
INTRODUCTION
When solid tumors grow, the oxygen demand increases rapidly while there is insufficient vascularization. This causes the tumor to become hypoxic at the tumor core and the edges of the invasive front [1]. Due to the lack of oxygen, ATP is mostly produced via a high rate of anaerobic glycolysis; this is called the Warburg effect. The Warburg effect leads to lactic acid fermentation in the cytosol and increased oxidative stress in the form of H2O2 and other radicals. H2O2 activates the transcription factor Nrf2 which further upregulates glycolysis-related genes and further contributes to the Warburg effect [2, 3]. ROS is important for tumor growth via the kinase AMPK. AMPK can suppress cell proliferation via cell cycle arrest [4] and its activation depends on reduction of cysteine residues by thioredoxin (Trx) at the catalytic subunit alpha [5, 6]. However, these sites can be oxidized by ROS to inactivate AMPK, promoting tumor cell proliferation. The increase in oxidative stress also translates to the tumor microenvironment (TME). The TME comprises the tumor cells itself and the stromal cell compartment directly surrounding it, containing blood vessels, cells from the immune system, fibroblasts and extracellular matrix [7, 8].
In cancer cells, there are multiple sources of ROS (Figure 1). Oncogenic mutations (e.g., in Ras, Myc and p53) can cause mitochondrial dysfunction and increased leakage of ROS. This leakage occurs mostly at complex 1 or complex 3 of the respiratory chain, where electrons from NAD(P)H or FADH2 react with oxygen to form superoxide anion. Second, ROS is formed enzymatically by NAD(P)H oxidases (NOX), which can be activated by various growth factors, such as VEGF and angiopoietin which are often upregulated in cancer [9, 10]. Third, exogenous radiation (e.g., UV light, radiotherapy) can cause the production of ROS [11]. Fourth, ROS are by-products of cellular metabolism such as from protein folding and beta-oxidation of fatty acids in the mitochondria and peroxisomes [2, 12]. Last, intracellular enzymes, such as xanthine oxidase, cyclooxygenases, cytochrome p450 enzymes, lipoxygenases and nitric oxide synthetase, generate ROS as a metabolic byproduct [11, 13]. Tumor cells thus have to cope with comparatively high intracellular levels of ROS and they do this by upregulating antioxidative systems such as Trx, peroxiredoxin (Prx), catalase, superoxide dismutases (SOD) and generation of NAD(P)H [13]. Against superoxide anion specifically, dismutation to H2O2 is the primary protective mechanism and this can occur enzymatically by SOD or spontaneously in acidic environments. Subsequently, H2O2 can then be degraded into water and oxygen by catalase [14].
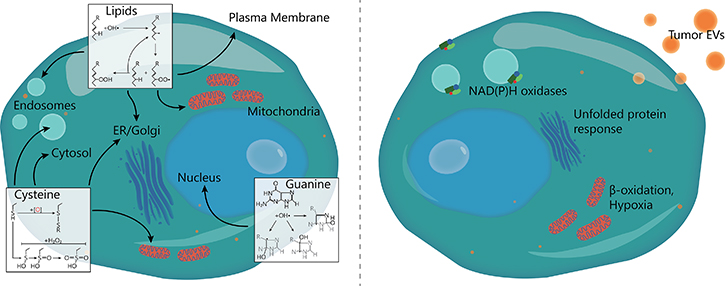
Figure 1: Targets and sources of ROS in DCs. (Left) ROS can attack both mono- and poly-unsaturated lipids in membranes, causing endosomal leakage and loss of pH and electron gradients. Cysteine residues on proteins can oxidize, resulting in the formation of disulfide bridges or a stepwise oxidation to sulfonic acid. This can activate redox-sensitive signaling factors, but also block enzymatic activity or cause protein misfolding. Finally, both free and DNA-helix incorporated guanine nucleotides can oxidize, leading to GC-TA or GC-CG transversion mutations. (Right) Sources of ROS for DCs in the TME: increased NOX (NAD(P)H oxidase) activity, ER stress due to the unfolded protein response, β-oxidation of fatty acids, abrogated electron transfer in mitochondria and uptake of ROS-containing tumor-derived EVs.
In addition to cancer cells, ROS-producing immune cells of myeloid origin are present in the TME [15]. These include DCs (see below), but are mainly macrophages that have differentiated from circulating monocytes [16] and, depending on the stimulus, can develop towards more cytotoxic or immunosuppressive phenotypes [17]. However, tumor-associated macrophages in the TME are usually immunosuppressive, as they produce IL-10 and transforming growth factor β [18] and recruit regulatory T cells via CCL22 [19]. ROS produced by these cells were found to be detrimental for cancer progression, as increased H2O2 secretion by macrophages was shown to be a sufficient trigger for both tumor initiation and development of epithelial cancer [20]. This was further exacerbated by H2O2-mediated induction of TNF-α and TNF receptor 1 transcription, which lead to recruitment of more H2O2-secreting macrophages. Additionally, ROS production by the monocyte precursors of these macrophages was shown to be a strong determinant for development towards an immunosuppressive phenotype [21].
Hypoxia and oxidative stress influence multiple functions of the cancer cells, such as angiogenesis, cell proliferation and apoptosis, and thereby can promote tumorigenesis. H2O2 promotes angiogenesis by activating the transcription factors NF-κB and AP-1, leading to activation of VEGF transcription factors NF-κB and AP1 [22], VEGF secretion [23] as well as VEGF receptor 2 transcription [24]. In addition, H2O2 can also activate VEGF receptor 2 in a ligand-independent manner via Src kinases [25]. VEGF activates NOX and thereby leads to the generation of more ROS, forming a positive feedback loop, and making NOX a potential therapeutic target for inhibition of angiogenesis [2, 9]. A major mechanism by which ROS affects physiological processes is by the formation of disulfide bonds. For example, H2O2 modifies the thiolates of cysteine residues in redox sensitive proteins [13] and particularly zinc-bound cysteines perform oxidative stress sensing [26]. Zinc-bound cysteines are present in zinc finger transcription factors, for example the GATA family of transcription factors and Krüppel-like Factor 2, both of which are involved in ROS-mediated signaling pathways [27–30]. Lastly, ROS influence apoptosis induced by the dimerization of the kinase ASK1 with TRAF2, which causes apoptosis via activation of the kinases JNK and p38 MAPK. This dimerization is induced by H2O2, but blocked by thioredoxin (Trx) [31, 32]. Trx is bound to ASK1 by a disulfide bond at the N-terminal domain of ASK1, leading to ubiquitination and proteosomal degradation of ASK1. High levels of H2O2 counteract the functioning of Trx and cause the release of Trx from ASK1, because of the formation of an intramolecular disulfide bond [33].
It is not completely understood how hypoxia and ROS in the TME influence infiltrating immune cells, which is the focus of this review. Especially DCs play a major role in generating anti-tumor immunity, due to their ability to activate naive T cells. After encountering an antigen, DCs can maturate and migrate to the lymph nodes to present processed antigens to T cells. The ability of DCs to (cross-)present antigens and activate T cells is influenced by the inflammatory environments that the cells encounter [34]. Since DCs encounter a variety of environments that differ in oxygen tension and ROS levels during antigen uptake and migration to the lymph nodes, it is likely that these environments affect the final immune response. The aim of this review is to provide a comprehensive overview of the effects of TME-associated oxidative stress on DCs.
ROS entry in DCs
Solid tumors are frequently invaded by DCs and other immune cells, which hence are exposed to the hypoxia and radicals in the TME. ROS may affect the plasma membrane composition of invading immune cells through oxidation of both the lipid bilayer and of membrane proteins [35]. However, to affect intracellular processes, ROS have to traverse the plasma membrane. Many species of ROS, such as superoxide anion, carry a free electron and cannot efficiently traverse the apolar lipid bilayers. However, either spontaneously or catalyzed by the abundant SODs, superoxide anion can dismutate to H2O2 which is uncharged and does not carry a free electron. H2O2 has a lower reactivity compared to ROS species such as superoxide anion and hydroxyl radicals and this makes H2O2 relatively stable and also allows it to diffuse through membranes and to enter the nucleus to cause DNA damage [11]. Its relatively high stability even allows H2O2 to signal between different cells [13]. These properties allow H2O2 to increase the redox potential of the TME. H2O2 cannot only passively diffuse through lipid membranes, but also enter cells through the aquaporin channel AQP8, as shown by heterologous expression in yeast [36], and through AQP1, 3 and 8 in a human leukemia cell line [37]. Both immature and monocyte-derived dendritic cells express AQP3, 7 and 9 and mature DCs express AQP7 and 9 [38, 39], suggesting that H2O2 can enter DCs via these channels. Other aquaporins might be involved as well, as the homologs AQP7, AQP9, AQP10, AQP12A and AQP12B are all expressed by human immune cells [40]. Superoxide anion can enter endothelial cells by diffusion through the Cl- channel-3 (Clc3) [41] and might also enter immune cells via this channel, as it is expressed in human macrophages and peripheral blood mononuclear cells [42, 43]. However, there is no experimental evidence yet that these channels mediate entry of H2O2 and superoxide anion in DCs.
ROS affect DC maturation
ROS are directly implicated in DC maturation by the activation of p38-MAPK and ERK1/2. DCs treated with 1-fluoro-2,4-dinitrobenzene, a skin sensitizer which is perceived as a danger signal by DCs, showed increased ROS production and activation of p38-MAPK via an unknown mechanism [44]. In line with ROS promoting DC activation, H2O2-treated human peripheral blood DCs were more efficient in promoting T cell proliferation and showed an upregulation of cell surface molecules MHC-II, CD40 and CD86, all important components of T cell activation [45]. The mechanism by which ROS promote DC maturation is not known.
ROS can also reduce DC maturation via ER stress. Danger signals such as 1-fluoro-2,4-dinitrobenzene cause accumulation of misfolded proteins in the cell, leading to ER stress and an increase in mitochondrial ROS production [44]. Oxidative stress can also affect ER function by disturbing Ca2+ homeostasis [44]. The accumulation of misfolded proteins activates the unfolded protein response, aimed at restoring normal cell function by halting translation, degradation of misfolded proteins, and increasing expression of chaperone proteins. This study observed that ROS affected the PERK-eIF2α-ATF4 axis of the unfolded protein response, which led to a short-term block of CD86, IL-1β and IL-12B expression in THP-1 monocytes. However, these genes were upregulated at later time points, indicating a pro-inflammatory response. ER stress is also caused by lipid peroxidation products that follow increased ROS production, such as 4-hydroxynonenal (4-HNE). This aldehyde readily forms protein adducts due to its reactivity with thiols and amine groups [46], which can trigger the unfolded protein response. Additionally, 4-HNE was shown to form adducts with ER-resident chaperone proteins [47], leading to increased activation of ER stress transcription factor XBP1 in ovarian cancer-associated DCs [48]. This in turn inhibited anti-tumor immunity via accumulation of lipid bodies in the DC, which blocks translocation of MHC-I to the cell surface [48, 49]. As 4-HNE is relatively stable and able to diffuse through membranes, DCs may even internalize 4-HNE from the TME [50, 51]. Likewise, malondialdehyde, another common lipid peroxidation product, also forms protein adducts which are shown to be strongly auto-immunogenic, which may hamper specific anti-tumor responses [52–54].
Effects of ROS on antigen presentation
ROS influence the ability of DCs to cross-present antigens to CD8+ cells [55]. Upon activation of Toll-like receptors (TLRs), NOX2 is activated in the DCs and produces large amounts of superoxide anion in endo/phagosomes [56–58]. This increases the endo/phagosomal pH through the consumption of protons by the dismutation of superoxide anion to H2O2 [55, 56]. The increased pH impacts cross-presentation through antigen conservation, as lysosomal hydrolases with acidic pH optima are less activated [55]. In addition, ROS can affect the activity of the endosomal V-ATPase by formation of a disulphide bond between cysteine residues located within the nucleotide-binding subunits, leading to its inactivation and reduced acidification of the endosomal lumen [56, 59]. Endo/phagosomal proteases like cathepsins are modified in a similar fashion, leading to altered epitopes for both MHC-I and MHC-II [60–62]. ROS can also induce the release of antigen from phagosomes into the cytosol by causing leakage of antigens through lipid peroxidation of the endo/phagosomal membranes, and this can promote antigen cross-presentation [57, 63].
However, the effect on antigen presentation depends on the cellular site of ROS production, as a study in aged mice suggested an inhibitory role for mitochondrial ROS in cross-presentation by bone marrow-derived DCs [64]. DCs from aged mice (16–20 months) show signs of mitochondrial dysfunction, leading to increased ROS production compared to DCs from young mice (2–3 months). Scavenging ROS partially improved the cross-presentation efficiency of the DCs from aged mice [64]. This finding indicates that, although DCs actively use endo/phagosomal ROS to enable cross-presentation, a general increase in the environmental redox potential could also hamper cross-presentation.
Extracellular vesicle release by tumor cells
A recently identified source of ROS in the TME are EVs released by tumor cells. EVs contain many different compounds, including ROS as shown by flow cytometry with a fluorescent ROS probe [58]. ROS was also found in EVs derived from hypoxic/reoxygenated human umbilical vein endothelial cells [65]. However, the source of EVs in the TME is still controversial, and it is debated whether cancer cells can dictate their content or they are only membrane blebs or necrotic cell bodies [66]. There is some evidence for controlled release of EVs in a process involving the endosomal sorting complex [67]. This complex generates the intraluminal vesicles of multivesicular bodies by bulging the membrane inwards onto the lumen of late endosomes. When multivesicular bodies fuse with the plasma membrane, these intraluminal vesicles are released as EVs [68, 69]. However, other mechanisms of EV formation have also been proposed, such as direct shedding from the plasma membrane [70, 71]. EVs of intracellular origin are often called exosomes, while EVs shed from the plasma membrane are called microvesicles. However, this nomenclature and the methods to discriminate between various sources of EVs are not yet standardized, therefore we will use the general term EVs in this review [66].
Several studies showed that hypoxia increases the release of EVs by various types of tumor cells, thereby suppressing the immune response [72, 73]. In a study on breast cancer cells, it was found that the transcription factor HIF-1α is responsible for this increase in EV production, since the release of EVs was blocked upon silencing of HIF-1α [73]. A second study on breast cancer cells came to a similar conclusion, and found that cells exposed to hypoxia overexpressed the small GTPase RAB22A in a HIF dependent manner, leading to increased formation of EVs in breast cancer [74]. The cargo of EVs is possibly influenced by hypoxia as well, as the level of the micro-RNA miR-210 is elevated in EVs upon hypoxia [73]. Transcription of miR-210 is mediated by HIFs and it has target genes involved in cell survival, angiogenesis and metabolism [75].
Effects of extracellular vesicles on DCs
EVs influence immune cells and for instance may control macrophage differentiation towards an immunosuppressive phenotype [76], but also might exert effects on DC function. Multiple mechanisms are suggested for the uptake of EVs by recipient cells: fusion, receptor-ligand interactions and endocytosis. The uptake of EVs by DCs is mediated by several factors, including CD11a, intracellular adhesion molecule 1, phosphatidylserine and milk fat globule E8 on DCs, and tetraspanins CD9 and CD81 on EVs [77]. Glycosylation is also involved in targeting EVs, as uptake of EVs derived from Jurkat T cells by myeloid DCs was inhibited by blocking Siglec-1 [78]. Siglec-1 preferentially binds to α2,3-linked sialic acids which decorate proteins on the surface of EVs. Furthermore, EV uptake by DCs involves interaction between LFA-1 and C-type lectin receptors like DEC205 on DCs with CD54 or various glycoproteins on the EVs [79].
After uptake, the ROS present in EVs might affect DC function. EVs derived from ovarian cancer cells promote antigen cross-presentation in DCs via ROS-mediated phagosomal alkalization [58], although in this study the effects of other EV components cannot be completely excluded. Moreover, it is unclear whether EV-derived ROS are directly responsible for ROS accumulation inside the phagosomes, or whether this is the result of ROS producing enzymes carried by the EVs [80] or other ROS-inducing components [81]. Besides changing the phagosomal pH, it is proposed that EVs induce antigen-specific tolerance in DCs. Tumor EVs contain antigens and EVs taken up by immature DCs are shown to inhibit the maturation of DCs [82]. In this study, EVs were internalized by mouse CD11c+ cells, which subsequently downregulated expression of the maturation markers MHC-II and CD86, while levels of the anti-inflammatory cytokine transforming growth factor β1 were elevated in these DCs. This steered CD4+ T cells towards a regulatory phenotype, capable of suppressing B cell responses. The uptake of EVs by DCs can however also promote CD8+ T cell responses, benefitting antitumor immunity, because mature DCs pulsed with EVs expressed MHC-I, MHC-II, CD40, CD54 and CD80 at higher levels than controls, while immature DCs pulsed with EVs did not respond [79]. In fact, mice bearing BL6-10OVA tumor cells were able to clear their tumors following adoptive transfer with mature DCs pulsed with OVA-containing EVs, while DCs pulsed with soluble OVA only reduced tumor growth temporarily [79]. These EVs might potentially contain ROS and tumor material which induces ROS and danger-associated molecular pathogen signaling via pattern recognition receptors. This would explain the more efficient cross-presentation of EV-derived antigens compared to soluble antigens [58], as ROS are a major factor in upregulating cross-presentation [55, 57, 60, 61, 83]. However, the effects of EV-containing ROS on antigen presentation are difficult to discern from the effects of other EV components, such as micro-RNAs.
The molecular mechanisms by which EVs affect DC maturation and the role of EV-encapsulated ROS in this process are still largely unknown. In murine CD11b+ myeloid DC precursors, tumor EVs inhibited the differentiation of DCs via enhanced expression of interleukin 6 (IL-6) [84]. After EV uptake, CD11c expression was significantly lowered and IL-6 expression was higher than in the control cultures. Precursor DCs were able to differentiate in the CD11c+ phenotype, but these DCs were less able to mature as measured by analysis of the expression of the co-stimulatory molecules CD86 and CD80. The expression of CD86 and CD80 was significantly lower and correlated with the concentration of EVs added to the culture [84]. When DC precursors were isolated from IL-6 knockout mice, DC maturation was not inhibited, suggesting that the inhibition is mediated by IL-6. The authors observed similar effects on differentiation of human CD14+ monocytes into monocyte-derived DCs after stimulation with isolated EVs from the MDA-MB-231 breast cancer cell line. In conclusion, several immunologically relevant effects of EVs have been described, many of which are likely to depend at least partly on EVs containing ROS. However, the effects of other compounds present in EVs cannot be excluded and this requires further investigation.
The TME is hypoxic
As mentioned above, a major cause for the generation of ROS in the TME is a disturbed metabolism of the cancer cells. Since ROS generation consumes molecular oxygen, this increases the hypoxic conditions caused by the poor vascularization frequently associated with tumors [85]. A major signaling component downstream of hypoxia is the HIF family of transcription factors, consisting of HIF-1α, HIF-2α and HIF-1β. At normoxic conditions, the alpha subunit is targeted for degradation via hydroxylation by prolyl hydroxylase domain enzymes and factor inhibiting HIF-1α. Upon hydroxylation, HIF-1α and HIF-2α bind to the von Hippel–Lindau tumor-suppressor protein, allowing ubiquitination and ultimately proteasomal degradation [86–88]. The hydroxylation reaction requires oxygen and therefore cannot efficiently occur under hypoxic conditions, resulting in the accumulation of the HIF-1α and/or HIF-2α subunits, allowing them to dimerize with HIF-1β and translocate to the nucleus. Here, the heterodimer binds to the hypoxia responsive element (HRE) in the promotor region of target genes [89–91]. Many of the HIF target genes are involved in angiogenesis and in erythropoiesis, and HIF also promotes glycolysis by upregulating expression of plasma membrane glucose transporters and inhibiting pyruvate dehydrogenase kinase, which blocks the translocation of pyruvate to the tricarboxylic acid cycle [92, 93].
Hypoxia alters DC differentiation and maturation
The effects of hypoxia on the differentiation and maturation of DCs are quite well studied, although there is little consensus between studies. For example, expression of MHC-II is mostly reported to decrease in hypoxic environments [94–99], but the opposite [100] or no effect [101, 102] have been reported as well. The same holds for DC maturation markers like CD80, CD83 or CD86, where several studies found no effects [99, 101, 102], but upregulation [100] and downregulation [95–97, 103] were reported as well. Hypoxic alteration of MHC-I expression is less well studied, however HIF-1α activity is implied in upregulating MHC-I expression [104]. These contradicting results likely arise from the complex interplay of ROS and hypoxia signaling with immune cell activation pathways. In particular, hypoxia is capable of altering TLR signaling [105] and subsequently leads to altered cytokine secretion patterns [95, 100]. Expression of TLR4 [106] and its downstream kinase MAP3K8 (also known as Cot or TPL-2) are upregulated by hypoxia [90, 107]. This leads to an hypoxic potentiation of TLR4-mediated secretion of TNF-α in human monocyte-derived DCs [107]. In line with this, hypoxia has also been found to increase secretion of other pro-inflammatory cytokines such as IL-6, IL-8 and IL-1β in primary human macrophages and osteoclasts [95, 108–110]. So, while hypoxia itself does not cause ROS formation, it can trigger inflammation which in turn promotes ROS formation. Since ROS formation consumes oxygen, it causes additional hypoxia, resulting in a feedback loop.
Hypoxia skews immature DCs towards a highly mobile phenotype by upregulating genes involved in cell motility [111]. The chemokine receptors CCR2, CCR3, CCR5, CX3CR1, C5R1 and FPR3 are upregulated upon hypoxia, while expression of chemokines CCL26, CCL24 and CCL14 are inhibited by hypoxia [98, 112]. This suggests that immature DCs differentiated in a hypoxic TME migrate away towards normoxic tissues, potentially suppressing anti-tumor immunity. Moreover, a variety of tumors secrete elevated levels of prostaglandin E2, which is a strong migratory stimulus for immature DCs [112]. Prostaglandin E2 is generated by cyclooxygenases, many of which are HIF target genes [90, 108]. In contrast, mature DCs downregulate expression of CCR7 in hypoxic conditions. CCR7 is the chemokine receptor which signals DCs to migrate to draining lymph nodes in response to lymph node derived chemokines [98]. This observation is in line with the poor chemotactic ability of hypoxic mature DCs in response to the lymph node chemokine MIP-3β. Thus, evidence suggests that the hypoxic TME promotes the expulsion of immature DCs from the tumor, whereas the migration of mature DCs to the lymph nodes is reduced.
The effects of hypoxia on DC maturation might well be transient. Murine DCs cultured under low oxygen tensions expressed lower levels of MHC-II, CD80 and CD86 compared to DCs under normoxic conditions [96]. However, reoxygenation of hypoxia-differentiated DCs restored the levels of these maturation markers, suggesting that once these DCs migrate towards the lymph nodes they can regain full functionality [96]. Finally, hypoxia affects antigen uptake as it decreased the phagocytic capacity of immature DC compared to DCs cultured under normoxic conditions [98]. In conclusion, hypoxia can both increase and decrease DC maturation, likely depending on the presence of ROS and other inflammatory stimuli in the TME. Moreover, hypoxia can promote immune tolerance, as it stimulates migration of immature DCs out of the TME while restricting migration of mature DCs to prevent T cell activation in draining lymph nodes.
Hypoxia skews T helper cell differentiation by DCs
T cell priming is an essential function of DCs and this process is affected by the TME as well. Immature blood monocyte-derived DCs cultured at hypoxic conditions were found to be biased towards a Th2-stimulatory phenotype by switching to secretion of IL-4 instead of IFN-γ [98]. These T cells mostly stimulate a humoral immune response and suppress DC activation via IL-10 secretion. In addition, DCs cultured under hypoxia secreted increased amounts of osteopontin, which strongly promotes tumor cell migration [98]. However, osteopontin might also promote an immune response, as it promotes IFN-α production via TLR9 signaling in plasmacytoid DCs, which upregulates the expression of MHC-I [113]. The TME also affects T cell recruitment to the tumor, as long-term hypoxia for multiple days was shown to increase the expression cytokines CCL3, CCL5 and CCL20 in mature DCs [114]. These cytokines are chemotactic for both activated and memory T cells, monocytes and immature DCs. Finally HIF-1α activates transcription of retinoic acid receptor-related orphan nuclear receptor gamma (RORγt) and together these two factors regulate the transcription of IL-17 [115]. Therefore, hypoxia increases the differentiation of T cells towards regulatory T helper 17 (Th17) cells via IL-6 in a uniquely TGF-β independent fashion [116]. Regulatory Th17 cells are associated with host defense and autoimmune inflammatory responses [117, 118] and promote tumor growth [115].
DISCUSSION
In this review, we discussed how the hypoxic, oxidative environment of the TME influences invading immune cells. Figure 2 shows an overview of the major effects that have been found so far. The effects of the cytokines and chemokines found in the TME have been quite extensively studied. In contrast, comparatively little is known about the effects on tumor invading immune cells of localized ROS and hypoxia that frequently hallmark the TME. Both the tumor cells and the immune cells produce various forms of ROS, that directly affect the cell physiology but can also target neighboring cells, either by diffusion of ROS or by ROS encapsulated in EVs (Figure 1). ROS generation consumes oxygen and is therefore often paired with local hypoxia, whereas hypoxia can promote formation of ROS, making it difficult to differentiate the effects of ROS and hypoxia from each other. As described in this review, the effects of hypoxia on DC phenotype and maturation are under debate. However, it is becoming increasingly clear that hypoxia stimulates migration of immature DCs but prevents migration of mature DCs. This might result in the TME becoming an immunosuppressive “DC trap”, with limited influx and maturation of immature DCs while the egress of activated DCs is prevented. However, in contrast to this, hypoxia seems to upregulate secretion of various pro-inflammatory anti-tumorigenic cytokines. Maybe these contradicting effects are the result of ROS signaling.
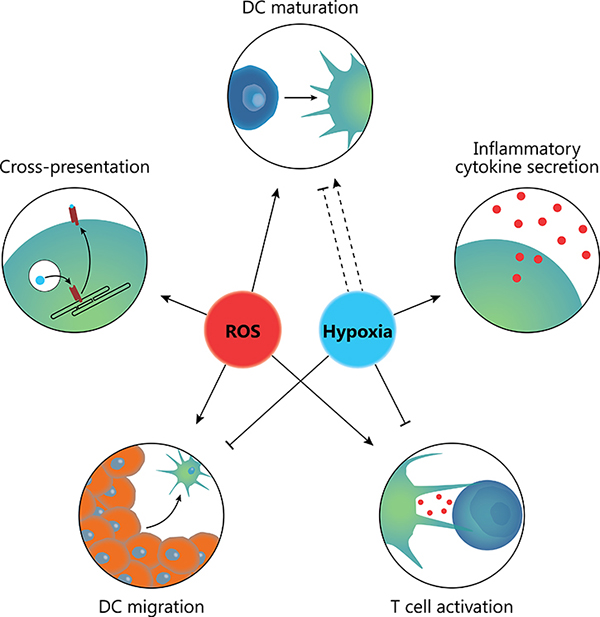
Figure 2: Combined effects of ROS and hypoxia in the TME. ROS promote DC maturation, antigen cross-presentation, DC migration and CD8+ T cell responses needed for anti-tumor immunity. In contrast, hypoxia can both lower or increase DC maturation and skews T cell responses towards a tolerogenic Th17 response. Moreover, hypoxia can inhibit ingress of immature DCs into the tumor whilst blocking migration of mature DCs from the tumor to draining lymph nodes. Hypoxia does however promote secretion of various inflammatory cytokines such as TNF-α.
A major question is whether tumor cells use EV release as a protective mechanism from oxidative damage as a result of their high anaerobic metabolic activity, or as an active defense mechanism to prevent immune responses. However, due to the small size and heterogeneity of EVs, it is difficult to study their content. In vitro approaches using artificial membranes carrying ROS might help to overcome this problem. Another problem is that resolving the physiological effects of specific sources and types of ROS remains challenging, due to their highly transient nature and the lack of specific probes that offer adequate spatiotemporal resolution. Controlling specific redox signaling and antioxidant pathways would be a valid approach to this problem, since these parameters can be modified with genetic techniques. In addition, ROS can be induced with organellar precision using fusion constructs of proteins with known cellular location with photosensitizer proteins like SuperNova [119]. Likewise, culture media can be supplemented with a wide range of antioxidants or radical-generating systems.
Another key question is whether ROS can be used to treat cancer. A possible avenue would be local administration of pro-oxidants in the TME. Tumor cells often display a defective Nrf2 pathway, rendering them more susceptible to oxidative stress [120], while DC maturation can be enhanced by ROS as described above. In a xenograft mouse model of chronic lymphocyte leukemia, pro-oxidative treatment strongly reduced tumor burden [120]. However, since ROS also has pro-tumorigenic effects, the opposite approach of administrating anti-oxidants is also possible. There have been several randomized controlled trials in which prophylactic effects of such antioxidant supplementation was investigated. However, for incidence of prostate and total cancer in men, supplemental vitamin E had no effects [121–123] and in one study even significantly increased prostate cancer incidence [124].
Since the effects of ROS on cancer and immune cells are complex and dependent on the site of ROS generation and the interplay with hypoxia and immune signaling, targeting ROS by simply administering pro- or antioxidants on might not be sufficient. Targeting ROS or antioxidants to a specific cell type may provide a more successful strategy to combat cancer. For instance, promoting ROS formation in the lumen of endo/phagosomes of DCs could be a strategy to promote antigen cross-presentation [55–57, 60, 61, 63, 125], whereas blockage of mitochondrial ROS formation might increase T cell activation in the lymph nodes [64]. In the paper by Dingjan et al., the photosensitizer protein KillerRed [126] was employed to increase endosomal ROS in DCs by transfecting a plasmid encoding a KillerRed fusion protein targeted to phagosomes. However, transfection of tumor-associated DCs in vivo is still very challenging. An alternative approach would be to target DCs with nanoparticles carrying a ROS-inducer [127–129], for example an iron core that promotes generation of highly reactive hydroxyl radicals through Fenton chemistry [130, 131].
In a similar fashion, cancer cells might be specifically targeted with antioxidants to block the pro-tumorigenic effects of ROS. While, as described above, systemic antioxidant therapy proved unsuccessful in cancer, localized interventions are still worth considering. Endosomal NOX2 activity was recently shown to play an important role in progression of prostate cancer [132], which could be targeted (for instance with antibodies) with antioxidant-carrying small particles for exclusive uptake via endocytosis by tumor cells [133]. Another interesting targeting approach is ROS-responsive nanoparticles for targeted delivery of hydrophilic and cationic drugs in ROS-producing cells [134]. In this study, Meng et al. showed that MnO2-based nanoparticles selectively release the HIF-1 inhibitor acriflavine in tumor cells after oxidation by H2O2 in vitro and in a mouse model of colon cancer. Although the authors did not investigate uptake by phagocytic cells, it is likely that this method is also capable of releasing compounds in phagosomes. Finally, it might be highly beneficial to sequester lipid peroxidation products such as MDA and 4-HNE due to their negative impact on DC function, as described above. Doing so would protect DCs against these effects without interfering with ROS-induced cross-presentation and DC maturation. Several potential compounds have been identified recently that warrant further investigation, of which histidine-containing dipeptides are currently the most promising [135–137].
Given that cancer cells use hypoxia and ROS to reprogram immune and stromal cells in the TME to prevent an immune response and augment tumor progression, while at the same time the immune system uses ROS to signal inflammation and combat infection, ROS have huge therapeutic potential for combating cancer. Given this duality, the timing and localization of pro- or antioxidant interventions is likely highly critical. Understanding the intricate pathways of the production, signalling and effector responses of hypoxia and ROS is essential for designing such therapies.
Abbreviations
4-HNE, 4-hydroxynonenal; AMPK, 5’ adenosine monophosphate-activated protein kinase; AP-1, activator protein 1; AQP, aquaporin; ASK1, apoptosis signal-regulating kinase 1; CCL, C-C Motif Chemokine Ligand; DC, dendritic cell; EV, extracellular vesicle; HIF, hypoxia-induced factor; IFN, interferon; JNK, c-Jun N-terminal kinase; LFA-1, lymphocyte function-associated antigen 1; MAPK, mitogen-associated protein kinase; NF-κb, nuclear factor kappa B; NOX, NAD(P)H oxidase; Nrf2, Nuclear factor-erythroid 2 p45-related factor 2; Prx, peroxiredoxin; ROS, reactive oxygen species; Siglec-1, sialic-acid-binding immunoglobin lectin 1; SOD, superoxide dismutase; TLR, Toll-like receptor; TME, tumor microenvironment; TNF-α, tumor necrosis factor alpha; Trx, thioredoxin; VEGF, vascular endothelial growth factor.
Author contributions
L.M.P. and W.V. wrote the manuscript, G.v.d.B. supervised the writing of the manuscript.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was funded by a Career Development Award from the Human Frontier Science Program and the Netherlands Organization for Scientific Research (NWO-ALW VIDI 864.14.001 and Gravitation 2013 ICI-024.002.009).
REFERENCES
1. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324:1029–33. https://doi.org/10.1126/science.1160809.
2. Lennicke C, Rahn J, Lichtenfels R, Wessjohann LA, Seliger B. Hydrogen peroxide - production, fate and role in redox signaling of tumor cells. Cell Commun Signal. 2015; 13:39. https://doi.org/10.1186/s12964-015-0118-6.
3. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, Motohashi H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012; 22:66–79. https://doi.org/10.1016/j.ccr.2012.05.016.
4. Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005; 18:283–93. https://doi.org/10.1016/j.molcel.2005.03.027.
5. Holmgren A. Thioredoxin structure and mechanism: conformational changes on oxidation of the active-site sulfhydryls to a disulfide. Structure. 1995; 3:239–43. https://doi.org/10.1016/S0969-2126(01)00153-8.
6. Holmgren A. Thioredoxin and glutaredoxin systems. J Biol Chem. 1989; 264:13963–66.
7. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646-74. https://doi.org/10.1016/j.cell.2011.02.013.
8. Policastro LL, Ibañez IL, Notcovich C, Duran HA, Podhajcer OL. The tumor microenvironment: characterization, redox considerations, and novel approaches for reactive oxygen species-targeted gene therapy. Antioxid Redox Signal. 2013; 19:854–95. https://doi.org/10.1089/ars.2011.4367.
9. Ushio-Fukai M, Nakamura Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008; 266:37–52. https://doi.org/10.1016/j.canlet.2008.02.044.
10. Łuczak K, Balcerczyk A, Soszyński M, Bartosz G. Low concentration of oxidant and nitric oxide donors stimulate proliferation of human endothelial cells in vitro. Cell Biol Int. 2004; 28:483–86. https://doi.org/10.1016/j.cellbi.2004.03.004.
11. Brieger K, Schiavone S, Miller FJ Jr, Krause KH. Reactive oxygen species: from health to disease. Swiss Med Wkly. 2012; 142:w13659. https://doi.org/10.4414/smw.2012.13659.
12. Speijer D, Manjeri GR, Szklarczyk R. How to deal with oxygen radicals stemming from mitochondrial fatty acid oxidation. Philos Trans R Soc Lond B Biol Sci. 2014; 369:20130446. https://doi.org/10.1098/rstb.2013.0446.
13. Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011; 194:7–15. https://doi.org/10.1083/jcb.201102095.
14. Sheng Y, Abreu IA, Cabelli DE, Maroney MJ, Miller AF, Teixeira M, Valentine JS. Superoxide dismutases and superoxide reductases. Chem Rev. 2014; 114:3854–918. https://doi.org/10.1021/cr4005296.
15. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013; 14:1014–22. https://doi.org/10.1038/ni.2703.
16. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005; 5:953–64. https://doi.org/10.1038/nri1733.
17. Nahrendorf M, Swirski FK. Abandoning M1/M2 for a Network Model of Macrophage Function. Circ Res. 2016; 119:414–17. https://doi.org/10.1161/CIRCRESAHA.116.309194.
18. Quatromoni JG, Eruslanov E. Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am J Transl Res. 2012; 4:376–89.
19. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004; 10:942–49. https://doi.org/10.1038/nm1093.
20. Canli Ö, Nicolas AM, Gupta J, Finkelmeier F, Goncharova O, Pesic M, Neumann T, Horst D, Löwer M, Sahin U, Greten FR. Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer Cell. 2017; 32:869–883.e5. https://doi.org/10.1016/j.ccell.2017.11.004.
21. Zhang Y, Choksi S, Chen K, Pobezinskaya Y, Linnoila I, Liu ZG. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. 2013; 23:898–914. https://doi.org/10.1038/cr.2013.75.
22. Chua CC, Hamdy RC, Chua BH. Upregulation of vascular endothelial growth factor by H2O2 in rat heart endothelial cells. Free Radic Biol Med. 1998; 25:891–97. https://doi.org/10.1016/S0891-5849(98)00115-4.
23. Cho M, Hunt TK, Hussain MZ. Hydrogen peroxide stimulates macrophage vascular endothelial growth factor release. Am J Physiol Circ Physiol; 2001; 280: H2357–H2363. https://doi.org/10.1152/ajpheart.2001.280.5.H2357.
24. González-Pacheco FR, Deudero JJ, Castellanos MC, Castilla MA, Álvarez-Arroyo MV, Yagüe S, Caramelo C. Mechanisms of endothelial response to oxidative aggression: protective role of autologous VEGF and induction of VEGFR2 by H2O2. Am J Physiol Circ Physiol; 2006; 291: H1395-401. https://doi.org/10.1152/ajpheart.01277.2005.
25. Warren CM, Ziyad S, Briot A, Der A, Iruela-Arispe ML. A ligand-independent VEGFR2 signaling pathway limits angiogenic responses in diabetes. Sci Signal. 2014; 7:ra1. https://doi.org/10.1126/scisignal.2004235.
26. Isaac M, Latour JM, Sénèque O. Nucleophilic reactivity of Zinc-bound thiolates: subtle interplay between coordination set and conformational flexibility. Chem Sci (Camb). 2012; 3:3409. https://doi.org/10.1039/c2sc21029k.
27. Schieber M, Chandel NS. TOR signaling couples oxygen sensing to lifespan in C. elegans. Cell Reports. 2014; 9:9–15. https://doi.org/10.1016/j.celrep.2014.08.075.
28. Murray TV, Smyrnias I, Shah AM, Brewer AC. NADPH oxidase 4 regulates cardiomyocyte differentiation via redox activation of c-Jun protein and the cis-regulation of GATA-4 gene transcription. J Biol Chem. 2013; 288:15745–59. https://doi.org/10.1074/jbc.M112.439844.
29. Long Y, Wang G, Li K, Zhang Z, Zhang P, Zhang J, Zhang X, Bao Y, Yang X, Wang P. Oxidative stress and NF-κB signaling are involved in LPS induced pulmonary dysplasia in chick embryos. Cell Cycle. 2018; 17:1757–71. https://doi.org/10.1080/15384101.2018.1496743.
30. Marin T, Gongol B, Chen Z, Woo B, Subramaniam S, Chien S, Shyy JY. Mechanosensitive microRNAs-role in endothelial responses to shear stress and redox state. Free Radic Biol Med. 2013; 64:61–68. https://doi.org/10.1016/j.freeradbiomed.2013.05.034.
31. Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012; 24:981–90. https://doi.org/10.1016/j.cellsig.2012.01.008.
32. Katagiri K, Matsuzawa A, Ichijo H. Regulation of apoptosis signal-regulating kinase 1 in redox signaling. Methods Enzymol. 2010; 474:277–88. https://doi.org/10.1016/S0076-6879(10)74016-7.
33. Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998; 17:2596–606. https://doi.org/10.1093/emboj/17.9.2596.
34. Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012; 12:557–69. https://doi.org/10.1038/nri3254.
35. Stark G. Functional consequences of oxidative membrane damage. J Membr Biol. 2005; 205:1–16. https://doi.org/10.1007/s00232-005-0753-8.
36. Bienert GP, Møller AL, Kristiansen KA, Schulz A, Møller IM, Schjoerring JK, Jahn TP. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J Biol Chem. 2007; 282:1183–92. https://doi.org/10.1074/jbc.M603761200.
37. Vieceli Dalla Sega F, Zambonin L, Fiorentini D, Rizzo B, Caliceti C, Landi L, Hrelia S, Prata C. Specific aquaporins facilitate Nox-produced hydrogen peroxide transport through plasma membrane in leukaemia cells. Biochim Biophys Acta. 2014; 1843:806–14. https://doi.org/10.1016/j.bbamcr.2014.01.011.
38. de Baey A, Lanzavecchia A. The role of aquaporins in dendritic cell macropinocytosis. J Exp Med. 2000; 191:743–48. https://doi.org/10.1084/jem.191.4.743.
39. Song MG, Hwang SY, Park JI, Yoon S, Bae HR, Kwak JY. Role of aquaporin 3 in development, subtypes and activation of dendritic cells. Mol Immunol. 2011; 49:28–37. https://doi.org/10.1016/j.molimm.2011.07.015.
40. Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Girón CG, Gil L, Gordon L, Haggerty L, et al. Ensembl 2018. Nucleic Acids Res. 2018; 46:D754–61. https://doi.org/10.1093/nar/gkx1098.
41. Fisher AB. Redox signaling across cell membranes. Antioxid Redox Signal. 2009; 11:1349–56. https://doi.org/10.1089/ars.2008.2378.
42. Schmidt T, Samaras P, Frejno M, Gessulat S, Barnert M, Kienegger H, Krcmar H, Schlegl J, Ehrlich HC, Aiche S, Kuster B, Wilhelm M. ProteomicsDB. Nucleic Acids Res. 2018; 46:D1271–81. https://doi.org/10.1093/nar/gkx1029.
43. Wilhelm M, Schlegl J, Hahne H, Gholami AM, Lieberenz M, Savitski MM, Ziegler E, Butzmann L, Gessulat S, Marx H, Mathieson T, Lemeer S, Schnatbaum K, et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014; 509:582–87. https://doi.org/10.1038/nature13319.
44. Luís A, Martins JD, Silva A, Ferreira I, Cruz MT, Neves BM. Oxidative stress-dependent activation of the eIF2α–ATF4 unfolded protein response branch by skin sensitizer 1-fluoro-2,4-dinitrobenzene modulates dendritic-like cell maturation and inflammatory status in a biphasic manner [corrected]. Free Radic Biol Med. 2014; 77:217–29. https://doi.org/10.1016/j.freeradbiomed.2014.09.008.
45. Rutault K, Alderman C, Chain BM, Katz DR. Reactive oxygen species activate human peripheral blood dendritic cells. Free Radic Biol Med. 1999; 26:232–38. https://doi.org/10.1016/S0891-5849(98)00194-4.
46. Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991; 11:81–128. https://doi.org/10.1016/0891-5849(91)90192-6.
47. Vladykovskaya E, Sithu SD, Haberzettl P, Wickramasinghe NS, Merchant ML, Hill BG, McCracken J, Agarwal A, Dougherty S, Gordon SA, Schuschke DA, Barski OA, O’Toole T, et al. Lipid peroxidation product 4-hydroxy-trans-2-nonenal causes endothelial activation by inducing endoplasmic reticulum stress. J Biol Chem. 2012; 287:11398–409. https://doi.org/10.1074/jbc.M111.320416.
48. Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K, Ellenson LH, Caputo T, Lee AH, et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell. 2015; 161:1527–38. https://doi.org/10.1016/j.cell.2015.05.025.
49. Veglia F, Tyurin VA, Mohammadyani D, Blasi M, Duperret EK, Donthireddy L, Hashimoto A, Kapralov A, Amoscato A, Angelini R, Patel S, Alicea-Torres K, Weiner D, et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat Commun. 2017; 8:2122. https://doi.org/10.1038/s41467-017-02186-9.
50. Negre-Salvayre A, Coatrieux C, Ingueneau C, Salvayre R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br J Pharmacol. 2008; 153:6–20. https://doi.org/10.1038/sj.bjp.0707395.
51. Halliwell B, Gutteridge JM. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J. 1984; 219:1–14. https://doi.org/10.1042/bj2190001.
52. Wållberg M, Bergquist J, Achour A, Breij E, Harris RA. Malondialdehyde modification of myelin oligodendrocyte glycoprotein leads to increased immunogenicity and encephalitogenicity. Eur J Immunol. 2007; 37:1986–95. https://doi.org/10.1002/eji.200636912.
53. Weismann D, Binder CJ. The innate immune response to products of phospholipid peroxidation. Biochim Biophys Acta. 2012; 1818:2465–75. https://doi.org/10.1016/j.bbamem.2012.01.018.
54. Wang G, Li H, Firoze Khan M. Differential oxidative modification of proteins in MRL+/+ and MRL/lpr mice: increased formation of lipid peroxidation-derived aldehyde-protein adducts may contribute to accelerated onset of autoimmune response. Free Radic Res. 2012; 46:1472–81. https://doi.org/10.3109/10715762.2012.727209.
55. Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon-Duménil AM, Seabra MC, Raposo G, Amigorena S. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell. 2006; 126:205–18. https://doi.org/10.1016/j.cell.2006.05.035.
56. Kotsias F, Hoffmann E, Amigorena S, Savina A. Reactive oxygen species production in the phagosome: impact on antigen presentation in dendritic cells. Antioxid Redox Signal. 2013; 18:714–29. https://doi.org/10.1089/ars.2012.4557.
57. Dingjan I, Verboogen DR, Paardekooper LM, Revelo NH, Sittig SP, Visser LJ, Mollard GF, Henriet SS, Figdor CG, Ter Beest M, van den Bogaart G. Lipid peroxidation causes endosomal antigen release for cross-presentation. Sci Rep. 2016; 6:22064. https://doi.org/10.1038/srep22064.
58. Battisti F, Napoletano C, Rahimi Koshkaki H, Belleudi F, Zizzari IG, Ruscito I, Palchetti S, Bellati F, Benedetti Panici P, Torrisi MR, Caracciolo G, Altieri F, Nuti M, Rughetti A. Tumor-Derived Microvesicles Modulate Antigen Cross-Processing via Reactive Oxygen Species-Mediated Alkalinization of Phagosomal Compartment in Dendritic Cells. Front Immunol. 2017; 8:1179. https://doi.org/10.3389/fimmu.2017.01179.
59. Feng Y, Forgac M. Inhibition of vacuolar H(+)-ATPase by disulfide bond formation between cysteine 254 and cysteine 532 in subunit A. J Biol Chem. 1994; 269:13224–30.
60. Rybicka JM, Balce DR, Chaudhuri S, Allan ER, Yates RM. Phagosomal proteolysis in dendritic cells is modulated by NADPH oxidase in a pH-independent manner. EMBO J. 2012; 31:932–44. https://doi.org/10.1038/emboj.2011.440.
61. Allan ER, Tailor P, Balce DR, Pirzadeh P, McKenna NT, Renaux B, Warren AL, Jirik FR, Yates RM. NADPH oxidase modifies patterns of MHC class II-restricted epitopic repertoires through redox control of antigen processing. J Immunol. 2014; 192:4989–5001. https://doi.org/10.4049/jimmunol.1302896.
62. Nagaoka Y, Otsu K, Okada F, Sato K, Ohba Y, Kotani N, Fujii J. Specific inactivation of cysteine protease-type cathepsin by singlet oxygen generated from naphthalene endoperoxides. Biochem Biophys Res Commun. 2005; 331:215–23. https://doi.org/10.1016/j.bbrc.2005.03.146.
63. Dingjan I, Paardekooper LM, Verboogen DR, von Mollard GF, Ter Beest M, van den Bogaart G. VAMP8-mediated NOX2 recruitment to endosomes is necessary for antigen release. Eur J Cell Biol. 2017; 96:705–14. https://doi.org/10.1016/j.ejcb.2017.06.007.
64. Chougnet CA, Thacker RI, Shehata HM, Hennies CM, Lehn MA, Lages CS, Janssen EM. Loss of Phagocytic and Antigen Cross-Presenting Capacity in Aging Dendritic Cells Is Associated with Mitochondrial Dysfunction. J Immunol. 2015; 195:2624–32. https://doi.org/10.4049/jimmunol.1501006.
65. Zhang Q, Shang M, Zhang M, Wang Y, Chen Y, Wu Y, Liu M, Song J, Liu Y. Microvesicles derived from hypoxia/reoxygenation-treated human umbilical vein endothelial cells promote apoptosis and oxidative stress in H9c2 cardiomyocytes. BMC Cell Biol. 2016; 17:25. https://doi.org/10.1186/s12860-016-0100-1.
66. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013; 200:373–83. https://doi.org/10.1083/jcb.201211138.
67. Subra C, Laulagnier K, Perret B, Record M. Exosome lipidomics unravels lipid sorting at the level of multivesicular bodies. Biochimie. 2007; 89:205–12. https://doi.org/10.1016/j.biochi.2006.10.014.
68. Harding C, Heuser J, Stahl P. Endocytosis and intracellular processing of transferrin and colloidal gold-transferrin in rat reticulocytes: demonstration of a pathway for receptor shedding. Eur J Cell Biol. 1984; 35:256–63.
69. Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D, Ricciardi-Castagnoli P, Raposo G, Amigorena S. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med. 1998; 4:594–600. https://doi.org/10.1038/nm0598-594.
70. Holme PA, Solum NO, Brosstad F, Røger M, Abdelnoor M. Demonstration of platelet-derived microvesicles in blood from patients with activated coagulation and fibrinolysis using a filtration technique and western blotting. Thromb Haemost. 1994; 72:666–71.
71. György B, Szabó TG, Pásztói M, Pál Z, Misják P, Aradi B, László V, Pállinger E, Pap E, Kittel A, Nagy G, Falus A, Buzás EI. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci. 2011; 68:2667–88. https://doi.org/10.1007/s00018-011-0689-3.
72. Wendler F, Favicchio R, Simon T, Alifrangis C, Stebbing J, Giamas G. Extracellular vesicles swarm the cancer microenvironment: from tumor-stroma communication to drug intervention. Oncogene. 2017; 36:877–84. https://doi.org/10.1038/onc.2016.253.
73. Webber J, Yeung V, Clayton A. Extracellular vesicles as modulators of the cancer microenvironment. Semin Cell Dev Biol. 2015; 40:27–34. https://doi.org/10.1016/j.semcdb.2015.01.013.
74. Wang T, Gilkes DM, Takano N, Xiang L, Luo W, Bishop CJ, Chaturvedi P, Green JJ, Semenza GL. Hypoxia-inducible factors and RAB22A mediate formation of microvesicles that stimulate breast cancer invasion and metastasis. Proc Natl Acad Sci USA. 2014; 111:E3234–42. https://doi.org/10.1073/pnas.1410041111.
75. Qin Q, Furong W, Baosheng L. Multiple functions of hypoxia-regulated miR-210 in cancer. J Exp Clin Cancer Res. 2014; 33:50. https://doi.org/10.1186/1756-9966-33-50.
76. Baj-Krzyworzeka M, Mytar B, Szatanek R, Surmiak M, Węglarczyk K, Baran J, Siedlar M. Colorectal cancer-derived microvesicles modulate differentiation of human monocytes to macrophages. J Transl Med. 2016; 14:36. https://doi.org/10.1186/s12967-016-0789-9.
77. Morelli AE, Larregina AT, Shufesky WJ, Sullivan ML, Stolz DB, Papworth GD, Zahorchak AF, Logar AJ, Wang Z, Watkins SC, Falo LD Jr, Thomson AW. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood. 2004; 104:3257–66. https://doi.org/10.1182/blood-2004-03-0824.
78. Yáñez-Mó M, Siljander PR, Andreu Z, Zavec AB, Borràs FE, Buzas EI, Buzas K, Casal E, Cappello F, Carvalho J, Colás E, Cordeiro-da Silva A, Fais S, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015; 4:27066. https://doi.org/10.3402/jev.v4.27066.
79. Hao S, Bai O, Li F, Yuan J, Laferte S, Xiang J. Mature dendritic cells pulsed with exosomes stimulate efficient cytotoxic T-lymphocyte responses and antitumour immunity. Immunology. 2007; 120:90–102. https://doi.org/10.1111/j.1365-2567.2006.02483.x.
80. Wen Z, Shimojima Y, Shirai T, Li Y, Ju J, Yang Z, Tian L, Goronzy JJ, Weyand CM. NADPH oxidase deficiency underlies dysfunction of aged CD8+ Tregs. J Clin Invest. 2016; 126:1953–67. https://doi.org/10.1172/JCI84181.
81. Liu ML, Scalia R, Mehta JL, Williams KJ. Cholesterol-induced membrane microvesicles as novel carriers of damage-associated molecular patterns: mechanisms of formation, action, and detoxification. Arterioscler Thromb Vasc Biol. 2012; 32:2113–21. https://doi.org/10.1161/ATVBAHA.112.255471.
82. Yang C, Kim SH, Bianco NR, Robbins PD. Tumor-derived exosomes confer antigen-specific immunosuppression in a murine delayed-type hypersensitivity model. PLoS One. 2011; 6:e22517. https://doi.org/10.1371/journal.pone.0022517.
83. Hari A, Ganguly A, Mu L, Davis SP, Stenner MD, Lam R, Munro F, Namet I, Alghamdi E, Fürstenhaupt T, Dong W, Detampel P, Shen LJ, et al. Redirecting soluble antigen for MHC class I cross-presentation during phagocytosis. Eur J Immunol. 2015; 45:383–95. https://doi.org/10.1002/eji.201445156.
84. Yu S, Liu C, Su K, Wang J, Liu Y, Zhang L, Li C, Cong Y, Kimberly R, Grizzle WE, Falkson C, Zhang HG. Tumor exosomes inhibit differentiation of bone marrow dendritic cells. J Immunol. 2007; 178:6867–75. https://doi.org/10.4049/jimmunol.178.11.6867.
85. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012; 148:399–408. https://doi.org/10.1016/j.cell.2012.01.021.
86. Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity. 2014; 41:518–28. https://doi.org/10.1016/j.immuni.2014.09.008.
87. Kietzmann T, Mennerich D, Dimova EY. Hypoxia-Inducible Factors (HIFs) and Phosphorylation: Impact on Stability, Localization, and Transactivity. Front Cell Dev Biol. 2016; 4:11. https://doi.org/10.3389/fcell.2016.00011.
88. Wang GL, Semenza GL. Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J Biol Chem. 1993; 268:21513–18. http://www.ncbi.nlm.nih.gov/pubmed/8408001.
89. Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005; 2005:re12. https://doi.org/10.1126/stke.3062005re12.
90. Fang HY, Hughes R, Murdoch C, Coffelt SB, Biswas SK, Harris AL, Johnson RS, Imityaz HZ, Simon MC, Fredlund E, Greten FR, Rius J, Lewis CE. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood. 2009; 114:844–59. https://doi.org/10.1182/blood-2008-12-195941.
91. Bhandari T, Olson J, Johnson RS, Nizet V. HIF-1α influences myeloid cell antigen presentation and response to subcutaneous OVA vaccination. J Mol Med (Berl). 2013; 91:1199–205. https://doi.org/10.1007/s00109-013-1052-y.
92. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006; 3:177–85. https://doi.org/10.1016/j.cmet.2006.02.002.
93. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006; 3:187–97. https://doi.org/10.1016/j.cmet.2006.01.012.
94. Goth SR, Chu RA, Pessah IN. Oxygen tension regulates the in vitro maturation of GM-CSF expanded murine bone marrow dendritic cells by modulating class II MHC expression. J Immunol Methods. 2006; 308:179–91. https://doi.org/10.1016/j.jim.2005.10.012.
95. Mancino A, Schioppa T, Larghi P, Pasqualini F, Nebuloni M, Chen IH, Sozzani S, Austyn JM, Mantovani A, Sica A. Divergent effects of hypoxia on dendritic cell functions. Blood. 2008; 112:3723–34. https://doi.org/10.1182/blood-2008-02-142091.
96. Wang Q, Liu C, Zhu F, Liu F, Zhang P, Guo C, Wang X, Li H, Ma C, Sun W, Zhang Y, Chen W, Zhang L. Reoxygenation of hypoxia-differentiated dentritic cells induces Th1 and Th17 cell differentiation. Mol Immunol. 2010; 47:922–31. https://doi.org/10.1016/j.molimm.2009.09.038.
97. Fliesser M, Wallstein M, Kurzai O, Einsele H, Löffler J. Hypoxia attenuates anti-Aspergillus fumigatus immune responses initiated by human dendritic cells. Mycoses. 2016; 59:503–08. https://doi.org/10.1111/myc.12498.
98. Yang M, Ma C, Liu S, Sun J, Shao Q, Gao W, Zhang Y, Li Z, Xie Q, Dong Z, Qu X. Hypoxia skews dendritic cells to a T helper type 2-stimulating phenotype and promotes tumour cell migration by dendritic cell-derived osteopontin. Immunology. 2009 (Suppl ); 128:e237–49. https://doi.org/10.1111/j.1365-2567.2008.02954.x.
99. Qu X, Yang MX, Kong BH, Qi L, Lam QL, Yan S, Li P, Zhang M, Lu L. Hypoxia inhibits the migratory capacity of human monocyte-derived dendritic cells. Immunol Cell Biol. 2005; 83:668–73. https://doi.org/10.1111/j.1440-1711.2005.01383.x.
100. Jantsch J, Chakravortty D, Turza N, Prechtel AT, Buchholz B, Gerlach RG, Volke M, Gläsner J, Warnecke C, Wiesener MS, Eckardt KU, Steinkasserer A, Hensel M, Willam C. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol. 2008; 180:4697–705. https://doi.org/10.4049/jimmunol.180.7.4697.
101. Zhao W, Darmanin S, Fu Q, Chen J, Cui H, Wang J, Okada F, Hamada J, Hattori Y, Kondo T, Hamuro J, Asaka M, Kobayashi M. Hypoxia suppresses the production of matrix metalloproteinases and the migration of human monocyte-derived dendritic cells. Eur J Immunol. 2005; 35:3468–77. https://doi.org/10.1002/eji.200526262.
102. Elia AR, Cappello P, Puppo M, Fraone T, Vanni C, Eva A, Musso T, Novelli F, Varesio L, Giovarelli M. Human dendritic cells differentiated in hypoxia down-modulate antigen uptake and change their chemokine expression profile. J Leukoc Biol. 2008; 84:1472–82. https://doi.org/10.1189/jlb.0208082.
103. Bosseto MC, Palma PV, Covas DT, Giorgio S. Hypoxia modulates phenotype, inflammatory response, and leishmanial infection of human dendritic cells. APMIS. 2010; 118:108–14. https://doi.org/10.1111/j.1600-0463.2009.02568.x.
104. Gupta P, Singh A, Gowda P, Ghosh S, Chatterjee A, Sen E. Lactate induced HIF-1α-PRMT1 cross talk affects MHC I expression in monocytes. Exp Cell Res. 2016; 347:293–300. https://doi.org/10.1016/j.yexcr.2016.08.008.
105. Zhang X, Li S, Li M, Huang H, Li J, Zhou C. Hypoxia-inducible factor-1α mediates the toll-like receptor 4 signaling pathway leading to anti-tumor effects in human hepatocellular carcinoma cells under hypoxic conditions. Oncol Lett. 2016; 12:1034–40. https://doi.org/10.3892/ol.2016.4705.
106. Kim SY, Choi YJ, Joung SM, Lee BH, Jung YS, Lee JY. Hypoxic stress up-regulates the expression of Toll-like receptor 4 in macrophages via hypoxia-inducible factor. Immunology. 2010; 129:516–24. https://doi.org/10.1111/j.1365-2567.2009.03203.x.
107. Paardekooper LM, Bendix MB, Ottria A, de Haer LW, Ter Beest M, Radstake TR, Marut W, van den Bogaart G. Hypoxia potentiates monocyte-derived dendritic cells for release of tumor necrosis factor α via MAP3K8. Biosci Rep. 2018; 38:BSR20182019. https://doi.org/10.1042/BSR20182019.
108. Xing Y, Wang R, Chen D, Mao J, Shi R, Wu Z, Kang J, Tian W, Zhang C. COX2 is involved in hypoxia-induced TNF-α expression in osteoblast. Sci Rep. 2015; 5:10020. https://doi.org/10.1038/srep10020.
109. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013; 496:238–42. https://doi.org/10.1038/nature11986.
110. Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V. Cutting edge: essential role of hypoxia inducible factor-1α in development of lipopolysaccharide-induced sepsis. J Immunol. 2007; 178:7516–19. https://doi.org/10.4049/jimmunol.178.12.7516.
111. Ricciardi A, Elia AR, Cappello P, Puppo M, Vanni C, Fardin P, Eva A, Munroe D, Wu X, Giovarelli M, Varesio L. Transcriptome of hypoxic immature dendritic cells: modulation of chemokine/receptor expression. Mol Cancer Res. 2008; 6:175–85. https://doi.org/10.1158/1541-7786.MCR-07-0391.
112. Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SA, Sahai E, Reis e Sousa C. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell. 2015; 162:1257–70. https://doi.org/10.1016/j.cell.2015.08.015.
113. Shinohara ML, Lu L, Bu J, Werneck MB, Kobayashi KS, Glimcher LH, Cantor H. Osteopontin expression is essential for interferon-α production by plasmacytoid dendritic cells. Nat Immunol. 2006; 7:498–506. https://doi.org/10.1038/ni1327.
114. Blengio F, Raggi F, Pierobon D, Cappello P, Eva A, Giovarelli M, Varesio L, Bosco MC. The hypoxic environment reprograms the cytokine/chemokine expression profile of human mature dendritic cells. Immunobiology. 2013; 218:76–89. https://doi.org/10.1016/j.imbio.2012.02.002.
115. Bailey SR, Nelson MH, Himes RA, Li Z, Mehrotra S, Paulos CM. Th17 cells in cancer: the ultimate identity crisis. Front Immunol. 2014; 5:276. https://doi.org/10.3389/fimmu.2014.00276.
116. Brucklacher-Waldert V, Ferreira C, Stebegg M, Fesneau O, Innocentin S, Marie JC, Veldhoen M. Cellular Stress in the Context of an Inflammatory Environment Supports TGF-β-Independent T Helper-17 Differentiation. Cell Reports. 2017; 19:2357–70. https://doi.org/10.1016/j.celrep.2017.05.052.
117. Stockinger B, Veldhoen M. Differentiation and function of Th17 T cells. Curr Opin Immunol. 2007; 19:281–86. https://doi.org/10.1016/j.coi.2007.04.005.
118. Matsui-Hasumi A, Sato Y, Uto-Konomi A, Yamashita S, Uehori J, Yoshimura A, Yamashita M, Asahara H, Suzuki S, Kubo M. E3 ubiquitin ligases SIAH1/2 regulate hypoxia-inducible factor-1 (HIF-1)-mediated Th17 cell differentiation. Int Immunol. 2017; 29:133–43. https://doi.org/10.1093/intimm/dxx014.
119. Takemoto K, Matsuda T, Sakai N, Fu D, Noda M, Uchiyama S, Kotera I, Arai Y, Horiuchi M, Fukui K, Ayabe T, Inagaki F, Suzuki H, Nagai T. SuperNova, a monomeric photosensitizing fluorescent protein for chromophore-assisted light inactivation. Sci Rep. 2013; 3:2629. https://doi.org/10.1038/srep02629.
120. Agathanggelou A, Weston VJ, Perry T, Davies NJ, Skowronska A, Payne DT, Fossey JS, Oldreive CE, Wei W, Pratt G, Parry H, Oscier D, Coles SJ, et al. Targeting the Ataxia Telangiectasia Mutated-null phenotype in chronic lymphocytic leukemia with pro-oxidants. Haematologica. 2015; 100:1076–85. https://doi.org/10.3324/haematol.2014.115170.
121. Fleshner NE, Kapusta L, Donnelly B, Tanguay S, Chin J, Hersey K, Farley A, Jansz K, Siemens DR, Trpkov K, Lacombe L, Gleave M, Tu D, Parulekar WR. Progression from high-grade prostatic intraepithelial neoplasia to cancer: a randomized trial of combination vitamin-E, soy, and selenium. J Clin Oncol. 2011; 29:2386–90. https://doi.org/10.1200/JCO.2010.32.0994.
122. Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N Engl J Med. 1994; 330:1029–35. https://doi.org/10.1056/NEJM199404143301501.
123. Gaziano JM, Glynn RJ, Christen WG, Kurth T, Belanger C, MacFadyen J, Bubes V, Manson JE, Sesso HD, Buring JE. Vitamins E and C in the prevention of prostate and total cancer in men: the Physicians’ Health Study II randomized controlled trial. JAMA. 2009; 301:52–62. https://doi.org/10.1001/jama.2008.862.
124. Klein EA, Thompson IM Jr, Tangen CM, Crowley JJ, Lucia MS, Goodman PJ, Minasian LM, Ford LG, Parnes HL, Gaziano JM, Karp DD, Lieber MM, Walther PJ, et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2011; 306:1549–56. https://doi.org/10.1001/jama.2011.1437.
125. Mantegazza AR, Savina A, Vermeulen M, Pérez L, Geffner J, Hermine O, Rosenzweig SD, Faure F, Amigorena S. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood. 2008; 112:4712–22. https://doi.org/10.1182/blood-2008-01-134791.
126. Bulina ME, Chudakov DM, Britanova OV, Yanushevich YG, Staroverov DB, Chepurnykh TV, Merzlyak EM, Shkrob MA, Lukyanov S, Lukyanov KA. A genetically encoded photosensitizer. Nat Biotechnol. 2006; 24:95–99. https://doi.org/10.1038/nbt1175.
127. Jia J, Zhang Y, Xin Y, Jiang C, Yan B, Zhai S. Interactions Between Nanoparticles and Dendritic Cells: From the Perspective of Cancer Immunotherapy. Front Oncol. 2018; 8:404. https://doi.org/10.3389/fonc.2018.00404.
128. Cruz LJ, Tacken PJ, Bonetto F, Buschow SI, Croes HJ, Wijers M, de Vries IJ, Figdor CG. Multimodal imaging of nanovaccine carriers targeted to human dendritic cells. Mol Pharm. 2011; 8:520–31. https://doi.org/10.1021/mp100356k.
129. Liu L, He H, Liang R, Yi H, Meng X, Chen Z, Pan H, Ma Y, Cai L. ROS-Inducing Micelles Sensitize Tumor-Associated Macrophages to TLR3 Stimulation for Potent Immunotherapy. Biomacromolecules. 2018; 19:2146–55. https://doi.org/10.1021/acs.biomac.8b00239.
130. Bielski BH, Arudi RL, Sutherland MW. A study of the reactivity of HO2/O2- with unsaturated fatty acids. J Biol Chem. 1983; 258:4759–61.
131. Aruoma OI, Halliwell B. Superoxide-dependent and ascorbate-dependent formation of hydroxyl radicals from hydrogen peroxide in the presence of iron. Are lactoferrin and transferrin promoters of hydroxyl-radical generation? Biochem J. 1987; 241:273–78. https://doi.org/10.1042/bj2410273.
132. Harrison IP, Vinh A, Johnson IR, Luong R, Drummond GR, Sobey CG, Tiganis T, Williams ED, O’ Leary JJ, Brooks DA, Selemidis S. NOX2 oxidase expressed in endosomes promotes cell proliferation and prostate tumour development. Oncotarget. 2018; 9:35378–93. https://doi.org/10.18632/oncotarget.26237.
133. Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J. 2004; 377:159–69. https://doi.org/10.1042/bj20031253.
134. Meng L, Cheng Y, Tong X, Gan S, Ding Y, Zhang Y, Wang C, Xu L, Zhu Y, Wu J, Hu Y, Yuan A. Tumor Oxygenation and Hypoxia Inducible Factor-1 Functional Inhibition via a Reactive Oxygen Species Responsive Nanoplatform for Enhancing Radiation Therapy and Abscopal Effects. ACS Nano. 2018; 12:8308–22. https://doi.org/10.1021/acsnano.8b03590.
135. Vistoli G, Colzani M, Mazzolari A, Maddis DD, Grazioso G, Pedretti A, Carini M, Aldini G. Computational approaches in the rational design of improved carbonyl quenchers: focus on histidine containing dipeptides. Future Med Chem. 2016; 8:1721–37. https://doi.org/10.4155/fmc-2016-0088.
136. Colzani M, Regazzoni L, Criscuolo A, Baron G, Carini M, Vistoli G, Lee YM, Han SI, Aldini G, Yeum KJ. Isotopic labelling for the characterisation of HNE-sequestering agents in plant-based extracts and its application for the identification of anthocyanidins in black rice with giant embryo. Free Radic Res. 2018; 52:896–906. https://doi.org/10.1080/10715762.2018.1490735.
137. Xie Z, Baba SP, Sweeney BR, Barski OA. Detoxification of aldehydes by histidine-containing dipeptides: from chemistry to clinical implications. Chem Biol Interact. 2013; 202:288–97. https://doi.org/10.1016/j.cbi.2012.12.017.