorpi T, et al. The tumorigenic fgfr3-tacc3 gene fusion escapes mir-99a regulation in glioblastoma. J Clin Invest. 2013; 123:855–865.
48. Wu YM, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, Lonigro RJ, Vats P, Wang R, Lin SF, Cheng AJ, Kunju LP, Siddiqui J, et al. Identification of targetable fgfr gene fusions in diverse cancers. Cancer Discov. 2013; 3:636–647.
49. Chao BH, Briesewitz R, Villalona-Calero MA. Ret fusion genes in non-small-cell lung cancer. J Clin Oncol. 2012; 30:4439–4441.
50. Ju YS, Lee WC, Shin JY, Lee S, Bleazard T, Won JK, Kim YT, Kim JI, Kang JH, Seo JS. A transforming kif5b and ret gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res. 2012; 22:436–445.
51. Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, Sakamoto H, Tsuta K, Furuta K, Shimada Y, Iwakawa R, Ogiwara H, Oike T, et al. Kif5b-ret fusions in lung adenocarcinoma. Nat Med. 2012; 18:375–377.
52. Palanisamy N, Ateeq B, Kalyana-Sundaram S, Pflueger D, Ramnarayanan K, Shankar S, Han B, Cao Q, Cao X, Suleman K, Kumar-Sinha C, Dhanasekaran SM, Chen YB, et al. Rearrangements of the raf kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med. 2010; 16:793–798.
53. Zhang Y, Gong M, Yuan H, Park HG, Frierson HF, Li H. Chimeric transcript generated by cis-splicing of adjacent genes regulates prostate cancer cell proliferation. Cancer Discov. 2012; 2:598–607.
54. Mathur M, Das S, Samuels HH. Psf-tfe3 oncoprotein in papillary renal cell carcinoma inactivates tfe3 and p53 through cytoplasmic sequestration. Oncogene. 2003; 22:5031–5044.
55. Mathur M, Tucker PW, Samuels HH. Psf is a novel corepressor that mediates its effect through sin3a and the DNA binding domain of nuclear hormone receptors. Mol Cell Biol. 2001; 21:2298–2311.
56. Kim S, Lim B, Kim J. Ews-oct-4b, an alternative ews-oct-4 fusion gene, is a potent oncogene linked to human epithelial tumours. Br J Cancer. 2010; 102:436–446.
57. Lee J, Kim JY, Kang IY, Kim HK, Han YM, Kim J. The ews-oct-4 fusion gene encodes a transforming gene. Biochem J. 2007; 406:519–526.
58. Kalvala A, Gao L, Aguila B, Dotts K, Rahman M, Nana-Sinkam SP, Zhou X, Wang QE, Amann J, Otterson GA, Villalona-Calero MA, Duan W. Rad51c-atxn7 fusion gene expression in colorectal tumors. Mol Cancer. 2016; 15:47.
59. Li F, Fang Z, Zhang J, Li C, Liu H, Xia J, Zhu H, Guo C, Qin Z, Li F, Han X, Wang Y, Feng Y, et al. Identification of tra2b-dnah5 fusion as a novel oncogenic driver in human lung squamous cell carcinoma. Cell Res. 2016; 26:1149–1164.
60. Ciampi R, Knauf JA, Kerler R, Gandhi M, Zhu Z, Nikiforova MN, Rabes HM, Fagin JA, Nikiforov YE. Oncogenic akap9-braf fusion is a novel mechanism of mapk pathway activation in thyroid cancer. J Clin Invest. 2005; 115:94–101.
61. Dibb NJ, Dilworth SM, Mol CD. Switching on kinases: Oncogenic activation of braf and the pdgfr family. Nat Rev Cancer. 2004; 4:718–727.
62. Broeker PL, Super HG, Thirman MJ, Pomykala H, Yonebayashi Y, Tanabe S, Zeleznik-Le N, Rowley JD. Distribution of 11q23 breakpoints within the mll breakpoint cluster region in de novo acute leukemia and in treatment-related acute myeloid leukemia: Correlation with scaffold attachment regions and topoisomerase ii consensus binding sites. Blood. 1996; 87:1912–1922.
63. Cimino G, Rapanotti MC, Biondi A, Elia L, Lo Coco F, Price C, Rossi V, Rivolta A, Canaani E, Croce CM, Mandelli F, Greaves M. Infant acute leukemias show the same biased distribution of all1 gene breaks as topoisomerase ii related secondary acute leukemias. Cancer Res. 1997; 57:2879–2883.
64. Rossler T, Marschalek R. An alternative splice process renders the mll protein either into a transcriptional activator or repressor. Die Pharmazie. 2013; 68:601–607.
65. Wang J, Muntean AG, Wu L, Hess JL. A subset of mixed lineage leukemia proteins has plant homeodomain (phd)-mediated e3 ligase activity. J Biol Chem. 2012; 287:43410–43416.
66. Robinson DR, Wu YM, Kalyana-Sundaram S, Cao X, Lonigro RJ, Sung YS, Chen CL, Zhang L, Wang R, Su F, Iyer MK, Roychowdhury S, Siddiqui J, et al. Identification of recurrent nab2-stat6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat Genet. 2013; 45:180–185.
67. Bandopadhayay P, Ramkissoon LA, Jain P, Bergthold G, Wala J, Zeid R, Schumacher SE, Urbanski L, O’Rourke R, Gibson WJ, Pelton K, Ramkissoon SH, Han HJ, et al. Myb-qki rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet. 2016; 48:273–282.
68. Wu J, Zhou L, Tonissen K, Tee R, Artzt K. The quaking i-5 protein (qki-5) has a novel nuclear localization signal and shuttles between the nucleus and the cytoplasm. J Biol Chem. 1999; 274:29202–29210.
69. Ramsay RG, Gonda TJ. Myb function in normal and cancer cells. Nat Rev Cancer. 2008; 8:523–534.
70. Gonda TJ, Buckmaster C, Ramsay RG. Activation of c-myb by carboxy-terminal truncation: Relationship to transformation of murine haemopoietic cells in vitro. EMBO J. 1989; 8:1777–1783.
71. Friedrich VL Jr. The myelin deficit in quacking mice. Brain Res. 1974; 82:168–172.
72. Danan-Gotthold M, Golan-Gerstl R, Eisenberg E, Meir K, Karni R, Levanon EY. Identification of recurrent regulated alternative splicing events across human solid tumors. Nucleic Acids Res. 2015; 43:5130–5144.
73. Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA, Goodall GJ. The rna binding protein quaking regulates formation of circrnas. Cell. 2015; 160:1125–1134.
74. Hayakawa-Yano Y, Suyama S, Nogami M, Yugami M, Koya I, Furukawa T, Zhou L, Abe M, Sakimura K, Takebayashi H, Nakanishi A, Okano H, Yano M. An rna-binding protein, qki5, regulates embryonic neural stem cells through pre-mrna processing in cell adhesion signaling. Genes Dev. 2017; 31:1910–1925.
75. Levinson S, Cagan RL. Drosophila cancer models identify functional differences between ret fusions. Cell Rep. 2016; 16:3052–3061.
76. Chang H, Sung JH, Moon SU, Kim HS, Kim JW, Lee JS. Egf induced ret inhibitor resistance in ccdc6-ret lung cancer cells. Yonsei Med J. 2017; 58:9–18.
77. Croyle M, Akeno N, Knauf JA, Fabbro D, Chen X, Baumgartner JE, Lane HA, Fagin JA. Ret/ptc-induced cell growth is mediated in part by epidermal growth factor receptor (egfr) activation: Evidence for molecular and functional interactions between ret and egfr. Cancer Res. 2008; 68:4183–4191.
78. Vaishnavi A, Schubert L, Rix U, Marek LA, Le AT, Keysar SB, Glogowska MJ, Smith MA, Kako S, Sumi NJ, Davies KD, Ware KE, Varella-Garcia M, et al. Egfr mediates responses to small-molecule drugs targeting oncogenic fusion kinases. Cancer Res. 2017; 77:3551–3563.
79. Wei L, Wang J, Lampert E, Schlanger S, DePriest AD, Hu Q, Gomez EC, Murakam M, Glenn ST, Conroy J, Morrison C, Azabdaftari G, Mohler JL, et al. Intratumoral and intertumoral genomic heterogeneity of multifocal localized prostate cancer impacts molecular classifications and genomic prognosticators. Eur Urol. 2017; 71:183–192.
80. Lu H, Villafane N, Dogruluk T, Grzeskowiak CL, Kong K, Tsang YH, Zagorodna O, Pantazi A, Yang L, Neill NJ, Kim YW, Creighton CJ, Verhaak RG, et al. Engineering and functional characterization of fusion genes identifies novel oncogenic drivers of cancer. Cancer Res. 2017; 77:3502–3512.
81. Gao Q, Liang WW, Foltz SM, Mutharasu G, Jayasinghe RG, Cao S, Liao WW, Reynolds SM, Wyczalkowski MA, Yao L, Yu L, Sun SQ, Chen K, et al, and Fusion Analysis Working Group, and Cancer Genome Atlas Research Network. Driver fusions and their implications in the development and treatment of human cancers. Cell Rep. 2018; 23:227–238.
82. Cui YX, Kerby A, McDuff FK, Ye H, Turner SD. Npm-alk inhibits the p53 tumor suppressor pathway in an mdm2 and jnk-dependent manner. Blood. 2009; 113:5217–5227.
83. Wiederschain D, Kawai H, Gu J, Shilatifard A, Yuan ZM. Molecular basis of p53 functional inactivation by the leukemic protein mll-ell. Mol Cell Biol. 2003; 23:4230–4246.
84. Choi WI, Yoon JH, Kim MY, Koh DI, Licht JD, Kim K, Hur MW. Promyelocytic leukemia zinc finger-retinoic acid receptor alpha (plzf-raralpha), an oncogenic transcriptional repressor of cyclin-dependent kinase inhibitor 1a (p21waf/cdkn1a) and tumor protein p53 (tp53) genes. J Biol Chem. 2014; 289:18641–18656.
85. Slupianek A, Schmutte C, Tombline G, Nieborowska-Skorska M, Hoser G, Nowicki MO, Pierce AJ, Fishel R, Skorski T. Bcr/abl regulates mammalian reca homologs, resulting in drug resistance. Mol Cell. 2001; 8:795–806.
86. Chatterjee P, Choudhary GS, Alswillah T, Xiong X, Heston WD, Magi-Galluzzi C, Zhang J, Klein EA, Almasan A. The tmprss2-erg gene fusion blocks xrcc4-mediated nonhomologous end-joining repair and radiosensitizes prostate cancer cells to parp inhibition. Mol Cancer Ther. 2015; 14:1896–1906.
87. Lunardi A, Varmeh S, Chen M, Taulli R, Guarnerio J, Ala U, Seitzer N, Ishikawa T, Carver BS, Hobbs RM, Quarantotti V, Ng C, Berger AH, et al. Suppression of chk1 by ets family members promotes DNA damage response bypass and tumorigenesis. Cancer Discov. 2015; 5:550–563.
88. Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbé DP, Gomez EC, Wang J, Long HW, Xu B, Brown M, et al. Rb1 and trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017; 355:78–83.
89. Martin P, Liu YN, Pierce R, Abou-Kheir W, Casey O, Seng V, Camacho D, Simpson RM, Kelly K. Prostate epithelial pten/tp53 loss leads to transformation of multipotential progenitors and epithelial to mesenchymal transition. Am J Pathol 2011; 179:422–435.
90. Zou M, Toivanen R, Mitrofanova A, Floch N, Hayati S, Sun Y, Le Magnen C, Chester D, Mostaghel EA, Califano A, Rubin MA, Shen MM, Abate-Shen C. Transdifferentiation as a mechanism of treatment resistance in a mouse model of castration-resistant prostate cancer. Cancer Discov. 2017; 7:736–749.
91. Blee AM, He Y, Yang Y, Ye Z, Yan Y, Pan Y, Ma T, Dugdale J, Kuehn E, Kohli M, Jimenez R, Chen Y, Xu W, et al. Tmprss2-erg controls luminal epithelial lineage and antiandrogen sensitivity in pten and tp53-mutated prostate cancer. Clin Cancer Res. 2018; 24:4551–4565.
92. Salzman J, Marinelli RJ, Wang PL, Green AE, Nielsen JS, Nelson BH, Drescher CW, Brown PO. Esrra-c11orf20 is a recurrent gene fusion in serous ovarian carcinoma. PLoS Biol. 2011; 9:e1001156.
93. Shin DH, Lee D, Hong DW, Hong SH, Hwang JA, Lee BI, You HJ, Lee GK, Kim IH, Lee YS, Han JY. Oncogenic function and clinical implications of slc3a2-nrg1 fusion in invasive mucinous adenocarcinoma of the lung. Oncotarget. 2016; 7:69450–65. https://doi.org/10.18632/oncotarget.11913.
94. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, et al, and TCGA Research Network. The somatic genomic landscape of glioblastoma. Cell. 2013; 155:462–477.
95. Cheng Y, Wang Y, Li J, Chang I, Wang CY. A novel read-through transcript jmjd7-pla2g4b regulates head and neck squamous cell carcinoma cell proliferation and survival. Oncotarget. 2017; 8:1972–1982.
96. Graham RP, Jin L, Knutson DL, Kloft-Nelson SM, Greipp PT, Waldburger N, Roessler S, Longerich T, Roberts LR, Oliveira AM, Halling KC, Schirmacher P, Torbenson MS. Dnajb1-prkaca is specific for fibrolamellar carcinoma. Mod Pathol. 2015; 28:822–829.
97. Fink AL. Chaperone-mediated protein folding. Physiol Rev. 1999; 79:425–449.
98. Ermisch M, Firla B, Steinhilber D. Protein kinase a activates and phosphorylates roralpha4 in vitro and takes part in roralpha activation by camk-iv. Biochem Biophys Res Commun. 2011; 408:442–446.
99. Mehta SL, Mendelev N, Kumari S, Andy Li P. Overexpression of human selenoprotein h in neuronal cells enhances mitochondrial biogenesis and function through activation of protein kinase a, protein kinase b, and cyclic adenosine monophosphate response element-binding protein pathway. Int J Biochem Cell Biol. 2013; 45:604–611.
100. French CA, Miyoshi I, Aster JC, Kubonishi I, Kroll TG, Dal Cin P, Vargas SO, Perez-Atayde AR, Fletcher JA. Brd4 bromodomain gene rearrangement in aggressive carcinoma with translocation t(15;19). Am J Pathol. 2 001; 159:1987–92.
101. French CA, Ramirez CL, Kolmakova J, Hickman TT, Cameron MJ, Thyne ME, Kutok JL, Toretsky JA, Tadavarthy AK, Kees UR, Fletcher JA, Aster JC. Brd-nut oncoproteins: A family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene. 2008; 27:2237–2242.
102. Fehr A, Roser K, Heidorn K, Hallas C, Loning T, Bullerdiek J. A new type of maml2 fusion in mucoepidermoid carcinoma. Genes Chromosomes Cancer. 2008; 47:203–206.
103. Tonon G, Modi S, Wu L, Kubo A, Coxon AB, Komiya T, O’Neil K, Stover K, El-Naggar A, Griffin JD, Kirsch IR, Kaye FJ. T(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a notch signaling pathway. Nat Genet. 2003; 33:208–213.
104. Serra A, Schackert HK, Mohr B, Weise A, Liehr T, Fitze G. T(11;19)(q21;p12~p13.11) and mect1-maml2 fusion transcript expression as a prognostic marker in infantile lung mucoepidermoid carcinoma. J Pediatr Surg. 2007; 42:E23–29.
105. Lennerz JK, Perry A, Mills JC, Huettner PC, Pfeifer JD. Mucoepidermoid carcinoma of the cervix: Another tumor with the t(11;19)-associated crtc1-maml2 gene fusion. Am J Surg Pathol. 2009; 33:835–843.
106. Tirado Y, Williams MD, Hanna EY, Kaye FJ, Batsakis JG, El-Naggar AK. Crtc1/maml2 fusion transcript in high grade mucoepidermoid carcinomas of salivary and thyroid glands and warthin’s tumors: Implications for histogenesis and biologic behavior. Genes Chromosomes Cancer. 2007; 46:708–715.
107. Kazakov DV, Vanecek T, Belousova IE, Mukensnabl P, Kollertova D, Michal M. Skin-type hidradenoma of the breast parenchyma with t(11;19) translocation: Hidradenoma of the breast. Am J Dermatopathol. 2007; 29:457–461.
108. Davis IJ, Hsi BL, Arroyo JD, Vargas SO, Yeh YA, Motyckova G, Valencia P, Perez-Atayde AR, Argani P, Ladanyi M, Fletcher JA, Fisher DE. Cloning of an alpha-tfeb fusion in renal tumors harboring the t(6;11)(p21;q13) chromosome translocation. Proc Natl Acad Sci U S A. 2003; 100:6051–6056.
109. Tognon C, Knezevich SR, Huntsman D, Roskelley CD, Melnyk N, Mathers JA, Becker L, Carneiro F, MacPherson N, Horsman D, Poremba C, Sorensen PH. Expression of the etv6-ntrk3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell. 2002; 2:367–376.
110. Lannon CL, Sorensen PH. Etv6-ntrk3: A chimeric protein tyrosine kinase with transformation activity in multiple cell lineages. Semin Cancer Biol. 2005; 15:215–223.
111. Li Z, Tognon CE, Godinho FJ, Yasaitis L, Hock H, Herschkowitz JI, Lannon CL, Cho E, Kim SJ, Bronson RT, Perou CM, Sorensen PH, Orkin SH. Etv6-ntrk3 fusion oncogene initiates breast cancer from committed mammary progenitors via activation of ap1 complex. Cancer Cell. 2007; 12:542–558.
112. Xie H, Rachakonda PS, Heidenreich B, Nagore E, Sucker A, Hemminki K, Schadendorf D, Kumar R. Mapping of deletion breakpoints at the cdkn2a locus in melanoma: Detection of mtap-anril fusion transcripts. Oncotarget. 2016; 7:16490–504. https://doi.org/10.18632/oncotarget.7503.
113. Yao F, Kausalya JP, Sia YY, Teo AS, Lee WH, Ong AG, Zhang Z, Tan JH, Li G, Bertrand D, Liu X, Poh HM, Guan P, et al. Recurrent fusion genes in gastric cancer: Cldn18-arhgap26 induces loss of epithelial integrity. Cell Rep. 2015; 12:272–285.
114. Lei JT, Shao J, Zhang J, Iglesia M, Chan DW, Cao J, Anurag M, Singh P, He X, Kosaka Y, Matsunuma R, Crowder R, Hoog J, et al. Functional annotation of esr1 gene fusions in estrogen receptor-positive breast cancer. Cell Rep. 2018; 24:1434–1444.
115. Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. Fusion of a kinase gene, alk, to a nucleolar protein gene, npm, in non-hodgkin’s lymphoma. Science. 1994; 263:1281–1284.
116. Shiota M, Fujimoto J, Semba T, Satoh H, Yamamoto T, Mori S. Hyperphosphorylation of a novel 80 kda protein-tyrosine kinase similar to ltk in a human ki-1 lymphoma cell line, ams3. Oncogene. 1994; 9:1567–1574.
117. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, et al. Identification of the transforming eml4-alk fusion gene in non-small-cell lung cancer. Nature. 2007; 448:561–566.
118. Togashi Y, Soda M, Sakata S, Sugawara E, Hatano S, Asaka R, Nakajima T, Mano H, Takeuchi K. Klc1-alk: A novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue only. PLoS One. 2012; 7:e31323.
119. Lin E, Li L, Guan Y, Soriano R, Rivers CS, Mohan S, Pandita A, Tang J, Modrusan Z. Exon array profiling detects eml4-alk fusion in breast, colorectal, and non-small cell lung cancers. Mol Cancer Res. 2009; 7:1466–1476.
120. Debelenko LV, Raimondi SC, Daw N, Shivakumar BR, Huang D, Nelson M, Bridge JA. Renal cell carcinoma with novel vcl-alk fusion: New representative of alk-associated tumor spectrum. Mod Pathol. 2011; 24:430–442.
121. Du XL, Hu H, Lin DC, Xia SH, Shen XM, Zhang Y, Luo ML, Feng YB, Cai Y, Xu X, Han YL, Zhan QM, Wang MR. Proteomic profiling of proteins dysregulted in chinese esophageal squamous cell carcinoma. J Mol Med 2007; 85:863–875.
122. Wiesner T, He J, Yelensky R, Esteve-Puig R, Botton T, Yeh I, Lipson D, Otto G, Brennan K, Murali R, Garrido M, Miller VA, Ross JS, et al. Kinase fusions are frequent in spitz tumours and spitzoid melanomas. Nat Commun. 2014; 5:3116.
123. Martelli MP, Sozzi G, Hernandez L, Pettirossi V, Navarro A, Conte D, Gasparini P, Perrone F, Modena P, Pastorino U, Carbone A, Fabbri A, Sidoni A, et al. Eml4-alk rearrangement in non-small cell lung cancer and non-tumor lung tissues. Am J Pathol. 2009; 174:661–670.
124. Birchmeier C, Sharma S, Wigler M. Expression and rearrangement of the ros1 gene in human glioblastoma cells. Proc Natl Acad Sci U S A. 1987; 84:9270–9274.
125. Takeuchi K, Choi YL, Togashi Y, Soda M, Hatano S, Inamura K, Takada S, Ueno T, Yamashita Y, Satoh Y, Okumura S, Nakagawa K, Ishikawa Y, Mano H. Kif5b-alk, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for alk-positive lung cancer. Clin Cancer Res. 2009; 15:3143–3149.
126. Aisner DL, Nguyen TT, Paskulin DD, Le AT, Haney J, Schulte N, Chionh F, Hardingham J, Mariadason J, Tebbutt N, Doebele RC, Weickhardt AJ, Varella-Garcia M. Ros1 and alk fusions in colorectal cancer, with evidence of intratumoral heterogeneity for molecular drivers. Mol Cancer Res. 2014; 12:111–118.
127. Lee J, Lee SE, Kang SY, Do IG, Lee S, Ha SY, Cho J, Kang WK, Jang J, Ou SH, Kim KM. Identification of ros1 rearrangement in gastric adenocarcinoma. Cancer. 2013; 119:1627–1635.
128. Pan Y, Zhang Y, Li Y, Hu H, Wang L, Li H, Wang R, Ye T, Luo X, Zhang Y, Li B, Cai D, Shen L, et al. Alk, ros1 and ret fusions in 1139 lung adenocarcinomas: A comprehensive study of common and fusion pattern-specific clinicopathologic, histologic and cytologic features. Lung Cancer. 2014; 84:121–126.
129. Peraldo Neia C, Cavalloni G, Balsamo A, Venesio T, Napoli F, Sassi F, Martin V, Frattini M, Aglietta M, Leone F. Screening for the fig-ros1 fusion in biliary tract carcinomas by nested pcr. Genes Chromosomes Cancer. 2014; 53:1033–1040.
130. Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, Hu Y, Tan Z, Stokes M, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007; 131:1190–1203.
131. Mani RS, Chinnaiyan AM. Triggers for genomic rearrangements: Insights into genomic, cellular and environmental influences. Nat Rev Genet. 2010; 11:819–829.
132. Bartkova J, Hamerlik P, Stockhausen MT, Ehrmann J, Hlobilkova A, Laursen H, Kalita O, Kolar Z, Poulsen HS, Broholm H, Lukas J, Bartek J. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene. 2010; 29:5095–5102.
133. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre’ M, Nucifo ro PG, Bensimon A, Maestro R, Pelicci PG, d’Adda di Fagagna F. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006; 444:638–642.
134. Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011; 145:435–446.
135. Fugazzola L, Pilotti S, Pinchera A, Vorontsova TV, Mondellini P, Bongarzone I, Greco A, Astakhova L, Butti MG, Demidchik EP. Oncogenic rearrangements of the ret proto-oncogene in papillary thyroid carcinomas from children exposed to the chernobyl nuclear accident. Cancer Res. 1995; 55:5617–5620.
136. Strick R, Strissel PL, Borgers S, Smith SL, Rowley JD. Dietary bioflavonoids induce cleavage in the mll gene and may contribute to infant leukemia. Proc Natl Acad Sci U S A. 2000; 97:4790–4795.
137. Blanco JG, Dervieux T, Edick MJ, Mehta PK, Rubnitz JE, Shurtleff S, Raimondi SC, Behm FG, Pui CH, Relling MV. Molecular emergence of acute myeloid leukemia during treatment for acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2001; 98:10338–10343.
138. Pedersen-Bjergaard J, Andersen MT, Andersen MK. Genetic pathways in the pathogenesis of therapy-related myelodysplasia and acute myeloid leukemia. Hematology Am Soc Hematol Educ Program. 2007; 392–397.
139. Haffner MC, Aryee MJ, Toubaji A, Esopi DM, Albadine R, Gurel B, Isaacs WB, Bova GS, Liu W, Xu J, Meeker AK, Netto G, De Marzo AM, et al. Androgen-induced top2b-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet. 2010; 42:668–675.
140. Lin C, Yang L, Tanasa B, Hutt K, Ju BG, Ohgi K, Zhang J, Rose DW, Fu XD, Glass CK, Rosenfeld MG. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009; 139:1069–1083.
141. Malli T, Duba HC, Erdel M, Marschon R, Kranewitter W, Deutschbauer S, Kralik J, Diel E, Guenther B, Mueller D, Webersinke G. Disruption of the arid1b and adamts6 loci due to a t(5;6)(q12.3;q25.3) in a patient with developmental delay. Am J Med Genet Part A. 2014; 164A:3126–3131.
142. Pintarelli G, Dassano A, Cotroneo CE, Galvan A, Noci S, Piazza R, Pirola A, Spinelli R, Incarbone M, Palleschi A, Rosso L, Santambrogio L, Dragani TA, Colombo F. Read-through transcripts in normal human lung parenchyma are down-regulated in lung adenocarcinoma. Oncotarget. 2016; 7:27889–98. https://doi.org/10.18632/oncotarget.8556.
143. Li H, Wang J, Mor G, Sklar J. A neoplastic gene fusion mimics trans-splicing of rnas in normal human cells. Science. 2008; 321:1357–1361.
144. Koontz JI, Soreng AL, Nucci M, Kuo FC, Pauwels P, van Den Berghe H, Dal Cin P, Fletcher JA, Sklar J. Frequent fusion of the jazf1 and jjaz1 genes in endometrial stromal tumors. Proc Natl Acad Sci U S A. 2001; 98:6348–6353.
145. Vandepoele K, Philippe J, Denys B. The ypel5-ppp1cb fusion transcript is detected in different hematological malignancies and in normal samples. Leuk Res Rep. 2015; 4:51–54.
146. Muchardt C, Yaniv M. When the swi/snf complex remodels. The cell cycle. Oncogene. 2001; 20:3067–3075.