
INTRODUCTION
Lung cancer is one of the most lethal cancers worldwide. Epidermal growth factor receptor (EGFR) gene mutation is known as one of the driver mutations of lung adenocarcinoma (LUAD) and LUAD patients harboring EGFR mutation account for more than half of LUAD patients in Asia [1]. EGFR–tyrosine kinase inhibitors (EGFR–TKIs), such as gefitinib and erlotinib, have been demonstrated to be effective for the patients with EGFR mutation [2, 3]. However, acquired resistance to EGFR-TKIs is practically unavoidable and is generally considered to occur as a result of gatekeeper mutations including a threonine-to-methionine substitution on codon 790 (T790M) [4]. It is true that effective EGFR-TKI was recently developed for the patients with LUAD harboring T790M mutation [5], but other mechanisms of therapeutic resistance to this EGFR-TKI through acquiring further mutations have been also reported [6]. Cytotoxic anti-cancer drugs have been widely used for those with LUAD acquiring EGFR-TKI resistance. However, to the best of our knowledge, the possible influence of EGFR-TKI resistance on development of chemoresistance in LUAD has remained virtually unknown because the relationship between EGFR-TKI resistance and chemotherapy resistance has not necessarily been studied in LUAD [7]. Therefore, in this study, we first aimed to study this potential correlation above in LUAD.
It is also true that numerous factors associated with chemoresistance have been reported in the literature. For instance, hypoxia inducible factor-1α (HIF-1α) and epithelial-mesenchymal transition (EMT) were both reported to induce chemoresistance in various human malignancies [7, 8]. HIF-1α is a well-known transcription factor activated in hypoxia state. However, its activation was reported as a result of an activation of the phosphoinositide 3-kinase (PI3K) / Akt pathway [9] under normoxia and numerous examples of HIF-1α activation without hypoxia have been since reported. For instance, HIF-1α was demonstrated to induce chemotherapy resistance in lung cancer under both hypoxia and normoxia state [10]. In addition, an involvement of multiple drug resistance 1, BCL2 / adenovirus E1B 19 kDa protein-interacting protein 3 and EMT have been reported to be associated with HIF-1α-induced chemoresistance [11–13]. In particular, an induction of EMT was reported to decrease the therapeutic efficacy of the first generation EGFR-TKI [7] and cytotoxic anti-cancer drugs in lung cancer patients [8]. However, the possible involvement of HIF-1α and EMT in EGFR-TKI resistance in chemotherapy resistance has remained virtually unknown.
p22phox forms a heterodimer with nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), one of the main reactive oxygen species (ROS) generators, in order to stabilize the cell membrane [14]. p22phox was also recently reported to increase cell survival and proliferation in various human malignancies. For instance, in pancreatic cancer, upregulation of p22phox was reported to inhibit apoptosis by ROS [15]. In prostate cancer, upregulation of p22phox was also reported to activate PI3K/Akt pathway and extracellular signal-regulated kinase 1/2 (ERK1/2) pathway and subsequently induce HIF-1α via ROS signaling [16]. In oral squamous cell carcinoma, p22phox overexpression was reported to inhibit cisplatin (CDDP)-induced apoptosis. Results of these studies above all indicated that p22phox status was indeed correlated with chemoresistance in several human malignancies. In non-small cell lung cancer (NSCLC), an increased expression of p22phox was reported to cause EMT [17]. However, the possible roles of p22phox in lung cancer after acquired resistance to EGFR-TKI have not been known. In addition, overexpression of NOX2 was reported to induce ROS in NSCLC with EGFR-TKI resistance [18]. Therefore, in this study, we firstly studied whether p22phox could possibly contribute to the development of chemoresistance via HIF-1α and EMT in LUAD harboring acquired resistance to EGFR-TKI or not and elucidated the possible development of new treatments to overcome the therapeutic resistance in these patients.
RESULTS
p22phox was upregulated in chemoresistant LUAD after acquired resistance to EGFR-TKI
We first performed cell viability assays after CDDP or pemetrexed (PEM) treatment using the WST-8 Cell Counting Kit-8. Results of cell viability in CDDP (1 μM and 10 μM) and PEM (0.01 μM, 0.1 μM, 1μM and 10 μM) treatment for 3 days in both PC9/GR and PC9/ER were significantly higher than those in PC9/6m (PC9/GR: P = 0.0421, 0.0003, 0.0091, 0.0007, 0.0491, 0.0070, PC9/ER: P = 0.003, P < 0.0001, P = 0.0044, P < 0.0001, P < 0.0001, P < 0.0001, respectively) (Figure 1A and 1B, respectively). In addition, in the cells treated with CDDP (10 μM) and PEM (1 μM and 10μM), the cell viability in PC9/ER was significantly higher than that in PC9/GR (P = 0.003, 0.0004, P < 0.0001, respectively) (Figure 1A and 1B). Therefore, those results above indicated that PC9/ER was highly chemoresistant LUAD cell line following acquired resistance to EGFR-TKI. Therefore, we performed comprehensive gene analysis by microarray in order to further study gene profiling of PC9/ER. Among the factors associated with HIF-1α pathway or EMT induction, both of which are well known to induce chemoresistance in several human malignancies, the status of p22phox in PC9/ER was particularly higher than that in PC9/6m and PC9/GR. The amounts of p22phox expression at both mRNA and protein levels were significantly higher in PC9/ER than those in both PC9/6m and PC9/GR (mRNA; P = 0.0002, 0.0002, respectively) (Figure 1C and 1D).
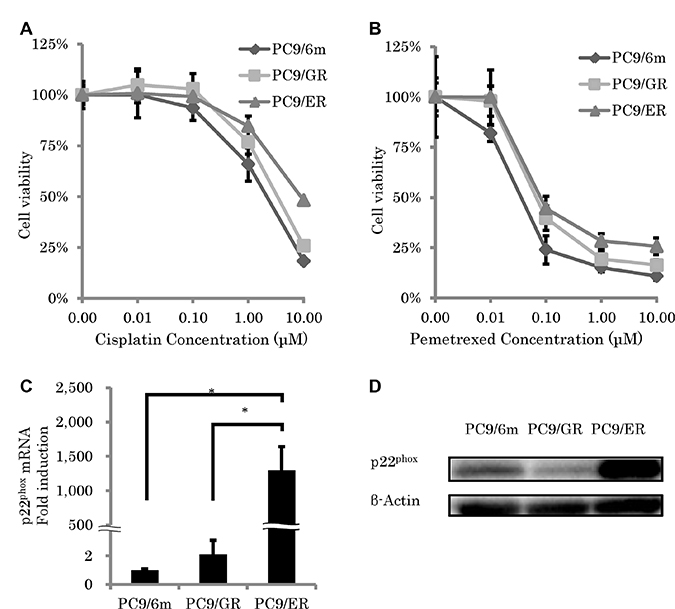
Figure 1: Chemosensitivity and expression of p22phox in epidermal growth factor receptor–tyrosine kinase inhibitor (EGFR-TKI)–resistant lung adenocarcinoma cells. (A, B) Cell viability was measured using WST-8 assay of control lung adenocarcinoma cell line (PC9/6m) and EGFR-TKI resistant lung adenocarcinoma cell lines (PC9/GR and PC9/ER) treated with cisplatin (A) and pemetrexed (B) for 72 h; N = 4. (C, D) Expression level of p22phox. mRNA levels (N = 3) (C) and protein expressions (D) of p22phox in high chemoresistant cell line (PC9/ER) were significantly higher than chemo-sensitive cell line (PC9/6m) and low chemoresistance cell line (PC9/GR). The significance of difference between indicated groups are calculated by Student’s t-test (*P < 0.05).
Knockdown of p22phox enhanced efficiency of chemotherapy in EGFR-TKI resistant LUAD cell lines harboring EGFR T790M mutation
To examine the roles of p22phox against chemoresistance in EGFR-TKI resistant LUAD, we performed knockdown of p22phox expression by using siRNA (mRNA; P = 0.0004) (Figure 2A and 2B). Results of the cell viability assay did demonstrate that knockdown of p22phox significantly enhanced efficiency of CDDP cytotoxicity (10 μM) in PC9/ER (P < 0.0001), but not in PC9/6m (P = 0.1704) (Figure 2C). Therefore, in order to further confirm whether the effects of p22phox on chemosensitivity was inherent to PC9/ER or not, we evaluated the effects of p22phox on other EGFR-TKI resistant LUAD cell lines. The amounts of p22phox expression at both mRNA and protein levels in H1975 and A549 were significantly higher than those in PC9 (mRNA; P < 0.0001, P = 0.0045, respectively) (Figure 2D and 2E). Knockdown of p22phox significantly enhanced efficiency of CDDP cytotoxicity (10 μM) in H1975 (P = 0.0202) but not in A549 (P = 0.0556) (Figure 2F).
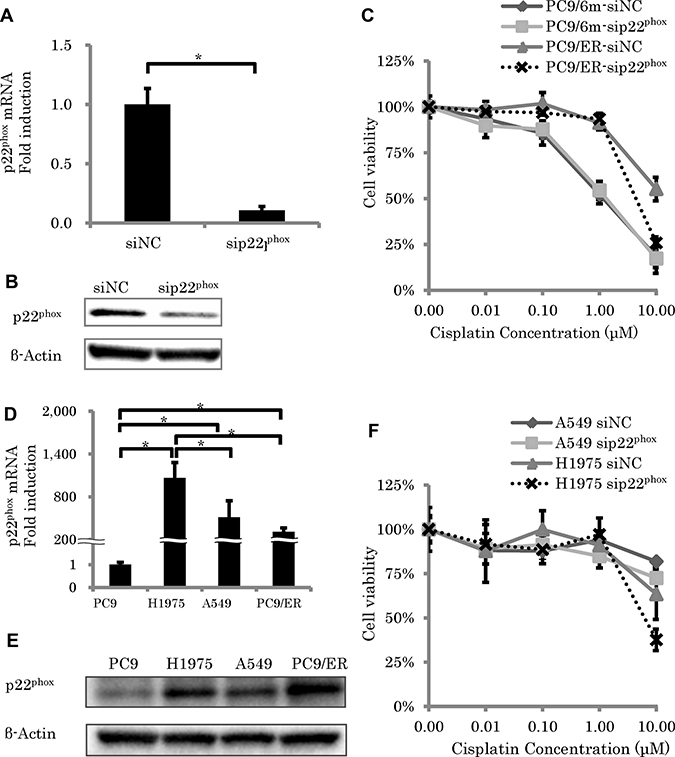
Figure 2: The effect of p22phox knockdown on sensitivity to cisplatin-induced cytotoxicity. (A, B) Expression level of p22phox mRNA (N = 3) (A) and protein expressions of p22phox (B) in EGFR-TKI and chemotherapy resistant lung adenocarcinoma cells (PC9/ER) were significantly knocked down by siRNA (5 nM). (C) Cell viability was measured using WST-8 assay in control lung adenocarcinoma cells (PC9/6m) and PC9/ER transfected with siRNA (5 nM) for 24h, followed by treatment with cisplatin for another 48 h (N = 4). (D, E) Expression level of p22phox. mRNA levels (N = 3) (D) and protein expressions (E) of p22phox in EGFR-TKI resistant cells (H1975) was significantly higher than PC9, A549 and PC9/ER. (F) Cell viability was measured using WST-8 assay in H1975 and A549 transfected with siRNA (5 nM) for 24h, followed by treatment with cisplatin for another 48 h (N = 4). The significance of difference between indicated groups are calculated by Student’s t-test (*P < 0.05).
p22phox regulated HIF-1α in EGFR-TKI resistant LUAD cell lines
We then focused on HIF-1α, because this protein was reported to contribute enormously to the development of chemoresistance [19] through an induction by p22phox via ROS [16], in order to further clarify the downstream of p22phox in the effect of chemoresistance. The amounts of HIF-1α expression at both mRNA and protein levels in PC9/ER were significantly higher than those in both PC9/6m and PC9/GR (mRNA; P = 0.0005, 0.0006, respectively) (Figure 3A and 3B, Supplementary Figure 1). We therefore performed knockdown of HIF-1α expression (P = 0.0003) (Figure 3C) in order to further examine the effects of HIF-1α on the development of chemoresistance in EGFR-TKI resistant LUAD. Results of the cell viability assay did demonstrate that the knockdown of HIF-1α significantly decreased the survival rate in PC9/ER (0.1 μM, 1 μM, 10 μM and 100 μM) (P = 0.0270, 0.0423, 0.0006, 0.0003, respectively) (Figure 3D). In addition, in order to clarify whether p22phox induced the expression of HIF-1α or not, we subsequently performed the knockdown of p22phox using its corresponding siRNA. Results did demonstrate that the knockdown of p22phox decreased the HIF-1α mRNA expression in PC9/ER (P = 0.0007) (Figure 3E).
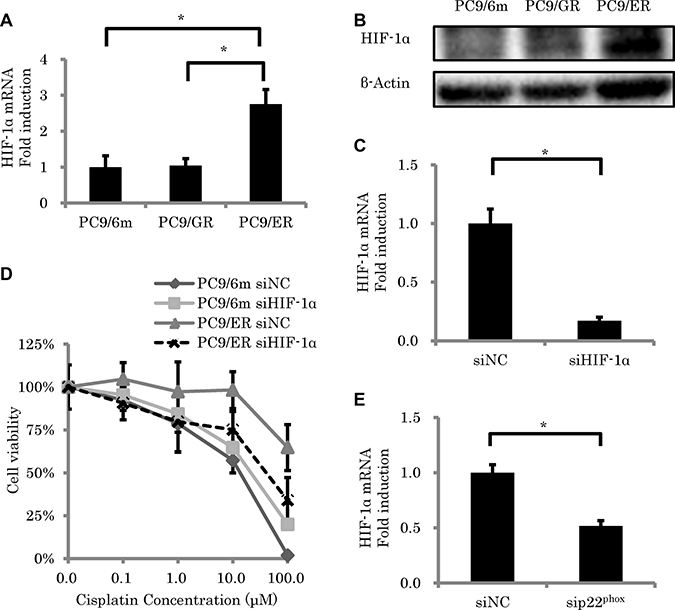
Figure 3: The effect of HIF-1α knockdown on sensitivity to cisplatin-induced cytotoxicity. (A, B) Expression level of HIF-1α. mRNA levels (N = 3) (A) and protein expressions (B) of HIF-1α in high chemoresistant cell line (PC9/ER) were significantly higher than in chemo-sensitive cells (PC9/6m) and in low chemoresistant cells (PC9/GR). (C) The expression of HIF-1α in PC9/ER was knocked down by siRNA (5 nM) (N = 3). (D) Cell viability was measured using WST-8 assay in control lung adenocarcinoma cell line (PC9/6m) and PC9/ER transfected with siRNA (5 nM) for 24h, followed by treatment with cisplatin for another 48 h (N = 4). (E) HIF-1α expression was significantly decreased by knockdown of p22phox in PC9/ER. The significance of difference between indicated groups are calculated by Student’s t-test (*P < 0.05).
p22phox induced EMT in EGFR-TKI resistant LUAD cell lines
The cells demonstrating spindle cell morphology were detected only in PC9/ER (Figure 4A). In addition, the expression of epithelial biomarker (E-cadherin) was significantly decreased (mRNA; P = 0.001) and the levels of mesenchymal proteins (Vimentin) and EMT inducible proteins, zinc finger E-box-binding homeobox 1 (ZEB1) and ZEB2, were also significantly increased in PC9/ER than PC9/6m (mRNA; P < 0.001, P = 0.0023, 0.0014, respectively) (Figure 4B and 4C). These results all indicated that EMT was indeed induced in PC9/ER. EMT was reported to contribute to the development of chemoresistance [8], we then studied the effects of p22phox and/or HIF-1α on EMT. Results demonstrated that the knockdown of p22phox by employing its corresponding siRNA significantly decreased the mRNA expression of ZEB1 (P = 0.0448) (Figure 4D). In addition, in order to further clarify the association of HIF-1α with EMT, we transfected plasmid of HIF-1α into PC9 and examined the expression of EMT-related genes. Both vimentin and ZEB1 levels were significantly increased by HIF-1α overexpression (P = 0.0144, 0.0282, respectively) (Figure 4E).
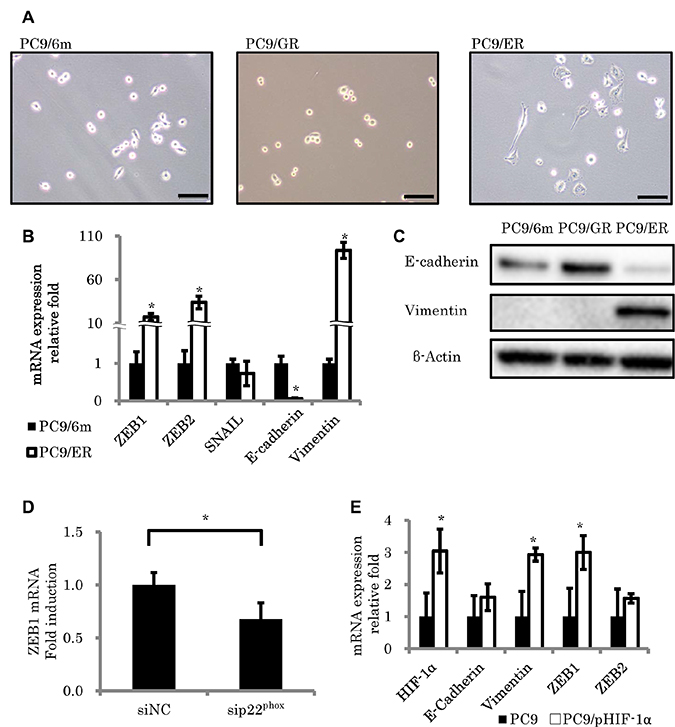
Figure 4: Morphologic changes consistent with epithelial to mesenchymal transition (EMT) in EGFR-TKI resistant lung adenocarcinoma cell lines. (A) Morphological features of control cells (PC9/6m) and EGFR-TKI resistant cell lines (PC9/GR, PC9/ER). PC9/ER demonstrated spindle cell appearance, which was consistent with EMT. (B, C) Expression of EMT-related factors. mRNA levels (N = 3) (B) and protein expressions (C) in EGFR-TKI resistant cells. (D) ZEB1 expression was significantly decreased by knockdown of p22phox in PC9/ER. (E) The effect of HIF-1α overexpression on EMT-related gene expressions in PC9. Both vimentin and ZEB1 levels were significantly increased in stable clones with high HIF-1α expression (PC9/pHIF-1α). The significance of difference between indicated groups are calculated by Student’s t-test (*P < 0.05).
Upregulation of p22phox expression in EGFR-TKI resistant LUAD tissue
We immunolocalized p22phox in clinical specimens of lung cancer in order to clarify the possible association of p22phox with clinicopathological factors of LUAD patients. Representative findings were presented in Figure 5A–5C. The great majority of T790M mutation negative LUAD were immunohistochemically negative for p22phox (Figure 5A) and only a few p22phox weakly positive carcinoma cells (Figure 5B) were detected in nine of 53 cases (Table 1A). No significant correlations between p22phox expression and any of the clinicopathological factors examined in T790M mutation negative 53 cases. A platinum based adjuvant chemotherapy was performed for nine patients, however, the tumor were resected completely in all nine cases. Therefore, we could not examine the association between p22phox expression and chemosensitivity. We additionally examined six cases which harbored T790M mutated EGFR. All of these T790M mutated LUAD cases were markedly immunopositive for p22phox (Figure 5C, Table 1B). These results did indicate the significant association between p22phox and T790M mutation in LUAD. In addition, two cases harboring primary T790M mutated EGFR did demonstrate rather weak E-cadherin immunoreactivity, although these two cases did not necessarily demonstrate the histological features of spindle cells and vimentin immunoreactivity. All four cases harboring acquired T790M mutated EGFR were immunohistochemically positive for vimentin. In addition, three of these four cases had foci of spindle-shaped tumor cells, and two of them were weakly positive for E-cadherin (Supplementary Figure 2). These six cases harboring EGFR-TKI resistance due to T790M mutated EGFR and p22phox immunoreactivities were therefore considered at the stage of progressing to EMT.
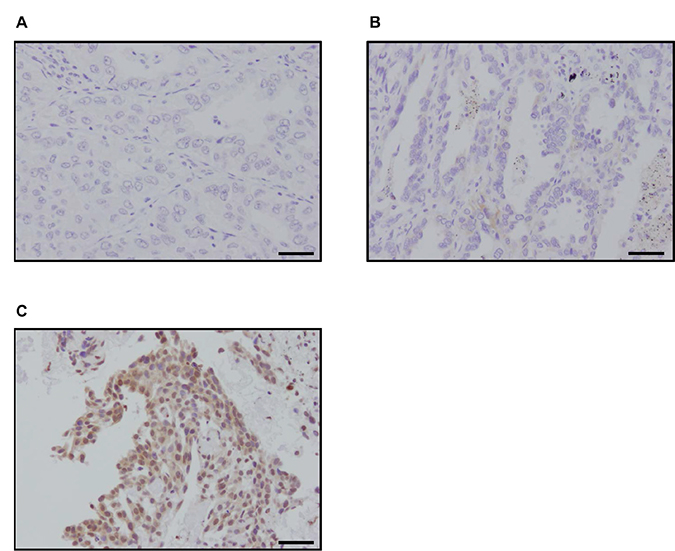
Figure 5: Immunohistochemistry of p22phox in patients with lung adenocarcinoma. (A–C) Representative findings of p22phox immunohistochemistry in patients with lung adenocarcinoma (bar: 50 μm). p22phox immunoreactivity was detected in the cytoplasm of carcinoma cells: (A) negative (T790M mutation negative), (B) weakly stained (T790M mutation negative), (C) strongly stained (T790M mutation positive).
Table 1:
(A) Association between p22phox immunoreactivity and clinicopathological factors in patients with lung adenocarcinoma (N = 53)
p22phox immunoreactivity |
|||
|---|---|---|---|
negative (N = 44) |
low (N = 9) |
P value |
|
Age |
68.2 ± 1.4 |
69.6 ± 2.5 |
0.692 |
Sex |
0.8662 |
||
Male |
22 |
7 |
|
Female |
22 |
2 |
|
Brinkman Index |
367.0 ± 477.7 |
298.8 ± 343.8 |
0.7025 |
pStage |
0.6091 |
||
I |
30 |
8 |
|
II |
6 |
0 |
|
III |
8 |
1 |
|
pT |
0.3854 |
||
1 |
29 |
4 |
|
2 |
14 |
5 |
|
3 |
1 |
0 |
|
pN |
0.6652 |
||
0 |
33 |
8 |
|
1–3 |
11 |
1 |
|
EGFR mutation |
0.456 |
||
positive |
16 |
5 |
|
negative |
28 |
4 |
|
Data are presented as average ± standard deviation, *P value < 0.05 significant. pT; pathological T factor; pN: pathological N factor; pStage: pathological Stage.
(B) Association between p22phox immunoreactivity and EGFR T790M mutation in lung adenocarcinoma patients harboring EGFR mutation (N =27)
p22phox immunoreactivity |
||||
|---|---|---|---|---|
negative (N = 16) |
low (N = 5) |
high (N = 6) |
P value |
|
T790M mutation |
< 0.001 |
|||
positive (N = 6) |
0 |
0 |
6 |
|
negative (N = 21) |
16 |
5 |
0 |
|
Data are presented as 0% (negative); 1–50% (low); 50% < (high), *P value < 0.05 significant.
DISCUSSION
To the best of our knowledge, this is the first study to evaluate the mechanisms of chemotherapy resistance induced as a result of acquired resistance to EGFR-TKI. Results of our present study did clearly demonstrate that p22phox influenced the status of chemosensitivity in EGFR-TKI resistant LUAD cell. In addition, p22phox abundant LUAD cells did acquire the features of chemoresistance via pathways such as HIF-1α and EMT. This study is the first report to fully investigate the significance of p22phox expression and its functions in LUAD acquiring EGFR-TKI resistance following EGFR-TKI therapy.
Results of comprehensive gene expression analysis by microarray also did demonstrate that p22phox was markedly increased in EGFR-TKI-resistant LUAD cell lines, which acquired therapeutic resistance to both EGFR-TKIs and cytotoxic anticancer drugs by long-term exposure of first generation EGFR-TKI (gefitinib and erlotinib). In addition, results of cell viability assays also demonstrated that p22phox knockdown by its corresponding siRNA increased therapeutic sensitivity to CDDP therapy in p22phox high LUAD cell lines. Therefore, these results all indicated that the increased expression of p22phox did contribute to the acquisition or development of chemoresistance in EGFR-TKI-resistant LUAD cells. In addition, the findings that the increased HIF-1α expression and the presence of EMT detected in p22phox abundant LUAD did demonstrate that p22phox could regulate HIF-1α pathway and EMT induction in EGFR-TKI-resistant LUAD, subsequently resulting in the process of acquisition of chemoresistance. These results were also consistent with the results of previous reports that increased p22phox contributed to carcinoma cell survival in some human malignancies. For instance, upregulation of p22phox was reported to activate both PI3K/Akt and ERK1/2 pathways and induce HIF-1α via ROS signaling in prostate cancer [16]; overexpression of p22phox was also reported to cause diminishing the entry of CDDP into the nuclei followed by an inhibition of apoptosis and drug resistance in oral squamous cell carcinoma [20]; EMT was also reported to be induced when p22phox expression was increased by exposure to tobacco smoke extract in NSCLC [17]. However, further comprehensive study to explore the possible correlation between p22phox and EMT is required for further clarification.
The A549 cell line exhibited high chemotherapy resistance and increased expression of p22phox mRNA in the same fashion as EGFR-TKI-resistant LUAD cell lines but p22phox knockdown did not attenuate chemoresistance in A549, in contrast to EGFR-TKI-resistant LUAD cell lines. This could be due to the difference of oncogene status. In detail, both EGFR-TKI-resistant LUAD cell lines, PC9/ER and H1975, had the common EGFR gene mutation (T790M and L858R mutation) [21], but A549 had wild-type EGFR. Therefore, some different mechanisms of chemoresistance in addition to p22phox pathway or EGFR gene mutation were reasonably postulated to occur in A549 but it awaits further investigations for clarification.
It was reported that when T790M mutation occurred in EGFR, it formed a complex with NOX [18, 22], which could then contribute to increased its expression and stability of p22phox protein but the association between EGFR pathway or minor mutations and p22phox may be required for confirmation.
We also immunolocalized p22phox in LUAD without EGFR-TKI treatment and T790M mutation. The detailed status of p22phox has not been studied in human LUAD. However, p22phox was reported to be more abundant in non-small cell lung carcinoma cells following the exposure to tobacco smoke extract [17], stress signal inducers such as smoking were considered to influence its expression. However further investigations are required to clarify the mechanisms of p22phox induction in lung cancer. In our present study, p22phox was expressed in only a few LUAD patients before acquisition of EGFR-TKI resistance and T790M mutation. In addition, even if p22phox was positive in these cases, the percentage of p22phox positive cells in the tumor cells was extremely low and the intensity was really weak. Therefore, the expressions of p22phox in naive LUAD tumor cells are relatively low. In addition, there were no significant differences of p22phox immunoreactivity between EGFR mutation positive and negative cases examined. As discussed above, other factors were considered to be associated with p22phox expression in naive LUAD. However, EGFR-TKI resistance or T790M mutation in carcinoma cells itself could induce p22phox and the status of p22phox in LUAD tissues without T790M mutation was by no means correlated with any of the clinicopathological factors examined. On the other hand, p22phox was significantly elevated in LUAD patients harboring T790M mutation, and these six cases demonstrated partial EMT status. Results of both in vivo and in vitro studies did indicate that p22phox expression could be correlated with development of chemoresistance and EMT in EGFR-TKI resistant LUAD possibly through T790M mutation. However, to our regret, the clinical specimens associated with EGFR-TKI resistant LUAD were not necessarily available in sufficient quantity in our present study. Therefore the correlation between T790M mutation and the potential roles of p22phox should be clarified by further studies.
In summary, this is the first study to demonstrate how chemoresistance could be induced in EGFR-TKI resistant LUAD in detail. Results did demonstrate that p22phox was an important factor influencing chemoresistance in EGFR-TKI resistant LUAD. p22phox also contributed to the development of chemoresistance partially via HIF-1α and EMT. These results will provide new insights into the development of therapeutic strategy for the patients with EGFR-TKI–resistant LUAD, for example, treatment targeting p22phox could possibly attenuate the chemotherapy resistance of LUAD which acquired resistance to EGFR-TKI.
MATERIALS AND METHODS
Reagents and antibodies
The following materials were commercially obtained: Gefitinib (Biaffin, Kassel, Germany); Erlotinib (kindly provided from Roche Diagnostics, Mannheim, Germany); Cisplatin (Wako Pure Chemical Industries, Osaka, Japan); Pemetrexed (Wako Pure Chemical Industries); TGF-β1 (PeproTech, Rocky Hill, USA).
Antibodies were obtained from the following sources: p22phox [E-11] (Santa Cruz Biotechnology, Santa Cruz, CA, USA); HIF-1α [h1alpha67] (Abcam, Cambridge, UK), E-cadherin [4A2C7] (Thermo Fisher Scientific, Rockford, USA), Vimentin [V9] (Acris, San Diego, USA), β-actin [AC-15] (Sigma-Aldrich, St. Louis, MO, USA).
Cell culture
Human LUAD cell lines used in this study are as follows: PC9 (Riken Cell Bank, Tsukuba, Japan), NCI-H1975 (H1975) and A549 (American Type Cell Culture Collection, Manassas, VA, USA) and PC9/6m, PC9/ER, and PC9/GR were established in our laboratory previously [23]. PC9 (Immuno-biological Laboratories, Gunma, Japan) was exposed to increasing concentrations of gefitinib and erlotinib to generate the gefitinib- and erlotinib-resistant cell lines (PC9/GR and PC9/ER, respectively). These doses were gradually increased to 10 nM (2 months), 1 μM (2 months), and 5 μM (2 months). PC9 cells were also cultured for 6 months (PC9/6m) in regular medium to eliminate the effects of long-term cell culture. These cells have EGFR mutation as follows: PC9, PC9/6m, and PC9/GR: exon 19 deletion; PC9/ER: exon 19 deletion, L858R mutation, and T790M mutation.
These cells were maintained in Roswell Park Memorial Institute media (RPMI) 1640 (Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS) (Nichirei, Tokyo, Japan) and 1% penicillin/streptomycin at 37° C in a humidified incubator containing 5% CO2.
Microarray
Total RNA was extracted with care from PC9/6m, PC9/GR, and PC9/ER as described below. Next, 44 K Whole Human genome arrays (G4112F) (Agilent Technologies, Santa Clara, CA, USA) were prepared and hybridized with linearly amplified and labeled total RNA, according to the manufacturer’s protocol. Complementary RNA (cRNA) probes were labeled using a Low Input Linear Amplification and Labeling Kit (Agilent Technologies). Fluorescently labeled probes were purified using an RNeasy Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. Results were extracted using Feature Extraction software (Agilent Technologies) and were analyzed using Genespring GX11 software (Agilent Technologies) to obtain gene expression ratios.
Cell viability assays
Two and/or three-day cell viability assays were carried out by plating 3000 cells per well of each cell lines into 96-well plates. The following day, each cell was treated with each drug across a 4-dose range from 1 nM to 10 μM. After 48h or 72 h of drug treatment, cell viability was measured using WST-8 Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto, Japan).
Real-Time RT-PCR
Total RNA was extracted carefully from cultured cells using TRI reagent (Molecular Research Center, Cincinnati, OH, USA) and was reverse transcribed to cDNA using a QuantiTect Reverse Transcription Kit (Qiagen). Levels of mRNA expression were semiquantified by performing real-time RT-PCR in a LightCycler System (Roche Diagnostics). The PCR mixture (20 μL) included 0.5 μM of p22phox primer or 1 μM of ribosomal protein L13a (RPL13A) primer and 2× QuantiTect SYBR Green PCR Master Mix (Qiagen). PCR protocol was as follows: initial denaturation at 95° C for 5 min, followed by 40 amplification cycles of 95° C for 10 s and annealing at 60° C for 30 s. The primers used for PCR were as follows: p22phox forward, 5′-CGCAGATCGGAGGCCACCATCAA-3′; p22phox reverse, 5′GCGAGGTCACACGACCTCGTCG3′; HIF1A forward, 5′-TGACCAGCAACTTGAGGAAGTACCATTAT-3′; HIF1A reverse, 5′-GGTGGGTAATGGAGACATTGCCAAATTT-3′; CDH1 forward, 5′-GCCTCCTGAAAAGAGAGTGGAAG-3′; CDH1 reverse, 5′-TGGCAGTGTCTCTCCAAATCCG-3′; VIM forward, 5′-TCAGAATATGAAGGAGGAAATGGC-3′; VIM reverse, 5′-GAGTGGGTATCAACCAGAGGGA GT-3′; ZEB1 forward, 5′-GCACCTGAAGAGGACCAG AG-3′; ZEB1 reverse, 5′-GTGTAACTGCACAGGGAG CA-3′; ZEB2 forward, 5′-TTCCTGGGCTACGACCAT AC-3′; ZEB2 reverse, 5′-GCCTTGAGTGCTCGATA AGG-3′; SNAIL1 forward 5′-AGCCTGGGTGCCCTCAA GATG-3′; SNAIL1 reverse 5′-CTTGGTGCTTGTGGAG CAGGGAC-3′; RPL13A forward, 5ʹ-CCTGGAGGAGAA GAGGAAAG-3ʹ; and RPL13A reverse, 5ʹ-TTGAGGACC TCTGTGTATTT-3ʹ. mRNA levels of each gene were expressed as the ratio of RPL13A mRNA levels.
Western blotting
Total protein was extracted using M-PER Mammalian Protein Extraction Reagent (Thermo Fisher Scientific) from cultured cells. Following measurement of protein concentration (Protein Assay Rapid Kit Wako; Wako Pure Chemical Industries, Osaka, Japan), the total protein was individually subjected to SDS-PAGE (SuperSep Ace; Wako, Japan). These proteins were transferred onto Hybond P polyvinylidene difluoride membrane (GE Healthcare, Buckinghamshire, UK). Then the proteins on the membrane were blocked in 5% nonfat dry skim milk powder (Wako) for over 1 h at room temperature and were incubated with primary antibodies for 24 h at 4° C using Immuno Shot (Cosmo Bio, Tokyo, Japan). The dilutions of primary antibodies used in this study were as follows: p22phox, 1:100;HIF-1α,1:200; E-Cadherin, 1:200; Vimentin, 1:200; β-actin, 1:1000. These antibody–protein complexes on the blot were detected using ECL-plus Western blotting detection reagents (GE Healthcare) following incubation with anti-rabbit or anti-mouse IgG horseradish peroxidase (GE Healthcare) at room temperature.
RNA interference
siRNA targeting p22phox used in this study was purchased from GeneDesign, Inc (Osaka, Japan). : sip22phox sense, 5ʹ- UAGUAAUUCCUGGUAAAGGTT-3ʹ; sip22phox anti-sense, 5ʹ- CCUUUACCAGGAAU UACUATT-3ʹ; siHIF-1α sense, 5′- GCCACUUCGA AGUAGUGCUTT-3′; and siHIF-1α antisense, 5′- AGCACUACUUCGAAGUGGCTT-3′. Silencer Select Negative Control 1 siRNA (Ambion, Austin, TX, USA) served as a negative control (siNC). siRNA 5 nM was transfected into cells (1 × 105 cells/mL) using Lipofectamine RNAi MAX reagent (Invitrogen, Carlsbad, CA, USA) for 48 or 72 h. Knockdown efficiency was assessed by RT–PCR or western blotting.
Plasmid transfection
HIF-1α expression construct was obtained by PCR amplification of the HIF-1α coding sequence using primer sequences: forward, 5′-AAAGCTAGCATTCACCATGGAGGGCGCC-3′ and reserve, 5′-AAAGGTACCCTCAGTTAACTTGATCCAAAGC-3′. The PCR fragments were digested with KpnI and NheI, inserted into KpnI/NheI site of pcDNA3.0 (-) vector (Thermo Fisher Scientific) and then sequenced. The PCR products were cloned into KpnI/NheI site of pcDNA3.0 vector, generating pcDNA3.0-HIF-1α construct. Generation of HIF-1α stable lines PC9 cells were seeded into 10 cm culture dishes at a density of 1 × 106 cells per dish. After attachment, the cells were transfected with HIF-1α (10 μg) construct or empty vector using Lipofectamine® 3000 (Thermo Fisher Scientific). At two days after transfection, the cells were re-plated in a 10 cm culture dish with medium containing 0.8 mg/ml G418. At two weeks later, the cells were sorted. The isolated colonies were seeded initially into each well of the 24-well plates, followed by progressive expansion of the cultures in large culture dishes. G418-resistant stable clones were established and maintained in medium with 0.8 mg/ml G418. Stable clones with high HIF-1α expression (PC9/pHIF-1α) were verified by qRT-PCR.
Reactive oxygen species assay
Intracellular ROS generation was measured using DCFDA / H2DCFDA - Cellular Reactive Oxygen Species Detection Assay Kit (Abcam). These assays were carried out by plating 25,000 cells per well of each cells, respectively, into black 96-well plates. The following day, each cells was incubated with DCFDA (25 μM) for 45 minutes at 37° C in the dark. After incubation, fluorescence was measured immediately using GLOMAX MULTI+ Detection system (Promega, Madison, WI).
Patients and tissue preparation
Surgical pathology specimens of 53 lung adenocarcinoma were retrieved from surgical patients from 2014 to 2015 at the department of Thoracic Surgery, Miyagi Cancer Center (Miyagi, Japan). The patients did neither receive chemotherapy nor radiation prior to surgery. In addition, we used six patients with lung adenocarcinoma having T790M mutation, of which four acquired EGFR-TKI resistance and underwent biopsy, and two showed primary T790M mutation in surgically resected specimens without EGFR-TKI treatment. All these six specimens were retrieved in Tohoku University Hospital (Miyagi, Japan) from 2017 to 2018. The study was conducted in accordance with the Declaration of Helsinki. The protocol was approved by the ethics committee at Tohoku University, and all patients provided informed consent.
Immunohistochemistry
We used mouse monoclonal antibody against p22phox [E-11] (Santa Cruz), Vimentin and E-cadherin. Histofine Kit (Nichirei) based on the streptavidin–biotin amplification method was used for p22phox in this study. Antigen retrieval for p22phox was performed using a microwave treatment with 1M citric acid buffer. The primary antibody was diluted by 1:1000. Antigen–antibody complexes were visualized using 3, 3′-diaminobenzidine solution (1 mM DAB, 50 mM Tris–HCl buffer (pH 7.6), and 0.006% H2O2) and counterstaining with hematoxylin. Human tonsil tissue was used as a positive control. Immunohistochemistry (Envision DAKO) was employed to detect the expressions of Vimentin and E-cadherin. The primary antibody was diluted by 1:200 for Vimentin and 1:500 for E-cadherin. Human normal stomach and breast tissue were used for positive control of Vimentin and E-cadherin, respectively.
Immunoreactivity of both p22phox and vimentin was evaluated in cytoplasm [20], and that of E-cadherin in cell membrane. We divided the cases into three groups according to the positive rate of p22phox: 0% (negative), 1-50% (low expression), 50% > (high expression).
Statistical analysis
Statistical analysis was performed using JMP Pro 14.0.0 (SAS Institute, Cary, NC, USA). Statistical differences between the two groups of immunohistochemical analyses were evaluated by t-test or Fisher tests. Statistical significance was defined as p < 0.05 in this study.
Abbreviations
EGFR, epidermal growth factor receptor; LUAD, lung adenocarcinoma; EGFR–TKIs, EGFR–tyrosine kinase inhibitors; T790M, threonine-to-methionine substitution on codon 790; HIF-1α, Hypoxia Inducible Factor-1α; EMT, Epithelial-Mesenchymal Transition; PI3K, phosphoinositide 3-kinase; NADPH, nicotinamide adenine dinucleotide phosphate; NOX, NADPH oxidase; ROS, reactive oxygen species; ERK1/2, extracellular signal-regulated kinase 1/2; CDDP, cisplatin; NSCLC, non-small cell lung cancer; PEM, pemetrexed; ZEB, Zinc finger E-box-binding homeobox.
Author contributions
MK and RS conceived and designed the study; MK performed most of the experiments and drafted the manuscript; RS, RN and CI assisted the experiments; JA, IS and YO provided clinical samples and interpreted results; YM and HS supervised all experiments; RS and HS edited the manuscript. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
The authors thank Mr. Katsuhiko Ono, Dr. Erina Iwabuchi (Tohoku University School of Medicine) and Ms. Kuniko Komuro (Miyagi Cancer Center) for their excellent technical support.
CONFLICTS OF INTEREST
We have no financial relationships to disclose.
FUNDING
This work was supported by JSPS KAKENHI Grant Numbers JP16K19073, JP18K15076.
REFERENCES
1. Shi Y, Au JS, Thongprasert S, Srinivasan S, Tsai CM, Khoa MT, Heeroma K, Itoh Y, Cornelio G, Yang PC. A Prospective, Molecular Epidemiology Study of EGFR Mutations in Asian Patients with Advanced Non–Small-Cell Lung Cancer of Adenocarcinoma Histology (PIONEER). J Thorac Oncol. 2014; 9:154–62. https://doi.org/10.1097/JTO.0000000000000033.
2. Fukuoka M. Multi-Institutional Randomized Phase II Trial of Gefitinib for Previously Treated Patients With Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2003; 21:2237–46. https://doi.org/10.1200/JCO.2003.10.038.
3. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, van Kooten M, Dediu M, Findlay B, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005; 353:123–32. https://doi.org/10.1056/NEJMoa050753.
4. Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib. N Engl J Med. 2005; 352:786–92. https://doi.org/10.1056/NEJMoa044238.
5. Jänne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, Haggstrom D, Felip E, Kim JH, et al. AZD9291 in EGFR Inhibitor–Resistant Non–Small-Cell Lung Cancer. N Engl J Med. 2015; 372:1689–99. https://doi.org/10.1056/NEJMoa1411817.
6. Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, Ercan D, Matthews SE, Cantarini M, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015; 21:560–2. https://doi.org/10.1038/nm.3854.
7. Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II, Yang SH, Kim CH, Lee JC. Epithelial to mesenchymal transition derived from repeated exposure to gefitinib determines the sensitivity to EGFR inhibitors in A549, a non-small cell lung cancer cell line. Lung Cancer. 2009; 63:219–26. https://doi.org/10.1016/j.lungcan.2008.05.017.
8. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, Schwabe RF, Vahdat LT, Altorki NK, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015; 527:472–6. https://doi.org/10.1038/nature15748.
9. Agani F, Jiang BH. Oxygen-independent Regulation of HIF-1: Novel Involvement of PI3K/ AKT/mTOR Pathway in Cancer. Curr Cancer Drug Targets. 2013; 13:245–51. https://doi.org/10.2174/1568009611313030003.
10. Sowa T, Menju T, Chen-Yoshikawa TF, Takahashi K, Nishikawa S, Nakanishi T, Shikuma K, Motoyama H, Hijiya K, Aoyama A, Sato T, Sonobe M, Harada H, et al. Hypoxia-inducible factor 1 promotes chemoresistance of lung cancer by inducing carbonic anhydrase IX expression. Cancer Med. 2017; 6:288–97. https://doi.org/10.1002/cam4.991.
11. Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002; 62:3387–94. https://doi.org/10.1158/0008-5472.can-04-1919.
12. Jianrui S, Zengqiang Q, Xianling G, Qiudong Z, Xue Z, Lu G, Kai S, Feng S, Mengchao W, Lixin W. Hypoxia-induced autophagy contributes to the chemoresistance of hepatocellular carcinoma cells. Autophagy. 2009; 5:1131–44. https://doi.org/10.4161/auto.5.8.9996.
13. Zhang W, Shi X, Peng Y, Wu M, Zhang P, Xie R, Wu Y, Yan Q, Liu S, Wang J. HIF-1α Promotes Epithelial-Mesenchymal Transition and Metastasis through Direct Regulation of ZEB1 in Colorectal Cancer. PLoS One. 2015; 10:e0129603. https://doi.org/10.1371/journal.pone.0129603.
14. Kawahara T, Ritsick D, Cheng G, Lambeth JD. Point mutations in the proline-rich region of p22phox are dominant inhibitors of Nox1- and Nox2-dependent reactive oxygen generation. J Biol Chem. 2005; 280:31859–69. https://doi.org/10.1074/jbc.M501882200.
15. Edderkaoui M, Nitsche C, Zheng L, Pandol SJ, Gukovsky I, Gukovskaya AS. NADPH oxidase activation in pancreatic cancer cells is mediated through Akt-dependent up-regulation of p22phox. J Biol Chem. 2011; 286:7779–87. https://doi.org/10.1074/jbc.M110.200063.
16. Li Q, Fu GB, Zheng JT, He J, Niu XB, Chen QD, Yin Y, Qian X, Xu Q, Wang M, Sun AF, Shu Y, Rui H, et al. NADPH oxidase subunit p22phox-mediated reactive oxygen species contribute to angiogenesis and tumor growth through AKT and ERK1/2 signaling pathways in prostate cancer. Biochim Biophys Acta. 2013; 1833:3375–85. https://doi.org/10.1016/j.bbamcr.2013.09.018.
17. Vaid M, Katiyar SK. Grape seed proanthocyanidins inhibit cigarette smoke condensate-induced lung cancer cell migration through inhibition of NADPH oxidase and reduction in the binding of p22(phox) and p47(phox) proteins. Mol Carcinog. 2015; 54:E61–71. https://doi.org/10.1002/mc.22173.
18. Leung ELH, Fan XX, Wong MP, Jiang ZH, Liu ZQ, Yao XJ, Lu LL, Zhou YL, Yau LF, Tin VPC, Liu L. Targeting Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer by Inducing Epidermal Growth Factor Receptor Degradation via Methionine 790 Oxidation. Antioxid Redox Signal. 2016; 24:263–79. https://doi.org/10.1089/ars.2015.6420.
19. Song X, Liu X, Chi W, Liu Y, Wei L, Wang X, Yu J. Hypoxia-induced resistance to cisplatin and doxorubicin in non-small cell lung cancer is inhibited by silencing of HIF-1α gene. Cancer Chemother Pharmacol. 2006; 58:776–84. https://doi.org/10.1007/s00280-006-0224-7.
20. Hung CC, Chien CY, Chiang WF, Lin CS, Hour TC, Chen HR, Wang LF, Ko JY, Chang CH, Chen JY. P22Phox Confers Resistance To Cisplatin, By Blocking Its Entry Into the Nucleus. Oncotarget. 2015; 6:4110–25. https://doi.org/10.18632/oncotarget.2893.
21. Cross DAE, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ, Hughes G, Rahi A, Jacobs VN, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014; 4:1046–61. https://doi.org/10.1158/2159-8290.CD-14-0337.
22. Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat Chem Biol. 2012; 8:57–64. https://doi.org/10.1038/nchembio.736.
23. Nihira K, Miki Y, Iida S, Narumi S, Ono K, Iwabuchi E, Ise K, Mori K, Saito M, Ebina M, Sato I, Maemondo M, Yamada-Okabe H, et al. An activation of LC3A-mediated autophagy contributes to de novo and acquired resistance to EGFR tyrosine kinase inhibitors in lung adenocarcinoma. J Pathol. 2014; 234:277–88. https://doi.org/10.1002/path.4354.