
INTRODUCTION
p73 is a transcription factor and a member of the p53-family [1–3]. p73 is transcribed in two alternative N-terminal isoforms: the use of an upstream promoter allows expression of transcriptional competent TAp73 isoforms; whereas transcription initiated from a downstream promoter results in the expression of N-terminal truncated isoforms missing the transactivation domain (ΔNp73) [1, 4]. Additional isoforms (α,β,γ,δ) are generated by C-terminal differential splicing [1, 4, 5]. Recently, we demonstrated that TAp73 acts as a tumor suppressor and its depletion results in a tumor prone phenotype [6]. On the contrary, ΔNp73 has oncogenic properties and transformed fibroblasts lacking ΔNp73 fail to grow when injected into immunocompromised mice [7]. The ability of TAp73 to suppress tumor formation depends on several mechanisms, including its ability to induce cell cycle arrest [8] and apoptosis [9–18] through regulation of target genes partially shared with p53 [19–23], such as p21 [24–27], PUMA [28–32] and BAX [33, 34]. These findings notwithstanding, data on humans show that, conversely to p53 [35–41], TAp73 is rarely mutated in cancer and its expression is often retained [1, 3, 42, 43], suggesting a more complex scenario for p73 in cancer.
Over the past decade, the metabolic adaptation of tumor cells has attracted increasing appreciation as main drivers of cancer growth. Cancer cells adopt their metabolism to sustain high rate of cell growth and proliferation and to survive a hostile environment. This alteration is necessary to satisfy metabolic demands including maintenance of elevated cellular ATP levels, active biosynthesis of macromolecules and preservation of the cellular redox equilibrium [44–48]. The change in glucose metabolism known as Warburg effect or aerobic glycolysis is probably the most characterized metabolic adaptation of cancer cells, which show sustained glucose consumption and glycolytic rate. Additional metabolic adaptations have been described for amino acid [49, 50] and lipid metabolism [51, 52], demonstrating an extensive metabolic rewiring that underpins tumorigenesis, and nurturing the idea that therapeutic interventions targeting metabolism could benefit cancer patients [53–58].
Of note, we have recently demonstrated that TAp73 regulates energetic and oxidative metabolism through modulation of mitochondrial function: we showed that depletion of TAp73 impairs activity of the complex IV of the electron transport chain, decreases mitochondrial respiration and intracellular ATP and increases ROS content [59, 60]. Moreover, we unveiled a role for TAp73 in regulating enzymes of serine-glycine metabolism in cancer cells [61] and lipid metabolism in hepatic cells [62]. Shortly after, Du and colleagues showed that glucose-6-phosphate dehydrogenase (G6PD), the rate-limiting enzyme in the pentose phosphate pathway (PPP) is a TAp73 target gene [63]. Though enhanced G6PD expression, TAp73 fuels the PPP leading to increase ribose and NADPH biosynthesis, with the final outcome of reducing cellular ROS and promoting cellular proliferation [63]. These last findings have challenged the common knowledge that TAp73 is a tumor suppressor [1] and suggested that it regulates metabolism in order to sustained cell replication.
Prompted by these results, we attempted to further elucidate the regulation of cellular metabolism by p73 performing high throughput metabolomics study upon ectopic expression of TAp73 in human p53-null osteosarcoma cell lines (SaOs-2). Here we report an interesting role for TAp73 in promoting anabolic metabolism, including increased synthesis of acetyl-CoA, polyamines, and membrane phospholipids. Moreover, our data suggest that TAp73 promotes glycolysis and enhances the Warburg effect. Finally, we uncovered additional regulations impinging on the methyl donor S-adenosyl-methionine (SAM), fatty acids and arginine-proline metabolism.
Overall, our findings demonstrate that TAp73 activates anabolic pathways compatible with proliferation and in line with the report from Du and colleagues. Notwithstanding, we deem the current data insufficient to question the tumor suppressive function of TAp73, which we consistently detect through robust activation of p21 and induction of apoptosis. Our findings should also be interpreted on the light of the multifaceted physiological activities of p73, including regulation of animal aging, senescence, neurodegerative diseases and fertility [59, 64, 65].
RESULTS
To investigate the effects of TAp73 expression on cellular metabolism, we used human p53/p73 null SaOs-2 osteosarcoma cell line, engineered to overexpress human HA-tagged TAp73 isoforms when cultured in the presence of the tetracycline analog doxycycline (Dox) [29] (Supplemental Figure 1). TAp73 has been reported to halt cell proliferation and to induce apoptosis [1, 16, 28]. Hence, to avoid unwanted effects on metabolic profiling caused by induction of cell cycle arrest or cell death, we carefully investigated the outcome of p73 induction over 72h time course. In our system, expression of both TAp73α C-terminal isoforms reached plateau after 16h of Dox treatment, without any discernible effect on cell cycle distribution (Supplemental Figure 1A and B). Of note, the p73 direct target p21 was also upregulated, indicating that p73 is transcriptionally active at the same time point (Supplemental Figure 1A). Next, we measured induction of cell death as sub-G1 peak. Expression of TAp73α isoform induced considerable apoptosis in late time points, starting at 24h after Dox addition. Importantly, similarly to cell cycle profile, early time points show no difference in cell survival when compared to untreated controls, whereas sustained cell death was observed at later time points (Supplemental Figure 1C).
Taken together, these data indicate that short time induction of TAp73 (8h and 16h) results in robust expression, engagement of transcriptional activity, with no overt effect on cell cycle and cell survival. Consequently, we decided to perform metabolomics profiling in SaOs-2 cell line at early time points (8h and 16h) after induction of TAp73β. We focused on the C-terminal β isoforms because of its stronger transcriptional activity, but affects cell cycle and survival similarly to TAp73α (not shown). The main predictable bias is that a short induction might not be sufficient for completion of elaborated metabolic pathways triggered or inhibited by TAp73. Regardless, we reasoned that changes in a subset of metabolites may suffice in identifying significant metabolic rearrangements triggered by p73.
We used GC-MS and LC-MS-MS platforms for metabolomic studies of TAp73β SaOs-2 Tet-on treated with vehicle or with 2μg/ml doxycycline for 8h and 16h. This approach enabled detection of 292 biochemical compounds, a fraction of which showed significant differences between the different groups (uninduced, 8h and 16h). 15% to 20% of all detected biochemicals displayed statistical significant differences (p < 0.05, Welch’s, two samples, t-test), between uninduced and induced samples (Table 1), whereas changes in 10% of the compounds approached statistical significance (0.05 < p < 0.1).
Table 1: Significantly metabolic alterations identified in SaOs-2 cells after TAp73 over expression.
SaOs-TAp73β |
Dox 8h vs CTRL |
Dox 16h vs CTRL |
Dox 16h vs Dox 8h |
Biochemicals p ≤ 0.05 |
55 (54/1) |
42 (34/8) |
17 (7/10) |
Biochemicals 0.05 ≤ p ≤ 0.10 |
30 (26/4) |
28 (15/13) |
17 (5/12) |
Significantly altered metabolites in SaOs-Tet-on. Red indicates number of upregulated compounds, whereas green indicates downregulation. Following log transformation and imputation with minimum observed values for each compound, Welch’s two-sample t-test was used to identify biochemicals that differed significantly between doxycycline-induced sample groups compared to control (no doxycycline), as well as identifying significant biochemical migration as induction progresses from eight to sixteen hours of doxycycline exposure. An estimate of the false discovery rate (q-value) is calculated to take into account the multiple comparisons that normally occur in metabolomic-based studies; as q-values were reasonable for p ≤ 0.05, no q-value cutoff was established.
Glucose metabolism through glycolysis assumes substantial relevance in cancer. One of the hallmarks of many cancer cells is the high rate of glucose consumption and its conversion into lactate even in the presence of oxygen (known as aerobic fermentation, or Warburg effect) [44]. Though still object of intense investigation and debate, the Warburg effect is important to increase energy production and to enrich the cellular milieu with glycolytic intermediates used in biosynthesis pathways necessary to sustain proliferation [44]. Strikingly, expression of p73 produced a decrease in intracellular glucose and pyruvate levels with a concomitant increase in lactate (Figure 1). This suggests that TAp73 is a positive regulator of glycolysis and enhances the Warburg effect. Consistent with enhanced glycolytic flux, we also detected increased amount of acetyl-CoA and citrate (Figure 1). Of note, citrate was the only TCA cycle metabolites to be consistently increased (data not shown). It is possible that 16h are not enough for the build-up of other TCA cycle intermediates, or anaplerotic reactions may contribute to stabilize the TCA cycle activity. Alternatively, citrate could be shunt to fatty acid biosynthesis instead of entering Krebs’s cycle, although we did not observed increased free fatty acids at 16h post-induction. On the other hand, the increase in acetyl-CoA, correlated with an overall increase in the biosynthesis of coenzyme A (CoA), as suggested by the slight decrease in its biochemical precursors in treated cells (Supplemental Figure 2). CoA is a pivotal cofactor essential for anabolic pathways involved in lipid, amino acid and carbohydrate metabolism: its augmented levels suggest increased metabolic rate in TAp73 expressing cells.
Another relevant observation, and one of the most consistent changes, was a significant higher level of amino acids and amino acid metabolites in Dox-treated samples (Figure 2). This can be interpreted as increased uptake of metabolites from the culture media and suggests increased metabolic activity as a result of p73 induction.
Moreover, we detected elevated levels of compounds involved in membrane biosynthesis in induced cells, including choline phosphate and phosphoethanolamine (Figure 3).
TAp73 also regulates polyamines metabolism, inducing their synthesis. Indeed, ornithine, putrescine and spermidine were significantly upregulated 8h after TAp73 induction (Figure 4) and 5-methylthioadenosine, the byproduct of spermidine and spermine biosynthesis, was also consistently upregulated. Polyamines are essential for cell proliferation and their metabolism is increased by oncogenes [66]. Indeed, targeting polyamines biosynthesis is regarded as a potential anticancer therapy [66]. Thus, these data reinforce the idea that TAp73-mediated metabolic rewiring sustains anabolic pathways.
Arginine metabolism through Urea cycle leads to proline, whose hydroxylation is an important step in collagen synthesis. We observed a significant increase in hydroxyproline and proline-hydroxyproline in induced cells, suggesting extracellular matrix remodeling triggered by TAp73 (Supplemental Figure 3).
Of note, our data also suggest that TAp73 might be involved in epigenetic changes. On one hand, we have described increased levels of acetyl-CoA (Figure 2 and Supplemental Figure 2), by far the main acetyl donor in the cell. On the other hand, we also detected a significant increment in the methyl-donor SAM, which correlated with a more modest increase in its demethylated byproducts S-adenosylhomocysteine (SAH) and cystathionine (Figure 5). Cystathionine is further metabolized to the amino acid cysteine, which shows a robust accumulation in induced cells (Figure 5). Although we cannot exclude that such increase could result from enhanced cellular uptake. Finally, an upward trend in methylated compounds such as 5-methyltetrahydrofolate (5MeTHF) and methylphosphate, supports an overall surge in methylation triggered by TAp73 (Figure 5).
TAp73-expressing cells exhibit a significant accumulation of carnitine conjugates of fatty acids (Supplemental Figure 4). Fatty acids are imported in the mitochondria as acyl conjugates and formation of acyl-carnitines is a limiting step in fatty acids oxidation. The reason for this build-up is unclear. Possibly, TAp73 induction is blocking β-oxidation, as suggested by the observed increment in carnitine β-oxidation intermediates. Alternatively, carnitine conjugates of fatty acids may be used for different metabolic processes (for example membrane biosynthesis).
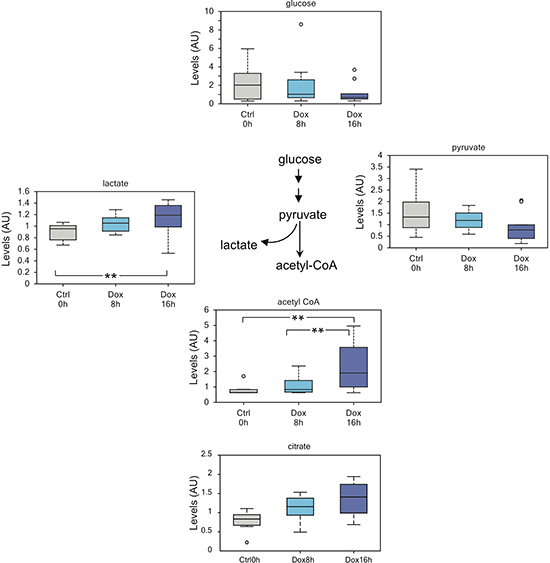
Figure 1: TAp73 enhances glycolytic flux. Increased glycolytic flux as suggested by reduced intracellular glucose and concomitant increased lactate and acetyl-CoA. This result suggests that TAp73 expression sustain Warburg effect and might help explaining while its expression is retained in many cancers. The decrease pyruvate levels probably reflect channeling into lactate and acetyl-CoA. Of note, the augmented acetyl-CoA levels may also stem for increased biosynthesis. **p < 0.05; *0.05 < p < 0.1.
To predict the possible gene/metabolite network, underlining the metabolomics effects of TAp73, we implemented a statistical procedure based on our current knowledge of signalling and metabolic pathways. We used all the metabolites resulted upregulated in our analysis, plus KEGG and Reactome databases to construct integral gene-metabolite network [67–69]. We used the integral network to compute the distance between a metabolite and p73. The distance was computed as a minimal number of steps on the network needed to get from p73 to the metabolite. Only 2 measured metabolites (glutamine and glutamate) were located within 2 steps from p73 and both are unregulated (Table 2, and Figure 6). Nine up-regulated metabolites (out of 33) resulted located within 4 steps, while only 7 (out of 160) of not up-regulated metabolites are so closed to p73 (Table 2). Interestingly this computational analysis highlighted the importance in the overall p73-dependent metabolic changes of glutaminolysis. TAp73 regulation on GLS-2 [70, 71], indeed, might widely modulate the entire aminoacids metabolism, beside the simple conversion of glutamine in glutamate (Figure 6). As consequences of GLS-2 regulation, TAp73 appears to indirectly affect aspartate, alanine, valine, leucine and isoleucine, up to 4–5 nodes of distance (Figure 6). However, this prediction is based on the current limited knowledge of p73-metabolic enzyme connections (Figure 6). In conclusion, this analysis confirms p73 effects on cell metabolism and provides a potential explanation at least for part of these effects.
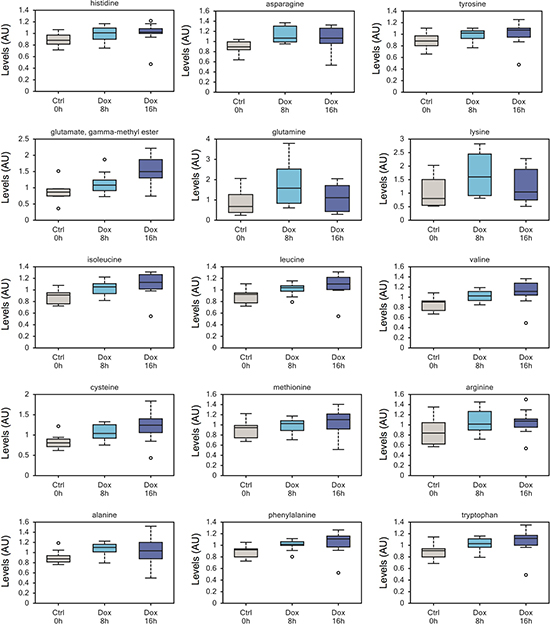
Figure 2: Increased amino acid uptake in TAp73 expressing cells. Increased intracellular contents of numerous amino acids after short-time induction of TAp73β, probably reflecting increased uptake from culture medium. This upward trend on intracellular amino acid content reflect an overall increase in anabolism upon induction of TAp73. All metabolomics data are represented as box plot, with upper and lower quartile, max and min distribution values and median value.
DISCUSSION
Oncogenes reprogram metabolism to sustain cell growth, whereas tumor suppressors halt malignancy also by mean of metabolic regulation. The awareness of the intense metabolic rewiring in cancer has stimulated research to unravel its origin and regulation. Recent work has also demonstrated that the p53 family plays an important role in regulating metabolism in different settings [59, 61, 63, 70, 72–76] and suggests that metabolic regulation contributes to the vast complexity of the p53 family functions, including cell death [77–81], redox homeostasis [82], development [83–87], senescence [88–90], aging [91–94] and fertility [95–97]. For this reason, we sought to investigate whether TAp73 regulates metabolism and how this could relate to physiological roles. With this in mind, we performed a high throughput metabolic study of osteosarcoma cells expressing tetracycline-inducible TAp73β. Through careful cell cycle and survival analysis, we were able to establish that induction of TAp73 for 8h and 16h resulted in abundant protein expression and engagement of transcriptional activity without any significant effect on cell proliferation and apoptosis. Hence, we exclude that our results are biased by p73 pro-apoptotic and anti-proliferative activities evident at later time points after TAp73 induction.
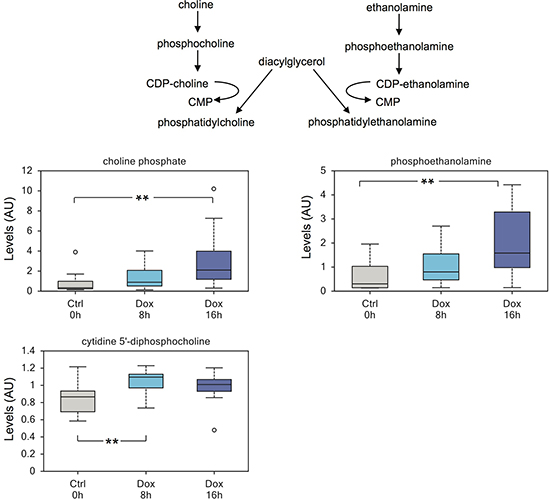
Figure 3: TAp73 regulates biosynthesis of phospholipids. Expression of TAp73β induces biosynthesis of membrane lipids, including a substantial increase in phosphoethanolamine. The data also suggest that increased cytidine 5’-monophosphate may partially results from enhanced lipid membrane biosynthesis, through conversion of diacylglycerol to phosphatidylcholine. **p < 0.05.
Interestingly, we found that expression of TAp73 modulates numerous metabolites with the net result of increasing cellular biosynthetic pathways and promoting glucose consumption and Warburg effect, a metabolic hallmark of cellular transformation [44, 98]. This last effect appears to be in striking contrast to p53-mediated inhibition of glycolytic flux [99]. Moreover, TAp73-expressing cells show increased amino acid uptake and increased levels of acetyl-CoA indicative of increased metabolic rate. Similarly, we detected increased biosynthesis of polyamines, as observed in proliferating and cancerous cells.
Therefore, it appears that, despite TAp73 is able to trigger a molecular program to arrest cell proliferation and induce apoptosis (i.e. upregulation of PUMA and p21) [28], it does promote a metabolic rearrangement that is reminiscent of an opposite, pro-growth and pro-proliferative outcome. This possibility was also suggested by a recently published manuscript reporting that TAp73 sustains proliferation through regulation of the PPP [63]. Du and colleagues show that TAp73-mediated expression of G6PD diverts glucose metabolism towards PPP and production of NADPH, which is required for ROS detoxification [100–104] and ribose for nucleotide biosynthesis and proliferation. Despite we report an increase rate of glucose metabolism, we did not observe evident differences in PPP. We did indeed record a striking increase in nucleotides levels in p73 expressing Saos-2 (manuscript in press), somehow in line with the pro-proliferative activity of TAp73 reported by Du and colleagues [63]. However, different interpretations might be applied to TAp73-depedent nucleotide biosynthesis. Being relevantly involved in DNA-damage response [105–113], TAp73 might also promote nucleotide biosynthesis in order to provide sufficient substrates to the DNA-repair machinery. In their study, Du and colleagues observed a substantial reduction of tumour proliferation in mouse xenograft tumour models after TAp73 knockdown, however this observation was not confirmed in other xenograft models [114], and contradicted by the tumourigenesis of TAp73-/- mice [6].
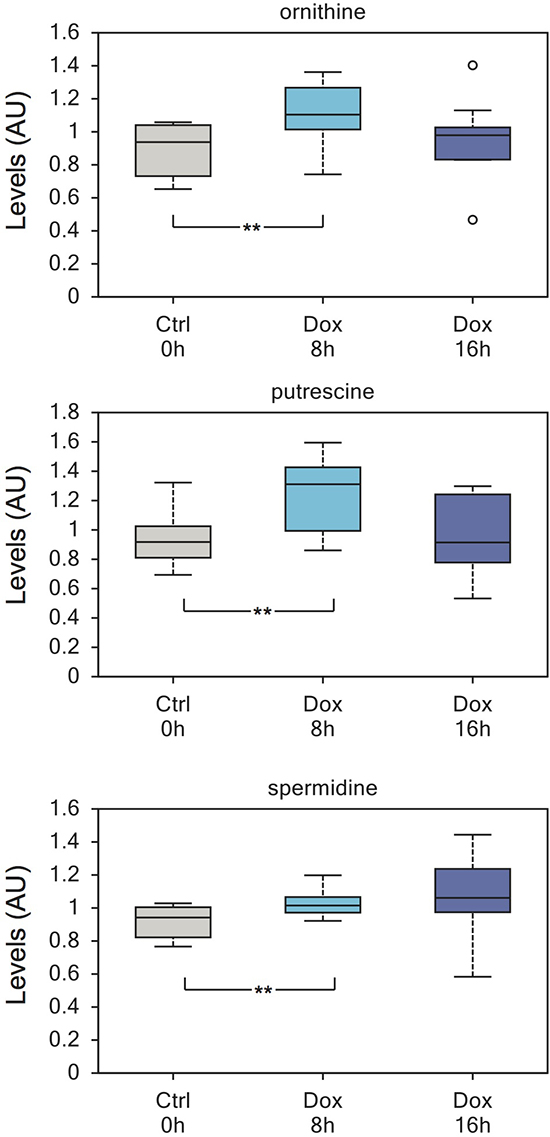
Figure 4: Enhanced polyamines biosynthesis induced by TAp73. Polyamines show a significant increase after 8h of TAp73β induction. Ornithine, putrescine and spermidine are all upregulated. Polyamine expression has been linked to proliferation and polyamine biosynthesis pathway is highly regarded for anticancer therapy. This increase is not maintained at 16h induction and the reason for this is currently unknown. **p < 0.05.
Overall, these findings bear several implications. Firstly, the operational definition of tumor suppressor might be misleading and might be unable to fully categorize genes. TAp73 regulation of metabolism should be interpreted on the light of its multifaceted activities, which include roles in fertility, development, neurodegeneration and aging [4, 22, 59, 64, 65, 115], beyond its function in cancer. Intriguingly, we and others have shown that lack of TAp73 compromise growth and maintenance of neural stem cells [64, 116]. The findings reported here prompt the intriguing possibility that impaired metabolism might underline the defect in stem cells maintenance after depletion of TAp73 and contribute to the neurodegerative outcome of p73 loss [117–119]. Moreover, we have provided evidence that loss of TAp73 leads to senescence and aging in knockout animals. We were able to correlate these phenotypes with decreased mitochondrial activity, enhanced ROS production and a marked sensitivity to oxidative stress. Nonetheless, the anabolic metabolism promoted by TAp73 might ultimately contribute to prevent accelerated senescence and aging, a possibility that warrants further investigation. This interpretation of the data would also reconcile TAp73 with its repeatedly proved role as tumor suppressor, a function that we have not failed to confirm in the present study, showing induction of p21 and apoptosis upon expression of TAp73.
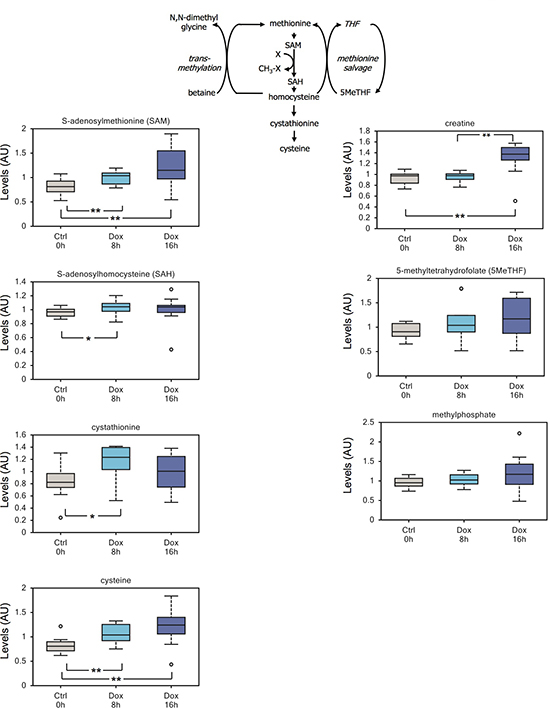
Figure 5: TAp73 expression affects the methionine cycle. A significant increment in the methyl-donor SAM correlates with an increase in its demethylated byproducts S-adenosylhomocysteine and cystathionine. Cysteine, the downstream metabolic product of cystathionine, also shows a robust accumulation in induced cells; although we cannot exclude that cysteine accumulation results from increased uptake from growth medium. Finally, we detected an upward trend in the methylated compound 5-methyltetrahydrofolate. Overall these changes suggest that TAp73 might regulate 1-carbon metabolism and epigenetic through availability of the methyl donor SAM. **p < 0.05; *0.05 < p < 0.1.
In this analysis we have identified additional metabolic changes. We report a sustained arginine metabolism in TAp73 expressing cells, which suggests a possible rearrangement of the extracellular matrix (ECM) environment. The recent discovery that p63 suppress cell motility and invasion [120–123], also suggests (i) that p73 might regulates cell motility through ECM rearrangement and (ii) that p63 might share a similar ability to regulate arginine metabolism and, possibly, ECM.
Also worth investigation is the finding of augmented SAM levels, which might link TAp73 to epigenetic rearrangement. If demonstrated, this possibility would reinforce the growing awareness that regulation of metabolism impacts cellular physiological processes, such as epigenetic and gene expression.
Further studies are necessary to address which genetic program underlines TAp73-mediated regulation of cellular metabolism and to understand how this applies to cancer and affects the phenotypes triggered by p73 depletion. Nonetheless, our work has unveiled an unexpected role for TAp73 in promoting anabolism and has clearly established a divergence between p53 and p73 in the regulation of cellular metabolism.
Table 2: Summary of metabolic network identified in SaOs-2 cells after TAp73 over expression.
Distance |
Odds Ratio |
Up regulated (33 is a number of upregulated) |
All other (160 is a number of all measured metabolites excluding 33 up regulated) |
p-value (fisher test) |
2 |
N/A (∞) |
2 (out of 33) |
0 (out of 160) |
~0.03 |
4 |
6.2 |
9 (out of 33) |
7 (out of 160) |
~ 0.001 |
5 |
4.5 |
13 (out of 33) |
14 (out of 160) |
~0.001 |
7 |
3.4 |
17 (out of 33) |
24 (out of 160) |
~ 0.01 |
10 |
1.2 |
33 (out of 33) |
126 (out of 160) |
0.23 |
The table 2 provides statistics on the number of metabolites that located on integral network within a fixed distance from p73. Odds ratio indicates enrichment of metabolites located within 4 steps to p73 among unregulated metabolites quantitatively: (9/33) / (7/160) ~ 6.2. Fisher test indicates that a probability to get similar or better enrichment randomly selecting 33 metabolites out of 193 measured is less than 1 in 1000.
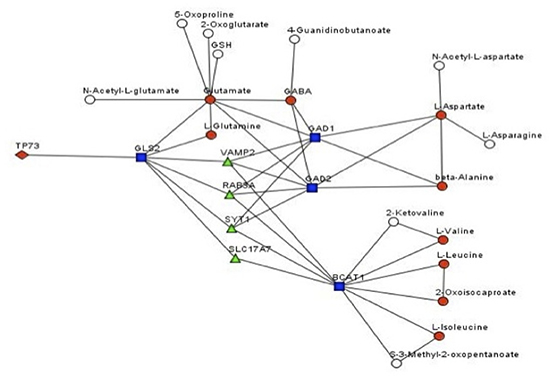
Figure 6: Genes/metabolites network affected by TAp73 Prediction of the gene/metabolite network affected by TAp73. In this network, a node is a gene or a metabolite. Edge between genes means that there is a signalling pathway where the genes are connected (transfer signal, interacts or a part of the same protein complex). Edge between a gene and a metabolite means that the gene product is enzyme known to catalyse the metabolite biotransformations.
MATERIALS AND METHODS
Cells culture
SaOs-2 Tet-On inducible for TAp73 were cultured at 37 °C in 5% CO2 in RPMI 1640 medium (Gibco), supplemented with 10% FCS, 250 mM L-glutamine, penicillin/streptomycin (1 U/ml), and 1 mM pyruvate (all from Life Technologies). TAp73 expression was induced by addition of doxycycline 2μg/ml for the indicated time.
Western blots
Proteins were extracted from cell pellets using RIPA buffer (25mM Tris/HCl pH 7.6, 150mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) supplemented with phosphatase and protease inhibitor cocktails (ROCHE). Quantification of protein extracts was performed using BCA protein assay from PIERCE. 40μg of protein were boiled for 6 minutes at 90°C and then separated using SDS-PAGE, transferred to nitrocellulose membranes using standard transfer techniques and blocked with 5% milk for 2h at room temperature. Primary antibodies were incubated O/N at 4°C in blocking with gentle agitation. We used rabbit HA (Santa Cruz, Y11), rabbit β-tubulin (Santa Cruz, H-135), rabbit p21 (H-164). Horseradish peroxidase (HRP)-conjugated secondary antibodies (BioRad) and ECL chemoluminescence substrate (PIERCE) were used for final detection.
Metabolic analysis
TAp73 SaOs-2 Tet-On cell lines were cultured in growing medium and treated for 8h and 16h with doxycycline 2μg/ml to induce TAp73β expression. Control cells were treated with vehicle (PBS) for 16h. Thirty million cells were spun down and pellets were washed once with cold PBS before being frozen in dry ice. All the samples were extracted using standard metabolic solvent extraction methods and analyzed through GC/MS and LS/MS as previously described [124].
Cell cycle and survival
For cell cycle analysis 500,000 cells were treated for the indicated time with doxycycline 2μg/ml, collected and fixed with ice cold 70% ethanol. After O/N fixing at -20°C, cells were washed in PBS, resuspended in 50μl of 10μg/ml RNase solution (SIGMA) and incubated for 10 minutes at 37°C. 500μl of staining solution (50μg/ml propidium iodide in PBS) was added to the cells, followed by additional incubation 30 minutes at 37°C. Stained cells were analyzed by flow cytometry and at least 10,000 cells per sample were collected. Data were analyzed using CELLQuest acquisition/analysis software.
REFERENCES
1. Rufini A, Agostini M, Grespi F, Tomasini R, Sayan BS, Niklison-Chirou MV, Conforti F, Velletri T, Mastino A, Mak TW, Melino G, Knight RA. p73 in Cancer. Genes & Cancer. 2011; 2:491–502.
2. Conforti F, Sayan AE, Sreekumar R, Sayan BS. Regulation of p73 activity by post-translational modifications. Cell death & disease. 2012; 3:e285.
3. Conforti F, Yang AL, Agostini M, Rufini A, Tucci P, Nicklison-Chirou MV, Grespi F, Velletri T, Knight RA, Melino G, Sayan BS. Relative expression of TAp73 and DeltaNp73 isoforms. Aging (Albany NY). 2012; 4:202–205.
4. Levrero M, De Laurenzi V, Costanzo A, Gong J, Wang JY, Melino G. The p53/p63/p73 family of transcription factors: overlapping and distinct functions. Journal of cell science. 2000; 113:1661–1670.
5. Grespi F, Amelio I, Tucci P, Annicchiarico-Petruzzelli M, Melino G. Tissue-specific expression of p73 C-terminal isoforms in mice. Cell cycle. 2012; 11:4474–4483.
6. Tomasini R, Tsuchihara K, Wilhelm M, Fujitani M, Rufini A, Cheung CC, Khan F, Itie-Youten A, Wakeham A, Tsao MS, Iovanna JL, Squire J, Jurisica I, Kaplan D, Melino G, Jurisicova A, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes & development. 2008; 22:2677–2691.
7. Wilhelm MT, Rufini A, Wetzel MK, Tsuchihara K, Inoue S, Tomasini R, Itie-Youten A, Wakeham A, Arsenian-Henriksson M, Melino G, Kaplan DR, Miller FD, Mak TW. Isoform-specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway. Genes & development. 2010; 24:549–560.
8. Dixit D, Sharma V, Ghosh S, Mehta VS, Sen E. Inhibition of Casein kinase-2 induces p53-dependent cell cycle arrest and sensitizes glioblastoma cells to tumor necrosis factor (TNFalpha)-induced apoptosis through SIRT1 inhibition. Cell death & disease. 2012; 3:e271.
9. Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature. 1991; 352:345–347.
10. Liebermann DA, Hoffman B, Steinman RA. Molecular controls of growth arrest and apoptosis: p53-dependent and independent pathways. Oncogene. 1995; 11:199–210.
11. Montero J, Dutta C, van Bodegom D, Weinstock D, Letai A. p53 regulates a non-apoptotic death induced by ROS. Cell death and differentiation. 2013; 20:1465–1474.
12. Guerrieri F, Piconese S, Lacoste C, Schinzari V, Testoni B, Valogne Y, Gerbal-Chaloin S, Samuel D, Brechot C, Faivre J, Levrero M. The sodium/iodide symporter NIS is a transcriptional target of the p53-family members in liver cancer cells. Cell death & disease. 2013; 4:e807.
13. Fan YH, Cheng J, Vasudevan SA, Dou J, Zhang H, Patel RH, Ma IT, Rojas Y, Zhao Y, Yu Y, Zhang H, Shohet JM, Nuchtern JG, Kim ES, Yang J. USP7 inhibitor P22077 inhibits neuroblastoma growth via inducing p53-mediated apoptosis. Cell death & disease. 2013; 4:e867.
14. DeMicco A, Yang-Iott K, Bassing CH. Somatic inactivation of Tp53 in hematopoietic stem cells or thymocytes predisposes mice to thymic lymphomas with clonal translocations. Cell cycle. 2013; 12:3307–3316.
15. Chillemi G, Davidovich P, D'Abramo M, Mametnabiev T, Garabadzhiu AV, Desideri A, Melino G. Molecular dynamics of the full-length p53 monomer. Cell cycle. 2013; 12:3098–3108.
16. Liu T, Roh SE, Woo JA, Ryu H, Kang DE. Cooperative role of RanBP9 and P73 in mitochondria-mediated apoptosis. Cell death & disease. 2013; 4:e476.
17. Nayak G, Cooper GM. p53 is a major component of the transcriptional and apoptotic program regulated by PI 3-kinase/Akt/GSK3 signaling. Cell death & disease. 2012; 3:e400.
18. Esposito F, Tornincasa M, Federico A, Chiappetta G, Pierantoni GM, Fusco A. High-mobility group A1 protein inhibits p53-mediated intrinsic apoptosis by interacting with Bcl-2 at mitochondria. Cell death & disease. 2012; 3:e383.
19. Lee SH, Woo TG, Lee SJ, Kim JS, Ha NC, Park BJ. Extracellular p53 fragment re-enters K-Ras mutated cells through the caveolin-1 dependent early endosomal system. Oncotarget. 2013; 4:2523–2531.
20. Kim H, Lee SH, Lee MN, Oh GT, Choi KC, Choi EY. p53 regulates the transcription of the anti-inflammatory molecule developmental endothelial locus-1 (Del-1). Oncotarget. 2013; 4:1976–1985.
21. Tomasini R, Mak TW, Melino G. The impact of p53 and p73 on aneuploidy and cancer. Trends in cell biology. 2008; 18:244–252.
22. Levine AJ, Tomasini R, McKeon FD, Mak TW, Melino G. The p53 family: guardians of maternal reproduction. Nature reviews Molecular cell biology. 2011; 12:259–265.
23. Billon N, Terrinoni A, Jolicoeur C, McCarthy A, Richardson WD, Melino G, Raff M. Roles for p53 and p73 during oligodendrocyte development. Development. 2004; 131:1211–1220.
24. Jost CA, Marin MC, Kaelin WG Jr. p73 is a simian [Correction of human] p53-related protein that can induce apoptosis. Nature. 1997; 389:191–194.
25. Sachweh MC, Drummond CJ, Higgins M, Campbell J, Lain S. Incompatible effects of p53 and HDAC inhibition on p21 expression and cell cycle progression. Cell death & disease. 2013; 4:e533.
26. Lee MS, Seo J, Choi DY, Lee EW, Ko A, Ha NC, Yoon JB, Lee HW, Kim KP, Song J. Stabilization of p21 (Cip1/WAF1) following Tip60-dependent acetylation is required for p21-mediated DNA damage response. Cell death and differentiation. 2013; 20:620–629.
27. Neise D, Sohn D, Stefanski A, Goto H, Inagaki M, Wesselborg S, Budach W, Stuhler K, Janicke RU. The p90 ribosomal S6 kinase (RSK) inhibitor BI-D1870 prevents gamma irradiation-induced apoptosis and mediates senescence via RSK- and p53-independent accumulation of p21WAF1/CIP1. Cell death & disease. 2013; 4:e859.
28. Ramadan S, Terrinoni A, Catani MV, Sayan AE, Knight RA, Mueller M, Krammer PH, Melino G, Ci E. p73 induces apoptosis by different mechanisms. Biochemical and biophysical research communications. 2005; 331:713–717.
29. Melino G, Bernassola F, Ranalli M, Yee K, Zong WX, Corazzari M, Knight RA, Green DR, Thompson C, Vousden KH. p73 Induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. The Journal of biological chemistry. 2004; 279:8076–8083.
30. Adlakha YK, Saini N. miR-128 exerts pro-apoptotic effect in a p53 transcription-dependent and -independent manner via PUMA-Bak axis. Cell death & disease. 2013; 4:e542.
31. Ray RM, Bhattacharya S, Johnson LR. Mdm2 inhibition induces apoptosis in p53 deficient human colon cancer cells by activating p73- and E2F1-mediated expression of PUMA and Siva-1. Apoptosis : an international journal on programmed cell death. 2011; 16:35–44.
32. Garrison SP, Phillips DC, Jeffers JR, Chipuk JE, Parsons MJ, Rehg JE, Opferman JT, Green DR, Zambetti GP. Genetically defining the mechanism of Puma- and Bim-induced apoptosis. Cell death and differentiation. 2012; 19:642–649.
33. Dewson G, Ma S, Frederick P, Hockings C, Tan I, Kratina T, Kluck RM. Bax dimerizes via a symmetric BH3:groove interface during apoptosis. Cell death and differentiation. 2012; 19:661–670.
34. Ozaki T, Naka M, Takada N, Tada M, Sakiyama S, Nakagawara A. Deletion of the COOH-terminal region of p73alpha enhances both its transactivation function and DNA-binding activity but inhibits induction of apoptosis in mammalian cells. Cancer research. 1999; 59:5902–5907.
35. Haupt S, Mitchell C, Corneille V, Shortt J, Fox S, Pandolfi PP, Castillo-Martin M, Bonal DM, Cordon-Cardo C, Lozano G, Haupt Y. Loss of PML cooperates with mutant p53 to drive more aggressive cancers in a gender-dependent manner. Cell cycle. 2013; 12:1722–1731.
36. Blandino G, Deppert W, Hainaut P, Levine A, Lozano G, Olivier M, Rotter V, Wiman K, Oren M. Mutant p53 protein, master regulator of human malignancies: a report on the Fifth Mutant p53 Workshop. Cell death and differentiation. 2012; 19:180–183.
37. Hill R, Rabb M, Madureira PA, Clements D, Gujar SA, Waisman DM, Giacomantonio CA, Lee PW. Gemcitabine-mediated tumour regression and p53-dependent gene expression: implications for colon and pancreatic cancer therapy. Cell death & disease. 2013; 4:e791.
38. Jung YS, Lee SJ, Lee SH, Chung JY, Jung YJ, Hwang SH, Ha NC, Park BJ. Loss of VHL promotes progerin expression, leading to impaired p14/ARF function and suppression of p53 activity. Cell cycle. 2013; 12:2277–2290.
39. Mello SS, Attardi LD. Not all p53 gain-of-function mutants are created equal. Cell death and differentiation. 2013; 20:855–857.
40. Haupt S, Gamell C, Wolyniec K, Haupt Y. Interplay between p53 and VEGF: how to prevent the guardian from becoming a villain. Cell death and differentiation. 2013; 20:852–854.
41. Silva N, Adamo A, Santonicola P, Martinez-Perez E, La Volpe A. Pro-crossover factors regulate damage-dependent apoptosis in the Caenorhabditis elegans germ line. Cell death and differentiation. 2013; 20:1209–1218.
42. Melino G, De Laurenzi V, Vousden KH. p73: Friend or foe in tumorigenesis. Nature reviews Cancer. 2002; 2:605–615.
43. Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Molecular cancer research : MCR. 2004; 2:371–386.
44. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nature reviews Cancer. 2011; 11:85–95.
45. Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012; 491:364–373.
46. Munoz-Pinedo C, El Mjiyad N, Ricci JE. Cancer metabolism: current perspectives and future directions. Cell death & disease. 2012; 3:e248.
47. Lyssiotis CA, Vander-Heiden MG, Munoz-Pinedo C, Emerling BM. Emerging concepts: linking hypoxic signaling and cancer metabolism. Cell death & disease. 2012; 3:e303.
48. Ferber EC, Peck B, Delpuech O, Bell GP, East P, Schulze A. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell death and differentiation. 2012; 19:968–979.
49. Amelio I, Cutruzzola F, Antonov A, Agostini M, Melino G. Serine and glycine metabolism in cancer. Trends in biochemical sciences. 2014; 39:191–198.
50. Tedeschi PM, Markert EK, Gounder M, Lin H, Dvorzhinski D, Dolfi SC, Chan LL, Qiu J, DiPaola RS, Hirshfield KM, Boros LG, Bertino JR, Oltvai ZN, Vazquez A. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell death & disease. 2013; 4:e877.
51. Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular fatty acid metabolism and cancer. Cell metabolism. 2013; 18:153–161.
52. Liu K, Czaja MJ. Regulation of lipid stores and metabolism by lipophagy. Cell death and differentiation. 2013; 20:3–11.
53. Finley LW, Zhang J, Ye J, Ward PS, Thompson CB. SnapShot: cancer metabolism pathways. Cell metabolism. : e462. 2013; 17:466–466.
54. Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nature reviews Drug discovery. 2011; 10:671–684.
55. Shi WY, Xiao D, Wang L, Dong LH, Yan ZX, Shen ZX, Chen SJ, Chen Y, Zhao WL. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell death & disease. 2012; 3:e275.
56. Goldberg L, Israeli R, Kloog Y. FTS and 2-DG induce pancreatic cancer cell death and tumor shrinkage in mice. Cell death & disease. 2012; 3:e284.
57. Ganapathy-Kanniappan S, Kunjithapatham R, Geschwind JF. Glyceraldehyde-3-phosphate dehydrogenase: a promising target for molecular therapy in hepatocellular carcinoma. Oncotarget. 2012; 3:940–953.
58. Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell death & disease. 2013; 4:e532.
59. Rufini A, Niklison-Chirou MV, Inoue S, Tomasini R, Harris IS, Marino A, Federici M, Dinsdale D, Knight RA, Melino G, Mak TW. TAp73 depletion accelerates aging through metabolic dysregulation. Genes & development. 2012; 26:2009–2014.
60. Bhagatte Y, Lodwick D, Storey N. Mitochondrial ROS production and subsequent ERK phosphorylation are necessary for temperature preconditioning of isolated ventricular myocytes. Cell death & disease. 2012; 3:e345.
61. Amelio I, Markert EK, Rufini A, Antonov AV, Sayan BS, Tucci P, Agostini M, Mineo TC, Levine AJ, Melino G. p73 regulates serine biosynthesis in cancer. Oncogene. 2013.
62. He Z, Liu H, Agostini M, Yousefi S, Perren A, Tschan MP, Mak TW, Melino G, Simon HU. p73 regulates autophagy and hepatocellular lipid metabolism through a transcriptional activation of the ATG5 gene. Cell death and differentiation. 2013; 20:1415–1424.
63. Du W, Jiang P, Mancuso A, Stonestrom A, Brewer MD, Minn AJ, Mak TW, Wu M, Yang X. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nature cell biology. 2013.
64. Killick R, Niklison-Chirou M, Tomasini R, Bano D, Rufini A, Grespi F, Velletri T, Tucci P, Sayan BS, Conforti F, Gallagher E, Nicotera P, Mak TW, Melino G, Knight RA, Agostini M. p73: a multifunctional protein in neurobiology. Molecular neurobiology. 2011; 43:139–146.
65. Inoue S, Tomasini R, Rufini A, Elia AJ, Agostini M, Amelio I, Cescon D, Dinsdale D, Zhou L, Harris IS, Lac S, Silvester J, Li WY, Sasaki M, Haight J, Brustle A, et al. TAp73 is required for spermatogenesis and the maintenance of male fertility. Proceedings of the National Academy of Sciences of the United States of America. 2014; 111:1843–1848.
66. Gerner EW, Meyskens FL, Jr. Polyamines and cancer: old molecules, new understanding. Nature reviews Cancer. 2004; 4:781–792.
67. Antonov AV, Schmidt EE, Dietmann S, Krestyaninova M, Hermjakob H. R spider: a network-based analysis of gene lists by combining signaling and metabolic pathways from Reactome and KEGG databases. Nucleic acids research. 2010; 38:W78–83.
68. Antonov AV. BioProfiling.de: analytical web portal for high-throughput cell biology. Nucleic acids research. 2011; 39:W323–327.
69. Dietmann S, Lee W, Wong P, Rodchenkov I, Antonov AV. CCancer: a bird's eye view on gene lists reported in cancer-related studies. Nucleic acids research. 2010; 38:W118–123.
70. Velletri T, Romeo F, Tucci P, Peschiaroli A, Annicchiarico-Petruzzelli M, Niklison-Chirou MV, Amelio I, Knight RA, Mak TW, Melino G, Agostini M. GLS2 is transcriptionally regulated by p73 and contributes to neuronal differentiation. Cell cycle. 2013; 12:3564–3573.
71. Amelio I, Markert EK, Rufini A, Antonov AV, Sayan BS, Tucci P, Agostini M, Mineo TC, Levine AJ, Melino G. p73 regulates serine biosynthesis in cancer. Oncogene. 2014; 33:5039–5046.
72. Grabow S, Waring P, Happo L, Cook M, Mason KD, Kelly PN, Strasser A. Pharmacological blockade of Bcl-2, Bcl-x(L) and Bcl-w by the BH3 mimetic ABT-737 has only minor impact on tumour development in p53-deficient mice. Cell death and differentiation. 2012; 19:623–632.
73. Blagosklonny MV. Tumor suppression by p53 without apoptosis and senescence: conundrum or rapalog-like gerosuppression? Aging (Albany NY). 2012; 4:450–455.
74. D'Alessandro A, Amelio I, Berkers CR, Antonov A, Vousden KH, Melino G, Zolla L. Metabolic effect of TAp63alpha: enhanced glycolysis and pentose phosphate pathway, resulting in increased antioxidant defense. Oncotarget. 2014; 5:7722–7733.
75. Berkers CR, Maddocks OD, Cheung EC, Mor I, Vousden KH. Metabolic regulation by p53 family members. Cell metabolism. 2013; 18:617–633.
76. Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012; 149:1269–1283.
77. Selvarajah J, Nathawat K, Moumen A, Ashcroft M, Carroll VA. Chemotherapy-mediated p53-dependent DNA damage response in clear cell renal cell carcinoma: role of the mTORC1/2 and hypoxia-inducible factor pathways. Cell death & disease. 2013; 4:e865.
78. Bergeaud M, Mathieu L, Guillaume A, Moll UM, Mignotte B, Le Floch N, Vayssiere JL, Rincheval V. Mitochondrial p53 mediates a transcription-independent regulation of cell respiration and interacts with the mitochondrial F(1)F0-ATP synthase. Cell cycle. 2013; 12:2781–2793.
79. Cheung EC, Athineos D, Lee P, Ridgway RA, Lambie W, Nixon C, Strathdee D, Blyth K, Sansom OJ, Vousden KH. TIGAR is required for efficient intestinal regeneration and tumorigenesis. Developmental cell. 2013; 25:463–477.
80. Marouco D, Garabadgiu AV, Melino G, Barlev NA. Lysine-specific modifications of p53: a matter of life and death?. Oncotarget. 2013; 4:1556–1571.
81. Huang BH, Zhuo JL, Leung CH, Lu GD, Liu JJ, Yap CT, Hooi SC. PRAP1 is a novel executor of p53-dependent mechanisms in cell survival after DNA damage. Cell death & disease. 2012; 3:e442.
82. Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, Vousden KH. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature. 2013; 493:542–546.
83. Paris M, Rouleau M, Puceat M, Aberdam D. Regulation of skin aging and heart development by TAp63. Cell death and differentiation. 2012; 19:186–193.
84. Hadjal Y, Hadadeh O, Yazidi CE, Barruet E, Binetruy B. A p38MAPK-p53 cascade regulates mesodermal differentiation and neurogenesis of embryonic stem cells. Cell death & disease. 2013; 4:e737.
85. Candi E, Rufini A, Terrinoni A, Giamboi-Miraglia A, Lena AM, Mantovani R, Knight R, Melino G. DeltaNp63 regulates thymic development through enhanced expression of FgfR2 and Jag2. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104:11999–12004.
86. Lena AM, Cipollone R, Amelio I, Catani MV, Ramadan S, Browne G, Melino G, Ci E. Skn-1a/Oct-11 and DeltaNp63alpha exert antagonizing effects on human keratin expression. Biochemical and biophysical research communications. 2010; 401:568–573.
87. Rufini S, Lena AM, Cadot B, Mele S, Amelio I, Terrinoni A, Desideri A, Melino G, Ci E. The sterile alpha-motif (SAM) domain of p63 binds in vitro monoasialoganglioside (GM1) micelles. Biochemical pharmacology. 2011; 82:1262–1268.
88. Burnley P, Rahman M, Wang H, Zhang Z, Sun X, Zhuge Q, Su DM. Role of the p63-FoxN1 regulatory axis in thymic epithelial cell homeostasis during aging. Cell death & disease. 2013; 4:e932.
89. Alexandrova EM, Moll UM. Generation of p53-deficient induced pluripotent stem cells from mouse embryo fibroblasts. Methods in molecular biology. 2013; 962:157–164.
90. Candi E, Amelio I, Agostini M, Melino G. MicroRNAs and p63 in epithelial stemness. Cell death and differentiation. 2014.
91. Lin SC, Taatjes DJ. DeltaNp53 and aging. Aging (Albany NY). 2013; 5:717–718.
92. Migliaccio E, Giorgio M, Pelicci PG. p53 and aging: role of p66Shc. Aging (Albany NY). 2013; 5:488–489.
93. Alexandrova EM, Petrenko O, Nemajerova A, Romano RA, Sinha S, Moll UM. DeltaNp63 regulates select routes of reprogramming via multiple mechanisms. Cell death and differentiation. 2013; 20:1698–1708.
94. Richardson RB. p53 mutations associated with aging-related rise in cancer incidence rates. Cell cycle. 2013; 12:2468–2478.
95. Kim SY, Cordeiro MH, Serna VA, Ebbert K, Butler LM, Sinha S, Mills AA, Woodruff TK, Kurita T. Rescue of platinum-damaged oocytes from programmed cell death through inactivation of the p53 family signaling network. Cell death and differentiation. 2013; 20:987–997.
96. Hutt K, Kerr JB, Scott CL, Findlay JK, Strasser A. How to best preserve oocytes in female cancer patients exposed to DNA damage inducing therapeutics. Cell death and differentiation. 2013; 20:967–968.
97. Amelio I, Grespi F, Annicchiarico-Petruzzelli M, Melino G. p63 the guardian of human reproduction. Cell cycle. 2012; 11:4545–4551.
98. Markert EK, Levine AJ, Vazquez A. Proliferation and tissue remodeling in cancer: the hallmarks revisited. Cell death & disease. 2012; 3:e397.
99. Madan E, Gogna R, Bhatt M, Pati U, Kuppusamy P, Mahdi AA. Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget. 2011; 2:948–957.
100. Weyemi U, Lagente-Chevallier O, Boufraqech M, Prenois F, Courtin F, Caillou B, Talbot M, Dardalhon M, Al Ghuzlan A, Bidart JM, Schlumberger M, Dupuy C. ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene. 2012; 31:1117–1129.
101. Chen K, Kirber MT, Xiao H, Yang Y, Keaney JF Jr. Regulation of ROS signal transduction by NADPH oxidase 4 localization. The Journal of cell biology. 2008; 181:1129–1139.
102. Eisenberg-Lerner A, Kimchi A. PKD is a kinase of Vps34 that mediates ROS-induced autophagy downstream of DAPk. Cell death and differentiation. 2012; 19:788–797.
103. Robert G, Puissant A, Dufies M, Marchetti S, Jacquel A, Cluzeau T, Colosetti P, Belhacene N, Kahle P, Da Costa CA, Luciano F, Checler F, Auberger P. The caspase 6 derived N-terminal fragment of DJ-1 promotes apoptosis via increased ROS production. Cell death and differentiation. 2012; 19:1769–1778.
104. Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A, Agostinis P. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell death and differentiation. 2012; 19:1880–1891.
105. Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, Jacks T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002; 416:560–564.
106. Urist M, Tanaka T, Poyurovsky MV, Prives C. p73 induction after DNA damage is regulated by checkpoint kinases Chk1 and Chk2. Genes & development. 2004; 18:3041–3054.
107. Yuan ZM, Shioya H, Ishiko T, Sun X, Gu J, Huang YY, Lu H, Kharbanda S, Weichselbaum R, Kufe D. p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature. 1999; 399:814–817.
108. Mazza D, Infante P, Colicchia V, Greco A, Alfonsi R, Siler M, Antonucci L, Po A, De Smaele E, Ferretti E, Capalbo C, Bellavia D, Canettieri G, Giannini G, Screpanti I, Gulino A, et al. PCAF ubiquitin ligase activity inhibits Hedgehog/Gli1 signaling in p53-dependent response to genotoxic stress. Cell death and differentiation. 2013; 20:1688–1697.
109. Evangelou K, Bartkova J, Kotsinas A, Pateras IS, Liontos M, Velimezi G, Kosar M, Liloglou T, Trougakos IP, Dyrskjot L, Andersen CL, Papaioannou M, Drosos Y, Papafotiou G, Hodny Z, Sosa-Pineda B, et al. The DNA damage checkpoint precedes activation of ARF in response to escalating oncogenic stress during tumorigenesis. Cell death and differentiation. 2013; 20:1485–1497.
110. Blasius M, Bartek J. ATM targets hnRNPK to control p53. Cell cycle. 2013; 12:1162–1163.
111. Bennetzen MV, Larsen DH, Dinant C, Watanabe S, Bartek J, Lukas J, Andersen JS. Acetylation dynamics of human nuclear proteins during the ionizing radiation-induced DNA damage response. Cell cycle. 2013; 12:1688–1695.
112. Kleiblova P, Shaltiel IA, Benada J, Sevcik J, Pechackova S, Pohlreich P, Voest EE, Dundr P, Bartek J, Kleibl Z, Medema RH, Macurek L. Gain-of-function mutations of PPM1D/Wip1 impair the p53-dependent G1 checkpoint. The Journal of cell biology. 2013; 201:511–521.
113. Morita A, Ariyasu S, Ohya S, Takahashi I, Wang B, Tanaka K, Uchida T, Okazaki H, Hanaya K, Enomoto A, Nenoi M, Ikekita M, Aoki S, Hosoi Y. Evaluation of zinc (II) chelators for inhibiting p53-mediated apoptosis. Oncotarget. 2013; 4:2439–2450.
114. Hong B, Prabhu VV, Zhang S, van den Heuvel AP, Dicker DT, Kopelovich L, El-Deiry WS. Prodigiosin rescues deficient p53 signaling and antitumor effects via upregulating p73 and disrupting its interaction with mutant p53. Cancer research. 2014; 74:1153–1165.
115. Lu F, Li YQ, Aubert I, Wong CS. Endothelial cells regulate p53-dependent apoptosis of neural progenitors after irradiation. Cell death & disease. 2012; 3:e324.
116. Alexandrova EM, Talos F, Moll UM. p73 is dispensable for commitment to neural stem cell fate, but is essential for neural stem cell maintenance and for blocking premature differentiation. Cell death and differentiation. 2013; 20:368.
117. Agostini M, Tucci P, Killick R, Candi E, Sayan BS, Rivetti di Val Cervo P, Nicotera P, McKeon F, Knight RA, Mak TW, Melino G. Neuronal differentiation by TAp73 is mediated by microRNA-34a regulation of synaptic protein targets. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108:21093–21098.
118. Grespi F, Melino G. P73 and age-related diseases: is there any link with Parkinson Disease? Aging (Albany NY). 2012; 4:923–931.
119. Wetzel MK, Naska S, Laliberte CL, Rymar VV, Fujitani M, Biernaskie JA, Cole CJ, Lerch JP, Spring S, Wang SH, Frankland PW, Henkelman RM, Josselyn SA, Sadikot AF, Miller FD, Kaplan DR. p73 regulates neurodegeneration and phospho-tau accumulation during aging and Alzheimer's disease. Neuron. 2008; 59:708–721.
120. Neilsen PM, Noll JE, Suetani RJ, Schulz RB, Al-Ejeh F, Evdokiou A, Lane DP, Callen DF. Mutant p53 uses p63 as a molecular chaperone to alter gene expression and induce a pro-invasive secretome. Oncotarget. 2011; 2:1203–1217.
121. Melino G. p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell death and differentiation. 2011; 18:1487–1499.
122. Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo V, Parenti AR, Rosato A, Bicciato S, Balmain A, Piccolo S. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 2009; 137:87–98.
123. Celardo I, Antonov A, Amelio I, Annicchiarico-Petruzzelli M, Melino G. p63 transcriptionally regulates the expression of matrix metallopeptidase 13. Oncotarget. 2014; 5:1279–1289.
124. Tucci P, Porta G, Agostini M, Dinsdale D, Iavicoli I, Cain K, Finazzi-Agro A, Melino G, Willis A. Metabolic effects of TiO2 nanoparticles, a common component of sunscreens and cosmetics, on human keratinocytes. Cell death & disease. 2013; 4:e549.