
INTRODUCTION
Basal cell carcinomas (BCCs) of the skin represent nearly one half of all cancers diagnosed in the United States [1, 2]. Although these tumors rarely metastasize and are an infrequent cause of cancer mortality [3], they are associated with significant morbidity due to local invasion and tissue destruction. The basal cell nevus syndrome (BCNS), also known as Gorlin syndrome or the nevoid basal cell carcinoma syndrome, is an autosomal dominant disorder characterized by multiple BCCs, occasional childhood malignancies, and developmental defects including palmar pits, jaw cysts, coarse facial appearance, malformations of the ribs, spine and brain, macrocephaly, and generalized overgrowth [4–6].
The majority of BCNS cases can be attributed to heterozygous germline mutations in PTCH1; the human homolog of the Drosophila gene, patched [7]. Patched (PTCH1) encodes the receptor for hedgehog (HH), a secreted morphogen involved in establishing the basic framework of developing embryos in Drosophila and in other organisms, including vertebrates [8, 9]. In the canonical pathway, binding of HH to PTCH releases inhibition on SMO, which then signals through a series of steps to zinc finger trans-activators that ultimately convey the signal to the nucleus. In humans, the trans-activator function is performed by GLI1, GLI2 and GLI3. Genes directly regulated by GLI include PTCH1, HIP (hedgehog interacting protein), and GLI1 itself, in many tissues. A host of other potential target genes in normal cells have been identified by expression microarrays where it appears that HH signaling is influenced by the stage of development and tissue-specific factors [8, 9], leading to different global expression patterns in different organs [10].
Appropriately, tumor suppressor genes and their effector pathways have been identified in several dominantly heritable cancers, providing insights about cancer initiation and potential intervention [11–13]. Analogous to retinoblastoma [11, 12] and other autosomal dominant forms of heritable cancer [13–18], BCCs in BCNS patients, arise through a two-hit mechanism in which “one hit” is an inherited, inactivating mutation in PTCH1, and the second hit is a somatically derived mutation in the remaining PTCH1 allele [7]. The great majority of sporadic BCCs have two somatically derived mutations in PTCH1, or less often, an activating mutation in SMO that mimics loss of PTCH1 [7, 19–23]. Although additional molecular changes are probably necessary for the development of BCCs, it is likely that loss of PTCH1 function and consequent activation of the hedgehog pathway is a necessary early step [24, 25].
This “one-hit model” for BCNS is supported by many features of this disorder such as developmental defects that have a generalized or symmetric manifestation which would be difficult to reconcile with a two-hit mechanism. Generalized overgrowth, acromegalic bone structure, and relative macrocephaly, for example, suggest that heterozygous loss of function leads to cellular hyper-proliferation in bone or cartilage [26]. In this regard, expression analysis of BCNS fibroblasts and fibroblasts from unaffected individuals [27] implicated genes that were hypothesized to contribute to a growth advantage in BCNS fibroblasts.
BCNS patients can develop thousands of skin tumors over their lifetimes, and treating this multiplicity of tumors with conventional surgical or other ablative therapies is a challenge. Preventive or curative medical therapy with an effective targeted agent that is known to have limited adverse, off-target effects would represent a great advance. Although several compounds are known to inhibit hedgehog signaling [28–30], their overall preventative efficacy of BCC remains to be established, and side effects limit their long-term use. In this regard, it is important to note that cell identity switch allows residual BCC to survive hedgehog pathway inhibition, necessitating the use of an additional agent(s) [31].
Rapamycin, also known as sirolimus, is an antibiotic isolated from Streptomyces hygroscopicus [32] that blocks mitogenic signaling by growth factor receptors and is a promising anti-neoplastic agent for a variety of tumors [33–37]. The best studied pathway affected by rapamycin is that conducted by the mTOR complex 1 (mTORC1), which consists of the mTOR protein (a member of the (PI(3)K) family) DEPTOR, PRAS40, RAPTOR, mLST8, and TTI1–TEL2 [33–37]. Importantly, rapamycin has also been shown to have an effect on the canonical HH pathway wherein crosstalk between mTOR and HH pathways has been indicated [38–40]. Thus, while blocking mTOR may take away a signaling component that is necessary but not sufficient for transformation, the mTOR and HH pathways coincidentally may target an overlapping set of genes that are likely to inhibit BCC carcinogenesis. Incidentally, rapamycin has been shown to inhibit BCC development in kidney transplant patients [41, 42] and has also been shown in a case report to inhibit BCCs, advocating its use when surgical intervention is counter-indicated [43].
The first aim of this study was to profile baseline global gene expression in cells obtained from histologically normal skin of BCNS subjects and compare these with skin cells from unaffected individuals to detect “single-hit” effects caused by PTCH1 (+/-). Since hedgehog activation has been proposed to function in carcinogenesis through paracrine activity on stroma as well as cell autonomous activity [44, 45], both keratinocytes and fibroblasts were analyzed in this study. Our second aim was to determine if a particular set of genes was differentially influenced by rapamycin in tissues that might have been “sensitized” to its effect by loss of one copy of PTCH1.
RESULTS
Exploratory analysis at baseline reveals no clear association of samples based on PTCH1 mutation status
The first goal of this study was to explore changes in gene expression that can be attributed to mutations in PTCH1 and can possibly contribute to molecular abnormalities of basal cell carcinomas. Here we profiled gene expression patterns of PTCH1 (+/-) keratinocytes and PTCH1 (+/-) fibroblasts derived from histologically normal tissues of BCNS subjects and compared them to cells derived from normal individuals at baseline and following treatment with rapamycin (Table 1). A principal components analysis (PCA) (Supplementary Figure 1) revealed that although the two treatments clustered separately, i.e., with and without rapamycin, the differences in gene expression due to PTCH1 mutation did not result in a significant clustering and separation of BCNS from normal samples, indicating that the PTCH1 mutation itself, whether truncating or missense, does not alter global gene expression of keratinocytes or fibroblasts enough to provide clear separation between BCNS and normal specimens. The lack of clear delineation of samples at baseline can be attributed to an attenuated canonical HH pathway in PTCH1 (+/-) cells grown in culture [46, 47]. However, rapamycin treatment caused significant changes in global gene expression pattern that provided a clear separation of treated and untreated samples derived from either BCNS patients or normal subjects (Figure 1).
Table 1: Characteristics of cases and controls
Participant ID |
Gender |
Age |
PTCH1 Sequencing |
Mutation Type |
Clinical features |
|||
|---|---|---|---|---|---|---|---|---|
BCCs |
Jaw Cysts |
Pits |
||||||
Cases |
||||||||
04 |
F |
55 |
W278X |
Truncating |
+ |
+ |
+ |
|
11 |
F |
60 |
W844G1 |
Missense |
+ |
+ |
+ |
|
12 |
F |
51 |
W926L2 |
Missense |
+ |
+ |
+ |
|
14 |
F |
30 |
Relative of 15, 16 |
Q853X3 |
Truncating |
+ |
+ |
+ |
07 |
M |
20 |
Codon 1124delC |
Truncating |
+ |
+ |
+ |
|
13 |
M |
25 |
Relative of 12 |
W926L2 |
Missense |
+ |
+ |
+ |
17 |
M |
44 |
C1043X |
Truncating |
+ |
+ |
+ |
|
18 |
M |
46 |
IVS15+9 G>C4 |
Missense |
+ |
+ |
+ |
|
22 |
M |
50 |
Exon 1E splice acceptor5 |
Missense |
+ |
+ |
- |
|
Controls |
||||||||
02 |
F |
41 |
Not done |
- |
- |
- |
- |
|
08 |
F |
21 |
Relative of 07 |
Wild type |
- |
- |
- |
- |
09 |
F |
27 |
Not done |
- |
- |
- |
- |
|
15 |
F |
54 |
Relative of 14 |
Wild type |
- |
- |
- |
- |
01 |
M |
37 |
Not done |
- |
- |
- |
- |
|
03 |
M |
51 |
Not done |
- |
- |
- |
- |
|
05 |
M |
49 |
Relative of 04 |
Wild type |
- |
- |
- |
- |
16 |
M |
53 |
Relative of 14 |
Wild type |
- |
- |
- |
- |
1Trp at amino acid 844 is 100% conserved in vertebrates and invertebrates. Codon numbering is according to NP_000255.
2Trp at amino acid 926 is 100% conserved in vertebrates and invertebrates; W926R reported previously in BCNS [67].
3Truncating mutation has been reported [67].
4Variant of unknown significance; however, PTCH1 mutations p.W926X/R have been reported in 5 cases [68].
5Creates an abnormal acceptor site predicted to be highly efficient at position 32 of exon 1E (G to A change).
The PCA results were further supported by unsupervised hierarchical clustering of genes (coefficient of variation of more than 0.1 in the microarray data) at baseline and after rapamycin treatment (Figure 1). While control samples distinctly cluster apart from rapamycin treated samples for both the keratinocytes (Figure 1A) and fibroblasts (Figure 1B), the clustering pattern was independent of PTCH1 mutation status or rapamycin concentration. Together, these results reaffirm our observation that mono-allelic PTCH1 mutation did not affect the global gene expression pattern to distinguish BCNS from normal samples in vitro. Secondly, the analysis suggests that the effect of rapamycin at a more stringent level of statistical scrutiny (FDR<0.05) is dependent on the intrinsic gene expression pattern unique to the sample derived from each individual.
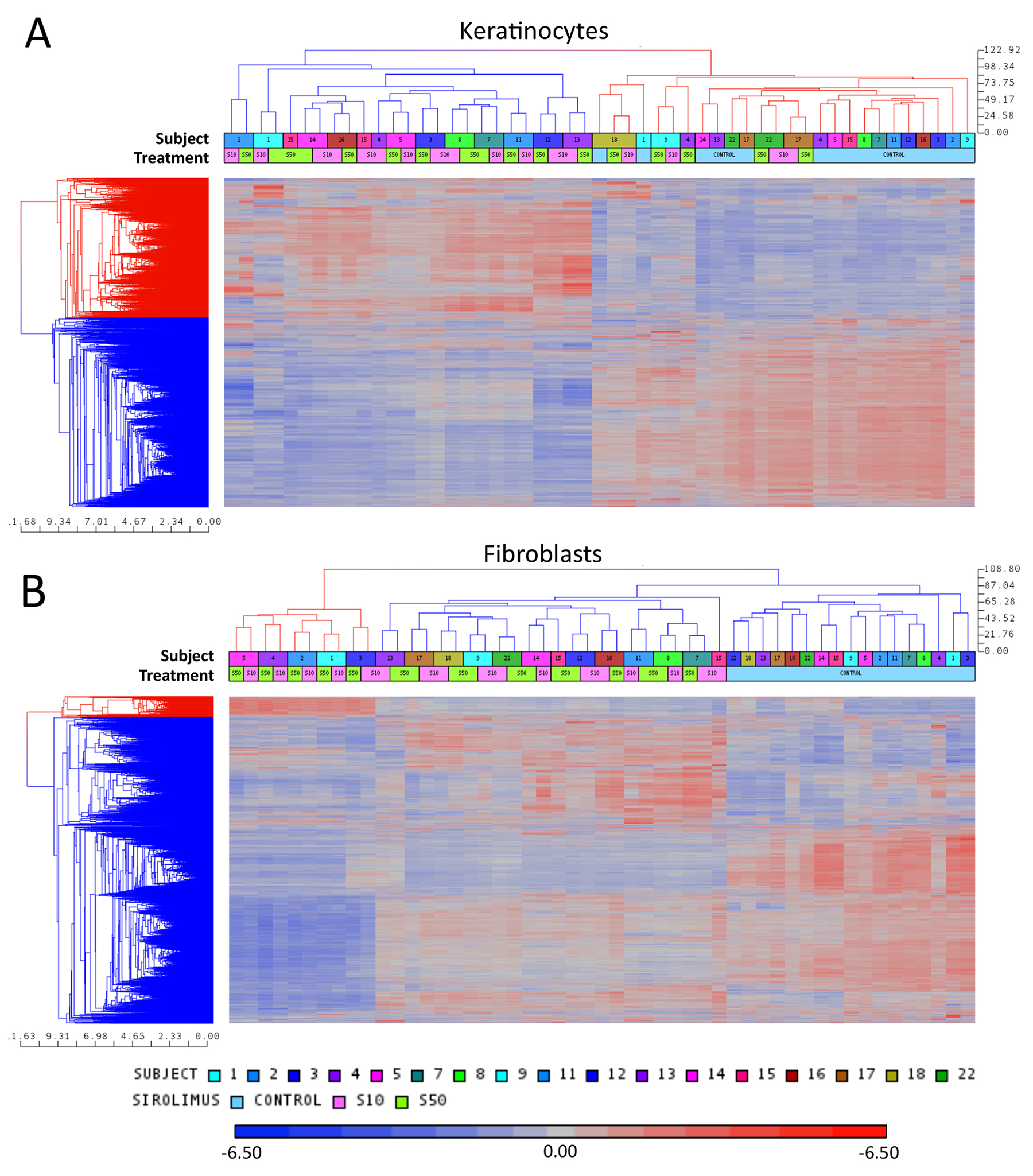
Figure 1: Unsupervised hierarchical clustering of microarray data of keratinocytes (A) and fibroblasts (B) for each subject with or without (control) rapamycin treatment. Coefficient of variation of more than 0.1. The subject numbers match the participant ID numbers in Table 1. S10 indicates sirolimus/rapamycin low dose and S50 indicates sirolimus/rapamycin high dose.
Differentially regulated genes in BCNS and normal samples at baseline under more relaxed statistical constrains
Following 5-way mixed-model ANOVA-REML (FC >1.3 and <-1.3; unadjusted p<0.05), we found several gene sets that were differentially expressed. Briefly, the baseline comparison of microarray data of keratinocytes and fibroblasts showed 585 and 857, respectively of differentially expressed genes between BCNS and normal subjects. Hence, relaxing the strict FDR cut-off, effected the unmasking of candidate genes that are differentially expressed at baseline in this hypothesis-generating analysis. The top up-regulated genes at baseline in keratinocytes derived from BCNS patients were NFIA-AS2, RBMY3AP, SEC16B, CROCC, SYN1, MKL1, FAM71A, LOC101928973, SIM2, GNG8 in comparison to normal keratinocytes, whereas GSTT1, STEAP4, NHLH2, MAST4, KLHL24, MALAT1, ARRDC3, LOC101928100, PELI, SLC28A3 were the most down-regulated genes (Table 2). The top up-regulated genes at baseline in BCNS fibroblasts were TMEM155, ABAT, BEX1, PDE4DIP, CPM, HNMT, SFRP2, AKR1C3, RAB27B, PLAC8, whereas GOLGA8A/GOLGA8B, NEAT1, MALAT1, MEG3, COL4A1, BCAT1, E2F7, FN1, LOC100190986, HELLS were the most down-regulated (Table 2). The ANOVA design and the comprehensive gene lists used for baseline comparison are provided in Supplementary File 1.
Table 2: List of top genes differentially regulated in keratinocytes and fibroblasts derived from BCNS subjects compared to normal individuals
Gene Symbol |
Gene title (HGNC approved) |
Fold change |
p-value |
|---|---|---|---|
Genes up-regulated in BCNS keratinocytes |
|||
NFIA-AS2 |
Nuclear factor I/A antisense RNA 2 |
1.67846 |
0.000241 |
RBMY3AP |
RNA binding motif protein, Y-linked, family 3, member A pseudogene |
1.61282 |
0.000475 |
SEC16B |
SEC16 homolog B (S. cerevisiae) |
1.59121 |
9.05E-05 |
CROCC |
ciliary rootlet coiled-coil, rootletin |
1.58479 |
0.000707 |
SYN1 |
synapsin I |
1.54555 |
0.000235 |
MKL1 |
Megakaryoblastic leukemia (translocation) 1 |
1.54463 |
0.004594 |
FAM71A |
family with sequence similarity 71, member A |
1.53406 |
0.001157 |
LOC101928973 |
uncharacterized |
1.53286 |
0.000580 |
SIM2 |
single-minded homolog 2 (Drosophila) |
1.52543 |
0.000744 |
GNG8 |
guanine nucleotide binding protein (G protein), gamma 8 |
1.52347 |
0.001798 |
Genes down-regulated in BCNS keratinocytes |
|||
GSTT1 |
glutathione S-transferase theta 1 |
-2.84883 |
0.014633 |
STEAP4 |
STEAP family member 4 |
-2.51858 |
0.011631 |
NHLH2 |
nescient helix loop helix 2 |
-2.21331 |
0.001196 |
MAST4 |
Microtubule Associated Serine/Threonine Kinase Family Member 4 |
-2.06517 |
0.016778 |
KLHL24 |
kelch-like 24 (Drosophila) |
-2.05221 |
0.010495 |
MALAT1 |
metastasis associated lung adenocarcinoma transcript 1 (non-protein coding) |
-1.95113 |
0.033836 |
ARRDC3 |
arrestin domain containing 3 |
-1.84582 |
0.032566 |
LOC101928100 |
uncharacterized |
-1.83053 |
0.013417 |
PELI1 |
Pellino homolog 1 (Drosophila) |
-1.77824 |
0.045639 |
SLC28A3 |
solute carrier family 28 (sodium-coupled nucleoside transporter), member 3 |
-1.77746 |
0.035360 |
Genes up-regulated in BCNS fibroblasts |
|||
TMEM155 |
transmembrane protein 155 |
2.61437 |
0.008088 |
ABAT |
4-aminobutyrate aminotransferase |
2.55331 |
0.011422 |
BEX1 |
brain expressed, X-linked 1 |
2.3517 |
0.027068 |
PDE4DIP |
phosphodiesterase 4D interacting protein |
2.32946 |
0.035918 |
CPM |
carboxypeptidase M |
2.27972 |
0.027836 |
HNMT |
histamine N-methyltransferase |
2.25939 |
0.035685 |
SFRP2 |
secreted frizzled-related protein 2 |
2.25661 |
0.036160 |
AKR1C3 |
aldo-keto reductase family 1, member C3 (3-alpha hydroxysteroid dehydrogenase, type II) |
2.23228 |
0.046084 |
RAB27B |
RAB27B, member RAS oncogene family |
2.14117 |
0.004020 |
PLAC8 |
placenta-specific 8 |
2.13321 |
0.011749 |
Genes down-regulated in BCNS fibroblasts |
|||
GOLGA8A |
golgin A8 family, member A |
-2.88122 |
0.013547 |
NEAT1 |
Nuclear Paraspeckle assembly transcript 1 (non-protein coding) |
-2.37719 |
0.032147 |
MALAT1 |
metastasis associated lung adenocarcinoma transcript 1 (non-protein coding) |
-2.36124 |
0.023835 |
MEG3 |
maternally expressed 3 (non-protein coding) |
-2.34455 |
0.019488 |
COL4A1 |
collagen, type IV, alpha 1 |
-2.26454 |
0.022223 |
BCAT1 |
branched chain amino-acid transaminase 1, cytosolic |
-2.16931 |
0.018688 |
E2F7 |
E2F transcription factor 7 |
-2.16325 |
0.029640 |
FN1 |
fibronectin 1 |
-2.1232 |
0.027185 |
LOC100190986 |
hypothetical LOC100190986 |
-2.09273 |
0.022763 |
HELLS |
helicase, lymphoid-specific |
-2.08964 |
0.009022 |
GSEA specifies baseline differences between BCNS and normal keratinocytes or fibroblasts
Next, we performed baseline gene set enrichment analyses (GSEA) of both keratinocytes and fibroblasts samples derived from BCNS patients and compared them to the control group (Figure 2). The results indicated a positive correlation of BCNS keratinocytes with predefined pancreatic β cell hallmark gene set that comprises 40 genes, specifically upregulated in pancreatic beta cells and known to be involved in glucose metabolism and dysglycemia (Figure 2A). Interestingly, BCNS keratinocytes, in contrast to normal keratinocytes, correlated negatively with the oncogenic gene signature mTOR_UP.N4.V1_DN in the MSigDb (Figure 2A and Supplementary File 2), comprising genes shown to be down-regulated upon treatment with rapamycin [48]. In the case of BCNS fibroblasts, compared with normal fibroblasts, GSEA pointed to a negative correlation with three oncogenic signatures in the MsigDb that comprised genes down-regulated in response to activated CTNNB1 overexpression (BCAT_BILD_ET_AL_DN) and RNAi-mediated JAK2 knockdown (JAK2_DN.V1_DN), and genes up-regulated upon RNAi-mediated knockdown of PCGF2 (MEL18_DN.V1_UP) (Figure 2B).
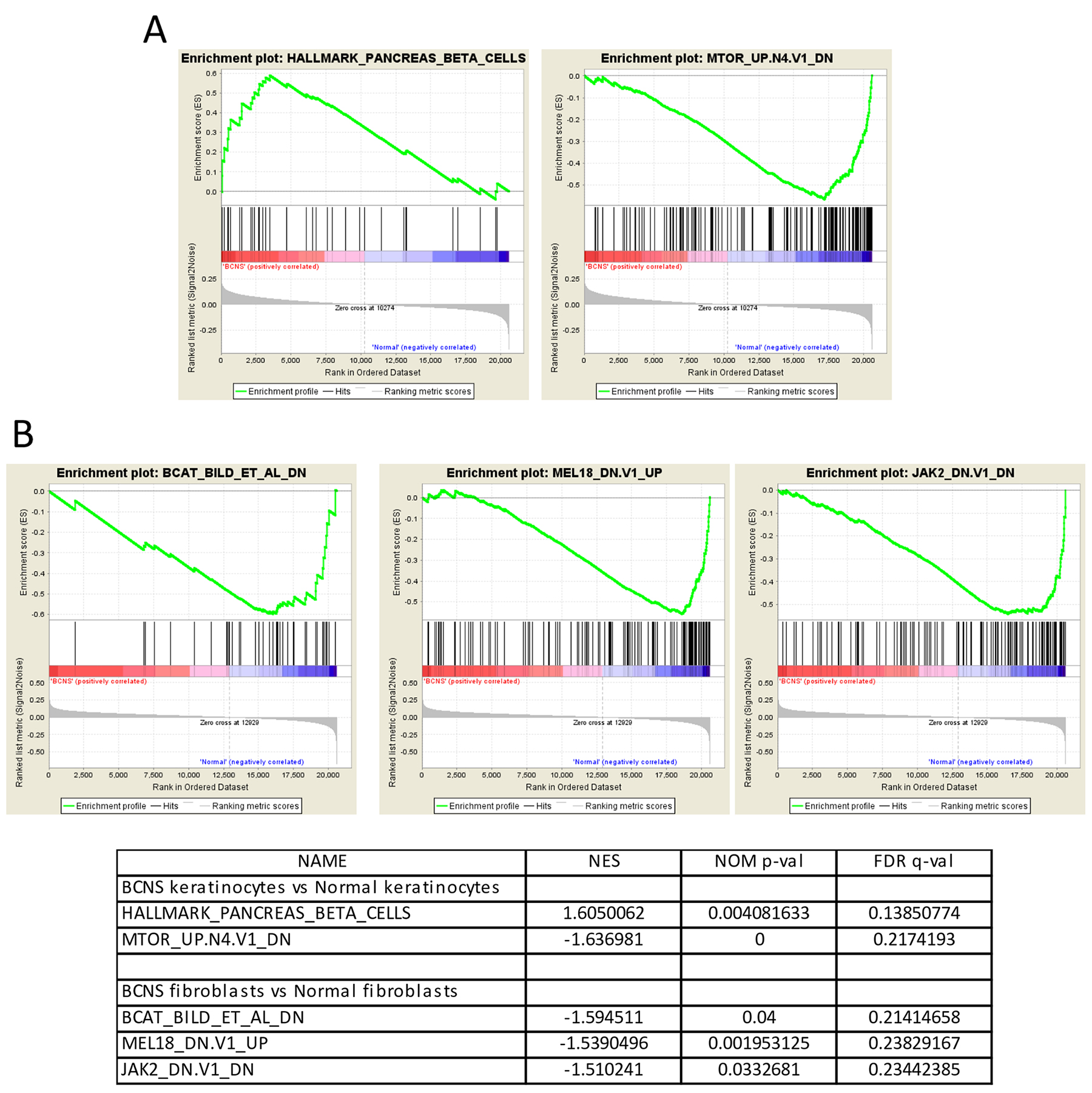
Figure 2: Gene set enrichment analyses (GSEA) on BCNS keratinocytes and fibroblasts compared to normal samples. (A) GSEA shows that the ‘HALLMARK_PANCREAS_BETA_CELLS’ gene-set comprising of genes specifically up-regulated in pancreatic beta cells is enriched in BCNS keratinocytes as compared to the normal keratinocyte samples. The GSEA analysis also indicates that the ‘MTOR_UP.N4.V1_DN’ gene set containing down-regulated genes post rapamycin treatment is enriched in the normal keratinocytes (A). (B) For the fibroblasts, GSEA shows that the ‘BCAT_BILD_ET_AL_DN’ gene set comprising of genes down-regulated due to activated CTNNB1 overexpression; the ‘MEL18_DN.V1_UP’ gene set comprising of up-regulated genes in medullablastoma cells after PCGF2 knockdown; and the ‘JAK2_DN.V1_DN’ gene set containing the genes down-regulated by JAK2 knock-down are all enriched in the normal fibroblast samples. For the enrichment plots, profile of running ES score and position of GeneSet members on the rank ordered list are denoted for HALLMARK_PANCREAS_BETA_CELLS (A), MTOR_UP.N4.V1_DN (A), BCAT_BILD_ET_AL_DN (B), MEL18_DN.V1_UP (B), JAK2_DN.V1_DN (B), and the respective normalized enrichment scores (NES), nominal p-values (NOM p-val), and q-values (FDR q-val) are summarized in table below the plots.
Biological differences attributed to rapamycin effects in BCNS and normal keratinocytes
Here we used GSEA algorithm on comprehensive microarray dataset without preprocessing to determine an a priori defined gene sets that exhibit statistically significant and biologically relevant differences between untreated and rapamycin treated BCNS keratinocytes and fibroblasts. The GSEA results indicated several significant pathways that were altered by rapamycin treatment in both BCNS keratinocytes and BCNS fibroblasts (Figure 3, Supplementary Files 2–4).
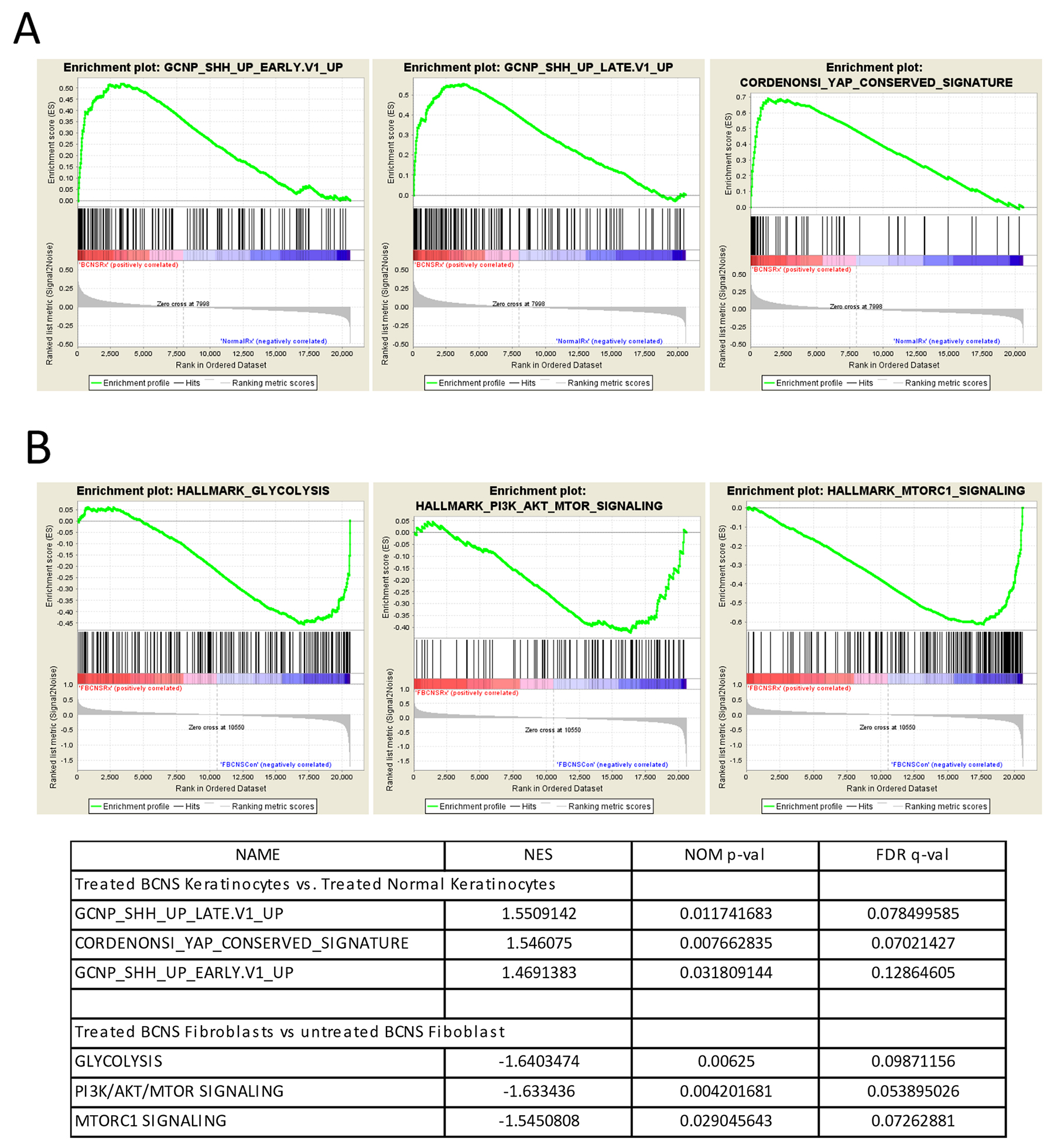
Figure 3: Gene set enrichment analyses (GSEA) on rapamycin-treated BCNS keratinocytes and fibroblasts compared to normal samples. (A) GSEA shows that the ‘GCNP_SHH_UP_EARLY.V1_UP’ gene set consisting of genes up-regulated in granule cell neuron precursors (GCNP) after Shh stimulation for 3h are positively correlated and enriched in BCNS keratinocytes treated with rapamycin. Interestingly, GSEA also indicates the enrichment of a YAP conserved signature in the rapamycin treated BCNS keratinocytes. (B) For the fibroblasts, GSEA indicates that normal fibroblasts treated with rapamycin correlate with the hallmark gene sets namely ‘GLYCOLYSIS’, ‘PI3K/AKT/MTOR SIGNALING’, and ‘MTORC1 SIGNALING’ as compared to rapamycin treated BCNS fibroblasts. For the enrichment plots, profile of running ES score and position of GeneSet members on the rank ordered list for are denoted for oncogenic signatures ‘GCNP_SHH_UP_LATE.V1_UP’ (A), CORDENONSI_YAP_CONSERVED_SIGNATURE’ (A), ‘GCNP_SHH_UP_EARLY.V1_UP’ (A), and the hallmark gene sets namely ‘GLYCOLYSIS’ (B), PI3K/AKT/MTOR SIGNALING (B), MTORC1 SIGNALING (B) and the respective normalized enrichment scores (NES), nominal p-values (NOM p-val), and q-values (FDR q-val) are summarized in table below the plots.
In BCNS keratinocytes, enriched signatures in the presence of rapamycin signal the involvement of and regulation by MYC and G2/M checkpoint genes, cell cycle related targets of E2F transcription factor and mitotic spindle assembly [49]. The oncogenic signatures enriched in BCNS keratinocyte treated with rapamycin showed sets of genes up-regulated in neuronal precursors after Shh stimulation, PIGF treatment, genes governed by overexpression of E2F1 and E2F3, and genes up-regulated in primary keratinocytes from RB1 and RBL1 skin-specific knockout mice. Interestingly as well, the YAP conserved signature [49–51], was also shown to be concordant with rapamycin treated BCNS keratinocytes (Figure 3A, Supplementary Files 2–4). The oncogenic signatures comprising of down-regulated genes leading to CTNNB1 overexpression, SRC overexpression, oncogenic KRAS expression in context of TBK1 knockdown, RNAi mediated knockdown of genes such as EIF4G1, HOXA9 and RPS14, and genes down-regulated after VEGFA treatment, positively correlated with BCNS keratinocytes after rapamycin treatment (data not shown).
Biological differences attributed to rapamycin effects in BCNS and normal fibroblasts
The GSEA pointed to genes upregulated by activation of PI3K/AKT/mTOR pathway and mTORC1 complex, as well as genes involved in mitotic spindle assembly, DNA repair, G2/M checkpoint, and genes regulated by MYC which appear to be in agreement with the transcriptomic profiles of BCNS fibroblasts post rapamycin treatment (Figure 3B, Supplementary Files 3–4). Additionally, genes upregulated by APC knockout, RNAi mediated ELK3 knockdown in response to hypoxia, MYC and IRF4 targets, along with genes down regulated in lesional skin biopsies from mycosis fungoides patients compared to normal skin samples positively correlated with the results on treated BCNS fibroblasts. Oncogenic signatures comprising of up-regulated genes in primary keratinocytes from RB1 and RBL2 skin specific knockout, genes upregulated upon EED or EZH2 knockdown, MYC overexpression, stimulation with Shh, and genes upregulated by everolimus are found to be correlated with treated BCNS fibroblasts. Also, significant immunologic signatures were enriched in the treated BCNS fibroblasts.
Furthermore, GSEA indicated genes involved in glycogenesis and gluconeogenesis and genes upregulated by PI3K/AKT/mTOR pathway and mTORC1 complex activation, genes important for and involved in mitotic spindle assembly, DNA repair, G2/M checkpoint, genes responding to estrogen and genes regulated by MYC [49] were negatively correlated with the transcriptomic profiles of BCNS fibroblasts post rapamycin treatment. Additionally, genes expressed in multipotent progenitors, genes upregulated by APC or BCL3 knockout or RNAi mediated ELK3 knockdown in response to hypoxia, MYC and IRF4 targets, along with genes downregulated in lesional skin biopsies from mycosis fungoides patients compared to normal skin samples and genes upregulated in peripheral blood monocytes of Sezary syndrome, were negatively correlated with rapamycin-treated BCNS fibroblasts. Also, oncogenic signatures comprising of YAP [50, 51] conserved signature, up-regulated genes in RB1 and RBL2 skin specific knockout primary keratinocytes, genes upregulated upon EED or EZH2 knockdown, MYC overexpression, stimulation with Shh, and genes upregulated by everolimus, were found to be negatively correlated with treated BCNS fibroblasts. Gene sets consisting of downregulated genes upon SRC overexpression, RPS14 knockdown, HOXA9 knockdown, and VEGFA treatment were also negatively correlated with rapamycin treated BCNS fibroblasts. On other hand, significant immunologic signatures were enriched in untreated BCNS fibroblasts.
Ingenuity Pathway Analysis (IPA) shows distinct signaling pathways enriched in BCNS samples treated with rapamycin
To study whether rapamycin treatment affected cells derived from BCNS patients with PTCH1 mutation differently than normal samples, we generated a gene list of the top differentially regulated and statistically significant genes (cut-off p-value with FDR <0.05, FC>1.3 or <-1.3), including the top genes as listed in Tables 3 and 4 (the entire gene list is available in Supplementary File 4). Thus, a total of 4797 genes were differentially expressed in normal fibroblasts and expression of 3214 genes were significantly altered in normal keratinocytes after rapamycin treatment. In the BCNS group, a total of 4959 genes and 1411 genes were differentially expressed in fibroblasts and keratinocytes, respectively after treatment with rapamycin. To tease out genes that were usually altered in both normal and BCNS groups following rapamycin treatment, we used Venn diagrams (Figure 4) to generate gene lists for further analysis.
Table 3: List of top genes differentially regulated after rapamycin treatment in keratinocytes and fibroblasts derived from normal individuals
Gene Symbol |
Gene title (HGNC approved) |
Fold change |
p-value |
|---|---|---|---|
Genes up-regulated in normal keratinocytes after rapamycin treatment |
|||
KLHL24 |
kelch-like family member 24 |
11.2986 |
0.000701 |
C5orf41 |
CREB3 regulatory factor |
7.59837 |
0.000914 |
IRF6 |
interferon regulatory factor 6 |
7.09155 |
0.003863 |
ATF3 |
activating transcription factor 3 |
6.42457 |
6.31E-05 |
GPNMB |
glycoprotein (transmembrane) nmb |
6.19196 |
0.000485 |
NEAT1 |
nuclear paraspeckle assembly transcript 1 (non-protein coding) |
5.539 |
0.001790 |
MALAT1 |
metastasis associated lung adenocarcinoma transcript 1 (non-protein coding) |
5.28172 |
0.000797 |
IRS2 |
insulin receptor substrate 2 |
4.58514 |
0.000667 |
DUSP10 |
dual specificity phosphatase 10 |
4.47873 |
0.000829 |
S100P |
S100 calcium binding protein P |
4.22571 |
3.44E-09 |
Genes down-regulated in normal keratinocytes after rapamycin treatment |
|||
RRM2 |
ribonucleotide reductase M2 |
-13.9567 |
0.001023 |
DTL |
denticleless E3 ubiquitin protein ligase homolog (Drosophila) |
-12.2969 |
0.003649 |
CCNE2 |
cyclin E2 |
-10.7273 |
2.43E-05 |
UHRF1 |
ubiquitin-like with PHD and ring finger domains 1 |
-10.2451 |
0.000128 |
MCM10 |
minichromosome maintenance complex component 10 |
-9.9609 |
0.002114 |
CDC6 |
cell division cycle 6 |
-9.89019 |
0.002154 |
THBS1 |
thrombospondin 1 |
-8.83678 |
6.24E-05 |
SHCBP1 |
SHC SH2-domain binding protein 1 |
-7.3742 |
4.93E-06 |
DHFR |
dihydrofolate reductase |
-7.32771 |
0.004707 |
ZWINT |
ZW10 interactor |
-7.20764 |
0.001275 |
Genes up-regulated in normal fibroblasts after rapamycin treatment |
|||
ADH1B |
alcohol dehydrogenase 1B (class I), beta polypeptide |
17.4006 |
7.18E-06 |
ITGB8 |
integrin, beta 8 |
6.98181 |
0.000417 |
DIO2 |
deiodinase, iodothyronine, type II |
6.75995 |
5.31E-05 |
FGF7 |
fibroblast growth factor 7 |
5.35228 |
0.001493 |
EFEMP1 |
EGF containing fibulin-like extracellular matrix protein 1 |
5.18771 |
0.000186 |
GRIA1 |
glutamate receptor, ionotropic, AMPA 1 |
5.17074 |
9.47E-09 |
SLC40A1 |
solute carrier family 40 (iron-regulated transporter), member 1 |
5.06774 |
0.000937 |
C13orf15 |
regulator of cell cycle |
4.82343 |
7.65E-05 |
FMO2 |
flavin containing monooxygenase 4 (non-functional) |
4.74587 |
2.43E-07 |
SVEP1 |
sushi, von Willebrand factor type A, EGF and pentraxin domain containing 1 |
4.70169 |
1.93E-06 |
Genes down-regulated in normal fibroblasts after rapamycin treatment |
|||
ANLN |
anillin, actin binding protein |
-40.9022 |
6.14E-06 |
PBK |
PDZ binding kinase |
-38.6028 |
2.70E-06 |
DLGAP5 |
discs, large (Drosophila) homolog-associated protein 5 |
-35.7893 |
7.76E-08 |
RRM2 |
ribonucleotide reductase M2 |
-34.5474 |
2.81E-06 |
HMMR |
hyaluronan-mediated motility receptor (RHAMM) |
-30.7856 |
1.68E-07 |
TOP2A |
topoisomerase (DNA) II alpha 170kDa |
-30.641 |
5.36E-07 |
SHCBP1 |
SHC SH2-domain binding protein 1 |
-30.4333 |
7.90E-09 |
CEP55 |
centrosomal protein 55kDa |
-29.9081 |
1.91E-09 |
TTK |
TTK protein kinase |
-29.053 |
1.60E-08 |
NDC80 |
NDC80 kinetochore complex component |
-28.6074 |
5.35E-08 |
Table 4: List of top genes differentially regulated after rapamycin treatment in keratinocytes and fibroblasts derived from BCNS individuals
Gene Symbol |
Gene title (HGNC approved) |
Fold change |
p-value |
|---|---|---|---|
Genes up-regulated in BCNS keratinocytes after rapamycin treatment |
|||
KLHL24 |
kelch-like family member 24 |
7.63805 |
0.000303 |
GPNMB |
glycoprotein (transmembrane) nmb |
5.38456 |
0.000811 |
ATF3 |
activating transcription factor 3 |
4.94199 |
0.000227 |
GABARAPL1 |
GABA(A) receptor-associated protein like 1 |
3.95871 |
0.000281 |
IRS2 |
insulin receptor substrate 2 |
3.88798 |
0.001592 |
JMY |
junction mediating and regulatory protein, p53 cofactor |
3.36795 |
1.90E-06 |
ADCK3 |
aarF domain containing kinase 3 |
3.21844 |
9.14E-05 |
BIRC3 |
baculoviral IAP repeat containing 3 |
3.11232 |
0.000889 |
ZBTB10 |
zinc finger and BTB domain containing 10 |
3.02502 |
0.002266 |
NCRNA00219 |
EPB41L4A antisense RNA 1 |
2.94874 |
4.48E-07 |
Genes down-regulated in BCNS keratinocytes after rapamycin treatment |
|||
THBS1 |
thrombospondin 1 |
-5.00466 |
0.001087 |
GJB2 |
gap junction protein, beta 2, 26kDa |
-3.99488 |
0.001130 |
NAV3 |
neuron navigator 3 |
-3.86258 |
0.000239 |
ABAT |
4-aminobutyrate aminotransferase |
-3.1763 |
1.41E-06 |
SHCBP1 |
SHC SH2-domain binding protein 1 |
-3.16137 |
0.001208 |
CDC25A |
cell division cycle 25A |
-3.08918 |
0.001806 |
CXCL14 |
chemokine (C-X-C motif) ligand 14 |
-2.93124 |
5.30E-06 |
SORL1 |
sortilin-related receptor, L(DLR class) A repeats containing |
-2.81784 |
0.001444 |
KRT1 |
keratin 1, type II |
-2.77983 |
0.001714 |
CDKN3 |
cyclin-dependent kinase inhibitor 3 |
-2.75698 |
0.000109 |
Genes up-regulated in BCNS fibroblasts after rapamycin treatment |
|||
ADH1B |
alcohol dehydrogenase 1B (class I), beta polypeptide |
14.5357 |
9.23E-06 |
TXNIP |
thioredoxin interacting protein |
6.7023 |
0.004666 |
ITGB8 |
integrin, beta 8 |
5.53155 |
0.001337 |
GLDN |
gliomedin |
5.28474 |
0.000403 |
DIO2 |
deiodinase, iodothyronine, type II |
5.08305 |
0.000173 |
GRIA1 |
glutamate receptor, ionotropic, AMPA 1 |
4.61537 |
1.38E-08 |
FGF9 |
fibroblast growth factor 9 |
4.57488 |
0.002351 |
C13orf15 |
regulator of cell cycle |
4.53467 |
7.38E-05 |
SVEP1 |
sushi, von Willebrand factor type A, EGF and pentraxin domain containing 1 |
4.33477 |
2.99E-06 |
SLC40A1 |
solute carrier family 40 (iron-regulated transporter), member 1 |
4.24931 |
0.002374 |
Genes down-regulated in BCNS fibroblasts after rapamycin treatment |
|||
ANLN |
anillin, actin binding protein |
-48.0347 |
4.14E-06 |
PBK |
PDZ binding kinase |
-45.1184 |
1.81E-06 |
KIF20A |
kinesin family member 20A |
-37.2298 |
2.83E-06 |
DLGAP5 |
discs, large (Drosophila) homolog-associated protein 5 |
-36.5714 |
7.41E-08 |
CCNB1 |
cyclin B1 |
-35.2177 |
3.43E-06 |
RRM2 |
ribonucleotide reductase M2 |
-29.9324 |
4.66E-06 |
NDC80 |
NDC80 kinetochore complex component |
-25.8672 |
8.81E-08 |
CEP55 |
centrosomal protein 55kDa |
-25.5206 |
2.31E-09 |
BIRC5 |
baculoviral IAP repeat containing 5 |
-25.4299 |
1.16E-07 |
HMMR |
hyaluronan-mediated motility receptor (RHAMM) |
-25.2382 |
4.16E-07 |
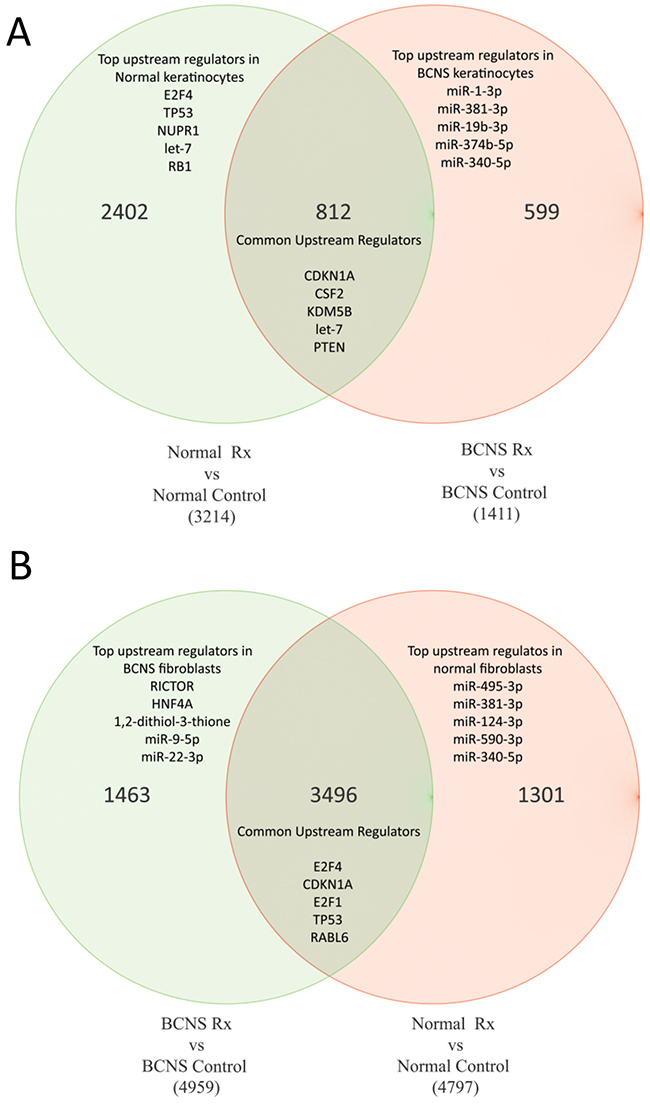
Figure 4: Venn diagrams of genes commonly altered in both normal and BCNS group, with or without rapamycin treatment. Normal treated (Rx) samples were compared to normal control samples, and these samples were then compared BCNS treated (Rx) samples to BCNS control samples. (A) Keratinocyte samples. (B) Fibroblast samples.
IPA core analysis indicates effect of rapamycin treatment on keratinocyte and fibroblasts
Here we explored the genes/signaling pathways differentially expressed in response to rapamycin. To this end, we performed core IPA analysis on comprehensive gene lists for fibroblasts and keratinocyte, including prioritized gene lists that were generated via the Venn diagram in Figure 4. The IPA core analysis showed enrichment of pathways that comprised mitotic role of Polo-like Kinases, role of CHK proteins in cell cycle checkpoint control, ATM signaling, cell cycle control of chromosomal replication and role of BRCA1 in DNA damage response as top canonical pathways, based on focus genes present among the 3214 genes that are differentially regulated in normal keratinocytes in response to rapamycin (Figure 5A, Table 5A). A Similar IPA core analysis of BCNS keratinocytes treated with rapamycin resulted in enrichment of PTEN signaling, Natural killer cell signaling, inhibition of angiogenesis by TSP1, p53 signaling and PI3K signaling in B lymphocytes as top canonical pathways, based on focus genes present in 1411 differentially regulated genes after rapamycin treatment (Figure 5B, Table 5B).
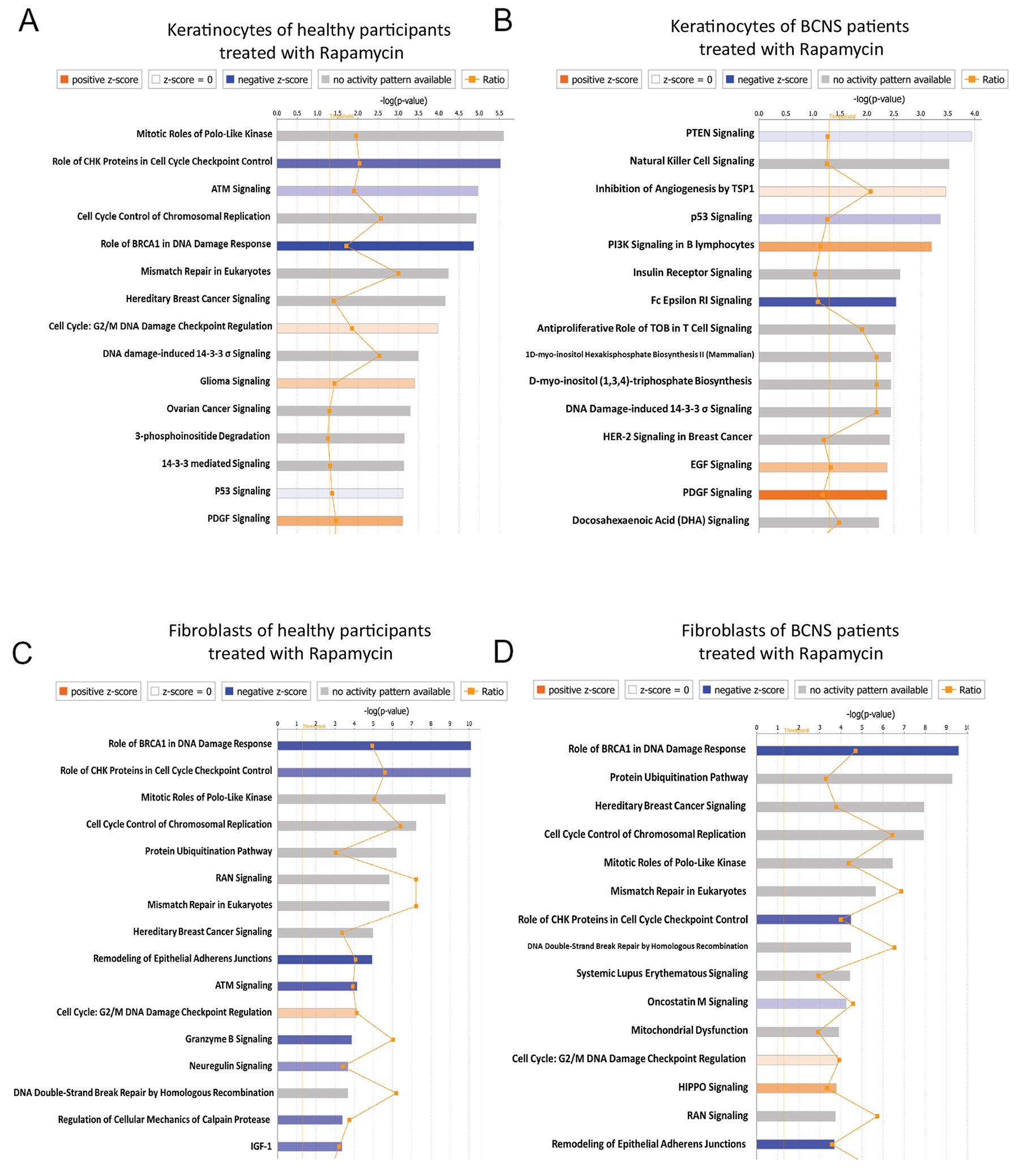
Figure 5: IPA core analysis indicates effect of rapamycin treatment on keratinocyte and fibroblasts. (A) Keratinocytes of healthy participants treated with rapamycin. (B) Keratinocytes of BCNS patients treated with rapamycin. (C) Fibroblasts of healthy participants treated with rapamycin. (D) Fibroblasts of BCNS patients treated with rapamycin.
Table 5A: Keratinocytes of healthy participants treated with rapamycin
Ingenuity Canonical Pathways |
-log(p-value) |
Molecules |
|---|---|---|
Mitotic Roles of Polo-Like Kinase |
5.6 |
PPP2R5E, KIF23, CHEK2, PPP2R4, CDC25A, PRC1, CDC20, PPP2R3A, CCNB2, KIF11, SMC3, PPP2R2A, WEE1, ANAPC11, PKMYT1, PLK4, ANAPC7, FZR1, CDC23, PLK1, CDK1, CCNB1, FBXO5 |
Role of CHK Proteins in Cell Cycle Checkpoint Control |
5.53 |
SLC19A1, PPP2R5E, E2F5, BRCA1, RFC3, CHEK2, PPP2R4, CDC25A, MDC1, PPP2R3A, PPP2R2A, RAD1, RAD9A, RFC5, CDKN1A, RFC2, RFC4, ATMIN, PLK1, CDK1, CHEK1 |
ATM Signaling |
4.97 |
JUN, BRCA1, CBX5, CREB1, CHEK2, CDC25A, GADD45B, MDC1, CCNB2, TDP1, SMC2, SMC3, FANCD2, RAD9A, CDKN1A, MAPK8, RAD51, GADD45G, CDK1, CCNB1, CHEK1 |
Cell Cycle Control of Chromosomal Replication |
4.92 |
CDK7, CHEK2, MCM7, ORC6, ORC1, CDC45, RPA3, MCM3, MCM6, CDK6, MCM4, MCM5, CDC6 |
Role of BRCA1 in DNA Damage Response |
4.87 |
RFC3, CHEK2, SMARCA4, MDC1, MSH6, RFC2, CDKN1A, FANCG, RBL1, RFC4, SLC19A1, E2F5, BRCA1, ATF1, SMARCE1, FANCM, FANCD2, BRD7, RFC5, ARID2, BRCA2, RAD51, PLK1, MSH2, CHEK1 |
Mismatch Repair in Eukaryotes |
4.24 |
SLC19A1, FEN1, RFC3, EXO1, MSH6, RFC5, RFC2, RFC4, MSH2 |
Hereditary Breast Cancer Signaling |
4.16 |
RFC3, AKT3, CHEK2, PIK3C2A, TUBG1, GADD45B, SMARCA4, PIK3CA, MSH6, RFC2, CDKN1A, FANCG, CDK6, RFC4, GADD45G, SLC19A1, BRCA1, PIK3R4, SMARCE1, FANCM, AKT2, WEE1, POLR2H, FANCD2, BRD7, RFC5, ARID2, BRCA2, RAD51, CDK1, CCNB1, MSH2, CHEK1 |
Cell Cycle: G2/M DNA Damage Checkpoint Regulation |
3.98 |
TOP2A, BRCA1, YWHAQ, CDK7, CHEK2, AURKA, PTPMT1, CCNB2, WEE1, PKMYT1, SKP2, CDKN1A, YWHAB, PLK1, CDK1, CCNB1, CHEK1 |
DNA damage-induced 14-3-3σ Signaling |
3.49 |
CCNE2, BRCA1, AKT3, RAD9A, CCNB2, AKT2, RAD1, CDK1, CCNB1 |
Glioma Signaling |
3.41 |
SOS2, AKT3, PIK3C2A, CALM1 (includes others), CAMK2D, PDGFB, PIK3CA, CDKN2D, CDKN1A, GRB2, SIN3A, PA2G4, RBL1, CDK6, E2F5, EGFR, PRKD3, PIK3R4, CAMK1D, AKT2, IGF1R, PRKCA, MAPK1, TFDP1, PRKCG |
Ovarian Cancer Signaling |
3.29 |
EDNRA, AKT3, PIK3C2A, WNT5A, FZD5, WNT11, PIK3CA, FZD1, MSH6, EDN1, SIN3A, PA2G4, WNT6, VEGFC, PRKAR2A, BRCA1, EGFR, TCF3, CGA, TCF4, PIK3R4, AKT2, CD44, BRAF, PTGS2, MAPK1, BRCA2, RAD51, TFDP1, MSH2, GJA1 |
3-phosphoinositide Degradation |
3.15 |
DUSP10, PPP2R5E, NUDT1, MTMR4, NUDT3, PPM1F, PPP2R4, PPP2R3A, MTMR3, STYXL1, OCRL, INPP5D, DUSP23, DUSP1, PPIP5K1, SYNJ2, MTMR6, PXYLP1, INPP5F, PTPMT1, PTPN2, CDC25A, PTPRO, PAWR, DUSP2, ACP1, TMEM55B, NUDT4, HACD2, PPP1R13B, NUDT15, IGBP1 |
14-3-3-mediated Signaling |
3.13 |
JUN, YWHAQ, AKT3, PIK3C2A, TUBB3, TUBG1, YAP1, TUBA1C, VIM, TUBA1A, SNCA, PIK3CA, GRB2, MAPK8, MAPT, YWHAB, FOXO1, TUBA3C/TUBA3D, PRKD3, PIK3R4, AKT2, TUBB4B, PRKCA, TUBA1B, MAPK1, TUBA4A, BAX, PRKCG |
p53 Signaling |
3.12 |
JUN, AKT3, TP53AIP1, CHEK2, PIK3C2A, GADD45B, THBS1, PIK3CA, CDKN1A, PLAGL1, MAPK8, GADD45G, BRCA1, TP63, PIK3R4, JMY, BIRC5, ST13, AKT2, CASP6, HIF1A, PPP1R13B, BAX, ADCK3, CHEK1 |
PDGF Signaling |
3.11 |
JUN, EIF2AK2, SOS2, PIK3C2A, INPP5F, CRKL, PIK3R4, CSNK2A2, JAK1, PDGFB, CSNK2A1, ABL2, OCRL, INPP5D, PIK3CA, ACP1, PRKCA, MAPK1, GRB2, SYNJ2, MAPK8 |
Table 5B: Keratinocytes of BCNS patients treated with rapamycin
Ingenuity Canonical Pathways |
-log(p-value) |
Molecules |
|---|---|---|
PTEN Signaling |
3.94 |
FGFR2, AKT3, INPP5F, BCL2L11, TGFBR3, CSNK2A2, AKT2, CSNK2A1, BMPR1B, H2BFM, OCRL, INPP5D, PIK3CA, CDKN1A, MAPK1, SYNJ2, ITGA4, TGFBR1 |
Natural Killer Cell Signaling |
3.52 |
PIK3R6, AKT3, INPP5F, LILRB1, AKT2, VAV2, FYN, OCRL, INPP5D, PIK3CA, PRKCA, TYROBP, SH3BP2, MAPK1, SYNJ2, NCK1 |
Inhibition of Angiogenesis by TSP1 |
3.46 |
JUN, FYN, AKT3, MMP9, MAPK1, TGFBR1, THBS1, AKT2 |
p53 Signaling |
3.36 |
JUN, PIK3R6, AKT3, TP53AIP1, JMY, BIRC5, ST13, THBS1, AKT2, PIK3CA, CDKN1A, PLAGL1, BAX, ADCK3, CHEK1 |
PI3K Signaling in B Lymphocytes |
3.2 |
JUN, AKT3, IRS2, CD79B, CHP1, ATF6, AKT2, PPP3CC, VAV2, ATF3, MALT1, FYN, INPP5D, PIK3CA, PLEKHA3, MAPK1, SH2B2 |
Insulin Receptor Signaling |
2.62 |
PIK3R6, AKT3, IRS2, INPP5F, SCNN1A, AKT2, H2BFM, FYN, OCRL, INPP5D, PIK3CA, GYS1, MAPK1, SYNJ2, SH2B2, NCK1 |
Fc Epsilon RI Signaling |
2.55 |
PIK3R6, AKT3, INPP5F, AKT2, VAV2, FYN, OCRL, INPP5D, PIK3CA, PRKCA, MAPK1, SYNJ2, PLA2G12A, PLA2G4A |
Antiproliferative Role of TOB in T Cell Signaling |
2.53 |
SMAD2, SKP2, MAPK1, CCNA2, TGFBR1, SMAD4 |
1D-myo-inositol Hexakisphosphate Biosynthesis II (Mammalian) |
2.44 |
OCRL, INPP5D, INPP5F, ITPKB, SYNJ2 |
D-myo-inositol (1,3,4)-trisphosphate Biosynthesis |
2.44 |
OCRL, INPP5D, INPP5F, ITPKB, SYNJ2 |
DNA damage-induced 14-3-3σ Signaling |
2.44 |
AKT3, CCNB2, AKT2, RAD1, CCNB1 |
HER-2 Signaling in Breast Cancer |
2.42 |
H2BFM, PIK3R6, AKT3, ITGB5, PIK3CA, ITGB6, PARD3, PRKCA, PARD6G, CDKN1A, AKT2 |
EGF Signaling |
2.37 |
JUN, PIK3R6, AKT3, PIK3CA, PRKCA, CSNK2A2, MAPK1, AKT2, CSNK2A1 |
PDGF Signaling |
2.37 |
JUN, PIK3R6, OCRL, INPP5D, PIK3CA, INPP5F, PRKCA, CSNK2A2, MAPK1, SYNJ2, CSNK2A1 |
Docosahexaenoic Acid (DHA) Signaling |
2.22 |
H2BFM, PIK3R6, AKT3, PIK3CA, APP, BAX, AKT2 |
In the case of the normal fibroblasts, an IPA core analysis of 4797 differentially regulated genes in response to rapamycin resulted in enrichment of role of BRCA1 in DNA damage response, role of CHK proteins in cell cycle checkpoint control, mitotic roles of Polo-like Kinases, cell cycle control of chromosomal replication and protein ubiquitination pathway as canonical signaling pathways (Figure 5C, Table 5C). On the other hand, a core analysis of 4959 genes enriched in rapamycin treated BCNS fibroblasts pointed to top canonical pathways that included those playing a role of BRCA1 in DNA damage response, protein ubiquitination pathway, hereditary breast cancer signaling, cell cycle control of chromosomal replication and mitotic roles of Polo-like Kinases (Figure 5D, Table 5D). Although it may appear that there was some overlap among canonical signaling pathways between normal and BCNS samples, the focus molecules in these pathways were different which would affect the direction of the pathway. BCNS keratinocytes had the most distinct canonical pathway signaling. The p-values and molecules for top canonical pathways are shown (Table 5A–5D); a comprehensive list is shown in Supplementary File 5.
Table 5C: Fibroblasts of healthy participants treated with rapamycin
Ingenuity Canonical Pathways |
-log(p-value) |
Molecules |
|---|---|---|
Role of BRCA1 in DNA Damage Response |
10.1 |
RFC3, FANCB, PBRM1, ATRIP, E2F1, BARD1, SMARCA4, MDC1, E2F3, NBN, TOPBP1, STAT1, MSH6, RFC2, FANCG, RPA1, RBL1, RFC4, MRE11A, SLC19A1, BRCA1, BRE, BRCC3, HLTF, FANCM, SMARCC2, RBBP8, BLM, BRIP1, RFC1, FANCD2, RFC5, FANCA, GADD45A, BRCA2, RAD51, ARID1A, PLK1, MSH2, CHEK1 |
Role of CHK Proteins in Cell Cycle Checkpoint Control |
10.1 |
RFC3, E2F1, PPP2R3B, CLSPN, MDC1, PPP2R3A, E2F3, PCNA, PPP2R5C, CDC25C, NBN, RAD9A, RFC2, RPA1, PPP2CB, RFC4, MRE11A, PPP2CA, SLC19A1, BRCA1, CDC25A, CDK2, PPP2R2A, RAD1, HUS1, RFC1, RFC5, PPP2R5D, RAD17, PLK1, CDK1, CHEK1 |
Mitotic Roles of Polo-Like Kinase |
8.76 |
KIF23, SMC1A, ESPL1, PPP2R3B, PPP2R3A, CCNB2, KIF11, ANAPC1, PPP2R5C, CDC25C, ANAPC5, PKMYT1, PPP2CB, HSP90AA1, PPP2CA, CDC27, CDC25A, PRC1, CDC20, PPP2R2A, CDC7, RAD21, PPP2R5D, PLK4, PLK2, HSP90AB1, PTTG1, FZR1, PLK1, CDK1, CCNB1, FBXO5, CDC25B |
Cell Cycle Control of Chromosomal Replication |
7.22 |
MCM7, MCM2, CDK2, ORC6, DBF4, CDC7, ORC1, CDC45, RPA1, RPA3, MCM3, MCM6, CDT1, CDK6, MCM4, MCM5, CDC6, ORC3 |
Protein Ubiquitination Pathway |
6.2 |
HSPA2, UBE2M, DNAJC5, PSMD6, SMURF2, USP7, ANAPC1, HSPB2, ANAPC5, USP1, USP46, PSMA7, DNAJC9, UBE2V2, UCHL5, PSMB2, PSMA1, TRAF6, TCEB2, UBE2L3, UBE2H, USP14, UBE2G2, PSMA6, UBE2V1, PSME1, PSMD12, DNAJC18, UBE2J2, PSMC5, SUGT1, PSMD1, USP13, DNAJB11, UBE2C, HLA-C, HLA-B, NEDD4L, USP47, USP19, DNAJA1, DNAJB13, DNAJC11, THOP1, HSPD1, DNAJC4, USP5, PSME2, CBL, PSMD11, UBE2S, PSMD9, TCEB1, PSMD14, PSMC1, USP24, PSMD2, HSP90AA1, UBE2R2, BRCA1, HSPB11, UCHL1, PSMC3, UBR1, UBE2N, CDC20, USP10, HSPA4, NEDD4, UBE2B,HLA-A, UBE2L6, UBE2I, UCHL3, PSMD13, DNAJB4, HSP90AB1, FZR1, USP53 |
RAN Signaling |
5.82 |
KPNA1, CSE1L, RANGAP1, KPNA2, XPO1, RCC1, KPNA3, IPO5, KPNA6, RAN, RANBP1, KPNB1 |
Mismatch Repair in Eukaryotes |
5.82 |
SLC19A1, RFC1, FEN1, RFC3, EXO1, MSH6, RFC5, RFC2, RPA1, RFC4, PCNA, MSH2 |
Hereditary Breast Cancer Signaling |
4.97 |
RFC3, FANCB, PBRM1, HDAC8, TUBG1, E2F1, BARD1, SMARCA4, PIK3R1, H2AFX, CDC25C, NBN, MSH6, RFC2, FANCG, RPA1, CDK6, RFC4, NPM1, MRE11A, SLC19A1, BRCA1, HDAC2, PIK3R3, POLR2E, HLTF, FANCM, SMARCC2, BLM, RFC1, FANCD2, POLR2D, RFC5, CCND1, FANCA, GADD45A, BRCA2, RAD51, HRAS, ARID1A, CDK1, CCNB1, MSH2, CHEK1 |
Remodeling of Epithelial Adherens Junctions |
4.94 |
IQGAP1, ACTR2, VCL, TUBB3, TUBB6, MAPRE1, TUBG1, HGS, TUBA1C, ARPC5L, APC, DNM1L, TUBA1A, MET, TUBA3C/TUBA3D, ARPC1A, CTNNA1, TUBB, ACTN1, NME1, TUBB4B, ARPC2, DNM2, RALA, TUBA1B, TUBA4A, ARF6 |
ATM Signaling |
4.15 |
JUN, BRCA1, CBX5, SMC1A, CDK2, CDC25A, MDC1, CCNB2, TDP1, SMC2, H2AFX, CDC25C, NBN, BLM, TRIM28, FANCD2, RAD9A, MAPK8, GADD45A, RAD51, MRE11A, CDK1, CCNB1, CHEK1 |
Cell Cycle: G2/M DNA Damage Checkpoint Regulation |
4.1 |
TOP2A, BRCA1, YWHAQ, YWHAG, AURKA, CKS2, CCNB2, YWHAH, CDC25C, BORA, PKMYT1, GADD45A, CKS1B, HIPK2, PRKDC, YWHAZ, PLK1, CDK1, CCNB1, CHEK1, CDC25B |
Granzyme B Signaling |
3.87 |
PARP1, APAF1, LMNB1, ENDOG, DFFA, PRKDC, CYCS, LMNB2, NUMA1, CASP8 |
Neuregulin Signaling |
3.68 |
SOS2, ERBB2IP, ITGA2, PIK3R1, PTPN11, STAT5A, GRB2, DCN, CRK, HSP90AA1, PSEN1, RPS6KB2, TGFA, HBEGF, EGFR, PIK3R3, NRG1, RNF41, SOS1, PLCG2, PDPK1, DLG4, PRKCA, MAPK1, ITGA4, PLCG1, HSP90AB1, STAT5B, HRAS, ITGA5 |
DNA dsBreak Repair by Homologous Recombination |
3.66 |
BRCA1, POLA1, GEN1, RPA1, LIG1, BRCA2, RAD51, MRE11A, NBN |
Regulation of Cellular Mechanics by Calpain Protease |
3.39 |
EGFR, TLN1, VCL, ITGA2, EZR, CDK2, PXN, ACTN1, TLN2, PTK2, CAPN5, CCND1, MAPK1, GRB2, ITGA4, CCNA2, CDK6, HRAS, ITGA5, CDK1, CAPN2 |
Table 5D: Fibroblasts of BCNS patients treated with rapamycin
Ingenuity Canonical Pathways |
-log(p-value) |
Molecules |
|---|---|---|
Role of BRCA1 in DNA Damage Response |
9.59 |
POU2F1, RFC3, FANCB, PBRM1, ATRIP, BARD1, SMARCA4, SMARCB1, NBN, DPF1, STAT1, MSH6, RFC2, FANCG, RPA1, RBL1, RFC4, MRE11A, SLC19A1, BRCA1, SMARCD1, BRE, MLH1, BABAM1, BRCC3, HLTF, SMARCE1, FANCM, SMARCC2, RBBP8, BRIP1, FANCD2, RFC5, FANCA, GADD45A, BRCA2, RAD51, PLK1, MSH2, CHEK1 |
Protein Ubiquitination Pathway |
9.27 |
HSPA2, HSPA1L, UBE2M, PSMD3, DNAJC5, PSMD6, SMURF2, PSMB5, ANAPC1, PSMC4, ANAPC5, USP1, USP46, PSMA7, DNAJC9, PSMB7, UBE2V2, UCHL5, PSMA2, PSMB2, UBE2A, PSMA1, USP32, TRAF6, UBE2L3, PSMA4, UBE2H, USP14, UBE2G2, PSMA6, HSPB8, UBE2V1, PSME1, DNAJB5, PSMC2, PSMD12, DNAJC18, UBE4A, UBE2J2, PSMC5, SUGT1, PSMD1, USP13, UBE2C, HLA-C, HLA-B, RBX1, USP47, DNAJA1, DNAJB13, PSMD7, MED20, THOP1, HSPD1, DNAJB12, DNAJC4, USP5, PSME2, CBL, PSMD11, UBE2S, PSMD4, PSMD9, TCEB1, PSMD14, PSMC1, PSMB3, PSMD2, PSMD5, UBE2R2, PSMA5, BRCA1, HSPB11, PSMC3, UBR1, UBE2N, CDC20, USP10, HSPA4, USP39, NEDD4, HLA-A, UBE2D2, UCHL3, PSMD13, DNAJB4, PSMD8, PAN2, FZR1, USP53 |
Hereditary Breast Cancer Signaling |
7.93 |
RFC3, HDAC8, TUBG1, POLR2F, BARD1, SMARCA4, POLR2K, SMARCB1, NBN, DPF1, RPA1, RFC4, MRE11A, HDAC1, SLC19A1, PIK3R3, SMARCD1, POLR2E, MLH1, HLTF, SMARCE1, SMARCC2, NRAS, FANCD2, POLR2D, CCND1, FANCA, GADD45A, BRCA2, MSH2, FANCB, PBRM1, PIK3R1, H2AFX, CDC25C, MSH6, RFC2, FANCG, CDK6, NPM1, BRCA1, HDAC2, SFN, RRAS2, FANCM, POLR2H, RFC5, RAD51, HRAS, CCNB1, CDK1, CHEK1 |
Cell Cycle Control of Chromosomal Replication |
7.92 |
MCM7, MCM2, CDK2, ORC6, CDK5, DBF4, CDC7, ORC1, CDC45, RPA1, RPA3, MCM3, MCM6, CDT1, CDK6, MCM4, MCM5, CDC6, ORC3 |
Mitotic Roles of Polo-Like Kinase |
6.44 |
KIF23, TGFB1, SMC1A, ESPL1, PPP2R4, PPP2R3A, CCNB2, KIF11, ANAPC1, PPP2R5C, CDC25C, ANAPC5, PKMYT1, ANAPC7, PPP2CA, CDC27, CDC25A, PRC1, CDC20, CDC7, RAD21, PPP2R5D, PLK4, PLK2, PTTG1, FZR1, PLK1, CDK1, CCNB1, FBXO5 |
Mismatch Repair in Eukaryotes |
5.64 |
SLC19A1, FEN1, RFC3, EXO1, MSH6, RFC5, RFC2, RPA1, MLH1, RFC4, PCNA, MSH2 |
Role of CHK Proteins in Cell Cycle Checkpoint Control |
4.48 |
SLC19A1, BRCA1, RFC3, PPP2R4, CDK2, CDC25A, CLSPN, PPP2R3A, PCNA, CDC25C, HUS1, PPP2R5C, NBN, RAD9A, RFC5, RFC2, PPP2R5D, RPA1, RFC4, MRE11A, PLK1, CDK1, PPP2CA, CHEK1 |
DNA dsBreak Repair by Homologous Recombination |
4.47 |
BRCA1, POLA1, RAD52, GEN1, RPA1, LIG1, BRCA2, RAD51, MRE11A, NBN |
Systemic Lupus Erythematosus Signaling |
4.42 |
SNRPB2, LSM2, SNRNP40, SNRPD1, HNRNPC, FCER1G, LSM12, PRPF19, SNRNP25, SNRPA, PIK3R3, LSM8, HLAG, PRPF3, LSM1, NRAS, PRPF38A, HLA-C, HLA-B, PRPF31, JUN, PIK3R1, CBL, IL37, SNRNP200, LSM5, SNRPA1, PRPF8, SNRNP27, GRB2, IGHG1, LSM14B, SNRPG, NFATC4, HLA-E, RRAS2, SNRPE, SNRPC, PRPF4, LSM4, SOS1, SART1, SNRPN, HLA-A, NHP2L1, SNRPF, TXNL4A, MAPK1, PRPF40A, EFTUD2, SNRPD3, HRAS, LSM3, ZCRB1, HNRNPA2B1, PPIH, SNRPB |
Oncostatin M Signaling |
4.24 |
MT2A, RRAS2, PLAU, SOS1, OSMR, NRAS, MMP3, STAT3, STAT1, STAT5A, TIMP3, MAPK1, GRB2, EPAS1, STAT5B, HRAS, MMP1 |
Mitochondrial Dysfunction |
3.89 |
VDAC3, ATP5F1, COX5A, MAPK10, MTND2, SDHB, NDUFA8, NDUFAB1, NDUFA9, SDHD, ATP5C1, BACE1, ATP5B, SDHC, UQCRC1, HTRA2, APP, NDUFB8, CPT1B, CYCS, VPS9D1, CYC1, SOD2, PRDX5, NDUFS6, AIFM1, MAOB, ATP5G3, COX11, VDAC1, COX7A1, CPT1A, GLRX2, NDUFV3, PRDX3, GPX4, NDUFA6, NDUFV2, MAOA, PSEN1, HSD17B10, ATP5G1, SURF1, PDHA1, NDUFS1, NDUFB6, CYB5A, NDUFA3, ATP5J2, COX7C |
Cell Cycle: G2/M DNA Damage Checkpoint Regulation |
3.86 |
TOP2A, BRCA1, YWHAQ, SFN, YWHAG, AURKA, YWHAE, CKS2, CCNB2, YWHAH, CDC25C, BORA, PKMYT1, GADD45A, CKS1B, PRKDC, YWHAZ, PLK1, CDK1, CCNB1, CHEK1 |
HIPPO signaling |
3.78 |
PPP1CC, YWHAQ, PARD3, PPP2R4, PPP2R3A, SMAD4, YWHAE, YWHAH, PPP2R5C, LLGL1, CSNK1D, PPP1R14B, ITCH, MOB1A, PPP2CA, CSNK1E, SFN, NF2, YWHAG, PPP1R12A, PPP1CA, TJP2, WWTR1, CD44, DLG4, RASSF1, PPP1CB, PPP2R5D, SAV1, YWHAZ, STK4 |
RAN Signaling |
3.73 |
KPNA1, CSE1L, RANGAP1, KPNA2, KPNA3, IPO5, KPNA6, RAN, RANBP1, KPNB1 |
In order to understand how canonical pathways that are uniquely enriched in normal or BCNS specimens respond differently to rapamycin due to differentially regulated genes, we dissected the gene list guided by the Venn diagram (Figure 4) and performed core analysis on each group. The canonical pathways enriched in both normal and BCNS keratinocytes, based on 812 common genes, were p53 signaling, DNA damage induced 14-3-3 signaling, EGF signaling, IL-3 signaling and PTEN signaling. The IPA core analysis of the 812 differentially regulated genes common to both BCNS and normal keratinocytes after rapamycin treatment predicted CDKN1A, KDM5B, let-7 and PTEN to be activated upstream regulators while CSF2 to be inhibited. The regulator networks associated with IDs correspond to diseases and function namely cell death of squamous cell carcinoma cell lines. The top up-regulated molecules were ATF3, GPNMB, KLHL24, IRS2, S100P, OTUD1, GABARAPL3, HIST3H2A, EPB41L4A-AS1, LIMA1, TIAM2, JMY and ADCK3. The top down-regulated molecules are THBS1, SHCBP1, CDC25A, NAV3, ZNF367, ABAT, NCAPG, PLAGL1, TTK, GJB2, CXCL14, SORL1, CDKN3, RAB7B.
The canonical pathways enriched in normal keratinocytes only, based on 2402 genes analyzed, were mitotic roles of polo-like kinases, mismatch repair in eukaryotes, role of CHK proteins in cell cycle checkpoint control, role of BRCA1 in DNA damage response and cell cycle control of chromosomal replication. The top up-regulated molecules only in normal keratinocytes are KLHL24, CREBRF, IRF6, NEAT1, MALAT1, DUSP10, TXNIP, DDX6, CITED2, ZMAT3. The top down-regulated molecules are RRM2, DTL, CCNE2, UHRF1, MCM10, CDC6, DHFR, ZWINT, GINS2, PTGS2 in normal keratinocytes in response to rapamycin. It is of interest that although the upstream regulator E2F4 has no activation prediction, TP53, NUPR1, let-7 and RB1 are predicted to be active upstream regulators in normal keratinocytes.
Hepatic fibrosis/hepatic stellate cell activation, HIPPO signaling, natural killer cell signaling, galactose degradation (Leloir Pathway) and ILK signaling were identified as the top canonical pathways unique to the 599 focus genes in the BCNS keratinocytes samples after rapamycin treatment. The top up-regulated molecules were BIRC3, ZBTB10, IFITM1, GART, CDK19, PLEKHM3, RSF1, FN1, ZCCHC7, TGFBR3. The top down-regulated molecules were KRT1, CLCA2, ARTN, TPM4, VPS53, S100A8, ACOT11, HDHD2, FKBP5, GNG8. The upstream regulators inhibited in the BCNS keratinocytes samples were microRNAs miR-1-3p (no activation score), miR-381-3p, miR-19b-3p, miR-19b-3p, miR-374b-5p, miR-340-5p. JNK signaling and miR-101-3p regulator network was predicted to be associated with the signaling response of BCNS keratinocytes to rapamycin.
Similar analysis of fibroblast samples that was based on 3496 differentially expressed genes common to both normal and BCNS fibroblasts resulted in enrichment of canonical pathways involving cell cycle control of chromosomal replication, role of BRCA1 in DNA damage response, mitotic roles of Polo-like kinase, hereditary breast cancer signaling and role of CHK proteins in Cell cycle checkpoint control. The top up-regulated molecules are ADH1B, ITGB8, DIO2, EFEMP1, GRIA1, SLC40A1, RGCC, FMO2, SVEP1, CCL2, GLDN, RGCC, STEAP4, DAPK1. The top down-regulated molecules were ANLN, PBK, DLGAP5, RRM2, HMMR, TOP2A, SHCBP1, CEP55, TTK, NDC80, KIF20A, CCNB1, BIRC5. In both normal and BCNS fibroblasts samples, upstream regulators CDKN1A and TP53 were activated whereas the E2F1 and RABL6 were predicted to be inhibited in response to rapamycin. It is of interest to note that RABL6 plays a role in cell growth and survival and its overexpression is associated with tumorigenesis.
The canonical pathways enriched in the normal fibroblast samples, based on 1301 unique genes, included CHK proteins in cell cycle checkpoint control, neuregulin signaling, PPAR/RXR activation, role of BRCA1 in DNA damage response and VEGF signaling. The top up-regulated molecules were FGF7, SPIDR, SFRP2, SYNPO2, FMO3, FAM198B, WISP1, IL6, CEBPD, JAM2. The top down-regulated molecules were HAS2, TMPO, C4orf46, USP1, MIS18BP1, HMGB2, NETO2, PSAT1, BLM, TRIM59. In the normal fibroblasts samples, upstream regulators miR-495-3p, miR-381-3p, miR-124-3p, miR-590-3p, miR-340-5p were predicted to be active.
Based on 1463 unique genes differentially regulated in the BCNS fibroblasts only, mitochondrial dysfunction, oxidative phosphorylation, agrin interactions at neuromuscular junction, regulation of actin-based motility by Rho and estrogen receptor signaling were enriched canonical pathways. The top up-regulated molecules were TXNIP, FGF9, WISP2, TMTC1, ZBTB16, NEAT1, SEMA5A, IGF2, LRRN4CL, KCND3. The top down-regulated molecules were THBD, PHLDA1, TJP2, PRPS1, SRSF3, C4orf32, MATN2, ATOH8, EGR1, MLLT11. In the BCNS fibroblasts samples, the upstream regulators RICTOR and miR-22-3p ((miRNAs w/seed AGCUGCC) were activated. The upstream regulator 1,2-dithiol-3-thione was predicted to be inhibited whereas no prediction for HNF4A and miR-9-5p (and other miRNAs w/seed CUUUGGU). The top regulator effects networks were associated with FLI1, MMP3, CD3 and NFE2L2. These analyses showed involvement of different focus molecules in different cell type that contribute to the same canonical pathways hinting to common signaling pathways (Supplementay File 5).
Networks enriched in BCNS samples after rapamycin treatment
Here we performed an IPA network analysis that revealed several important such networks that are significantly altered post-rapamycin treatment (Figures 6 and 7). The most notably altered pathway in BCNS keratinocytes compared with normal keratinocytes, based on 35 focus molecules (score 50), was linked to digestive system development and function (Figure 6A and 7A). Several genes such as APP, SPON1 (cell adhesion promotes neurite growth and axonal guidance), CISD2 (associated with neurodegenerative Wolfram syndrome 2), KLK9 (associated with collagen biosynthesis and modification), CDCA3 (involved in cell cycle regulation mediated by APC) were included in this network and are related to development, neuronal growth and function.
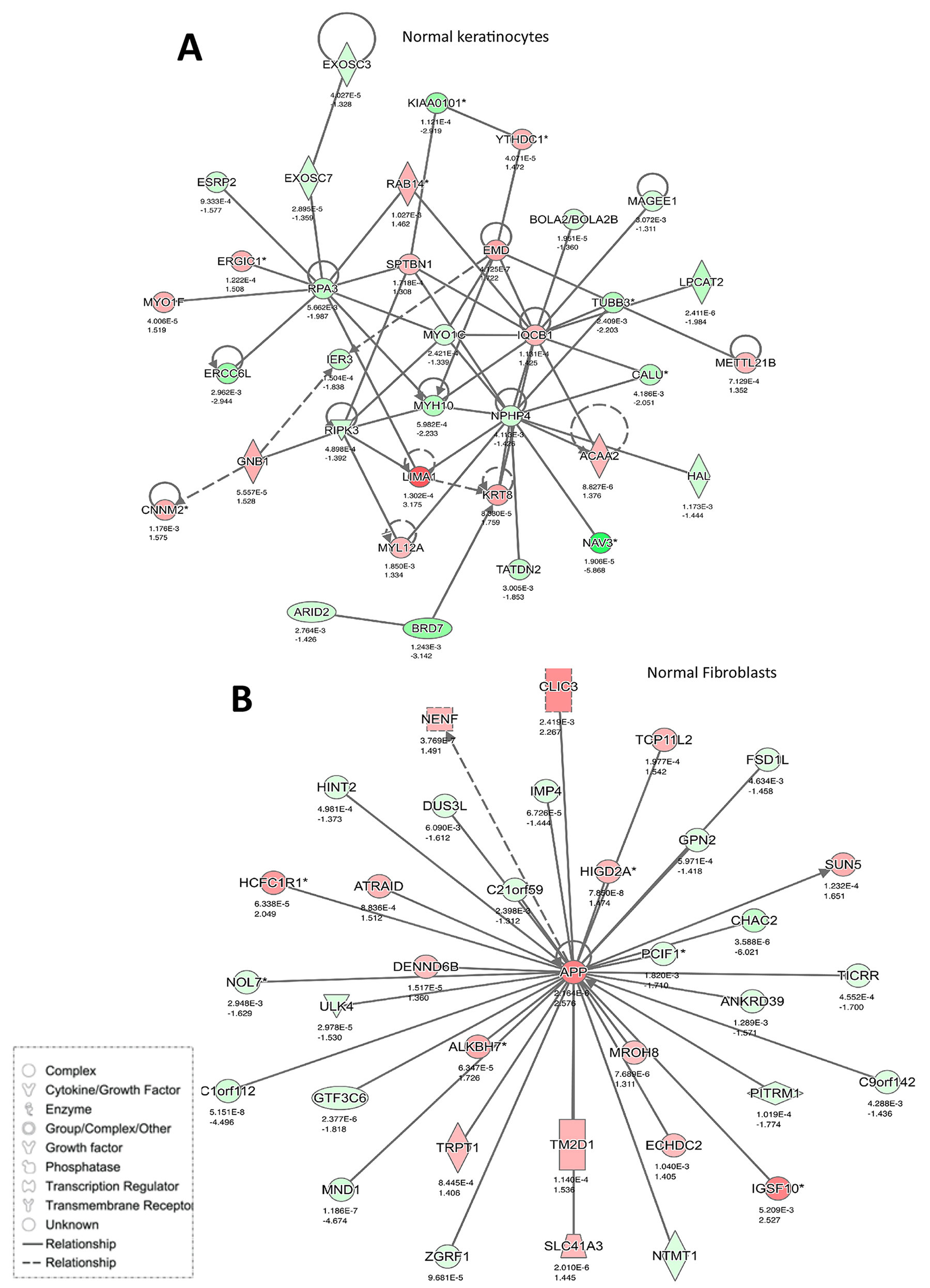
Figure 6: Networks enriched in normal cells after rapamycin treatment. (A) Keratinocytes and (B) fibroblasts. Gene expression variation by at least 2-fold is depicted by color (red, up-regulated; green, down-regulated; gray, no significant change).
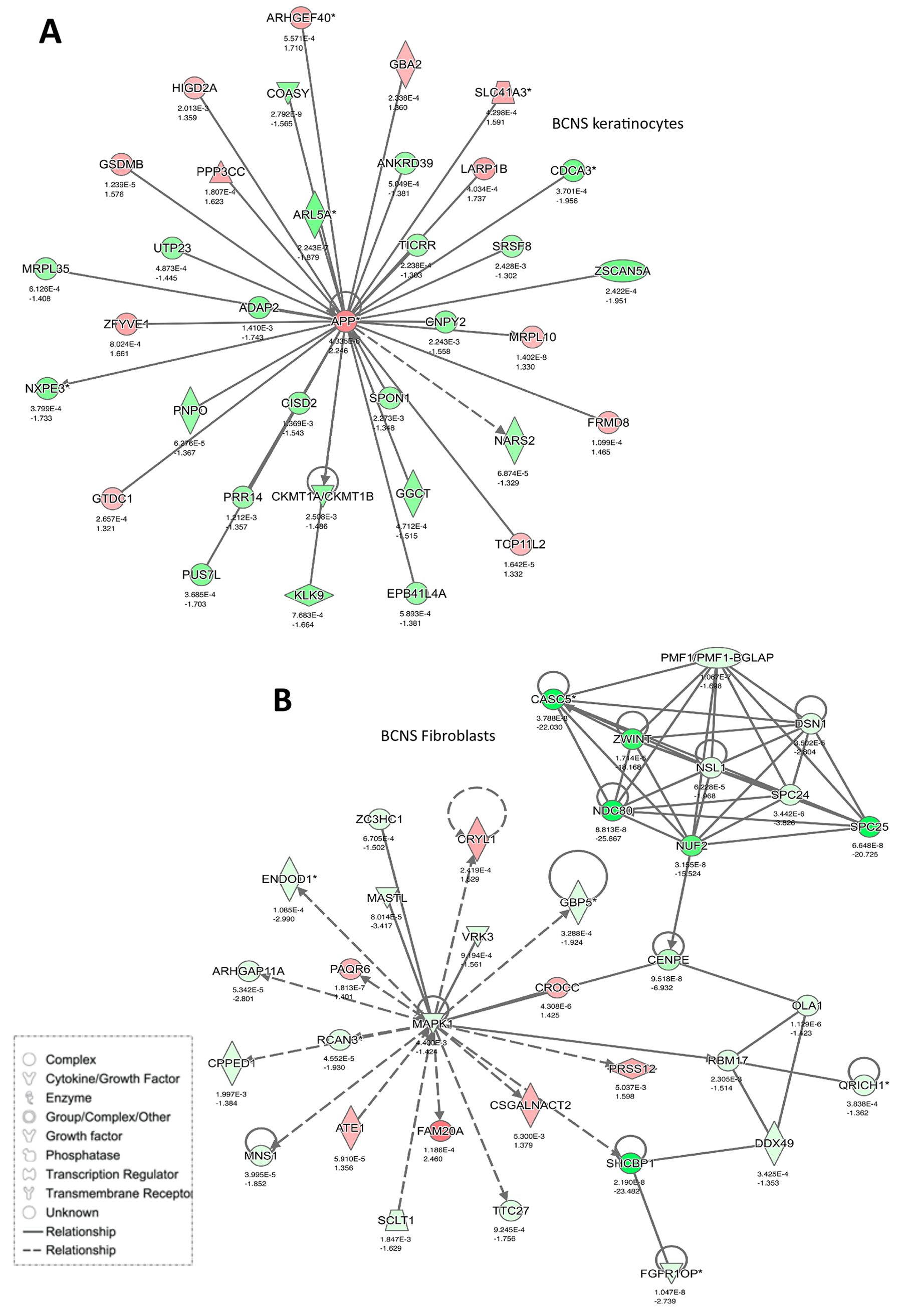
Figure 7: Networks enriched in BCNS cells after rapamycin treatment. (A) Keratinocytes and (B) fibroblasts. Gene expression variation by at least 2-fold is depicted by color (red, up-regulated; green, down-regulated; gray, no significant change).
Similar analyses of BCNS and normal fibroblasts, based on 35 focus molecules, showed enrichment of networks related to cell cycle, DNA replication, recombination and repair (Figure 6B and 7B). Notably, CASC5 (kinetochore-microtubule attachment and chromosome segregation, MAPK1 (involved in proliferation, differentiation, transcriptional regulation), and CROCC (centrosome cohesion before mitosis, associated with medulloblastoma) were included in this network.
DISCUSSION
The cellular mechanisms whereby hedgehog (HH) pathway activation leads to BCCs are yet to be established [29]. Excess HH signaling may directly cause proliferation of BCC precursors [9, 52]. Alternatively, the HH pathway may be involved primarily in stem cell maintenance as has been shown in tumor stem cells for some tissues [8, 53, 54]. As either a direct effector of proliferation or maintainer of stem cells, HH would be acting in a cell autonomous role in BCCs. An alternative model involves a paracrine function in which HH signaling acts upon stroma to generate a tumor-promoting microenvironment while having a modest role in the tumor cells themselves [44, 45]. This model is appealing in the case of BCCs because of evidence that conditioned stroma is necessary for maintenance of tumor viability and growth. There are data supporting all three of these biological roles in cancer, and it is not clear which may apply to epidermal cell transformation [29]. Likewise, the fine details of the molecular mechanisms by which HH pathway activation exerts its effects are not entirely understood. Most investigations have focused on downstream targets of the GLI transactivators. A number of specific candidate genes have been studied in human and mouse BCCs including PDGF receptor, BCL2, cyclin D1 and D2. However, unbiased attempts by global gene expression to determine key HH target genes important in BCCs have yielded inconsistent results (reviewed in [22]).
Using exploratory analysis at baseline, we were unable to identify gene expression changes that molecularly distinguish affected individuals from controls. The lack of clear delineation of BCNS subjects from normal controls is depicted by PCA and HC analyses of fibroblasts or keratinocytes (Supplementary Figure 1 and Figure 1). Our results showing lack of BCNS sample clustering on the basis of mutation type or gender are in line with previous observations demonstrating lack of evidence for genotype-phenotype correlation in BCNS cases, including no association between mutation type, age of onset, or number of BCCs developed in BCNS patients [55]. Conceivably, subtle gene expression changes caused by single copy loss of PTCH1 may enable the intact PTCH1 gene to exert negative feedback control, particularly under in vitro growth conditions. Along these lines, previous studies showed that the up-regulated HH-signaling in vivo was suppressed in BCC and medulloblastoma (MB) cells grown in vitro [46, 47]. Moreover, HH signaling of MB cells grown in vitro was not restored upon transplantation in nude mice, nor did they respond to treatment with HH antagonists [47]. It is also interesting to consider whether apart from the classical tumor suppressor ‘two-hit’ model, the ‘continuum model’ [56] that accounts for subtle dosage effects, ‘obligate haploinsufficiency’, is associated with the effect of PTCH1 tumor suppressor gene in BCNS.
Hence, our results at low stringency (p-value <0.05, no FDR cut-off; FC 1.3) have enabled us to unmask candidate baseline differences between BCNS and normal control cells that are likely to play a role in BCC growth under conditions in which the HH pathway has been suppressed [47]. Thus, a total of 585 and 857 genes differentially expressed in keratinocytes and fibroblasts samples from BCNS subjects, respectively were identified. For example, a Gene Set Enrichment Analysis (GSEA) of highly regulated candidate genes recognized pancreatic β cell hallmark genes and mTOR signaling genes in BCNS keratinocytes, whereas analyses of BCNS fibroblasts candidate genes showed gene signatures that affect pathways regulating pluripotency of stem cells, including the WNT pathway. Moreover, these differentially affected gene signatures included canonical and non-canonical HH pathways in BCNS keratinocytes and fibroblasts that are likely to play a role in BCC development. One such example that is likely relevant in this context is our finding that MKL1 is up-regulated in BCNS keratinocytes, since its activation within the non-canonical HH pathway has recently been shown to promote drug resistance of BCCs [57].
Importantly, the phenotypic features of BCNS patients shown in our study and elsewhere [4, 58–61], provide strong support for a two-hit mechanism wherein the first hit is associated with early molecular changes and a variety of developmental defects occurring concurrently. While some of these defects may in fact arise through a two-hit mechanism, it is difficult to imagine how such symmetric features as generalized overgrowth, macrocephaly, coarse facies, and hypertelorism could arise through any mechanism other than one invoking an effect of haploinsufficiency. Hence, gene expression patterns in tissues at some stages of development must almost certainly differ between BCNS subjects and controls. Accordingly, networks of differentially regulated genes at baseline shown here are of interest as they were found to be enriched in processes which might play important roles in the development of congenital phenotype (e.g., macrocephaly, skeletal abnormalities, bossing of forehead). For example, BEX1 (Brain Expressed, X-Linked 1) plays a role in cell cycle progression and neuronal differentiation, and is related to p75NTR/NGFR-mediated signaling and it inhibits neuronal differentiation in response to nerve growth factor (NGF). SOX4 (SRY-related HMG-box) is a member of SOX family of transcription factors that regulate pathways in embryonic development and cell fate determination and is related to ERK signaling. MEG3 (Maternally Expressed 3) may function as a lncRNA tumor suppressor as it inhibit tumor cell proliferation, interacts and regulates p53 gene expression and its down-regulation has been experimentally observed in cancer cell lines of various origins. SYN1 (Synapsin 1) is member of neuronal phosphoproteins that function to regulate axonogenesis and synaptogenesis. SIM2 (Drosophila single-minded gene homolog) encodes for transcription factor that regulates neurogenesis and may have pleiotropic effects in tissues during development. Thus, perturbations caused by loss of function PTCH1 mutations although subtle, seem sufficient to result in developmental anomalies and BCCs in BCNS patients. The occurrence of PTCH1 mutations that result in developmental anomalies concomitant with BCC expression serve as poignant reminder of the role of PTCH in normal tissue development and as a tumor suppressor gene (TSG), a basic tenet in cancer biology, including all TSGs identified to date [62].
Prior studies suggest that signaling through the hedgehog and mTOR pathways have separate, albeit synergistic effects on malignant transformation [33–35, 37]. Indeed, our present GSEA studies (vide supra) showed an activated mTOR pathway in BCNS PTCH1 (+/-) keratinocytes. Accordingly, mTOR/S6K1 and HH pathways coincidentally target an overlapping set of genes that are key modulators of HH-related carcinogenesis [38–40]. In this regard, a recent study has demonstrated the inhibition of human rhabdomyosarcoma xenografts by rapamycin through its effect on HH effector genes such as Gli1 and Gli 2 as well as PTCH1 and PATCH2 [63]. These effects were associated with over 80% reduction in cyclin D1, a downstream transcription target for both HH and mTOR signaling pathways [63].
To investigate the differential effect of rapamycin in BCNS, we compared the gene expression profiles of keratinocytes and fibroblasts derived from normal subjects and from unaffected skin cells of BCNS patients. Accordingly, rapamycin affected the expression of 1411 genes and 3214 genes which were altered in BCNS and normal keratinocytes, respectively (FC >1.3 and <-1.3; FDR <0.05), including 812 genes whose altered expression was shared by both group. A similar analysis of BCNS and normal fibroblasts resulted in 4959 genes and 4797 genes, respectively wherein expression in each group was altered following rapamycin treatment, including expression of 3496 genes that was common in both groups. The increased stringency in analyzing the rapamycin effect as compared with baseline’s lesser stringency was meant to focus on the most salient alterations seen due to rapamycin. We focused on these gene lists via an IPA analysis in order to discern differences in molecular functions and canonical signaling. For example, our results show that rapamycin treatment of BCNS keratinocytes affected Wnt/β-catenin Signaling, ILK signaling, epithelial adherens junction signaling, Sonic Hedgehog signaling, IGF-1 signaling, caveolar-mediated endocytosis signaling, and amyloid processing (Supplementary Data). On the other hand, pathways that were associated with response to rapamycin in BCNS fibroblasts include paxillin signaling, DNA damage-induced 13-3-3σ, integrin signaling, Cdc42 signaling, telomere extension by telomerase, HGF and VEGF signaling. The overlap between hedgehog targets and rapamycin targets provides evidence for a model in which the anti-neoplastic effect of rapamycin relates to suppression of HH signaling. Rapamycin is the subject of approximately 1500 clinical trials for cancer treatment and prevention listed on the NIH Clinical Trials web site (clinicaltrials.gov). Among these trials, five ongoing or recently completed studies relate to the use of this compound in prevention of skin cancer in transplant recipients.
This study underscores the need to catalog baseline molecular differences in BCNS cohorts in order to anticipate the numerous manifestations characteristic of BCNS, particularly the locally invasive but seldom metastatic nature of BCCs. Perturbations caused by loss of function of PTCH1 mutations unmasked herein, including alterations in canonical and noncanonical HH pathways, although subtle, seem sufficient to result in developmental anomalies and BCCs in BCNS patients. This is the first study involving skin derived keratinocytes and fibroblasts from BCNS patients wherein global gene profiling may be used to generate rationale hypotheses and design of future experiments, including noncanonical HH pathways.
MATERIALS AND METHODS
Ethics statement
The clinical protocol under which this study (https://clinicaltrials.gov/ct2/show/record/NCT00433485) was conducted was approved by Yale University’s Institutional Review Board, the Clinical Trials Protocol Review Committee of the Yale Comprehensive Cancer Center, and by the sponsoring agency, the Division of Cancer Prevention, National Cancer Institute. Informed consent was obtained to collect tissue from study participants meeting clinical diagnostic criteria for familial BCNS and normal individuals in accordance to the ethical standards and principles expressed in the Declaration of Helsinki.
Study participants
Four female and five male BCNS cases were ascertained through the clinical practices and previous research programs (AEB and DJL). Eight age- and sex- matched, unaffected controls included relatives of cases and normal volunteers at Yale University. The mean ages of cases and controls were virtually identical - 42.3+/-14.0 and 41.6+/-12.5 - although female cases were older on average than female controls while male cases were younger on average than male controls (Table 1).
Cases were chosen on the basis of meeting clinical criteria for BCNS [4]. In addition, PTCH gene sequencing was performed by the CLIA-certified DNA Diagnostics Laboratory in the Department of Genetics at Yale. Four affected subjects had a mutation predicted to truncate the PTCH protein. Three had missense alterations in codons conserved from humans to invertebrates and not seen in approximately 125,000 normal controls (gnomAD browser: http://gnomad.broadinstitute.org/gene/ENSG00000185920). One of these mutations was reported previously in BCNS [40]. Subject number 18 had no clearly deleterious mutation. However, the diagnostics methods used in this study would not have detected exon-size or larger deletions and duplications or other large-scale rearrangements, such as inversions. This subject was heterozygous for SNPs throughout the gene, ruling out “hemizygosity” (heterozygous deletion) of the whole gene. Subject 22 had an alteration in exon 1E predicted to create a highly efficient, abnormal splice acceptor site (http://www.fruitfly.org/seq_tools/splice.html). This variant was not seen in 125,000 normal controls. Among the controls, four were relatives of cases involved in this study, and all tested negative for the mutation found in their affected relative. Among the remaining four controls, none had any of the exclusion criteria listed below:
• One or more basal cell carcinomas
• Palmar or plantar pits typical of BCNS
• History of medulloblastoma
• History of odontogenic keratocyst or any jaw cyst for which a histopathologic diagnosis could not be ascertained
• History of ovarian or cardiac fibroma
• Polydactyly
• Macrocephaly determined after adjustment for height
• Craniofacial features of BCNS including cleft palate, frontal bossing, hypertelorism, iris coloboma or other developmental defects of the eye, or other typical facial features.
Specimen acquisition and tissue culture
Four 5 mm punch biopsies were obtained from unaffected skin of the upper inner arm of each case and control. Keratinocyte and fibroblast cultures were established as previously described [41]. Briefly, keratinocytes were harvested from epidermis by overnight exposure to Dispase at 4°C and seeded onto mitomycin C-treated feeders of 3T3 cells. Once colonies of keratinocytes were growing, they were expanded in low calcium MCDB medium. Fibroblasts were harvested from minced pieces of dermis obtained after dispase treatment of skin biopsies and expanded in DMEM plus 10% serum. Twenty-four vials of low-passage cells with approximately 5×105 cells/vial were frozen in liquid nitrogen from each subject.
For experiments examining the effects of rapamycin, primary keratinocyte and fibroblast cultures were plated at approximately 25% confluence in 60 mm plates. One day after seeding, they were treated with 0μM (vehicle only), 10μM or 50μM rapamycin in DMSO (Sigma, Inc.) for 48 hours. Cells were nearly confluent when harvested.
Gene expression and statistical analysis
RNA extraction
Total RNA was extracted from the keratinocyte and fibroblast cultures using the standard TRIzol® (Invitrogen) protocol and was further purified using the RNeasy cleanup procedure (Qiagen) according to manufacturer’s instructions. The quality of total RNA was assessed by Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA) for visual absence of genomic DNA contamination and integrity of 28S and 18S bands. Only samples with an A260/A280 ratio of at least 1.9 were used for microarray analysis. Samples were stored in 5μg aliquots at -70° C until use.
Microarray processing and data analysis
Gene expression profiling was performed on total 102 samples on Affymetrix Human Genome U133 Plus 2.0 GeneChip Array®, comprising of fibroblasts and keratinocytes treated with vehicle, low- and high-dose rapamycin. Microarray processing was performed at Yale. The cDNA and cRNA preparation, cRNA labeling and hybridization to Affymetrix Human Genome U133 Plus 2.0 GeneChip® microarrays (Affymetrix, Santa Clara, CA) was performed as per standard protocols according to manufacturer’s instructions. The arrays were scanned using Hewlett-Packard GeneArray Scanner and the scanned output files were visually inspected for hybridization artifacts. The data quality was checked for the presence of spiked control cRNAs, background values caused by array autofluorescence and non-specific binding, and Q value. The microarray gene expression data have been deposited in Gene Expression Omnibus (GSE#120242).
The microarray data in.cel files was imported and RMA normalization performed using Partek® Genomics Suite® software, version 6.6©; 2014 (Partek Inc., St. Louis, MO, USA) followed by QC metrics and preliminary exploratory analysis. Attributes were assigned to the microarray files to the random effects namely subject and scan dates, and the fixed effects namely the cell type, gender, PTCH mutation type and rapamycin treatment followed by exploratory principal components analysis to observed clustering based on various sample attributes.
For unsupervised analysis, first probe level data was collapsed to gene level data by retaining for each gene, amongst multiple possible probes, the probe that had maximum coefficient of variation. All other probes were discarded. After collapsing the probe level data to gene level data, a secondary filter to exclude genes with coefficient of variation less than 0.1 (exclude genes with CV<0.1) was applied to normalized data resulting in 5384 and 3920 genes in keratinocytes and fibroblasts respectively that were clustered using Euclidean and average linkage method as similarity measure to identify patterns of clustering in the samples. Batch effect arising due to samples being run at different time points was studied using the statistical batch effect removal tool as per the manual (Partek GS’s User Manual). To ensure that the batch effect is not the major source of variation, scan dates were included in the ANOVA model as well as sources of variation were studied. To identify differentially expressed genes, a mixed- model ANOVA with restricted maximum likelihood (REML) method to estimate variance components was used in this unbalanced, mixed model study that takes into account both ‘fixed’ experimental factors such as with disease status/mutation type, cell type, treatment and ‘random effects’ such as scan dates and subject. To generate gene lists for contrasts included in the ANOVA-REML design, significance of change cut-offs of FDR <0.05 and size of fold change >1.3 or <-1.3 were used.
Gene Set Enrichment Analysis (GSEA)
GSEA was performed on the comprehensive microarray datasets for control and treatment comparisons to determine differences and enriched gene sets in the normal and BCNS groups. GSEA was performed as per software instruction to estimate the association between predefined gene sets in the MSigDb and the phenotype defined by the gene expression profiles of BCNS and normal keratinocytes and fibroblasts before and after treatment with rapamycin [64, 65]. The comparison of the groups was performed using GSEA by permuting the phenotype for 1000 times, and selecting the weighted scoring scheme with signal to noise statistical metric to rank genes and complete the GSEA analysis.
Ingenuity Pathway Analysis (IPA®)
Following ANOVA, gene lists were created by applying false discovery rate (FDR), calculated using Benjamini’s method [66] cut-off of <0.05 before uploading to IPA. For pathway analysis, these datasets with p-values and fold changes for each logical comparison were analyzed through QIAGEN’s Ingenuity® Pathway Analysis® (IPA®, QIAGEN Redwood City, https://www.qiagen.com/ingenuity) using flexible file format, Affymetrix as identifier, and Human Genome U133 Plus 2.0 array as the platform used.
Abbreviations
AGRN, agrin; BCNS, Basal Cell Nevus Syndrome; BCC, basal cell carcinoma; DEPTOR, DEP domain-containing mTOR-interacting protein; FDR, false discovery rate; FC, fold change; GSEA, gene set enrichment analysis; HC, hierarchy clustering; HH, hedgehog; HIP, hedgehog interacting protein; IPA, Ingenuity Pathway Analysis; mTOR, mammalian target of rapamycin; mLST8, mTOR Associated Protein, LST8 Homolog; PI3K, Phosphoinositide 3-kinase; PTCH, Patched; PCA, principle component analysis; PRAS40, proline-rich Akt substrate of 40 kDa; RAPTOR, Regulatory-associated protein of mTOR; SMO, smoothened; TTi1-TEL2, Tel two interacting protein 1- telomere maintenance 2.
ACKNOWLEDGMENTS AND FUNDING
We are grateful to Drs Chirayu Goswami# and Yunlong Liu, The Center for Computational Biology and Bioinformatics and the center for Medical Genomics, Indiana University School of Medicine, Indianapolis, IN for their support in data analysis.
#Currently at Jefferson Hospital, Thomas Jefferson University, Philadelphia, PA; *
Financial Support: This work was supported by the National Cancer Institute N01 CN-43300 and INGEN, and supported in part by the Lilly Endowment, Inc. MA is supported by R01 ES026219.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Fears TR, Scotto J. Changes in skin cancer morbidity between 1971-72 and 1977-78. J Natl Cancer Inst. 1982; 69:365–70.
2. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009; 59:225–49. https://doi.org/10.3322/caac.20006.
3. Weinstock MA, Bogaars HA, Ashley M, Litle V, Bilodeau E, Kimmel S. Nonmelanoma skin cancer mortality. A population-based study. Arch Dermatol. 1991; 127:1194–7.
4. Kimonis VE, Goldstein AM, Pastakia B, Yang ML, Kase R, DiGiovanna JJ, Bale AE, Bale SJ. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997; 69:299–308.
5. Gorlin RJ. Nevoid basal cell carcinoma (Gorlin) syndrome. Genet Med. 2004; 6:530–9.
6. Klein RD, Dykas DJ, Bale AE. Clinical testing for the nevoid basal cell carcinoma syndrome in a DNA diagnostic laboratory. Genet Med. 2005; 7:611–9.
7. Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S, Negus K, Smyth I, Pressman C, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996; 85:841–51.
8. Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell. 2008; 15:801–12. https://doi.org/10.1016/j.devcel.2008.11.010.
9. Ingham PW, Placzek M. Orchestrating ontogenesis: variations on a theme by sonic hedgehog. Nat Rev Genet. 2006; 7:841–50. https://doi.org/10.1038/nrg1969.
10. Yu M, Gipp J, Yoon JW, Iannaccone P, Walterhouse D, Bushman W. Sonic hedgehog-responsive genes in the fetal prostate. J Biol Chem. 2009; 284:5620–9. https://doi.org/10.1074/jbc.M809172200.
11. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971; 68:820–3.
12. Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001; 1:157–62. https://doi.org/10.1038/35101031.
13. Kopelovich L, Shea-Herbert B. Heritable one-hit events defining cancer prevention? Cell Cycle. 2013; 12:2553–7. https://doi.org/10.4161/cc.25690.
14. Kopelovich L. Phenotypic markers in human skin fibroblasts as possible diagnostic indices of hereditary adenomatosis of the colon and rectum. Cancer. 1977; 40:2534–41.
15. Yeung AT, Patel BB, Li XM, Seeholzer SH, Coudry RA, Cooper HS, Bellacosa A, Boman BM, Zhang T, Litwin S, Ross EA, Conrad P, Crowell JA, et al. One-hit effects in cancer: altered proteome of morphologically normal colon crypts in familial adenomatous polyposis. Cancer Res. 2008; 68:7579–86. https://doi.org/10.1158/0008-5472.CAN-08-0856.
16. Herbert BS, Chanoux RA, Liu Y, Baenziger PH, Goswami CP, McClintick JN, Edenberg HJ, Pennington RE, Lipkin SM, Kopelovich L. A molecular signature of normal breast epithelial and stromal cells from Li-Fraumeni syndrome mutation carriers. Oncotarget. 2010; 1:405–22. https://doi.org/10.18632/oncotarget.175.
17. Peri S, Caretti E, Tricarico R, Devarajan K, Cheung M, Sementino E, Menges CW, Nicolas E, Vanderveer LA, Howard S, Conrad P, Crowell JA, Campbell KS, et al. Haploinsufficiency in tumor predisposition syndromes: altered genomic transcription in morphologically normal cells heterozygous for VHL or TSC mutation. Oncotarget. 2017; 8:17628–42. https://doi.org/10.18632/oncotarget.12192.
18. Bellacosa A, Godwin AK, Peri S, Devarajan K, Caretti E, Vanderveer L, Bove B, Slater C, Zhou Y, Daly M, Howard S, Campbell KS, Nicolas E, et al. Altered gene expression in morphologically normal epithelial cells from heterozygous carriers of BRCA1 or BRCA2 mutations. Cancer Prev Res (Phila). 2010; 3:48–61. https://doi.org/10.1158/1940-6207.CAPR-09-0078.
19. Gailani MR, Stahle-Backdahl M, Leffell DJ, Glynn M, Zaphiropoulos PG, Pressman C, Unden AB, Dean M, Brash DE, Bale AE, Toftgard R. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat Genet. 1996; 14:78–81. https://doi.org/10.1038/ng0996-78.
20. Gailani MR, Leffell DJ, Ziegler A, Gross EG, Brash DE, Bale AE. Relationship between sunlight exposure and a key genetic alteration in basal cell carcinoma. J Natl Cancer Inst. 1996; 88:349–54.
21. Gailani MR, Bale SJ, Leffell DJ, DiGiovanna JJ, Peck GL, Poliak S, Drum MA, Pastakia B, McBride OW, Kase R, Greene M, Mulvihill J, Bale AE. Developmental defects in Gorlin syndrome related to a putative tumor suppressor gene on chromosome 9. Cell. 1992; 69:111–7.
22. Epstein EH. Basal cell carcinomas: attack of the hedgehog. Nat Rev Cancer. 2008; 8:743–54. https://doi.org/10.1038/nrc2503.
23. Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, Bonifas JM, Lam CW, Hynes M, Goddard A, Rosenthal A, Epstein EH Jr, de Sauvage FJ. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998; 391:90–2. https://doi.org/10.1038/34201.
24. Sidransky D. Is human patched the gatekeeper of common skin cancers? Nat Genet. 1996; 14:7–8. https://doi.org/10.1038/ng0996-7.
25. Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 1997; 386:761, 763. https://doi.org/10.1038/386761a0.
26. Roush W. Putting the brakes on bone growth. Science. 1996; 273: 579.
27. Valin A, Barnay-Verdier S, Robert T, Ripoche H, Brellier F, Chevallier-Lagente O, Avril MF, Magnaldo T. PTCH1 +/- dermal fibroblasts isolated from healthy skin of Gorlin syndrome patients exhibit features of carcinoma associated fibroblasts. PLoS One. 2009; 4: e4818. https://doi.org/10.1371/journal.pone.0004818.
28. So PL, Tang JY, Epstein EH. Novel investigational drugs for basal cell carcinoma. Expert Opin Investig Drugs. 2010; 19:1099–112. https://doi.org/10.1517/13543784.2010.504714.
29. Bakshi A, Chaudhary SC, Rana M, Elmets CA, Athar M. Basal cell carcinoma pathogenesis and therapy involving hedgehog signaling and beyond. Mol Carcinog. 2017; 56:2543–57. https://doi.org/10.1002/mc.22690.
30. Yang L, Xie G, Fan Q, Xie J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene. 2010; 29:469–81. https://doi.org/10.1038/onc.2009.392.
31. Biehs B, Dijkgraaf GJP, Piskol R, Alicke B, Boumahdi S, Peale F, Gould SE, de Sauvage FJ. A cell identity switch allows residual BCC to survive Hedgehog pathway inhibition. Nature. 2018; 562:429–33. https://doi.org/10.1038/s41586-018-0596-y.
32. Sehgal SN, Baker H, Vezina C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J Antibiot (Tokyo). 1975; 28:727–32.
33. Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, Wang S, Ren P, Martin M, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012; 485:55–61. https://doi.org/10.1038/nature10912.
34. Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013; 493:338–45. https://doi.org/10.1038/nature11861.
35. Dunlop EA, Tee AR. Mammalian target of rapamycin complex 1: signalling inputs, substrates and feedback mechanisms. Cell Signal. 2009; 21:827–35. https://doi.org/10.1016/j.cellsig.2009.01.012.
36. Rao RD, Buckner JC, Sarkaria JN. Mammalian target of rapamycin (mTOR) inhibitors as anti-cancer agents. Curr Cancer Drug Targets. 2004; 4:621–35.
37. Beauchamp EM, Platanias LC. The evolution of the TOR pathway and its role in cancer. Oncogene. 2013; 32:3923–32. https://doi.org/10.1038/onc.2012.567.
38. Louro ID, McKie-Bell P, Gosnell H, Brindley BC, Bucy RP, Ruppert JM. The zinc finger protein GLI induces cellular sensitivity to the mTOR inhibitor rapamycin. Cell Growth Differ. 1999; 10:503–16.
39. Wang Y, Ding Q, Yen CJ, Xia W, Izzo JG, Lang JY, Li CW, Hsu JL, Miller SA, Wang X, Lee DF, Hsu JM, Huo L, et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell. 2012; 21:374–87. https://doi.org/10.1016/j.ccr.2011.12.028.
40. Brechbiel J, Miller-Moslin K, Adjei AA. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat Rev. 2014; 40:750–9. https://doi.org/10.1016/j.ctrv.2014.02.003.
41. Karayannopoulou G, Euvrard S, Kanitakis J. Differential expression of p-mTOR in cutaneous basal and squamous cell carcinomas likely explains their different response to mTOR inhibitors in organ-transplant recipients. Anticancer Res. 2013; 33:3711–4.
42. de Fijter JW. Cancer and mTOR inhibitors in transplant recipients. Transplantation. 2017; 101:45–55. https://doi.org/10.1097/TP.0000000000001447.
43. Eibenschutz L, Colombo D, Catricala C. Everolimus for compassionate use in multiple Basal cell carcinomas. Case Rep Dermatol Med. 2013; 2013: 604301. https://doi.org/10.1155/2013/604301.
44. Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, Marshall D, Fu L, Januario T, Kallop D, Nannini-Pepe M, Kotkow K, Marsters JC, et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008; 455:406–10. https://doi.org/10.1038/nature07275.
45. Theunissen JW, de Sauvage FJ. Paracrine Hedgehog signaling in cancer. Cancer Res. 2009; 69:6007–10. https://doi.org/10.1158/0008-5472.CAN-09-0756.
46. Aszterbaum M, Epstein J, Oro A, Douglas V, LeBoit PE, Scott MP, Epstein EH Jr. Ultraviolet and ionizing radiation enhance the growth of BCCs and trichoblastomas in patched heterozygous knockout mice. Nat Med. 1999; 5:1285–91. https://doi.org/10.1038/15242.
47. Sasai K, Romer JT, Lee Y, Finkelstein D, Fuller C, McKinnon PJ, Curran T. Shh pathway activity is down-regulated in cultured medulloblastoma cells: implications for preclinical studies. Cancer Res. 2006; 66:4215–22. https://doi.org/10.1158/0008-5472.CAN-05-4505.
48. Wei G, Twomey D, Lamb J, Schlis K, Agarwal J, Stam RW, Opferman JT, Sallan SE, den Boer ML, Pieters R, Golub TR, Armstrong SA. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell. 2006; 10:331–42. https://doi.org/10.1016/j.ccr.2006.09.006.
49. Bonilla X, Parmentier L, King B, Bezrukov F, Kaya G, Zoete V, Seplyarskiy VB, Sharpe HJ, McKee T, Letourneau A, Ribaux PG, Popadin K, Basset-Seguin N, et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat Genet. 2016; 48:398–406. https://doi.org/10.1038/ng.3525.
50. Zhang H, Pasolli HA, Fuchs E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc Natl Acad Sci U S A. 2011; 108:2270–5. https://doi.org/10.1073/pnas.1019603108.
51. Quan T, Xu Y, Qin Z, Robichaud P, Betcher S, Calderone K, He T, Johnson TM, Voorhees JJ, Fisher GJ. Elevated YAP and its downstream targets CCN1 and CCN2 in basal cell carcinoma: impact on keratinocyte proliferation and stromal cell activation. Am J Pathol. 2014; 184:937–43. https://doi.org/10.1016/j.ajpath.2013.12.017.
52. Athar M, Tang X, Lee JL, Kopelovich L, Kim AL. Hedgehog signalling in skin development and cancer. Exp Dermatol. 2006; 15:667–77. https://doi.org/10.1111/j.1600-0625.2006.00473.x.
53. Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, Devereux WL, Rhodes JT, Huff CA, Beachy PA, Watkins DN, Matsui W. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci U S A. 2007; 104:4048–53. https://doi.org/10.1073/pnas.0611682104.
54. Van Scott EJ, Reinertson RP. The modulating influence of stromal environment on epithelial cells studied in human autotransplants. J Invest Dermatol. 1961; 36:109–31.
55. Wicking C, Shanley S, Smyth I, Gillies S, Negus K, Graham S, Suthers G, Haites N, Edwards M, Wainwright B, Chenevix-Trench G. Most germ-line mutations in the nevoid basal cell carcinoma syndrome lead to a premature termination of the PATCHED protein, and no genotype-phenotype correlations are evident. Am J Hum Genet. 1997; 60:21–6.
56. Berger AH, Knudson AG, Pandolfi PP. A continuum model for tumour suppression. Nature. 2011; 476:163–9. https://doi.org/10.1038/nature10275.
57. Whitson RJ, Lee A, Urman NM, Mirza A, Yao CY, Brown AS, Li JR, Shankar G, Fry MA, Atwood SX, Lee EY, Hollmig ST, Aasi SZ, et al. Noncanonical hedgehog pathway activation through SRF-MKL1 promotes drug resistance in basal cell carcinomas. Nat Med. 2018; 24:271–81. https://doi.org/10.1038/nm.4476.
58. Boutet N, Bignon YJ, Drouin-Garraud V, Sarda P, Longy M, Lacombe D, Gorry P. Spectrum of PTCH1 mutations in French patients with Gorlin syndrome. J Invest Dermatol. 2003; 121:478–81. https://doi.org/10.1046/j.1523-1747.2003.12423.x.
59. Fan Z, Li J, Du J, Zhang H, Shen Y, Wang CY, Wang S. A missense mutation in PTCH2 underlies dominantly inherited NBCCS in a Chinese family. J Med Genet. 2008; 45:303–8. https://doi.org/10.1136/jmg.2007.055343.
60. Lo Muzio L, Pastorino L, Levanat S, Musani V, Situm M, Ponti G, Bianchi Scarra G. Clinical utility gene card for: Gorlin syndrome--update 2013. Eur J Hum Genet. 2013; 21. https://doi.org/10.1038/ejhg.2012.299.
61. Jones EA, Sajid MI, Shenton A, Evans DG. Basal cell carcinomas in gorlin syndrome: a review of 202 patients. J Skin Cancer. 2011; 2011: 217378. https://doi.org/10.1155/2011/217378.
62. Park S, Supek F, Lehner B. Systematic discovery of germline cancer predisposition genes through the identification of somatic second hits. Nat Commun. 2018; 9: 2601. https://doi.org/10.1038/s41467-018-04900-7.
63. Kaylani SZ, Xu J, Srivastava RK, Kopelovich L, Pressey JG, Athar M. Rapamycin targeting mTOR and hedgehog signaling pathways blocks human rhabdomyosarcoma growth in xenograft murine model. Biochem Biophys Res Commun. 2013; 435:557–61. https://doi.org/10.1016/j.bbrc.2013.05.001.
64. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005; 102:15545–50. https://doi.org/10.1073/pnas.0506580102.
65. Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011; 27:1739–40. https://doi.org/10.1093/bioinformatics/btr260.
66. Benjamini Y, Yekutieli D. The control of false discovery rate in multiple testing under dependency. Ann Stat. 2001; 29:1165–88.
67. Jayaraman SS, Rayhan DJ, Hazany S, Kolodney MS. Mutational landscape of basal cell carcinomas by whole-exome sequencing. J Invest Dermatol. 2014; 134:213–20. https://doi.org/10.1038/jid.2013.276.
68. Lindstrom E, Shimokawa T, Toftgard R, Zaphiropoulos PG. PTCH mutations: distribution and analyses. Hum Mutat. 2006; 27:215–9. https://doi.org/10.1002/humu.20296.