
INTRODUCTION
Castration-resistant prostate cancer (CRPC) occurs when androgen ablation therapy fails. Patients with CRPC have an average survival time of 16 to 18 months from identification of recurrence [1–3]. A variety of mechanisms have been proposed for progression that bypasses current therapies targeting the androgen receptor (AR), including production of intratumoral androgens, increased conversion of adrenal androgen to testosterone, and increased AR expression after hormone deprivation [4–7]. In addition, various cytokines and growth factors have been shown to activate AR through direct binding or by cross-talk mechanisms [8, 9]. Functionally, all of these mechanisms rely on continued activation of the AR through its ligand-binding domain (LBD).
However, the recent identification of androgen receptor splices variants (AR-Vs) provides an alternative explanation for the development of CRPC. AR-Vs have been identified by several independent groups in human prostate cancer cell lines, xenografts, metastases, and circulating tumor cells [10–15]. Most characteristically, these variants are devoid of the ligand binding domain (LBD) but retain the capability to engage transcriptional machinery and promote the regulation of AR-target genes. The potential role of AR-Vs in driving prostate cancer progression is supported by several independent correlative clinical studies describing the significant association of AR-Vs with advanced disease progression and a shorter survival period [15–18]. Among the constitutively active AR-Vs, AR-V7 (or AR3) and ARv567es are the two most commonly described in advanced disease [17, 19]. AR-V7 has been reported in many prostate tissues both benign and malignant, while ARv567es has only been seen in malignant prostate glands [10, 14, 18, 19]. Furthermore, the AR3/V7 and ARv567es transgenic mouse models demonstrated that expression of AR variant in mouse prostate induced high-grade prostatic intraepithelial neoplasia (PIN) [20] and/or invasive prostatic carcinoma [21].
The mechanism of AR-Vs in CRPC transcriptional regulation still remains unclear. Present evidence suggests, in addition to activation of the classical AR target genes, constitutively active AR splice variants are associated with a distinct transcription program in prostate cancer cells as well as in prostate cancer xenografts displaying treatment-induced AR-Vs expression [22]. Importantly, this distinct expression signature is enriched with cell cycle genes compared to the canonical AR-ligand dependent gene signature. Very interestingly, another study also described a similar transcription program comprised of upregulated cell-cycle genes in the androgen-independent prostate cancer (AIPC) cell line LNCaP-abl [23]. Although the latter research did not involve the role of AR variant, the ubiquitin-conjugating enzyme E2C (UBE2C) was the most up-regulated cell cycle gene in both studies. UBE2C is an anaphase-promoting complex/cyclosome (APC/C) E2 ubiquitin-conjugating enzyme. It can inactivate the M-phase check point and enhance cell growth. UBE2C has been shown to be a prominent oncogene in solid tumors, and it is found overexpressed in various types of solid tumors including late-stage prostate cancer [24–27]. Taken together, these studies indicate the presence of a distinct gene expression profile in CRPC that is different from the canonical AR-dependent transcriptome, one that might be associated with different transcriptional machinery of AR-Vs. The underlying mechanism on how AR-Vs induce a distinct transcriptional profile remains to be elucidated.
Modulation of androgen receptor (AR) co-regulators might play a pivotal role in CRPC [28, 29]. Previous studies have indicated that epigenetic markers and collaborating transcription factors can be ascribed to androgen-independent prostate cancer, including histone markers, [30] FoxA1 [31, 32], MED1(Mediator complex subunit 1) [33] and FOXO1. [34] Chen et al showed p-MED1 could drive CRPC cancer growth through a looping pattern on the UBE2C locus [35]. The chromatin looping functional pattern occurred uniquely in CRPC but not in androgen-dependent prostate cancer (ADPC). Given the critical role identified for MED1 in mediating UBE2C expression in CRPC, we asked whether co-activator MED1 could interact with AR-Vs to mediate the downstream events of transcriptional regulation.
RESULTS
MED1 is necessary for ARv567es induced UBE2C regulation and subsequent prostate cancer cell growth
We first confirmed the regulatory effect of ARv567es variant on UBE2C expression in the AR-dependent cell line LNCaP. After performing transient transfection of an ARv567es expression vector 3Flag-CMV-ARv567es, we assayed UBE2C protein and mRNA levels by Western blot and quantitative RT-PCR analysis. As shown in Figure 1A, the expression of UBE2C increased in the LNCaP-ARv567es cells grown in media without DHT, consistent with previous reports [22]. To investigate whether MED1 is involved in this regulatory activity, MED1 expression was silenced by RNAi (Figure S1). Subsequently, the up-regulation of UBE2C was significantly impaired at both the mRNA and protein level (Figure 1A). These data suggest MED1 is involved in ARv567es mediated UBE2C expression.
The expression of ARv567es in metastases portends a rapid progression of the tumor [18], which was also suggested by our in vitro study. The ARv567es expressing stable cell line LNCaP-ARv567es (see methods) grows faster than control LNCaP-Lenti cells in the absence, but not in the presence of DHT (Figure 1B). To investigate whether there is a functional interaction between MED1 and ARv567es, we tested the effect of MED1 silencing on ARv567es induced cell proliferation. Silencing of MED1 decreased proliferation of LNCaP-ARv567es cells compared with scramble control (Figure 1C and S2). This effect was more significant in the androgen-deprived condition. This phenomenon indicates MED1 is more involved with ARv567es transcriptional regulation when the ligand is absent, but it is not actively interacting with full-length AR (ARfl) when androgen is present. QPCR was further performed to investigate the UBE2C expression in the stable cell lines (Figure S3). Higher UBE2C expression was seen in the LNCaP-ARv567es cell line in the absence of DHT; both DHT and siMED1 significantly inhibited UBE2C expression. Collectively, these data suggest MED1 plays an essential role in the ARv567es induction of UBE2C and subsequent prostate cancer cell growth in an androgen-independent manner.
MED1 is recruited to ARv567es independent of full-length AR
As reported in a previous study we demonstrated that ARv567es binds to full-length AR (ARfl ) and increases the stability of ARfl protein [19]. Here, we investigated whether there is a physical interaction between MED1 and ARv567es, and whether this interaction is mediated by full-length AR. The co-immunoprecipitation assay was performed with the LNCaP cell line transiently transfected with Flag-tagged ARv567es. As shown in Figure 2A, the anti-Flag antibody could pull down p-MED1 as well as ARfl, indicating a physical association of ARv567es with p-MED1 and ARfl in the context of protein activity. Of note, Flag-tagged ARfl also pulled down p-MED1. However, with the same amount of input protein lysates (100 ug), ARv567es showed abundance of p-MED1 co-precipitation especially in the absence of DHT, but ARfl pulled down much less p-MED1 protein. This finding indicates ARv567 has more potency to recruit p-MED1 when androgen is depleted, which is exactly in accordance with the impaired cell growth by siMED1 in androgen-depleted condition shown in Figure 1C.
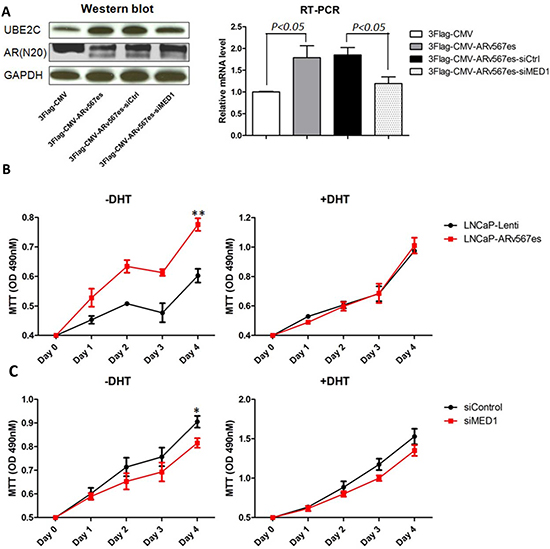
Figure 1: ARv567es increased UBE2C expression and improved prostate cancer cell proliferation, which was blocked by MED1 silencing. (A) Western blot and qRT-PCR showed significantly increased UBE2C expression in transiently transfected LNCaP cells with 3Flag-CMV-ARv567es expression vector. Up-regulation of UBE2C by ARv567es could be attenuated by MED1 siRNA at mRNA and protein level. (B) ARv567es stable expressing cell line, LNCaP-ARv567es showed significant higher proliferation rate (**p < 0.01) compared with Lenti virus empty vector control cell line in the absence, but not presence of DHT. (C) ARv567es induced LNCaP cell proliferation could be blocked by MED1 silencing (*p < 0.05) when DHT was not present, but not significant with DHT.
To rule out the possibility that ARfl might mediate the interaction between ARv567es and p-MED1, the AR-null M12 cell model was also used. As shown in Figure 2B, in the cumate inducible M12-ARv567es stable cell line, the reverse pull-down of ARv567es by p-MED1 antibody further demonstrated the physical interaction between these two molecules. The IgG controls showed the specificity of the co-IP binding (Figure S4A, B). In total, these data indicate ARv567es, p-MED1, and ARfl might form a ternary complex or bind separately, but ARv567es bound to p-MED1 independent of full-length AR.
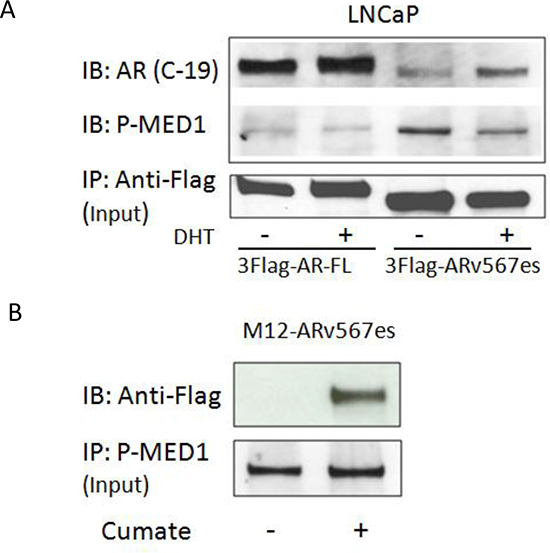
Figure 2: ARv567es could bind p-MED1 independent of full length AR. (A) In androgen-dependent LNCaP cells transiently transfected with Flag-tagged ARv567es, pulling-down of Flag could co-precipitate both ARfl and p-MED1. With the same input volumes of 100ug, ARv567es showed more abundant p-MED1 pull-down than that of full-length AR (ARfl). (B) Reversed pull-down with p-MED1 antibody could co-precipitate Flag-tagged ARv567es in the cumate-inducible M12-ARv567es prostate cancer cell line.
The binding of phospho-MED1 to UBE2C promoter and enhancer increased when ARv567es was present
Since p-MED1 was recruited by ARv567es, we further tested whether ARv567es could enhance p-MED1 binding to UBE2C transcriptional elements. As described by Chen et al, UBE2C promoter and enhancer regions can form a chromatin loop while triggering transcriptional initiation in CRPC cells [35]. Using ChIP assay, we tested p-MED1 binding capacity to the UBE2C promoter and all identified enhancers in LNCaP-Lenti and LNCaP-ARv567es cells under different conditions. As shown in Figure 3A, when DHT was absent (T+S media), more p-MED1 binding (5–8 folds) occurred at UBE2C transcriptional regions, but not at control regions, in the LNCaP-ARv567es cells compared to LNCaP-Lenti cells. Similar binding occurred when ARfl was inhibited by enzalutamide (MDV3100), a potent AR LBD inhibitor [36]. However, in the presence of DHT, and thus ARfl activation, the increased binding in LNCaP-ARv567es cells diminished to a level seen in the LNCaP-Lenti cells. When both DHT and MDV3100 were present (MDV+DHT), a combination that partially inhibits ARfl activity, the LNCaP-ARv567es cell showed stronger p-MED1 binding than the LNCaP-Lenti cells, but still lower levels of binding than the LNCaP-ARv567es T+S and MDV3100 treated groups (1–2 fold over IgG). These results strongly indicate that ARv567es endows p-MED1 higher binding ability to UBE2C transcriptional regions in a ligand-independent manner. Consistent with the ChIP assay results, the UBE2C mRNA level (Figure S5) exactly corresponded to the binding capacity of p-MED1 to UBE2C transcriptional elements. This indicates p-MED1 is the key mediator in ARv567es induced UBE2C transcription.
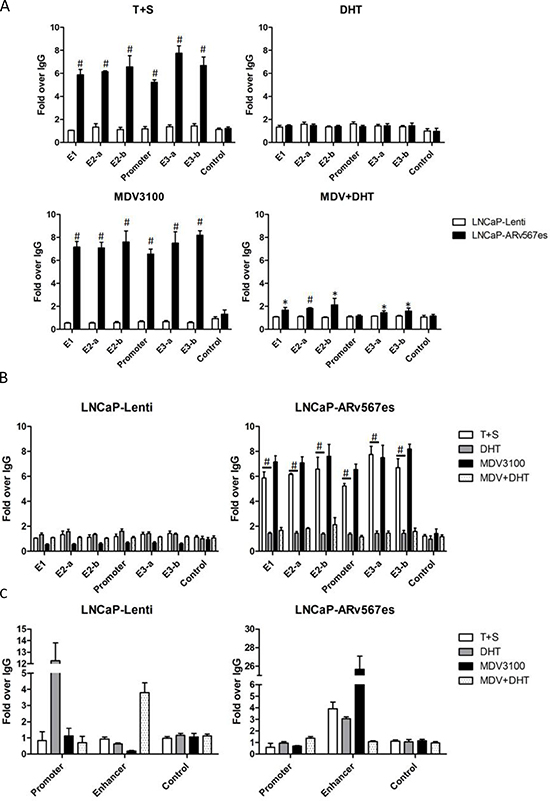
Figure 3: ARv567es recruited p-MED1 to UBE2C enhancer and promoter regions independent of androgen. (A) p-MED1 binding to UBE2C promoter and enhancers increased in LNCaP-ARv567es cells compared with LNCaP-Lenti cells, in the conditions of androgen deprivation (T+S) and full-length AR inhibition (MDV, MDV+DHT); ARfl activation by DHT abated the extra recruitment induced by ARv567es. (B) The reversed effect of DHT and MDV on the recruitment of p-MED1 to UBE2C promoter and enhancers in LNCaP-ARv567es cells and LNCaP-Lenti cells. (C) ARv567es also showed increased binding of p-MED1 to PSA enhancers but not in the promoter region especially when androgen receptor was inhibited (MDV3100). *p < 0.05 and #p < 0.01.
Figure 3A elucidates higher p-MED1 binding induced by ARv567es than ARfl, and Figure 3B more clearly shows the divergent effects of androgen deprivation (T+S), ARfl activation (DHT) and ARfl inhibition (MDV3100) on p-MED1/UBE2C binding in these two cell lines. While androgen deprivation and ARfl inhibition could abate p-MED1 binding to UBE2C in androgen-dependent LNCaP-Lenti cells, they could inversely and vigorously activate the p-MED1 binding to UBE2C in LNCaP-ARv567es cells. This finding could coincidently address the mechanism on how androgen deprivation treatment (ADT) induces growth inhibition of androgen-dependent cancer, and also on how ARv567es contributes to castration-resistance development. ARfl activation (DHT) also has a contrary effect on these two cell lines. While DHT slightly strengthened the recruitment of p-MED1in LNCaP-Lenti cells, it significantly blocked this action in LNCaP-ARv567es cells (p < 0.01). The negative regulation of DHT on ARv567es activated p-MED1 binding was very consistent with our previous findings: DHT led to decreased cell proliferation (Figure 1C) as well as lower amounts of p-MED1/ ARv567es precipitation (Figure 2A).
In summary, these data lead to the following conclusions: 1) Transactivation of ARfl by ligand confers low p-MED1 binding capacity to UBE2C transcriptional regions; 2) ARv567es is a potent effector driving p-MED1 to the UBE2C locus independent of androgen; 3) When ARfl and ARv567es coexist and DHT is present, ARfl takes priority, and recruits less p-MED1, thus facilitating low levels of binding to UBE2C transcriptional elements; 4) When androgens are depleted, ARv567es takes the stage and recruits more p-MED1 resulting in much higher levels of binding to UBE2C regulatory regions. These findings are consistent with the chromatin-looping hypothesis [35] and could explain why the UBE2C promoter-enhancer loop is more likely to occur in CRPC cells but not in androgen-dependent prostate cancer cells.
As a control, PSA (KLK3) promoter and enhancer regions were also investigated with p-MED1 ChIP assays in LNCaP-ARv567es cells (Figure 3C). Very similar to UBE2C regulatory regions, p-MED1 showed high levels of binding to the PSA enhancer element when ARfl was inhibited by MDV3100, and then next highest when DHT was absent (T+S). However, the p-MED1 binding to PSA promoter is not affected in all the treatment groups compared with IgG control, which is consistent with PSA mRNA outcomes (Figure S5). These findings indicate that ARv567es/p-MED1 might perform alternative transcriptional regulation of AR canonical genes, but not necessarily affect the final gene expression.
Phosphorylated MED1 mediates ARv567es induced UBE2C locus transcription
We further studied the role of MED1 on ARv567es induced transcriptional activity in the UBE2C locus. The 1.2 kb-long enhancer 1 (E1) fragment was used in a luciferase reporter assay (Figure 4A). E1 is located 20 kb 5’ of the UBE2C transcription start site (TSS) and has the highest activity in a 3C assay [35]. Three ~400bp regions in Enhancer 1 termed E1-1, E1-2 and E1-3 were systematically subcloned into the pGL4.10-E4TATA-Luc vector. The reporter activities were measured in M12-Lenti cells and M12-ARv567es cells. The M12 prostate cancer cell line is an AR negative line, so the effect of ARfl could be excluded. While E1-1 showed comparable transcriptional activities in M12-Lenti and M12-ARv567es cells (Figure S6A, B), E1-2 and E1-3 both induced significantly higher Luc signals when ARv567es was present (p < 0.01) (Figure 4B). After the expression of MED1 in M12-ARv567es cells was silenced by siRNA, the Luc expression was inhibited subsequently (Figure 4C). Similar results were seen in Chen et al’s study, with control groups showing higher Luc activities in the LNCaP cell line, but much lower levels in PC3 cells. The reason might be due to very active E4TATA basal promoter activity in M12 and LNCaP cell lines. However, it does not affect these data, which indicated that ARv567es enhanced UBE2C locus transcription and this enhancement was mediated by MED1.
ARv567es/p-MED1 complex interaction with the PI3K/AKT pathway
The PTEN tumor suppressor gene is mutated in 50% of human prostate cancers. In addition, 70% of late stage prostate cancers show alterations in the PTEN/PI3K/AKT pathway [37]. We investigated whether the PI3K/AKT pathway is involved in ARv567es/p-MED1/UBE2C transcriptional activity. The PI3K inhibitor LY294002 was used to examine the effect of PI3K/AKT pathway inhibition on MED1 phosphorylation and UBE2C expression in LNCaP-ARv567es and M12-ARv567es cells. As shown in Figure 5(A, B), LY294002 reduced AKT phosphorylation at S473 and MED1 phosphorylation at T1457 leading to reduced UBE2C protein expression. The involvement of MAPK/ERK pathway was also investigated. Inhibition of the MAPK pathway by PD98059 did not have any effect on MED1 phosphorylation and UBE2C expression in M12-ARv567es cells (Figure 5C). p-ERK protein was non-detectable in LNCaP-ARv567es cell by western blot, and UBE2C expression was not affected after treatment by PD98059 (data not shown). The involvement of PI3K/AKT, but not the MAPK pathway, in the proliferation of the LNCaP-ARv567es cells was further observed in MTT assays by inhibited cell growth of LY294002 treated cells, but not of PD98059 (Figure S7).
Another interesting finding we observed in this experiment was the decreased UBE2C protein expression in the LNCaP-ARv567es cells when DHT was added to T+S media (Figure 5A). This result not only addresses the direct mechanism of impaired LNCaP-ARv567es cell growth under DHT stimulation (shown in Figure 1C), but also consistent with the negative regulatory effects of DHT on p-MED1 co-immunoprecipitation with ARv567es (Figure 2A) and p-MED1 recruitment to the UBE2C locus (Figure 3A, B).
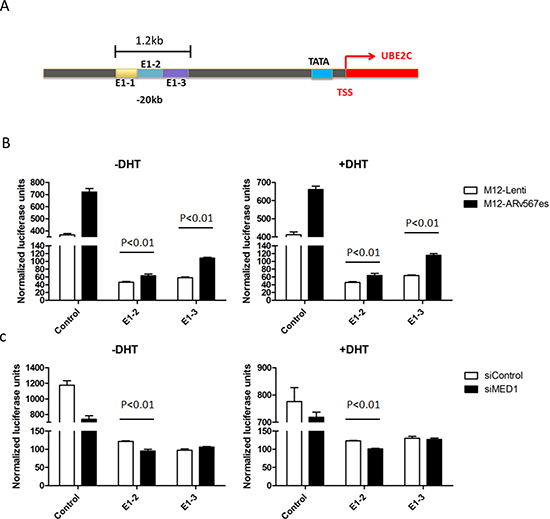
Figure 4: ARv567es Increased UBE2C enhancer transcriptional activities. (A) The diagrammatic view of the UBE2C Enhancer-1 region (20 kb upstream of the UBE2C transcriptional start site) used in the Luciferase reporter assay. (B) Even though lower than the E4TATA control, the luciferase activities of UBE2C E1-2 and E1-3 region were significantly higher (p < 0.01) in M12-ARv567es cell compared with LNCaP-Lenti cells. (C) MED1 silencing attenuated the transcriptional activation of ARv567es on UBE2C enhancer specially showing significance on E1-2 (p < 0.01).
ARv567es/p-MED1 transcriptional regulation is associated with FoxA1
As reported previously, phosphorylation of MED1 facilitates FoxA1, Pol II and TATA binding protein (TBP) recruitment and mediates their interactions on chromatin, leading to chromatin looping [35]. Here we performed co-IP with p-MED1 and Flag-tagged ARv567es to examine the recruitment of FoxA1, a key component in the loop complex. In cumate inducible LNCaP-ARv567es and M12-ARv567es cells, p-MED1 antibody and Flag antibody were used as the pull-down antibodies followed by western blotting with the immune precipitated protein. As shown in Figure 6A and 6C, FoxA1 is co-precipitated with p-MED1 and Flag in both variant cell lines, which indicates FoxA1 is associated with the transcriptional complex of p-MED1/ARv567es, and this process is not dependent on ARfl. RNAi against FoxA1 demonstrated decreased expression of UBE2C in both cell lines (Figure 6B, D), which further indicates the critical role of FoxA1 in ARv567es/p-MED1 transcriptional regulation. IgG controls were included in Figure S8 to show the specificity of the co-IP binding (Figure S8A, B).
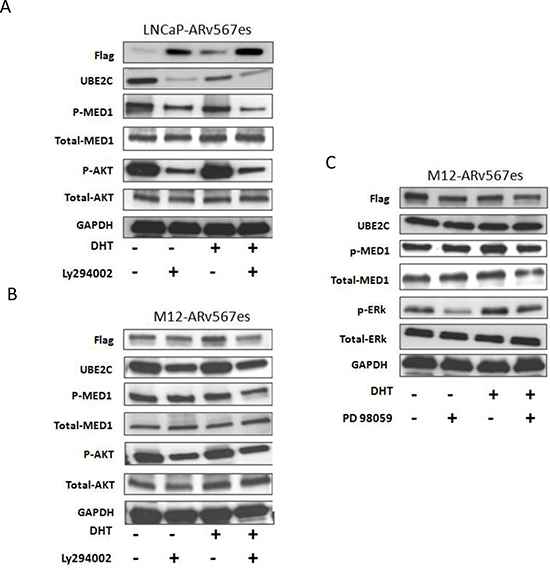
Figure 5: ARv567es/P-MED1 complex cross talks with PI3K-AKT pathway, but not with MAPK pathway. Inhibition of PI3K kinase by LY294002 led to decreased MED1 phosphorylation and UBE2C expression both in LNCaP-ARv567es cells (A) and M12-ARv567es cells (B). MAPK inhibitor PD98059 had no effect either on MED1 phosphorylation or UBE2C expression in M12-ARv567es cells (C).
DISCUSSION
The occurrence of ligand-independent, constitutively active androgen receptor splice variants in castration–resistant prostate cancer provides a conceptually simple explanation for the development of resistance to prostate cancer therapies that target the ligand-binding domain. The AR-Vs induce a distinct transcriptome in which overexpression of UBE2C and other cell cycle genes predominate, suggesting there might be different, or unique, transcriptional machinery used by AR variants compared to full-length AR.
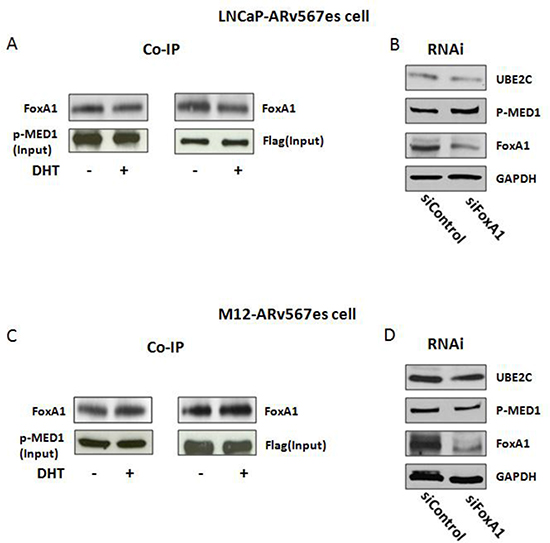
Figure 6: FoxA1 was involved in the ARv567es/p-MED1 transcriptional regulation. (A, C) Co-IP showed the binding of FoxA1 to p-MED1 and ARv567es in M12-ARv567es and LNCaP–ARv567es cell lines. (B, D) Silencing of FoxA1 decreased UBE2C expression in both cell lines.
MED1, also termed TRAP220, is a 220-kDa subunit of the human thyroid hormone receptor-associated protein (TRAP)-Mediator complex. TRAP Mediator has been described as a co-activator for a broad range of nuclear hormone receptors as well as other classes of transcriptional activators, including glucocorticoid receptor [38], estrogen receptor [39, 40] and androgen receptor [41]. Recently, the role of MED1 in prostate cancer oncogenesis and progression has gained recognition [33]. MED1 expression is upregulated in the epithelium of clinically localized human prostate cancer and in CRPC cell lines. Ectopic MED1 overexpression in prostate cancer xenografts significantly promoted tumor growth in nude mice [42]. Notably, MED1 overexpression in CRPC cells leads to upregulation of distinct AR target genes involving cell cycle progression, including UBE2C [23, 35]. Knockdown of MED1 resulted in cell-cycle arrest and decreased proliferation, which is also evident in our LNCaP-ARv567es cell lines.
It has been reported that phospho-MED1 mediates UBE2C locus looping in castration-resistant prostate cancer cells, but not in the androgen-dependent prostate cancer cells [35]. The chromatin looping model nicely elucidates the components in this complex, however it does not specify which transcription factor initiates looping formation given that MED1 interacts with multiple transcription factors as mentioned above. In the castrate-resistant cancer cells, there is low possibility for ARfl still functioning actively with ligand depletion, but another story for constitutive active AR-Vs. Herein, we have identified that ARv567es associates with p-MED1 as a key mediator in CRPC transcriptional activity. We found that: (i) MED1 is necessary for ARv567es induced UBE2C up-regulation and subsequent prostate cancer cell growth; (ii) p-MED1 is recruited to ARv567es independent of full-length AR; (iii) p-MED1 has higher recruitment to UBE2C promoter and enhancer regions in the presence of ARv567es, (iv) ARv567es enhanced UBE2C transcription could be blocked by silencing MED1; (v) ARv567es/p-MED1 signaling crosstalks with the PI3K/AKT pathway but not the MAPK pathway, and (vi) FoxA1 is involved in ARv567es/p-MED1 induced UBE2C long range chromatin interactions.
Previous data demonstrated that an interaction between MED1 and AR worked in a ligand-dependent manner in androgen responsive prostate cells. However, a recent study showed MED1 could functionally interact with androgen receptor in a non-canonical way via a newly discovered binding motif in the AR N-terminal Tau-1 domain. [43] This study is of high interest to us, as the ARv567es splice variant lacks the ligand-binding domain, its interaction with MED1 might be mediated by ARfl. However, based on this study we know that ARv567es, which contains the N-terminus, has the structural base to directly interact with MED1. Consistent with our data, we could see the binding of MED1 and ARv567es through co-IP in AR positive LNCaP cells, but also in the AR-null M12 cells transfected with ARv567es. However, whether ARv567es functionally interacts with MED1 through this particular Tau-1 domain needs to be further investigated.
The most significant finding in this study is the divergent mechanism of ARfl and ARv567es in regulating p-MED1 recruitment, driving p-MED1 to the UBE2C locus, and then affecting UBE2C expression and cancer cell growth. Here we raise a “p-MED1 switch” hypothesis (Figure 7) that could reasonably address this: when androgen/DHT is available (prior to ADT) in androgen dependent prostate cancer (ADPC), more p-MED1 is recruited to ARfl but no chromatin looping forms and ARfl has a low level of activation on UBE2C transcription (Figure 7A). After ADT therapy, the DHT is depleted and ARfl signaling is inhibited, AR splice variants are formed as a survival mechanism due to the stress on the cells; the variants have higher affinity to p-MED1, and therefore recruit more p-MED1 to the UBE2C promoter and enhancers with the assistance of FoxA1, resulting in chromatin looping. This strongly enhances UBE2C expression, leading to the CRPC stage, which has enhanced cell survival and increased cell proliferation (Figure 7B).
p-MED1 recruitment in castrate conditions to the UBE2C promoter/enhancer regions was very high in the presence of ARv567es. In the MDV+DHT treated LNCaP-ARv567es cells, however, MDV3100 was not able to fully attenuate the DHT effect (Figure 3B). The LNCaP-ARv567es cells also express ARfl. MDV3100 has lower affinity to the ARfl LBD and is not able to fully outcompete the ligand. Thus, the inability of MDV to overcome DHT’s suppressive effect on p-MED1 recruitment in this cell line suggests that when DHT is present, it will facilitate the “p-MED1 switch” to ARfl signaling leading to low levels of UBE2C transcription.
The recruitment of p-MED1 to AR canonical genes, such as PSA, differs from non-canonical genes, such as UBE2C. PSA ChIP in LNCaP-ARv567es cells demonstrated that p-MED1 is only recruited to PSA enhancer regions, but not to the promoter. In contrast, in the LNCaP-Lenti cells p-MED1 had the strongest recruitment to the PSA promoter in the presence of DHT. PSA transcript levels overall are significantly lower in the LNCaP-ARv567es cells compared with LNCaP-Lenti cells. Perhaps recruitment of p-MED1 to the enhancer regions of PSA is inhibitory whereas recruitment to the promoter region enhances transcription. These data indicate the “p-MED1 switch” hypothesis may not apply to canonical AR genes.
Multiple signaling pathways have been described leading to MED1 phosphorylation in a variety of conditions, including MAPK/ERK signaling [42, 44] [45, 46], AMP-activated protein kinase (AMPK) [47] and PI3K/AKT [35, 42]. In our study, we investigated ERK and AKT signaling pathways in LNCaP-ARv567es cells and M12-ARv567es cells, and found only the PI3K pathway was involved in phosphorylating MED1 in the context of AR variant. PTEN deletion with AKT pathway activation has long been recognized as one of the most important mechanisms of CRPC [48]. A previous study reported the activity of another AR variant, AR-V7, is regulated in a PTEN-PI3K-AKT-dependent way [49]; here we further confirmed the involvement of PI3K in AR-Vs’ function.
The concurrence of the presence of constitutively active AR splice variant, increased MED1 and UBE2C, as well as crosstalk with the PI3K-AKT pathway, signifies that CRPC has multiple factors synergistically contributing to the process. Identification of potent inhibitors for AR-Vs and combining agents that target MED1, UBE2C and phospho-PI3K/AKT would provide an effective therapeutic strategy in future clinical trials.
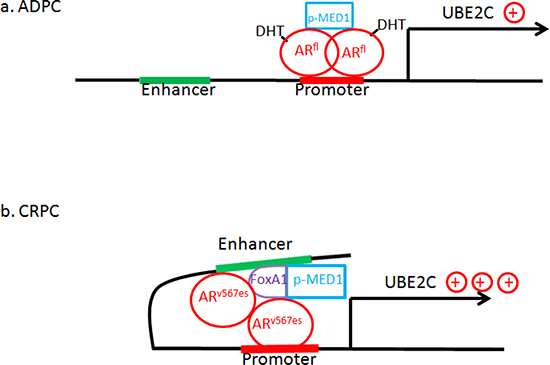
Figure 7: “The p-MED1 switch hypothesis”: Model of ARfl/ARv567es/p-MED1 transcriptional regulation on UBE2C in prostate cancer. (a) In ADPC cells with the presence of DHT, p-MED1 goes to ARfl but no chromatin looping forms and there is a low level of activation on UBE2C transcription; (b) In CRPC cells with DHT depleted, AR splice variants appear which have higher affinity to p-MED1, and recruit more p-MED1 to the UBE2C promoter and enhancers with the assistance of FoxA1, resulting in looping formation and vigorously activating UBE2C expression and leading to cell survival and increased cell proliferation.
MATERIALS AND METHODS
Cell culture, plasmid construction and transient transfection
LNCaP human prostate cancer cell line was obtained from the American Type Culture Collection. Early-passage cells were used in all experiments. These cells were grown in T-medium (Invitrogen, Grand Island NY, USA) supplemented with 10% FBS, 100 units/ml penicillin, and 100 ug/ml streptomycin at 37°C with 5% CO2. M12 cells are AR-negative and were obtained from Dr. J. Ware at the Medical College of Virginia. The generation and characterization of the M12 prostate cell line has been described previously [50–53]. M12 cells were cultured in RPMI 1640 medium (Invitrogen) containing 5% FBS, 10 ng/ml EGF, 0.02 mM dexamethasone, 5 ug/ml insulin, 5 ug/ml transfection, 5 ng/ml selenium, fungizone, and gentamicin at 37°C with 5% CO2. cDNA of the entire ARv567es variant and ARfl were cloned into p3xFlag-CMV-9 vector as described previously [19, 53]. The expression constructs were transfected into the human prostate cancer cell lines using TurboFect reagent according to the manufacturer’s protocol (Thermo Scientific, Pittsburgh PA, USA).
Generation of ARv567es expressing stable cell lines
LNCaP and M12 cell lines expressing cumate-inducible 3xflag tagged ARv567es, LNCaP-ARv567es and M12-ARv567es, were made using the SparQ™ cumate switch lentivector system (Systems Biosciences, Mountain View, CA). Briefly, the flag-tagged ARv567es sequence was first subcloned into the lentiviral expression vector pCDH-EF1-CymR-T2A-Puro and then packaged into lentiviral particles using pPACK™ packaging systems (System Biosciences). Finally, LNCaP and M12 cells were infected with 1 × 107 virus particles per 1 × 106 cells then selected with 1 ug/ml puromycin (Invitrogen) for 10 days. Cells infected by empty lentivectors, LNCaP-Lenti/M12-Lenti, were used as control cells.
Quantitative real-time PCR
Total RNA was isolated using Trizol reagent according to the manufacturer’s instructions (Invitrogen). cDNA was reverse transcribed from total RNA (1 ug) using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). Real-time polymerase chain reactions were performed using SYBR Green PCR Master Mix (Applied Biosystems, Grand Island, NY) on a ViiA 7 Real Time-PCR System (Applied Biosystems) following the manufacturer’s instructions. Primer sequences and Taqman probe sequences used in this study are listed in Table 1. The housekeeping gene RPL13A was used as an endogenous control.
Table 1: QPCR Primer sequences
Target gene |
Primer Sequence (5’ to 3’) |
UBE2C+ |
TGGTCTGCCCTGTATGATGT |
UBE2C- |
AAAAGCTGTGGGGTTTTTCC |
PSA+ |
CAACCTGCAAACCTAGGGAA |
PSA- |
TCAGGGTTGACAGGAGGAAC |
RPL13A+ |
CCTGGAGGAGAAGAGGAAAGAGA |
RPL13A- |
TTGAGGACCTCTGTGTATTTGTCAA |
Proliferation assay
100 ul of cells were seeded at the concentration of 10,000 per well in 96-well plates in serum free media containing transferrin and selenium (T+S). For cells with dihydrogestosterone (DHT) treatment, 10-9 M was added. Cells were allowed to proliferate for 24, 48, 72, and 96 hrs respectively. 20 ul/well of CellTiter 96 AQueous One Solution reagent was added and incubated at 37°C for two hours prior to reading (Promega, Madison WI, USA). The absorbance was then recorded using iMark Microplate Reader (Bio-Rad) at a wavelength of 490 nm. Each point represents the mean ± SD of 3 replicates.
RNA interference (RNAi)
Small interfering RNA (siRNA) duplexes for MED1 (siMED1) and FoxA1 (siFoxA1), as well as scrambled negative controls (siControl) were purchased from OriGene (Rockville, MD). The siRNA sequences are listed in Table 2. siRNA duplexes were transfected using TurboFect reagent according to the manufacturer’s instructions (Thermo Scientific).
Western blotting
Cells were washed with PBS and lysed with cold lysis buffer (50 Mm HEPES, 150 mM NaCl, 1.5 mM EGTA, 1% Triton X-100) containing protease inhibitors (Complete Mini Tablets) (Roche, USA) and Phosphatase Inhibitors Cocktail II (Sigma-Aldrich, USA). Protein concentration of cell extracts was determined by the BCA Protein assay (Thermo Scientific). Twenty micrograms of protein was electrophoresed through 4–15% gradient SDS-PAGE and subsequently transferred onto nitrocellulose membranes using Invitrogen iBlot Gel transfer system, and probed with respective antibodies at 4°C overnight. Antibodies used in this study and the working conditions are listed in Table 3. The blots were washed and incubated with a horseradish peroxidase-conjugated secondary antibodies (Pharmacia Biotech, Piscataway, NJ) for 1 hour at room temperature. Immunoreactive proteins were detected by ECL (Pharmacia Biotech). The membranes were stripped for 30 minutes in stripping buffer (Pierce, Rockford, IL) and reprobed with anti-GAPDH antibody as a loading control.
Co-Immunoprecipitation
Cells were lysed in cold lysis buffer using the aforementioned lysis buffer with complete protease and phosphatase inhibitors (Roche Applied Science). Precleared cell lysate was incubated with anti-Flag or anti-p-MED1 antibodies overnight and then with ultralink immobilized protein A/G plus beads (Thermo Scientific) for 24 hours. Beads were washed four times with cold lysis buffer, and then samples were degenerated by boiling. Lastly, immune complexes were applied in Western blotting as previously described.
Table 2: siRNA sequences
siMED1 |
(1) GGAUUAGACAGCAAACCAGGGAAGC |
siFoxA1 |
(1) GGAGGAGAGAUAAGUUAUAGGGAGC |
Table 3: Antibodies and reagents
1st/2ndAntibody/Reagent |
Company |
Anti-Flag |
Sigma |
Anti-AR (C-19) |
Santa Cruz |
Anti-AR (N20) |
Santa Cruz |
Anti-MED1 |
Santa Cruz |
Anti-p-MED1 |
Abcam |
Anti-UBE2C |
Boston Biochem |
Anti-AKT |
Cell Signaling |
Anti-p-AKT |
Cell Signaling |
Anti-ERK |
Cell Signaling |
Anti-p-ERK |
Cell Signaling |
Anti-H3K4ME1 |
Abcam |
Anti-H3K4ME3 |
Abcam |
Anti-FoxA1 |
Thermo Scientific |
Anti-AR-V7 |
Precision Antibody |
GADPH |
Cell Signaling |
Anti-Rabbit IgG |
Pharmacia Biotech |
Anti-Mouse IgG |
Pharmacia Biotech |
Dihydrotestosterone (DHT) |
Sigma |
MDV3100 (Enzalutamide) |
Selleck Chemicals |
LY294002 |
Sigma |
PD98059 |
Cell Signaling |
ChIP assay
ChIP assays were performed using MAGnigyTM Chromatin Immunoprecipitation System (Invitrogen) according to the manufacturer’s instructions. Briefly, chromatin was crosslinked for 10 min at room temperature with 1% formaldehyde. After sonication with Diagenode Bioruptor (Denville, NJ), chromatin was sheared into fragments of ~500bp and immunoprecipitated with Dynabeads coupled with anti-p-MED1 antibody or isotope control. After washing, the reversed ChIP DNA was purified and then analyzed by real-time PCR.The primers used are listed in Table 4.
Luciferase reporter assay
The pGL4.10-E4TATA-Luc vectors with UBE2C Enhancer-1 (E-1) sequences and pBEC22 (a Renilla luciferase vector) were kindly provided by Dr. Qianben Wang [35]. Three ~400 bp regions in Enhancer 1 called E1-1, E1-2 and E1-3 were systematically subcloned into pGL4.10-E4TATA-Luc vector. pGL4.10-E4TATA-Luc and pBEC22 were created by insertion of a 50-bp minimal E4 TATA promoter sequence, driving luciferase expression, into Bgl II to Hind III sites of vectors pGL4.10 and pGL4.70 (Promega) [54, 55]. For reporter assays, transfection was performed using Turbofect transfection reagent (Thermo Scientific). Cells were allowed to grow for another 24 hr before harvest. Luciferase activity was measured using a Dual-Luciferase Reporter Assay kit and GloMaxTM System (Promega).
Statistical analyses
All the experiments were performed at least three times. The data are displayed as mean ± SEM. When two groups were compared, 2-tailed Student’s t test was used (GraphPad Prism, version5.0d; GraphPad Software, La Jolla, CA). A p value of 0.05 or less was considered significant.
Table 4: ChIP Primer sequences
UBE2C |
|
E1+ |
TGCCATGTGCCCTAGAAACTG |
E1− |
CAAGCTCAGCAAAATGGTGAAA |
Promoter+ |
GCCCGAGGGAAATTGGAT |
Promoter− |
TTACTCCGCGTGGGAACACT |
E2-a+ |
GTGCGTGGTGGATCAAGTTATC |
E2-a− |
GGGTGCTCATCCCCATGA |
E2-b+ |
TCCTTTTTAGGGACATGGATGAAG |
E2-b− |
GTGTTTGGTTTTTTGTCCTTGTGAT |
E3-a+ |
CCCTGGTGGGCCTAGATGA |
E3-a− |
CAACTTCTCCCTTCCCCTGTCT |
E3-b+ |
CATCCCCCCACACGAAGTTA |
E3-b− |
TGGATAGGGAGGGTCTTGTATGA |
UBE2C |
|
Control+ |
CCACAAACTCTTCTCAGCTGGG |
Control− |
TTCTTTCCTTCCCTGTTACCCC |
PSA |
|
Enhancer+ |
GCCTGGATCTGAGAGAGATATCATC |
Enhancer− |
ACACCTTTTTTTTTCTGGATTGTTG |
Promoter+ |
CCTAGATGAAGTCTCCATGAGCTACA |
Promoter− |
GGAGGGAGAGCTAGCATTG |
ACKNOWLEDEGEMENTS
We would like to thank Dr. Qianben Wang for providing the UBE2C luciferase reporter constructs and p-MED1 antibody. This work was supported by the National Cancer Institute Pacific Northwest Prostate Cancer Specialized Program of Research Excellence (SPORE; 2 P50 CA 097186-07 Project 5), PO1 CA85859, P01 CA163227, US Army Medical Research and Materiel Command Prostate Cancer Research Programs (W81XWH-11-1-0551 and W81XWH-13-2-0093), and Department of Veterans Affairs, to S.R.P.
Conflict of interest
The authors declare no conflict of interest.
REFERENCES
1. Marques RB, Dits NF, Erkens-Schulze S, van Weerden WM, Jenster G. Bypass mechanisms of the androgen receptor pathway in therapy-resistant prostate cancer cell models. PloS one. 2010; 5:e13500.
2. Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nature clinical practice Urology. 2009; 6:76–85.
3. Attar RM, Takimoto CH, Gottardis MM. Castration-resistant prostate cancer: locking up the molecular escape routes. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009; 15:3251–3255.
4. Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer research. 2008; 68:4447–4454.
5. Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, Ettinger SL, Gleave ME, Nelson CC. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer research. 2008; 68:6407–6415.
6. Mostaghel EA, Page ST, Lin DW, Fazli L, Coleman IM, True LD, Knudsen B, Hess DL, Nelson CC, Matsumoto AM, Bremner WJ, Gleave ME, Nelson PS. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer research. 2007; 67:5033–5041.
7. Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, Febbo PG, Balk SP. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer research. 2006; 66:2815–2825.
8. Aaronson DS, Muller M, Neves SR, Chung WC, Jayaram G, Iyengar R, Ram PT. An androgen-IL-6-Stat3 autocrine loop re-routes EGF signal in prostate cancer cells. Molecular and cellular endocrinology. 2007; 270:50–56.
9. Wu JD, Haugk K, Woodke L, Nelson P, Coleman I, Plymate SR. Interaction of IGF signaling and the androgen receptor in prostate cancer progression. Journal of cellular biochemistry. 2006; 99:392–401.
10. Zhang X, Morrissey C, Sun S, Ketchandji M, Nelson PS, True LD, Vakar-Lopez F, Vessella RL, Plymate SR. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PloS one. 2011; 6:e27970.
11. Shafi AA, Cox MB, Weigel NL. Androgen receptor splice variants are resistant to inhibitors of Hsp90 and FKBP52, which alter androgen receptor activity and expression. Steroids. 2013.
12. Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocrine-related cancer. 2011; 18:R183–196.
13. Guo Z, Qiu Y. A new trick of an old molecule: androgen receptor splice variants taking the stage?! International journal of biological sciences. 2011; 7:815–822.
14. Hu R, Isaacs WB, Luo J. A snapshot of the expression signature of androgen receptor splicing variants and their distinctive transcriptional activities. The Prostate. 2011; 71:1656–1667.
15. Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Fedor HL, Lotan TL, Zheng Q, De Marzo AM, Isaacs JT, Isaacs WB, Nadal R, Paller CJ, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. The New England journal of medicine. 2014; 371:1028–1038.
16. Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, Kong X, Melamed J, Tepper CG, Kung HJ, Brodie AM, Edwards J, Qiu Y. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer research. 2009; 69:2305–2313.
17. Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, Bova GS, Luo J. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer research. 2009; 69:16–22.
18. Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, Bergh A, Wikstrom P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PloS one. 2011; 6:e19059.
19. Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, Page ST, Coleman IM, Nguyen HM, Sun H, Nelson PS, Plymate SR. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. The Journal of clinical investigation. 2010; 120:2715–2730.
20. Sun F, Chen HG, Li W, Yang X, Wang X, Jiang R, Guo Z, Chen H, Huang J, Borowsky AD, Qiu Y. Androgen receptor splice variant AR3 promotes prostate cancer via modulating expression of autocrine/paracrine factors. The Journal of biological chemistry. 2013.
21. Liu G, Sprenger C, Sun S, Epilepsia KS, Haugk K, Zhang X, Coleman I, Nelson PS, Plymate S. AR variant ARv567es induces carcinogenesis in a novel transgenic mouse model of prostate cancer. Neoplasia. 2013; 15:1009–1017.
22. Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, Edwards J, Isaacs WB, Nelson PS, Bluemn E, Plymate SR, Luo J. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer research. 2012; 72:3457–3462.
23. Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, Chen Z, Beroukhim R, Wang H, Lupien M, Wu T, Regan MM, Meyer CA, Carroll JS, Manrai AK, Janne OA, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009; 138:245–256.
24. Lin J, Raoof DA, Wang Z, Lin MY, Thomas DG, Greenson JK, Giordano TJ, Orringer MB, Chang AC, Beer DG, Lin L. Expression and effect of inhibition of the ubiquitin-conjugating enzyme E2C on esophageal adenocarcinoma. Neoplasia. 2006; 8:1062–1071.
25. Pallante P, Berlingieri MT, Troncone G, Kruhoffer M, Orntoft TF, Viglietto G, Caleo A, Migliaccio I, Decaussin-Petrucci M, Santoro M, Palombini L, Fusco A. UbcH10 overexpression may represent a marker of anaplastic thyroid carcinomas. British journal of cancer. 2005; 93:464–471.
26. Wagner KW, Sapinoso LM, El-Rifai W, Frierson HF, Butz N, Mestan J, Hofmann F, Deveraux QL, Hampton GM. Overexpression, genomic amplification and therapeutic potential of inhibiting the UbcH10 ubiquitin conjugase in human carcinomas of diverse anatomic origin. Oncogene. 2004; 23:6621–6629.
27. Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA, Shah RB, Chandran U, Monzon FA, Becich MJ, Wei JT, Pienta KJ, Ghosh D, Rubin MA, Chinnaiyan AM. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer cell. 2005; 8:393–406.
28. Yuan X, Balk SP. Mechanisms mediating androgen receptor reactivation after castration. Urologic oncology. 2009; 27:36–41.
29. Heinlein CA, Chang C. Androgen receptor (AR) coregulators: an overview. Endocrine reviews. 2002; 23:175–200.
30. Crea F, Clermont PL, Mai A, Helgason CD. HISTONE MODIFICATIONS, STEM CELLS and PROSTATE CANCER. Current pharmaceutical design. 2013.
31. Augello MA, Hickey TE, Knudsen KE. FOXA1: master of steroid receptor function in cancer. The EMBO journal. 2011; 30:3885–3894.
32. Bernardo GM, Keri RA. FOXA1: a transcription factor with parallel functions in development and cancer. Bioscience reports. 2012; 32:113–130.
33. Vijayvargia R, May MS, Fondell JD. A coregulatory role for the mediator complex in prostate cancer cell proliferation and gene expression. Cancer research. 2007; 67:4034–4041.
34. Mediwala SN, Sun H, Szafran AT, Hartig SM, Sonpavde G, Hayes TG, Thiagarajan P, Mancini MA, Marcelli M. The activity of the androgen receptor variant AR-V7 is regulated by FOXO1 in a PTEN-PI3K-AKT-dependent way. The Prostate. 2013; 73:267–277.
35. Chen Z, Zhang C, Wu D, Chen H, Rorick A, Zhang X, Wang Q. Phospho-MED1-enhanced UBE2C locus looping drives castration-resistant prostate cancer growth. The EMBO journal. 2011; 30:2405–2419.
36. Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, Rathkopf D, Shelkey J, Yu EY, Alumkal J, Hung D, Hirmand M, Seely L, Morris MJ, Danila DC, Humm J, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1–2 study. Lancet. 2010; 375:1437–1446.
37. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, et al. Integrative genomic profiling of human prostate cancer. Cancer cell. 2010; 18:11–22.
38. Hittelman AB, Burakov D, Iniguez-Lluhi JA, Freedman LP, Garabedian MJ. Differential regulation of glucocorticoid receptor transcriptional activation via AF-1-associated proteins. The EMBO journal. 1999; 18:5380–5388.
39. Zhu Y, Qi C, Jain S, Le Beau MM, Espinosa R. 3rd, Atkins GB, Lazar MA, Yeldandi AV, Rao MS, Reddy JK. Amplification and overexpression of peroxisome proliferator-activated receptor binding protein (PBP/PPARBP) gene in breast cancer. Proceedings of the National Academy of Sciences of the United States of America. 1999; 96:10848–10853.
40. Treuter E, Johansson L, Thomsen JS, Warnmark A, Leers J, Pelto-Huikko M, Sjoberg M, Wright AP, Spyrou G, Gustafsson JA. Competition between thyroid hormone receptor-associated protein (TRAP) 220 and transcriptional intermediary factor (TIF) 2 for binding to nuclear receptors. Implications for the recruitment of TRAP and p160 coactivator complexes. The Journal of biological chemistry. 1999; 274:6667–6677.
41. Wang Q, Sharma D, Ren Y, Fondell JD. A coregulatory role for the TRAP-mediator complex in androgen receptor-mediated gene expression. The Journal of biological chemistry. 2002; 277:42852–42858.
42. Jin F, Irshad S, Yu W, Belakavadi M, Chekmareva M, Ittmann MM, Abate-Shen C, Fondell JD. ERK and AKT signaling drive MED1 overexpression in prostate cancer in association with elevated proliferation and tumorigenicity. Molecular cancer research : MCR. 2013; 11:736–747.
43. Jin F, Claessens F, Fondell JD. Regulation of androgen receptor-dependent transcription by coactivator MED1 is mediated through a newly discovered noncanonical binding motif. The Journal of biological chemistry. 2012; 287:858–870.
44. Cui J, Germer K, Wu T, Wang J, Luo J, Wang SC, Wang Q, Zhang X. Cross-talk between HER2 and MED1 regulates tamoxifen resistance of human breast cancer cells. Cancer research. 2012; 72:5625–5634.
45. Misra P, Owuor ED, Li W, Yu S, Qi C, Meyer K, Zhu YJ, Rao MS, Kong AN, Reddy JK. Phosphorylation of transcriptional coactivator peroxisome proliferator-activated receptor (PPAR)-binding protein (PBP). Stimulation of transcriptional regulation by mitogen-activated protein kinase. The Journal of biological chemistry. 2002; 277:48745–48754.
46. Pandey PK, Udayakumar TS, Lin X, Sharma D, Shapiro PS, Fondell JD. Activation of TRAP/mediator subunit TRAP220/Med1 is regulated by mitogen-activated protein kinase-dependent phosphorylation. Molecular and cellular biology. 2005; 25:10695–10710.
47. Viswakarma N, Jia Y, Bai L, Gao Q, Lin B, Zhang X, Misra P, Rana A, Jain S, Gonzalez FJ, Zhu YJ, Thimmapaya B, Reddy JK. The Med1 Subunit of the Mediator Complex Induces Liver Cell Proliferation and Is Phosphorylated by AMP Kinase. The Journal of biological chemistry. 2013; 288:27898–27911.
48. Majumder PK, Sellers WR. Akt-regulated pathways in prostate cancer. Oncogene. 2005; 24:7465–7474.
49. Zhao Y, Tindall DJ, Huang H. Modulation of androgen receptor by FOXA1 and FOXO1 factors in prostate cancer. International journal of biological sciences. 2014; 10:614–619.
50. Jackson-Cook C, Bae V, Edelman W, Brothman A, Ware J. Cytogenetic characterization of the human prostate cancer cell line P69SV40T and its novel tumorigenic sublines M2182 and M15. Cancer genetics and cytogenetics. 1996; 87:14–23.
51. Plymate SR, Tennant M, Birnbaum RS, Thrasher JB, Chatta G, Ware JL. The effect on the insulin-like growth factor system in human prostate epithelial cells of immortalization and transformation by simian virus-40 T antigen. The Journal of clinical endocrinology and metabolism. 1996; 81:3709–3716.
52. Bae VL, Jackson-Cook CK, Maygarden SJ, Plymate SR, Chen J, Ware JL. Metastatic sublines of an SV40 large T antigen immortalized human prostate epithelial cell line. The Prostate. 1998; 34:275–282.
53. Plymate SR, Tennant MK, Culp SH, Woodke L, Marcelli M, Colman I, Nelson PS, Carroll JM, Roberts CT. Jr., Ware JL. Androgen receptor (AR) expression in AR-negative prostate cancer cells results in differential effects of DHT and IGF-I on proliferation and AR activity between localized and metastatic tumors. The Prostate. 2004; 61:276–290.
54. Lin YS, Carey MF, Ptashne M, Green MR. GAL4 derivatives function alone and synergistically with mammalian activators in vitro. Cell. 1988; 54:659–664.
55. Bolton EC, So AY, Chaivorapol C, Haqq CM, Li H, Yamamoto KR. Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes & development. 2007; 21:2005–2017.