
INTRODUCTION
The MAPK signaling pathway (i.e., RAS-RAF-MEK-ERK pathway) regulates cellular proliferation, survival, and differentiation and contributes to the pathogenesis of melanoma [1]. Multiple genetic changes can lead to hyperactivation of this pathway, and are involved in the pathogenesis of various solid tumor types, including melanoma and thyroid, colorectal, and ovarian cancer [2–5]. Constitutive MAPK pathway activation in cancer can occur through several mechanisms, most frequently via mutations in BRAF or RAS. Activating NRAS and BRAF mutations are present in approximately 20% [6, 7] and 50% [6, 8] of primary cutaneous melanomas, respectively.
A number of therapies that directly target the MAPK pathway have been approved for BRAF-mutated melanoma, including the BRAFV600- specific inhibitors vemurafenib (single agent), and dabrafenib (single agent or in combination with trametinib), and mitogen-activated protein kinase (MEK) inhibitors trametinib (single agent or in combination with dabrafenib) and cobimetinib (in combination with vemurafenib) [9–15]. The combination of BRAF and MEK inhibitor therapies has shown significantly improved benefit over BRAF inhibitor monotherapy in phase III clinical trials [16–18]. In addition to the nonspecific immunotherapies, three immune checkpoint modulators, ipilimumab (anti-cytotoxic T-lymphocyte associated antigen 4 [CTLA-4]), nivolumab (anti–programmed cell death protein 1 [PD-1]), and pembrolizumab (anti–PD-1), have been approved for the treatment of melanoma [19].
Therapies targeting either mutated BRAF or NRAS alone can encounter a number of challenges, including resistance to BRAF inhibitors, which occurs frequently, mostly through reactivation of the MAPK pathway; common paths to resistance include BRAF amplification or alternative splicing and mutations in RAS, MAP2K1, and CDKN2A [20, 21], among other mechanisms.
In contrast to BRAF-mutated melanoma, no approved targeted therapies exist for NRAS-mutated melanoma, but positive phase III data on PFS have been reported for a study comparing the efficacy of binimetinib (MEK162) versus dacarbazine in unresectable or metastatic NRAS-mutated melanoma (NEMO study) [22]. Binimetinib is an oral, selective, ATP-uncompetitive inhibitor of MEK 1 and MEK 2 [23].
Binimetinib has shown promising results in BRAF-mutated melanoma, both alone [24] and in combination [25]. In addition, preclinical MEK inhibitor activity has been shown in BRAF-mutated melanoma [26]. The safety profile of binimetinib and preliminary signs of antitumor activity were shown in a phase I trial in patients with advanced solid tumors [27, 28]. This open-label phase II study assessed the use of binimetinib in patients with BRAFV600- or NRAS-mutated advanced melanoma. The efficacy and safety results from an earlier data cut-off of February 29, 2012 (smaller subgroups of patients with NRAS- or BRAF-mutated melanoma) have been previously reported [24]. No patients had a complete response, and 6 of 30 patients (20%) with NRAS-mutated melanoma (3 confirmed) and 8 of 41 patients (20%) with BRAF-mutated melanoma (2 confirmed) had a partial response. Binimetinib was the first targeted therapy to show activity in patients with NRAS-mutated melanoma.
In this study (NCT01320085), biomarker data from binimetinib-treated patients with NRAS- and BRAF-mutated melanoma were analysed as prespecified secondary and exploratory objectives to investigate the extent of MAPK pathway inhibition and further genetic pathway alterations, in order to find potential predictive markers of response to binimetinib. Among others, dual-specificity phosphatase 6 (DUSP6) and phosphorylated extracellular signal-regulated kinase (pERK) are known/predicted biomarkers of MAPK inhibition and could potentially predict the extent of response to treatment with binimetinib [20, 29].
RESULTS
Patient disposition and characteristics
At the trial data cut-off date (7 January 2014), a total of 183 patients were enrolled. Sixty-six patients with BRAF mutations were treated: 41 received binimetinib 45 mg twice daily (BID), and 25 received binimetinib 60 mg BID (subsequently reduced to 45 mg BID). A total of 117 patients with NRAS mutations received binimetinib 45 mg BID (Table 1). Patient demographics and disease characteristics are shown in Supplementary Table 1.
Table 1: Patient disposition
BRAF-mutant |
NRAS-mutant |
All patients |
||
|---|---|---|---|---|
Binimetinib |
Binimetinib |
Binimetinib |
N = 183 |
|
Patients treated, n (%) |
||||
Treatment discontinued |
41 (100) |
23 (92.0) |
104 (88.9) |
168 (91.8) |
Treatment ongoinga |
0 |
2 (8.0) |
13 (11.1) |
15 (8.2) |
Primary reason for end of treatment, n (%) |
||||
Adverse event(s)b |
12 (29.3) |
5 (20.0) |
14 (12.0) |
31 (16.9) |
Patient withdrew consent |
2 (4.9) |
1 (4.0) |
4 (3.4) |
7 (3.8) |
Disease progression |
26 (63.4) |
16 (64.0) |
86 (73.5) |
128 (69.9) |
Protocol deviation |
1 (2.4) |
1 (4.0) |
0 |
2 (1.1) |
Duration of exposure, median (range), weeks |
9.6 (1.1–26.6) |
8.0 (2.0–102) |
15.9 (0.3–87.9) |
11.6 (0.3–102.0) |
BRAF mutation status, n (%)c |
||||
None (no wild-type mutation detected) |
0 |
0 |
3 (2.6) |
|
V600E |
34 (82.9) |
19 (76.0) |
0 |
|
V600K |
5 (12.2) |
1 (4.0) |
0 |
– |
Unknown mutation |
1 (2.4)f |
2 (8.0)f |
0 |
|
Other (mutations other than V600E/K)d |
1 (2.4)g |
0 |
0 |
|
Missing (no V600 BRAF mutation data)e |
0 |
3 (12.0)f |
114 (97.4) |
|
NRAS mutation status, n (%)c |
||||
None (no mutation detected) |
0 |
5 (20.0) |
4 (3.4)h |
|
Q61 |
0 |
0 |
100 (85.5) |
|
G12/13 |
0 |
0 |
2 (1.7) |
– |
Unknown mutationc |
1 (2.4) |
0 |
1 (0.9)i |
|
Missing (no NRAS mutation data) |
40 (97.6) |
20 (80.0) |
10 (8.5)j |
|
Clinical activityk [24] |
(n = 35) |
NR |
(n = 28) |
|
DCR, n (%) |
21 (60) |
NR |
19 (68) |
|
aTreatment ongoing at the time of the cut-off (Jan 7, 2014)
bIncludes fatal case with liver failure in the BRAF-mutated 60-mg treatment group;
cAny other mutation not known; known mutation includes all Q61, A59T, A11T, G12V, G13R;
dAny other known mutation (L597, D594, G606, K60);
eAnalysis results are missing;
fV600E according to local laboratory;
gV600R according to local laboratory;
hThree Q61R (1 result received after database lock) and one G12 (result received after database lock);
iQ61R according to local laboratory;
jThree Q61R, two Q61L, two Q61K, two Q61, and one G12 mutation according to local laboratory.
kData for clinical activity available for 63 patients for response-rate analysis set (Ascierto, 2013).
DCR, disease control rate; NR, not reported.
Efficacy and safety analysis
Efficacy and safety results for this study have been previously reported for the BRAF- and NRAS-mutated arms for two data cut-off points, 29 February 2012 [24] and 7 January 2014 [31]. Biomarker results presented herein are derived from the later cut-off date.
Biomarker analysis
Biomarkers were analyzed to evaluate MAPK pathway inhibition and analysis was undertaken to evaluate on-treatment biomarker expression and frequency of tumor genetic alterations at baseline. Results were compared against melanoma cases in The Cancer Genome Atlas (TCGA) database and in the context of clinical outcomes, where appropriate. Twenty-five fresh, paired (baseline and on Cycle 1, Day 15) tumor samples were collected for pharmacodynamic biomarker analysis. Fifteen pairs were evaluable for pERK analysis (three pairs in the BRAF-mutated 45 mg subgroup; four and eight pairs, respectively, in the BRAF-mutated 60 mg and NRAS-mutated arms). Fourteen pairs were evaluable for DUSP6 analysis (three pairs each in the BRAF-mutated 45 mg and 60 mg arms and eight pairs in the NRAS-mutated arm).
Pharmacodynamic analysis of postbaseline pERK and DUSP6 expression in patients with BRAF and NRAS mutations showed MAPK pathway inhibition (Figure 1). Decreased postbaseline cytoplasmic and nuclear pERK expression was observed in 11 of 15 and in 9 of 15 paired samples, respectively, and decreased total DUSP6 expression postbaseline was observed in 10 of 14 paired samples. Median reduction in pERK H-score was 47% and 70% in the cytoplasmic and nuclear compartments, respectively, and median DUSP6 reduction, in Δ Ct, was 36%. MAPK pathway inhibition was shown in both responders and nonresponders, with no apparent association between reduced expression of either pERK or DUSP6 with overall response rates.
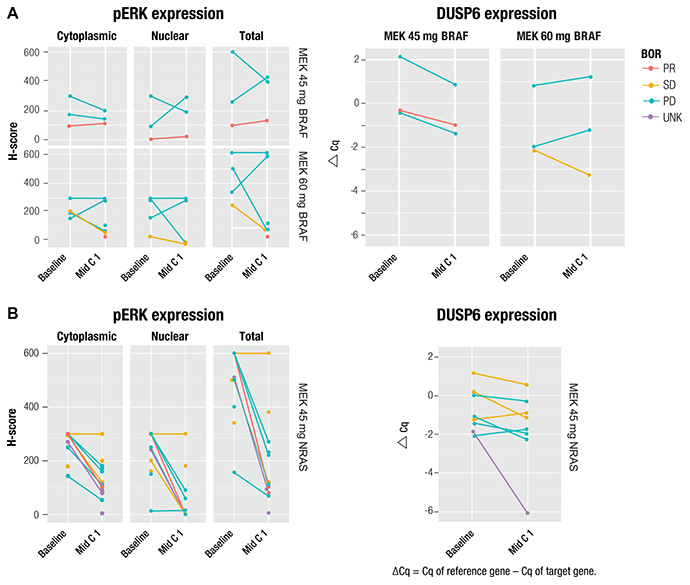
Figure 1: Change from baseline in pERK and DUSP6 expression in patients with (A) BRAF or (B) NRAS mutation and correlation with best overall response. Single dots represent unpaired biopsies. BOR, best overall response; Cq, quantification cycle; PD, progressive disease; PR, partial response; SD, stable disease; UNK, unknown.
Comparison of patients in the BRAF- and NRAS-mutated arms with TCGA melanoma cases showed overall concordance of the tumor genetic landscape with regard to the percentage of patients experiencing alterations in the most frequently mutated genes (Supplementary Figure 1A and 1B). Within the BRAF-mutated group, concordance was observed in PTEN, TRRAP, and TP53. Slightly more CDKN2A alterations and fewer CDKN2B alterations were observed in study patients compared with the cases in the TCGA database, possibly due to higher sequencing depth and more systematic annotation of variants in this study, respectively. Within the NRAS-mutated group, concordance was observed in CDKN2A/B, TP53, and NOTCH2. There were no trends showing any association between efficacy and the subtype of NRAS Q61 mutations (Supplementary Figure 2). Supplementary Table 2 provides additional context for the BRAF and NRAS mutations, including mutation type and presence or absence in the Catalogue of Somatic Mutations in Cancer (Supplementary Table 2).
A weak association between specific mutations or total number of mutations with either measure of efficacy was shown among patients with either BRAF- or NRAS-mutated melanoma (Figure 2A and 2B, respectively). Notable differences between BRAF- and NRAS-mutated melanomas at baseline included PTEN (21% vs 2.5% of patients, respectively) and P53 (13% vs 22%, respectively). Five BRAF-mutated tumors had broad amplifications on chromosome 7 and five others on chromosome 1, while three NRAS-mutated tumors exhibited amplifications on chromosome 11. Amplifications tended to be associated with shorter progression-free survival (PFS); for example, amplifications in CCND1 or CCND3 occurred only in five NRAS-mutated patients with a PFS shorter than the median (≤ 3.6 months).
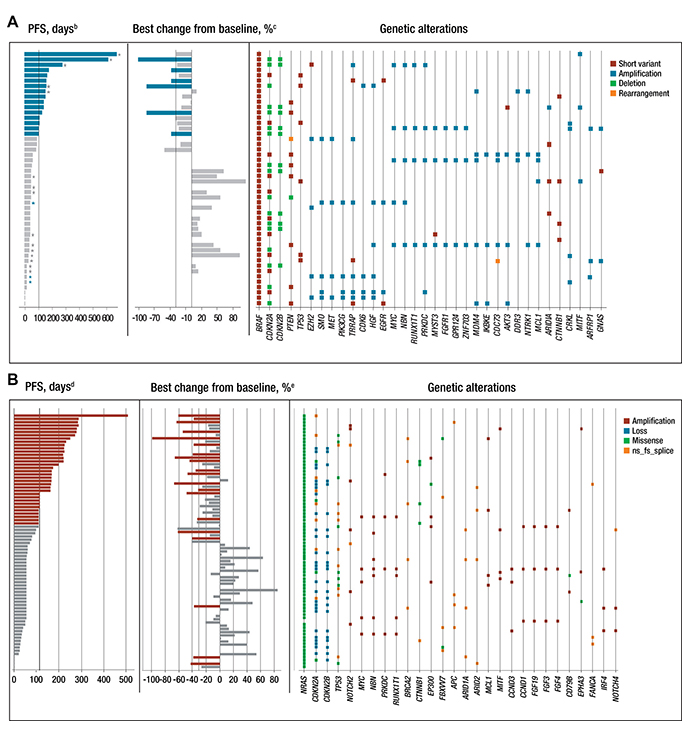
Figure 2: Genetic landscape of tumor samples and efficacy in patients with (A) BRAF mutationa and (B) NRAS mutation. aThe genetic landscape shows gene alterations that occurred in ≥ 3 patients in the BRAF-mutant population; bBlue indicates patients with PFS ≥ 3.5 months (median PFS in the 45 mg BRAFV600 arm); cBlue indicates patients with reduction in sum of the longest diameter from baseline ≥ 30%; dRed indicates patients with PFS ≥ 3.6 months (median PFS in the NRAS-mutated arm); eRed indicates patients with reduction in sum of the longest diameter from baseline ≥ 30%; *Patients in the BRAF-mutant group receiving binimetinib 60 mg BID who were dose reduced to 45 mg BID; *Patients in the BRAF-mutant group receiving binimetinib 60 mg BID who were not dose reduced. MET and HGF amplifications in the BRAF panel CCND3 and CCND1 amplifications in the NRAS panel are indicated with a black box. BID, twice daily; fs, frameshift; ns, nonsense; PFS, progression-free survival.
Beyond individual genes, pathways or coherent classes of genes were examined, namely the cell cycle and PI3K and P53 pathways as well as epigenetic regulators, transcription factors, and DNA damage response genes. The rationale for these groupings is that genes with parallel or related functions relevant to melanoma may harbor mutually exclusive mutations, as is the case for BRAF and NRAS, such that they would escape notice when examined individually. Among both BRAF- and NRAS-mutated arms, genetic pathway alterations were observed, predominantly within the cell cycle genes (Figure 2A and 2B; Supplementary Table 3). Within the BRAF-mutated group, these alterations were driven primarily by CDKN2A, which was altered in 27 of 48 patients (56.3%). Among the NRAS-mutated group, 45 of 78 patients (58%) had alterations in the cell cycle genes; 27 of 65 patients (42%) had both alterations within the cell cycle genes and a short PFS (≤ 110 days).
DISCUSSION
The biomarker analyses of this study aimed to describe the biological impact of MEK-targeted inhibition of the MAPK pathway with binimetinib, establish the BRAF- and NRAS-mutated tumor genetic landscape, and explore the link between genetic pathway alterations and the response to binimetinib. Understanding the biological impact of MEK inhibition, and the predictive value of tumor genetic markers and pathway alterations on response, could be invaluable for optimizing the efficacy of targeted therapy. Pharmacodynamic analyses showed MAPK pathway inhibition by binimetinib on Day 15 through decreased pERK levels and DUSP6 gene expression. These decreases were observed in both BRAF- and NRAS-mutated arms, and were moderate and consistent with observations in other cancer studies with MEK inhibitors, such as the observed suppression of pERK by cobimetinib (GDC-0973) both in vitro and in vivo [32]. However, no association between reduced pERK or DUSP6 levels with clinical efficacy was observed, likely due to limited data or sampling time points. Another consideration is that in certain genomic contexts, including some tumors with RAS mutations (ie, preclinical data of BRAF/RAS-WT tumor cells) [33], the MEK-ERK pathway may not be fundamental for tumor cell proliferation; thus, inhibition of the MEK-ERK pathway may not reduce the survival of certain tumor cells.
Overall, the tumor genetic landscape for patients with BRAF- and NRAS-mutated melanoma was concordant with that reported in TCGA melanoma cases. This observation demonstrated a consistency of genetic alterations in this patient subset with historical samples. This equivalence also contributes to the validation of these biomarker results, leading toward a better understanding of changes that occur within patients with BRAF- and NRAS-mutated melanoma and their predictive value.
Several patients with BRAF-mutated tumors had amplification of genes on the long arm of chromosome 7 (7q); seven patients exhibited MET and/or HGF amplifications, with coamplification occurring in three of them. Five of these seven patients (including the three patients with coamplification) had a PFS that was shorter than the median PFS of 3.5 months for the population with BRAF mutations. Particular genes of high interest on 7q include HGF, MET, EZH2, and SMO. The latter two have been associated with driving the progression of melanoma [34, 35]; HGF and MET form a functional pair since they are cognate ligand and receptor, respectively. Among patients in the NRAS-mutated group, CCND1 or CCND3 amplifications were exclusively seen in five patients with shorter PFS, indicating that constitutive CDK4/6 pathway signaling may lead to resistance. In this regard, a phase Ib/II study (NCT01781572) with binimetinib in combination with the CDK4/6 inhibitor ribociclib (LEE011) in patients with NRAS-mutant melanoma has recently completed, with preliminary data from the initial Phase 1b study suggesting a manageable safety profile and favourable efficacy [36]. Furthermore, positive phase III data have been reported for the NEMO study comparing the efficacy of binimetinib single agent versus dacarbazine in unresectable or metastatic NRAS-mutant melanoma [22]. We cannot rule out other genetic alterations that were observed in patients with progressive disease that may have had an effect on PFS.
Binimetinib showed activity in patients with BRAF- or NRAS-mutated melanoma through MAPK pathway inhibition, and genomic profiling highlighted genetic alterations of interest that could be used as potential predictive biomarkers of response to binimetinib. Although all comparisons with patient outcomes for these data are currently observational in nature, they are indicative of the potential predictive use of genetic data in future larger cohorts. Combined with interpretation of biomarker data from other ongoing studies of binimetinib, these data could provide context toward understanding the biological impact of and prediction of response to binimetinib. Of relevance, assessment of potential additional biomarkers of efficacy or safety was incorporated in the aforementioned NEMO trial, in the phase III COLUMBUS trial (NCT01909453) comparing binimetinib plus encorafenib with encorafenib or vemurafenib in patients with BRAF-mutated melanoma, and in the phase II LOGIC-2 trial (NCT02159066) investigating sequential encorafenib/binimetinib combination therapy followed by a combination with targeted agents after disease progression in patients with BRAFV600 -mutated melanoma.
MATERIALS AND METHODS
Patients
Details of the study have been published previously [24]. Briefly, baseline BRAF or NRAS status was assessed using archival or fresh tumor biopsies either at a local or central laboratory (MolecularMD) and analyzed by a semiquantitative polymerase chain reaction (BRAF) or bidirectional Sanger sequencing assay (NRAS). After patient enrollment, all tumor biopsies assessed at local laboratories were sent to the central laboratory for mutational status confirmation.
The study was designed, undertaken, and reported in accordance with the Declaration of Helsinki and the ICH Harmonised Tripartite Guideline for Good Clinical Practice. The protocol was approved by an institutional review board, independent ethics committee, or research ethics board at each institution. All patients provided written informed consent before screening and additional consent if participating in the exploratory biomarker analysis.
Study design and treatments
This study was a nonrandomized, open-label phase II study in which patients were divided into one of three treatment arms according to tumor NRAS or BRAF status: binimetinib 45 mg BID or 60 mg BID for BRAF-mutant tumors, or binimetinib 45 mg BID for NRAS-mutant tumors. The 60 mg BID dose of binimetinib for patients with BRAF-mutated tumors was subsequently reduced to 45 mg BID per a protocol amendment following two serious adverse events (grade 4 acute liver failure in 1 patient; grade 3 cardiomyopathy, decreased ejection fraction and tachycardia in a second patient). Treatment was administered in 28-day cycles. Binimetinib was administered orally (film-coated tablet) BID from Day 1 of Cycle 1 and continuously throughout the study.
The primary endpoint was the proportion of patients who achieved an objective response (complete response + partial response). Secondary endpoints included PFS, time to response, safety, tolerability, pharmacokinetics, and pharmacodynamics. Biomarker analyses, the focus of this manuscript, were prespecified secondary and exploratory objectives, and included: assessment of pharmacodynamic effects of binimetinib on MEK/MAPK signaling by analysis of pERK and DUSP6 gene expression in pre- versus post-dose tumor biopsies; examination of correlations between pERK, DUSP6 expression and efficacy; assessment of the baseline molecular status of the tumors and exploration of potential predictive biomarkers of response to binimetinib.
Study procedures
Safety and pharmacokinetic assessments and overall efficacy data from an earlier cut-off were reported previously [24]. Biomarker-related efficacy data are presented here. A whole blood sample (~6.0 mL) was taken from all patients (at Cycle 1 Day 1) to provide a non-tumorous tissue sample to perform genetic analysis (if compliant with local IRB requirements) This sample was analyzed to compare tumor-specific gene alterations in DNA from tumor samples with DNA from normal-non-tumor cells. Baseline and on-study (Cycle 1, Day 15) fresh tumor biopsy samples were collected from patients in all three treatment arms and analyzed for the pharmacodynamic markers pERK and DUSP6. Immunohistochemistry (IHC) data reported from the lab included quantitative data (eg, percent tumor and percent positive cells) or a semi quantitative measure of protein expression reported as 3 individual components, 1+ 2+ and 3+. The pathologist determined whether the staining in a cellular compartment was absent (0+), slight (1+), moderate (2+), or strong (3+). The H-Score used to assess pERK for each cellular compartment was then calculated as the sum of (the percentages of stained cells * their intensity), or (%1+) + (2 * %2+) + (3 * %3+) and ranged between 0 and 300. Δ Ct (cycle threshold), a relative measure of the concentration of target in the PCR reaction, was used to measure DUSP-6. Δ Ct is the normalization of Raw Ct that is calculated by subtracting the baseline (reference sample): (Δ Ct = Ct Gene of interest – Ct Internal control). Deep sequencing of formalin-fixed, paraffin-embedded tumor samples from enrolled patients was used to profile genomic alterations in 296 cancer-related genes in order to identify potential predictive markers of binimetinib sensitivity, as previously described [30]. Briefly, DNA was sequenced at high depth (median 744X) on an Illumina HiSeq 2500 sequencer following probe-based targeted exome capture.
Analyses were descriptive and exploratory in nature, and no inferential analysis was performed. Data were summarized with respect to demographic and baseline characteristics and all relevant pharmacodynamics and genetic alteration measurements.
Author contributions
All authors contributed to the collection and assembly of data, manuscript writing, and final approval of the manuscript. CvH, RRP, AD, VA, HN, AZ, and RD contributed to the study concept and design and analyses and interpreted the data.
ACKNOWLEDGMENTS
The authors thank the patients and their families and the staff of each site participating in this study. Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals and Array Biopharma. We thank Ori Bowen and Linda Feighery, who were both a part of Articulate Science at the time this manuscript was developed, as well as The Medicine Group, LLC for medical editorial assistance with this manuscript.
CONFLICTS OF INTEREST
CvH reports grants from Astra Zeneca, Merck, MSD, Novartis outside the submitted work. AH reports grants from Amgen, BMS, Celgene, Eisai, GSK, Merck Serono, MSD/Merck, Novartis, Roche, other from Amgen, BMS, MedImmune, MSD/Merck, Nektar Therapeutics, Novartis, Oncosec, Philogen, Provectus, Regeneron, Roche outside the submitted work; CB reports personal fees from Amgen, GSK, personal fees and non-financial support from Merck Sharp & Dohme, Novartis, BMS, and from Roche, personal fees from AstraZeneca, outside the submitted work; DS reports personal fees and other from Amgen, grants, personal fees and other from BMS, personal fees and other from Novartis, Roche, and Array, grants, personal fees and other from Merck/MSD, personal fees from Pfizer, outside the submitted work; PAA reports grants and personal fees from BMS, Roche-Genentech, Ventana, and Array, personal fees from Novartis, Amgen, and from Merck Sharp & Dohme, outside the submitted work; MCH reports grants and personal fees from Novartis, personal fees from MolecularMD, during the conduct of the study; grants and personal fees from Blueprint Medicines, personal fees from Pfizer, outside the submitted work; RRP, AD, VA, HN, AZ were employed by Novartis; RD reports research funding from Novartis, MSD, BMS, Roche, GSK, and has consultant or advisory board relationship with Novartis, MSD, BMS, Roche, GSK, and Amgen. All remaining authors have declared no conflicts of interest.
FUNDING
Funding for this study was provided by Novartis Pharmaceuticals Corporation and Array Biopharma. Editorial support was funded by the study sponsors.
REFERENCES
1. Vennepureddy A, Thumallapally N, Motilal Nehru V, Atallah JP, Terjanian T. Novel drugs and combination therapies for the treatment of metastatic melanoma. J Clin Med Res. 2016; 8:63–75.
2. Frémin C, Meloche S. From basic research to clinical development of MEK1/2 inhibitors for cancer therapy. J Hematol Oncol. 2010; 3:8.
3. Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A, Frederick DT, Barzily-Rokni M, Straussman R, Haq R, Fisher DE, Mesirov JP, Hahn WC, et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014; 4:816–827.
4. Pratilas CA, Solit DB. Targeting the mitogen-activated protein kinase pathway: physiological feedback and drug response. Clin Cancer Res. 2010; 16:3329–3334.
5. Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA, Bäsecke J, Stivala F, Donia M, Fagone P, Malaponte G, Mazzarino MC, Nicoletti F, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget. 2011; 2:135–64. https://doi.org/10.18632/oncotarget.240.
6. Jakob JA, Bassett RL Jr, Ng CS, Curry JL, Joseph RW, Alvarado GC, Rohlfs ML, Richard J, Gershenwald JE, Kim KB, Lazar AJ, Hwu P, Davies MA. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012; 118:4014–4023.
7. Lee JH, Choi JW, Kim YS. Frequencies of BRAF and NRAS mutations are different in histological types and sites of origin of cutaneous melanoma: A meta-analysis. Br J Dermatol. 2011; 164:776–784.
8. Colombino M, Capone M, Lissia A, Cossu A, Rubino C, De Giorgi V, Massi D, Fonsatti E, Staibano S, Nappi O, Pagani E, Casula M, Manca A, et al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J Clin Oncol. 2012; 30:2522–2529.
9. Zelboraf (vemurafenib), Prescribing Information. Genentech Inc., San Francisco, CA. 2017.
10. Zelboraf (vemurafenib), Summary of Product Characteristics. Roche Products, Limited; Welwyn Garden City, UK. 2018.
11. Tafinlar (dabrafenib), Prescribing Information. Novartis Pharmaceuticals Corporation, East Hanover, NJ. 2018.
12. Tafinlar (dabrafenib), Summary of Product Characteristics. Novartis EuroPharm Limited, Dublin. 2018.
13. Mekinist (trametinib), Prescribing Information. Novartis Pharmaceuticals Corporation, East Hanover, NJ. 2018.
14. Mekinist (trametinib). Summary of Product Characteristics. Novartis EuroPharm Limited, Dublin. 2018.
15. Cotellic (cobimetinib), Prescribing Information. Genentech Inc., San Francisco, CA. 2018.
16. Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, Chiarion Sileni V, Lebbe C, Mandalà M, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014; 371:1877–1888.
17. Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, Chiarion-Sileni V, Drucis K, Krajsova I, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015; 372:30–39.
18. Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, Maio M, Mandalà M, Demidov L, Stroyakovskiy D, Thomas L, de la Cruz-Merino L, Dutriaux C, Garbe C, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014; 371:1867–1876.
19. Dillon AB, Lin K, Kwong A, Ortiz S. Immunotherapy in melanoma, gastrointestinal (GI), and pulmonary malignancies. AIMS Public Health. 2015; 2:86–114.
20. Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, Kelley MC, Kefford RF, Chmielowski B, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014; 4:80–93.
21. Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, Hodis E, Rosenberg M, McKenna A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014; 4:94–109.
22. Dummer R, Schadendorf D, Ascierto PA, Arance A, Dutriaux C, Di Giacomo AM, Rutkowski P, Del Vecchio M, Gutzmer R, Mandala M, Thomas L, Demidov L, Garbe C, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017; 18:435–445.
23. Lee PA, Wallace E, Marlow A, Yeh T, Marsh V, Anderson D, Woessner R, Hurley B, Lyssikatos J, Poch G, Gross S, Rana S, Winski S, Koch K. Preclini cal development of ARRY-162, a potent and selective MEK 1/2 inhibitor. Cancer Res. 2010; 70:2515.
24. Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, Blank CU, Hauschild A, Beck JT, St-Pierre A, Niazi F, Wandel S, Peters M, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013; 14:249–256.
25. van Herpen C, Postow M, Carlino M, Kalkavan H, Weise A, Amaria RN, De Vos F, Carvajal RD, Matano A, Bhansali S, Lam L, Yerramilli-Rao P, Sosman JA. A phase 1b/2 study of ribociclib (LEE011; CDK4/6 inhibitor) in combination with binimetinib (MEK162; MEK inhibitor) in patients with NRAS-mutant melanoma. Eur J Cancer. 2015; 51:S663.
26. Winski S, Anderson D, Bouhana K, Impastato R, Woessner R, Zuzack J, Tunquist B, Garrus J, Pheneger T, Lee P. MEK162 (ARRY-162), a novel MEK 1/2 inhibitor, inhibits tumor growth regardless of KRas/Raf pathway mutations. Eur J Cancer. 2010; 8:56.
27. Bendell JC, Papadopoulos K, Jones SF, Barrett E, Guthrie K, Kass CL, Litwiler KS, Napier C, Patnaik A. Abstract B243: A phase I dose-escalation study of MEK inhibitor MEK162 (ARRY-438162) in patients with advanced solid tumors.. Mol Cancer Ther. 2011; 10:B243.
28. Finn R, Javle M, Tan B, Weekes C, Bendell J, Patnaik A, Khan G, Laheru D, Anderson L, Christy-Bittel J, Barrett E, Guthrie K, Litwiler K, Bekaii-Saab TS. A phase I study of MEK inhibitor MEK162 (ARRY-438162) in patients with biliary tract cancer. J Clin Oncol. 2012; 30:220.
29. Li W, Song L, Ritchie AM, Melton DW. Increased levels of DUSP6 phosphatase stimulate tumourigenesis in a molecularly distinct melanoma subtype. Pigment Cell Melanoma Res. 2012; 25:188–199.
30. Frampton G, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, Sun J, Juhn F, Brennan K, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013; 31:1023–1031.
31. van Herpen C, Agarwala SS, Hauschild A, Dummer R, Berking C, Beck JT, Schadendorf D, Gibney GT, Jansen R, Queirolo P, Ascierto PA, Blank CU, Nauwelaerts H, et al. Overall survival and biomarker results from a phase 2 study of MEK1/2 inhibitor binimetinib (MEK162) in patients with advanced NRAS-mutant melanoma. Ann Oncol. 2014; 25:S1–S41.
32. Hoeflich KP, Merchant M, Orr C, Chan J, Den Otter D, Berry L, Kasman I, Koeppen H, Rice K, Yang NY, Engst S, Johnston S, Friedman LS, et al. Intermittent administration of MEK inhibitor GDC-0973 plus PI3K inhibitor GDC-0941 triggers robust apoptosis and tumor growth inhibition. Cancer Res. 2012; 72:210–219.
33. Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, Ye Q, Lobo JM, She Y, Osman I, Golub TR, Sebolt-Leopold J, Sellers WR, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006; 439:358–62.
34. Zingg D, Debbache J, Schaefer SM, Tuncer E, Frommel SC, Cheng P, Arenas-Ramirez N, Haeusel J, Zhang Y, Bonalli M, McCabe MT, Creasy CL, Levesque MP, et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat Commun. 2015; 6:6051.
35. Santini R, Vinci MC, Pandolfi S, Penachioni JY, Montagnani V, Olivito B, Gattai R, Pimpinelli N, Gerlini G, Borgognoni L, Stecca B. Hedgehog-GLI signaling drives self-renewal and tumorigenicity of human melanoma-initiating cells. Stem Cells. 2012; 30:1808–1818.
36. Schuler MH, Ascierto PA, Leon De Vos FVF, Postow MA, Van Herpen CML, Carline MS, Sosman JA, Berking C, Long GV, Weise A, Gutzmer R, Kaatz M, et al. Phase 1b/2 trial of ribociclib+binimetinib in metastatic NRAS-mutant melanoma: Safety, efficacy, and recommended phase 2 dose (RP2D). J Clin Oncol. 2017; 15:9519.