
INTRODUCTION
Multiple myeloma (MM) is a plasma cell disorder characterized by multiple complex numerical and structural abnormalities [1]. Hyperdiploidy (47-57 chromosomes) is found in 50-60% of patients and is characterized by gains of whole chromosomes, consisting mostly of odd numbered chromosomes including 3, 5, 7, 9, 11, 15, 19, and 21. The remainder of patients have translocations involving the IGH locus on chromosome 14 and are associated with a hypodiploid karyotype and a poor prognosis.
Hyperhaploidy is defined as a karyotype with 24-34 chromosomes and is rare in MM [2, 3], but is seen in many cancer types, including acute lymphoblastic leukemia (ALL), myeloid leukemias, chondrosarcomas, and squamous cell carcinomas [4–6]. Hyperhaploidy in MM occurs with monosomy of many chromosomes, but those chromosomes associated with trisomies in hyperdiploidy remain disomic. In ALL, a different set of chromosomes retain disomy, namely 14, 18, and 21. Interestingly, in all cancers with hyperhaploidy, including MM, chromosome 18 is usually disomic, suggesting that disomy of chromosome 18 is required for a viable cell.
We have previously shown that this group of patients is associated with a poor prognosis, with a 5-year survival rate of only 23% [2], and this was assumed to be linked to deletions of 17p and amplification of 1q in these samples. However, no sequencing of hyperhaploid samples has been performed to determine if there is a mutational profile associated with this high risk group. Recent studies have shown that del17p is not solely responsible for the poor prognosis associated with MM, and that mutations in the tumor suppressor gene TP53 are more prognostic [7–9]. However, when mutations and deletions are taken into account there is a profound effect on prognosis when both alleles are affected, resulting in biallelic inactivation. We have recently shown that biallelic inactivation of TP53 is a feature of Double-Hit MM, defining 6.1% of newly diagnosed MM patients with a median progression free survival of 15.4 months [10].
Herein, we have performed sequencing on a set of hyperhaploid MM samples to determine the mutational background of this rare subgroup and the mechanism in which it is generated.
RESULTS
Copy number profiling reveals retention of heterodisomy in hyperhaploid samples
We performed exome sequencing on 5 patient samples who had been identified as hyperhaploid by karyotyping and identified one additional patient sample during routine targeted sequencing. Two additional samples were identified in the MGP study dataset, giving a total of 8 hyperhaploid samples. The data were analyzed for copy number and B allele frequency. Although different methodologies were used to identify these hyperhaploid samples we do not believe that it affects the results, and that it is better to use all datasets to characterize this small subset of MM patients. Hyperhaploidy was differentiated from hyperdiploidy in exome data using B allele frequency in the tumor compared to matched normal sample. Where consistent loss of heterozygosity (LOH) was seen the ploidy of the samples was re-normalized so that those chromosomes with LOH had a copy number of one, Figure 1A.
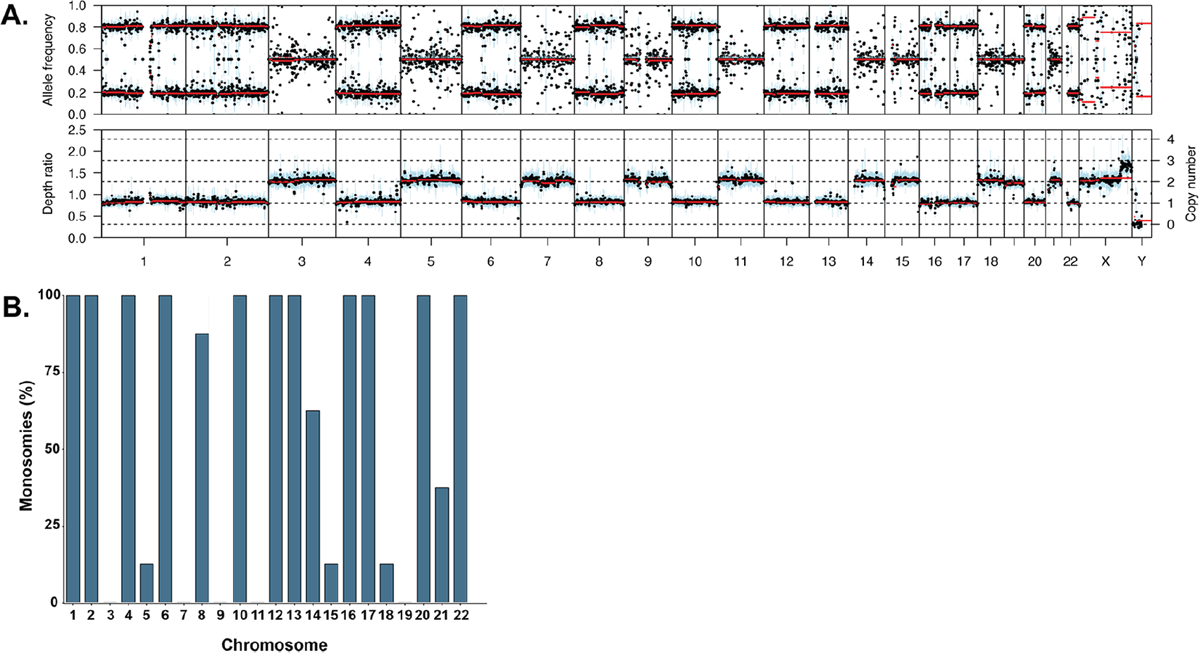
Figure 1: (A) Example B allele frequency plot showing retention of heterodisomy in the diploid chromosomes (upper) and the corresponding copy number plot (lower). (B) Percentage of monosomies per chromosome in the hyperhaploid samples (n=8).
The number of monosomies per sample varied from 12 to 14 and was more frequent on the chromosomes not associated with trisomies in hyperdiploid samples, namely chromosomes 1, 2, 4, 6, 10, 12, 13, 16, 17, 20, and 22, Figure 1B. Those chromosomes that are associated with trisomies in hyperdiploid myeloma were less likely to be monosomic, with the exception of chromosome 18. Chromosome 18 is not associated with hyperdiploidy but was disomic in 7/8 samples.
Hyperhaploidy is generated through sequential loss of chromosomes
By examining all samples and chromosomes it was apparent that hyperhaploidy does not occur randomly. There are three possible ways in which hyperhaploidy can be generated, Figure 2: 1) Halving of a diploid genome followed by gain of odd numbered chromosomes, 2) Gain of odd numbered chromosomes followed by loss of a haploid genome, and 3) Loss of chromosomes from a diploid state. The first method would result in genome-wide LOH, which we do not detect. The second method would result in LOH occurring on one-third of disomic chromosomes, due to loss of the “odd” allele. We do not see any LOH in the disomic chromosomes, so this mechanism is unlikely. The third method would result in retention of heterodisomy on the disomic chromosomes in all cases and is compliant with our data where all disomic chromosomes retain heterodisomy.
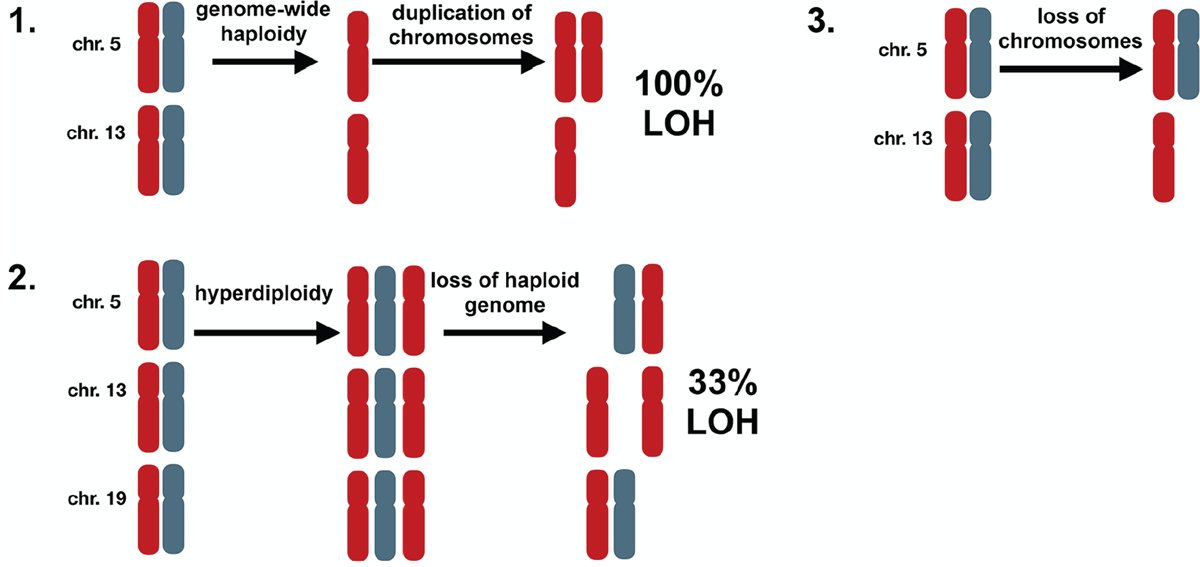
Figure 2: Possible mechanisms of generating hyperhaploidy. 1. Cells undergo genome-wide loss of chromosomes resulting in haploidy (n=1) followed by subsequent duplication of some chromosomes (n=1.4), resulting in loss of heterozygosity (LOH) on all chromosomes. 2. Cells first become hyperdiploid (n=2.4) before losing a haploid genome (n=1.4). In this instance paternal or maternal chromosomes are lost randomly resulting in diploid chromosomes with LOH 33% of the time. 3. Cells undergo loss of chromosomes resulting in hyperhaploid state (n=1.4) with retention of heterodisomy in all diploid chromosomes, which is consistent with our results.
In support of the third hypothesis, we sequenced serial samples from one patient which were taken 2 years apart. The first sample, taken from the left sacrum, had retention of autosomes 3, 5, 7, 9, 11, 15, 18, 19, and 21. However, the second sample taken from the iliac crest two years later had monosomy of chromosome 15, Figure 3, suggesting a gradual loss of chromosomes over time or between sites in the skeleton.
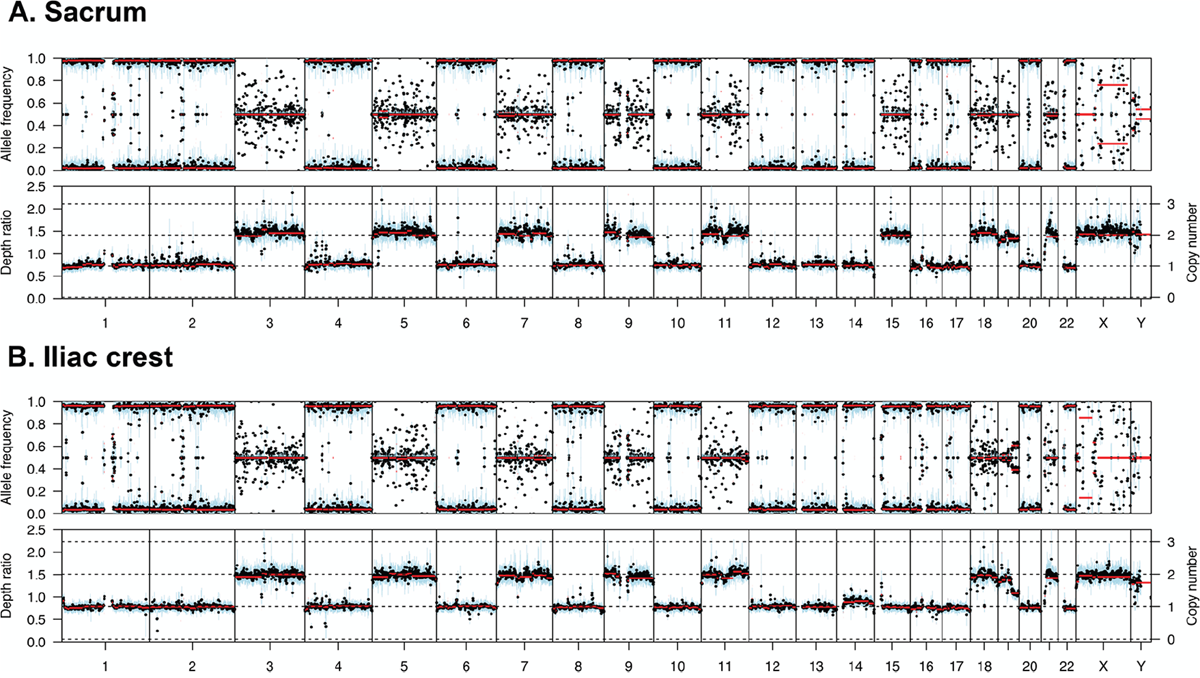
Figure 3: Copy number and B allele plots from two samples from the same patient. (A) A sample taken from the left sacrum shows retention of heterodisomy of chromosome 15. (B) A sample taken from the iliac crest two years after the sacrum sample shows monosomy of chromosome 15, indicating gradual loss of chromosomes over time.
Hyperhaploidy is associated with biallelic TP53 inactivation and NF-κB pathway activation
We examined the sequencing data for mutations that may be associated with hyperhaploidy. We found that all samples had a TP53 mutation, Table 1. The mutations were similar to those seen in other studies and were mostly clonal, with a median variant allele frequency of 0.92. Given that all the samples had monosomy of chromosome 17, the hyperhaploid samples all have biallelic inactivation of TP53, which is defining attribute of Double-Hit myeloma. Two of the samples had mutations in TP53 which were not clonal, with variant allele frequencies of 0.42 and 0.59 with deletion of chromosome 17. The presence of sub-clonal TP53 mutations with deletion of 17p suggests that deletion occurred first in these samples, followed by mutation.
Table 1: TP53 mutations detected in hyperhaploid samples
Patient ID |
Codon change |
cDNA position |
VAF |
|---|---|---|---|
13875 |
E287fs |
c.860-861insTCTTCCTCAGGTCCCCCCGGTGTAGGGA |
0.92 |
17188 |
V73fs |
c.216delC |
0.96 |
24138 |
LGFL111-114L |
c.334-342delGGCTTCTTG |
0.42 |
27082 |
F338fs |
c.1013delT |
0.95 |
27647 |
R273H |
c.818G>A |
0.92 |
35652 |
G244D |
c.731G>A |
0.59 |
MMRF_1364 |
K292fs |
c.873delG |
0.96 |
MMRF_1499 |
F134S |
c.401T>C |
0.89 |
We examined the samples for the presence of mutations in 63 previously identified driver genes in MM [12], and found a limited number of mutations. Out of the 63 genes, mutations were found in ATM, CDKN1B, CREBBP, CYLD, KMT2B, KRAS, MAX, TP53, and TRAF3. Mutations were highly clonal across all driver genes, with a median cancer cell fraction of 0.91, suggesting that mutations occurred early in disease pathogenesis. Apart from TP53, multiple mutations were seen in CYLD (n=4) and TRAF3 (n=2) implicating the NF-κB pathway as being important in hyperhaploid samples.
DISCUSSION
Here we have performed sequencing of a set of hyperhaploid MM patient samples, which define a high risk group with a poor prognosis and a 5-year survival rate of 23%, and account for approximately 0.25% of newly diagnosed myeloma cases [2]. Other genomic markers associated with poor prognosis include deletion of 1p32, 1q12, 17p, and gain of 1q21 [13]. Hyperhaploidy results in monosomies of chromosomes 1 and 17, potentially giving reason to the poor prognosis associated with this group. However, recent studies have also shown that biallelic inactivation of TP53, through either mutation or deletions, is also associated with a very poor prognosis [7, 9, 10].
We have recently described Double-Hit MM to include biallelic inactivation of TP53, and results in a median PFS of 15.4 months [10]. We have also shown that deletion of TP53 alone is not sufficient to result in poor outcome in several independent datasets [7, 10]. Here we have shown that hyperhaploid myeloma samples always have biallelic inactivation of TP53, which would explain the association with poor prognosis in this group, defining them as Double-Hit MM. The fact that the copy number abnormalities associated with hyperhaploidy include monosomy of chromosome 17 predisposes this group to a defined “first hit” upon which mutation of the remaining allele constitutes the “second hit”, driving pathogenesis and aggressive disease. This type of association between genetic markers can be described as an oncogenic dependency [12], in which the primary copy number changes prompt accrual of dependent secondary abnormalities, which in this case are mutations in TP53.
Hyperhaploidy is similar to hyperdiploidy in that the same set of autosomes have a higher copy number relative to the other chromosomes, with the exception of chromosome 18. Chromosome 18 retains disomy in hyperhaploid cells where we may expect monosomy based on predefined knowledge from hyperdiploid samples. The retention of chromosome 18 is seen in other hyperhaploid cancer types as well as MM, but the reason for this is unclear [4, 5, 14]. The similarity in the gain of odd numbered chromosomes between hyperhaploid and hyperdiploid states may suggest a common cell of origin, where hyperdiploid cells lose a haploid genome equivalent resulting in a hyperhaploid clone. However, previous studies have shown that no hyperdiploid clones were present in hyperhaploid samples [2]. Here we have shown retention of heterodisomy on all disomic chromosomes meaning that the hyperhaploid cells cannot have originated from a hyperdiploid cell, based on the chances of generating copy number neutral LOH. Therefore, we suggest that hyperhaploidy is generated through loss of chromosomes, which may happen as one catastrophic cell division or as successive loss of chromosomes. This method is similar to the proposed mechanism of gain of chromosomes in hyperdiploidy, in which samples may show a dominant clone where all cells have the same gains of chromosomes, or other samples may have subclones showing successive gains of chromosomes [15].
The NF-κB pathway is frequently activated in MM through inactivation of negative regulators of the pathway (e.g. CYLD, TRAF3, BIRC2, BIRC3) or over-expression of positive regulators (e.g. NIK) [16–18]. In this set of hyperhaploid MM samples we identified mutations in CYLD or TRAF3 in three patients (37.5%). We have previously shown that mutations in CYLD and TRAF3 are associated with non-hyperdiploid karyotypes with a t(4;14). In this respect, the hyperhaploid mutational spectra does not resemble hyperdiploidy, but has more in common with the high risk t(4;14) group, with more NF-κB mutations, and deletions of 1p, 13q, and 17p.
In conclusion, we have shown that hyperhaploid MM is a subgroup of Double-Hit MM, with biallelic inactivation of TP53, which results in a poor prognosis. The hyperhaploid karyotype results from loss of chromosomes, either in one catastrophic cell division or successive losses, and does not originate from a hyperdiploid clone.
MATERIALS AND METHODS
Patient sample selection
Samples previously identified as hyperhaploid by karyotyping were selected for whole exome sequencing. An additional hyperhaploid sample was identified through routine targeted sequencing. Samples had undergone CD138+ cell selection by either AutoMACS (Miltenyi) or RoboSep (Stem Cell Technologies) and DNA was extracted. Patient matched control DNA was also isolated from peripheral blood stem cell harvest samples.
Exome and targeted sequencing
DNA was prepped for sequencing using previously described protocols [11]. Briefly, 100 ng of DNA was fragmented, end-repaired, and adapters ligated using the HyperPlus kit (KAPA Biosystems). After PCR amplification the libraries were hybridized with probes against either the entire exome (MedExome, Nimblegen) or a targeted panel of 140 genes using SeqCap reagents (Nimblegen). Hybridized libraries underwent further amplification before being sequenced on a NextSeq500 (Illumina).
Sequence analysis
Targeted panel and Myeloma Genome Project (MGP) samples were analyzed in the manner described previously [12]. For MedExome samples, FastQC (v0.11.5) was used for basic quality control of Illumina paired-end sequencing data. Sequences were aligned to reference genome hg38 using BWA (v.0.7.17). Samples were de-duplicated using Picard Tools (v.1.85). Variants were called using Strelka (v2.8.3), variant annotation was provided by Variant Effect Predictor (v85), and filtered using fpfilter (https://github.com/ckandoth/variant-filter). Copy-number alterations were determined using Sequenza (v2.1.2.14). The median coverage for MedExome samples was 115x (range: 108-143x) and for the targeted panel sample was 213x. Data have been deposited at the European Genome-Phenome Archive under accession number EGAS00001003203.
Author contributions
BAW and CA were responsible for study design, data analysis, and manuscript preparation. CA, MAB, CPW, and YW were responsible for bioinformatic analysis and processing. RGT prepared and sequenced samples. CS, ST, MZ, FvR, FED, GJM, NW, SKJ, JRS provided input and/or patient material. All authors approved the final manuscript.
ACKNOWLEDGMENTS AND FUNDING
The authors would like to acknowledge the assistance of the Myeloma Center administration staff, including David Ashmore, Philip Farmer, Michael Rutherford, Nathan Petty, Sandy Mattox, Kristen Reynolds, and Blake Pond. Funding support for the CoMMpass dataset was provided by the Myeloma Genome Project. The CoMMpass dataset was generated by the Multiple Myeloma Research Foundation in collaboration with the Multiple Myeloma Research Consortium.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
REFERENCES
1. Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012; 12:335-48. https://doi.org/10.1038/nrc3257.
2. Sawyer JR, Tian E, Shaughnessy JD, Epstein J, Swanson CM, Stangeby C, Hale CL, Parr L, Lynn M, Sammartino G, Lukacs JL, Stein C, Bailey C, et al. Hyperhaploidy is a novel high-risk cytogenetic subgroup in multiple myeloma. Leukemia. 2017; 31:637-44. https://doi.org/10.1038/leu.2016.253.
3. Hoctor VT, Campbell LJ. Hyperhaploid plasma cell myeloma. Cancer Genet. 2012; 205:414-8. https://doi.org/10.1016/j.cancergen.2012.05.004.
4. Holzmann C, Löning T, Bullerdiek J. Hyperhaploid uterine mesenchymal tumors-a novel genetic subgroup? Cancer Genet. 2016; 209:278-81. https://doi.org/10.1016/j.cancergen.2016.02.005.
5. Mandahl N, Johansson B, Mertens F, Mitelman F. Disease-associated patterns of disomic chromosomes in hyperhaploid neoplasms. Genes Chromosomes Cancer. 2012; 51:536-44. https://doi.org/10.1002/gcc.21947.
6. Harrison CJ, Johansson B. Acute lymphoblastic leukemia. In: Heim S and Mitelman F, eds. Cancer cytogenetics: Wiley-Blackwell. 2009; pp. 233-96.
7. Thanendrarajan S, Tian E, Qu P, Mathur P, Schinke C, van Rhee F, Zangari M, Rasche L, Weinhold N, Alapat D, Bellamy W, Ashby C, Mattox S, et al. The level of deletion 17p and bi-allelic inactivation of TP53 has a significant impact on clinical outcome in multiple myeloma. Haematologica. 2017; 102:e364-e7. https://doi.org/10.3324/haematol.2017.168872.
8. Chavan SS, He J, Tytarenko R, Deshpande S, Patel P, Bailey M, Stein CK, Stephens O, Weinhold N, Petty N, Steward D, Rasche L, Bauer M, et al. Bi-allelic inactivation is more prevalent at relapse in multiple myeloma, identifying RB1 as an independent prognostic marker. Blood Cancer J. 2017; 7:e535. https://doi.org/10.1038/bcj.2017.12.
9. Weinhold N, Ashby C, Rasche L, Chavan SS, Stein C, Stephens OW, Tytarenko R, Bauer MA, Meissner T, Deshpande S, Patel PH, Buzder T, Molnar G, et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood. 2016; 128:1735-44. https://doi.org/10.1182/blood-2016-06-723007.
10. Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies F, Rosenthal A, Wang H, Qu P, Hoering A, Samur M, Towfic F, Ortiz M, et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia. 2019; 33:159-170. https://doi.org/10.1038/s41375-018-0196-8.
11. Walker BA. Whole Exome Sequencing in Multiple Myeloma to Identify Somatic Single Nucleotide Variants and Key Translocations Involving Immunoglobulin Loci and MYC. Methods Mol Biol. 2018; 1792:71-95. https://doi.org/10.1007/978-1-4939-7865-6_6.
12. Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies FE, Rosenthal A, Wang H, Qu P, Hoering A, Samur M, Towfic F, Ortiz M, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood. 2018; 132:587-97. https://doi.org/10.1182/blood-2018-03-840132.
13. Walker BA, Leone PE, Chiecchio L, Dickens NJ, Jenner MW, Boyd KD, Johnson DC, Gonzalez D, Dagrada GP, Protheroe RK, Konn ZJ, Stockley DM, Gregory WM, et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood. 2010; 116:e56-65. https://doi.org/10.1182/blood-2010-04-279596.
14. Nord KH, Paulsson K, Veerla S, Wejde J, Brosjo O, Mandahl N, Mertens F. Retained heterodisomy is associated with high gene expression in hyperhaploid inflammatory leiomyosarcoma. Neoplasia. 2012; 14:807-12.
15. Pawlyn C, Melchor L, Murison A, Wardell CP, Brioli A, Boyle EM, Kaiser MF, Walker BA, Begum DB, Dahir NB, Proszek P, Gregory WM, Drayson MT, et al. Coexistent hyperdiploidy does not abrogate poor prognosis in myeloma with adverse cytogenetics and may precede IGH translocations. Blood. 2015; 125:831-40. https://doi.org/10.1182/blood-2014-07-584268.
16. Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, Van Wier S, Tiedemann R, Shi CX, Sebag M, Braggio E, Henry T, Zhu YX, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007; 12:131-44.
17. Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, Lenz G, Hanamura I, Wright G, Xiao W, Dave S, Hurt EM, Tan B, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007; 12:115-30.
18. Morgan GJ, He J, Tytarenko R, Patel P, Stephens OW, Zhong S, Deshpande S, Bauer M, Weinhold N, Schinke C, Rasche L, Bailey M, Ali S, et al. Kinase domain activation through gene rearrangement in multiple myeloma. Leukemia. 2018; 32:2435-2444. https://doi.org/10.1038/s41375-018-0108-y.