
INTRODUCTION
Microsatellite instability (MSI) colorectal cancer is a hypermutable subclass due to mismatch repair (MMR)-deficiency. It includes two types; Lynch syndrome (LS) caused by germline mutations in the MMR protein genes, and sporadic disease without hereditary background but with somatic mutations and/or epigenetic changes in the responsible gene [1]. These mutations result from unrepaired replication errors including repetitive DNA sequences, causing short insertion-deletion (indel) mutations in mono- and di-nucleotide repeats, as well as other types of mutations [2, 3]. Therefore, MMR-deficient colorectal cancer subclass gives a strong indication for immune checkpoint therapy using anti–PD-1 or –PD-L1 antibodies, with remarkable clinical efficacy [4].
While this subclass takes up 5–15% of colorectal cancer cases, this proportion can vary not only among geographic populations, but also depending on the accuracy of diagnostic methods [5]. Currently, two major methods are employed. One is immunohistochemistry (IHC) of MMR proteins as adopted by many institutions as the standard test [5, 6]. It aims to detect lack of MMR protein(s) as MLH1, MSH2, MSH6, and/or PMS2 [7, 8], which can be misleading partly because the diagnosis depends on ‘loss’ of staining in cancer cells among co-existing stromal cells that show normal levels of expression.
The other is based on the analyses of tumor genome that shows microsatellite instability (MSI), increased mutational burden and mononucleotide repeat frameshift mutations in some key coding genes [9, 10]. In these DNA analyses, tumor DNA is usually extracted from either frozen or, more commonly, formalin-fixed paraffin-embedded (FFPE) tumor tissues resected surgically [11]. While laser-capture microdissection of frozen or FFPE samples can isolate cancer cell-enriched areas, this technique is labor-intensive, and may not be suitable for clinical screening service. In practice, macrodissection of cancer cells is widely used [4]. However, the purity of cancer DNA thus isolated can be compromised because the tumor tissues contain not only cancer epithelial cells, but also non-cancer stromal cells of which wild-type DNA may confuse the diagnosis [12]. Furthermore, DNA samples from FFPE tissues are chemically damaged significantly, often making the analysis difficult.
To overcome this problem of mixed cell population that composes the cancer tissue, it would be ideal if we can isolate only the cancer epithelial cells. To this end, it has become possible recently to culture and propagate the tumor-initiating cells (TICs, or cancer stem cells) in 3D matrix in vitro [13, 14]. This technique has provided not only cancer- but also normal epithelial-stem cells under rapidly growing conditions. Besides, it eliminates the stromal cells in the culturing process. Our current success rate for spheroid establishment in cancer- and normal epithelial-stem cells are ~90% and 100%, respectively [14]. Because the majority of colorectal cancer patients undergo resection operations of the primary tumors, fresh tissue samples are usually available except for very early or inoperable late stages. Cancer spheroid cultures offer us excellent test materials for genomic and expression analyses as well as for immunohistochemical staining of MMR proteins. Furthermore, we have just reported that chemosensitivity of spheroid-derived xenografts accurately reflects that of the clinical response [15]. To prepare for prospective studies, we are currently culturing colorectal cancer spheroids of all resected stage III/IV primary tumors at Kyoto University Hospital. This particular report shows one of these efforts that take the advantage of available spheroid cultures. In the present study, we have investigated the feasibility and reliability of exploiting cultured spheroids as the source material for MSI diagnosis.
RESULTS
Detection of microsatellite instability (MSI) in colorectal cancer using on-chip electrophoresis of satellite marker PCR products from spheroid-derived DNA
We recently established spheroid cultures of colorectal cancer tumor-initiating cells (TICs, or cancer stem cells) as well as those of their normal epithelial stem cells from 111 specimens surgically resected at Kyoto University Hospital according to the protocol reported previously [13]. Clinicopathological characteristics of the patients are summarized in Table 1.
Table 1: Clinicopathological characteristics of patients
n = 110a |
||
|---|---|---|
Sex |
Male |
65 |
Female |
45 |
|
Age, years |
Median (MIN–MAX) |
69 (36–89) |
Amsterdam criteria |
0 |
|
Bethesda criteria |
0 |
|
Past history |
Lynch associated cancers of the colon, rectum stomach, uterine endometrium, small bowel and/or urinary tract |
10 |
Familial adenomas polyposis |
0 |
|
Tumor location |
Right side |
38 |
Left side |
43 |
|
Rectum |
30 |
|
Tumor stage |
1 |
11 |
2 |
48 |
|
3 |
36 |
|
4 |
16 |
|
Histological grade |
Low grade |
102 |
High grade |
4 |
|
Mucinous |
5 |
|
No. of passages from spheroid establishment to extract DNA samples |
||
Median (MIN–MAX) |
5 (1–22) |
|
aOne patient had synchronous double cancer in sigmoid and transverse colon.
To improve molecular diagnosis of microsatellite instability (MSI) colorectal cancer, we employed an on-chip MSI test [16], and examined the above 111 cases using DNA samples purified from cultured spheroid cells. To this end, we first extracted DNA from both cancer and normal epithelial spheroids of the same patients, and amplified by PCR five MMR-target microsatellite markers recommended in the Bethesda panel [1]; BAT25, BAT26, D2S123, D5S346, and D17S250. We then analyzed the PCR products by microfluidics-based on-chip electrophoresis in an Agilent Bioanalyzer (see Materials and Methods), and compared their electropherogram overlays regarding the peak positions between the cancer and normal cell DNA. This on-chip assay provides a higher resolution because of the shorter running time than the conventional fluorochrome-based assay (i.e., capillary electrophoresis) widely used in MSI test [16]. As indel mutations generated by DNA strand slippage during replication remained unrepaired in the genomic DNA of MMR-deficient cancer cells, the peak positions of their electropherograms often shifted from those of the normal epithelial cell DNA of the same patient (Figure 1A, HC26T and HC4T). In microsatellite-stable (MSS) cancer cells, the peak positions of the PCR products were identical between cancer and normal cells, as expected (Figure 1A, HC51T). We also show better resolution and lower noise obtained by the on-chip MSI electrophoresis of spheroid DNA in Figure 1A. For all satellite markers tested, the peak heights of PCR products amplified from spheroid-derived DNA were significantly higher than those of FFPE tissue-derived DNA (Figure 1B and 1C), which helped identify the difference between cancer and normal cell peaks.
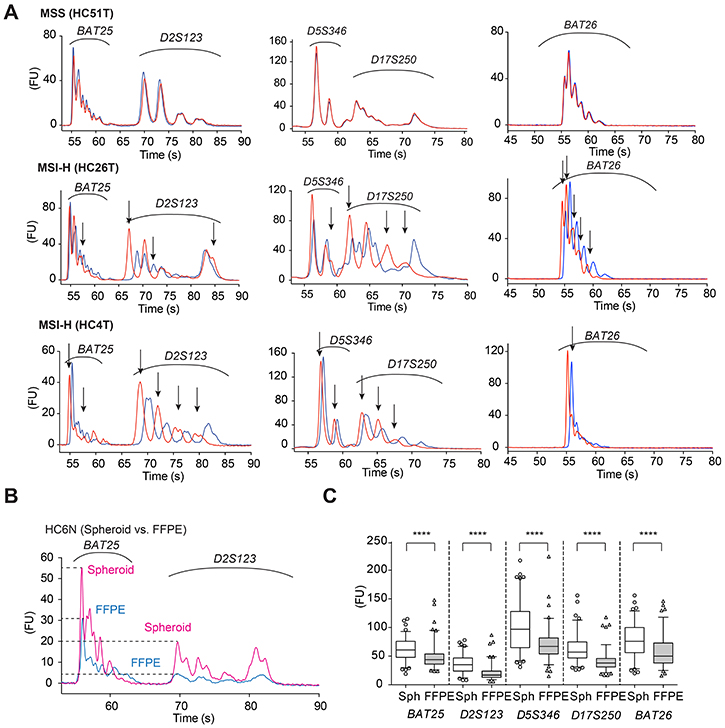
Figure 1: Electropherograms of on-chip MSI analysis using spheroid-derived DNA samples. (A) HC51T. Representative electropherograms of an MSS case for five microsatellite markers of the Bethesda panel. Red lines show the PCR products amplified from the cancer cell spheroid DNA samples, whereas blue lines indicate those from the normal epithelial stem cell DNA of the same patient. HC26T and HC4T. Representative electropherograms of MSI-H cases. The peak patterns between tumor-initiating cells (red) and the normal epithelial stem cells (blue) are separated for all loci tested. The ordinate shows fluorescence intensity in arbitrary unit (FU). (B) On-chip electropherogram of a representative case in which FFPE tumor-derived DNA sample (blue) showed much lower peaks with poorer resolution than spheroid-derived DNA (red) of the same patient normal mucosal stem cells (HC6N). The ordinate shows fluorescence intensity in arbitrary unit (FU). (C) Maximum electrophoretic peak-heights of PCR-amplified MSI markers (shown at bottom) compared between spheroid- and FFPE tumor-derived DNA samples. Note that spheroid-derived (Sph) DNA gave taller peaks than FFPE tumor-derived (FFPE) DNA for all five Bethesda panel markers. ****P < 0.0001, two-tailed Mann-Whitney U test (n = 99).
We diagnosed the MSI status according to the Bethesda criteria that recommended calling MSI-high (MSI-H) when more than 30% of cancer cell microsatellite markers showed different peaks from normal cell markers [2]. Namely, in the set of five markers above, we designated MSI-H when two or more had shifted cancer peaks (e.g., Figure 1A, HC26T and HC4T). When only dinucleotide repeat markers as D2S123, D5S346 and/or D17S250 were mutated, a secondary panel of markers with mononucleotide repeats (BAT40 and MYCL) was tested as recommended by the revised Bethesda criteria [1]. In some exceptional cases such as HC106T, only one marker (D2S123) showed clear shifts on peaks that reminded us of typical indel mutations. Accordingly we then tested two more satellite markers, and finally diagnosed the case as MSI-H (Supplementary Figure 1). Based on these results, we detected total of seven (6.3%) MSI-H cases and 104 MSI-L or MSS cases (30 MSI-L and 74 MSS).
Mutational burden assessed by next generation sequencing (NGS) of spheroid-derived DNA
In MMR-deficient colorectal cancer, it is known that genomic mutational burden increases not only because of dinucleotide and mononucleotide repeats described above, but also due to increased mutations of various kinds [17, 18]. One of the methods to assess such a condition is to estimate the density of mutations. It has been reported that the whole genome mutational burden can be estimated in coding sequence subsets [19–21]. Thus, we sequenced the coding regions of 409 cancer-related genes spanning 1.29 Mb using cancer spheroid DNA samples (see Materials and Methods).
As described in Materials and Methods, however, this analysis was not so simple practically as it appeared in the concept. Although it was ideal to use spheroid DNA from the matched normal colonic epithelial cells of the same patients as references for cancer cells, this doubled the sequencing cost. As a more cost-efficient alternative, we referred to a database and eliminated polymorphic variants commonly found in the geographic population (see Materials and Methods, and Supplementary Figure 2). Just in case, we also sequenced the matched normal epithelial spheroid DNA for two MSI-H cases, and used the data as additional references. These results were similar to those obtained by the alternative method using a normal population variant database (Supplementary Figure 3).
Using the variant-filtering method (Supplementary Figure 2), we searched for cancer-driving mutations in seven MSI-H and 11 MSI-L/MSS cases (Supplementary Tables 1 and 2, respectively). Consistent with an earlier report [4], the results showed significantly higher mutational densities in the MSI-H tumor genome than in the MSS, regarding both total mutations (median, 65 vs. 8; p < 0.0001) and indel mutations (median, 11 vs. 1; p < 0.0001) (Figure 2A, left and right, respectively). Specific indel mutations found in the sequenced genes are shown in Figure 2B and 2C. In the MSI-L or MSS cases, mutations of tumor DNA were found typically in APC and/or TP53 whereas, in MSI-H tumors, they were in a variety of cancer-related genes.
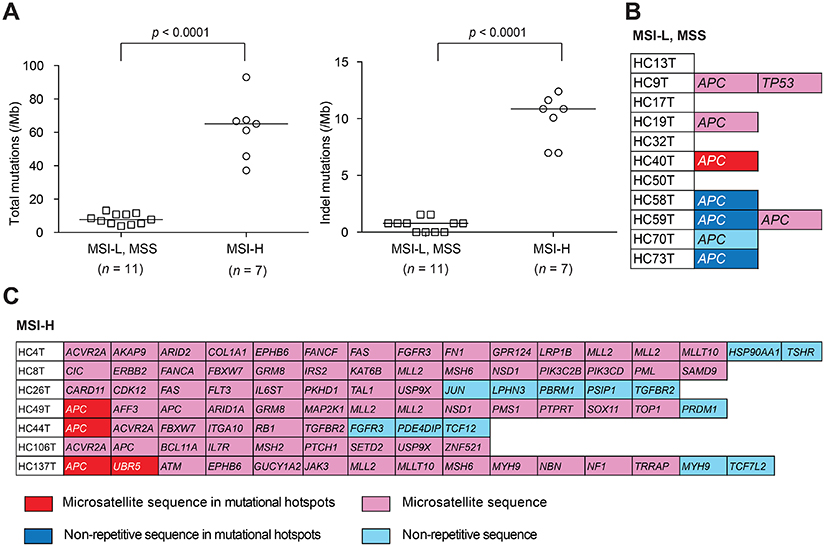
Figure 2: Mutational burden estimation. (A) Total (left) and indel (right) mutational densities of MSI-H colorectal cancer spheroid DNA samples compared with those of MSI-L or MSS spheroid determined by NGS for 409 cancer-related genes spanning 1.29 Mb of the coding sequences. (B, C) List of indel mutations detected in the coding regions of 409 cancer-related genes in 11 MSI-L or MSS colorectal cancer spheroid DNA samples (B) and in seven MSI-H cases (C). Color codes in (B, C) indicate the locations of mutations: Red, in microsatellite sequences of mutational hotspots; Pink, in microsatellite sequences; Dark blue, in non-repetitive sequences of mutational hotspots; Light blue, in non-repetitive sequences.
In addition, we sequenced FFPE cancer tissue-derived DNA from two MSI-H cases, HC4T and HC49T for 409 cancer related genes (Supplementary Figure 4). The quality of the FFPE tumor DNA data were significantly lower than those of spheroid DNA, although the overall numbers of mutations/variants appeared similar. For example, in colon cancer HC4T, we found 12 of 79 variants were specific to FFEP DNA in addition to 67 common variants including three key mutations (in BRAF, MTOR and RUNX1; Supplementary Figure 4). However, the allelic frequencies for 11 of the 12 were < 40%. Such low frequencies were likely caused by the co-existing stromal cells in the FFPE tumor tissues. This is in a strong contrast to 20 (23%) variants that were specific to the spheroid DNA of which allelic frequencies for 16 of the 20 variants were higher than 40%, suggesting that they represent the majority of cancer cells. Accordingly, the same ambiguity can remain even if frozen cancer tissues are used as the DNA source instead of FFEP tumors. In this regard, some recent studies reported poor tumor cell purity in cancer tissue samples [12, 22, 23], one of which demonstrated low tumor cell contents that reduced the mutation frequency for key cancer mutations to < 5% [12]. Another study reported the discordance of sequencing data between cultured patient-derived cancer cells and primary tumors [23]. The low tumor purity samples reduced mutational frequency below the variant-calling limit in high-throughput DNA sequencing. Therefore, it appears that the difference in the DNA source can affect the accuracy of cancer diagnosis significantly.
Direct sequencing of spheroid-derived DNA for MMR-target key coding genes
While mutations in the non-coding satellite markers of MMR targets can be detected by on-chip electrophoresis, most key functional targets that are affected in cancer by MMR deficiency reside in mononucleotide repeats in the coding sequences, often leaving frameshift mutations unrepaired in MSI-H cancer cells [5, 18]. To investigate whether above on-chip electrophoresis results were consistent with the mutational status in such MMR targets, we selected four representative genes; TGFBR2 (A10), BAX (G8), IGF2R (G8), and CASP5 (A10). After amplifications by PCR on cancer spheroid DNA, we directly sequenced the products for mutations in the mononucleotide repeats (see Materials and Methods).
Because of the high purity of spheroid DNA both chemically and cell population-wise, it allowed us to detect not only homozygous but also heterozygous mutations in the MMR-target repeats by Sanger sequencing of the PCR products. For example, A10/A10 of TGFBR2 mutated into A10/A9 is ending as an A/G superimposed peak (Figure 3A, TGFBR2). Beyond this position, all nucleotide peaks of the wild-type DNA strand superimposed with those of the mutated strand down to the first frameshift termination codon. Other representative sequence data for BAX, IGF2R and CASP5 are also shown (Figure 3A). We noted that these mutations were often eliminated by variant calling software although they had remained in the raw data (Supplementary Figure 2).
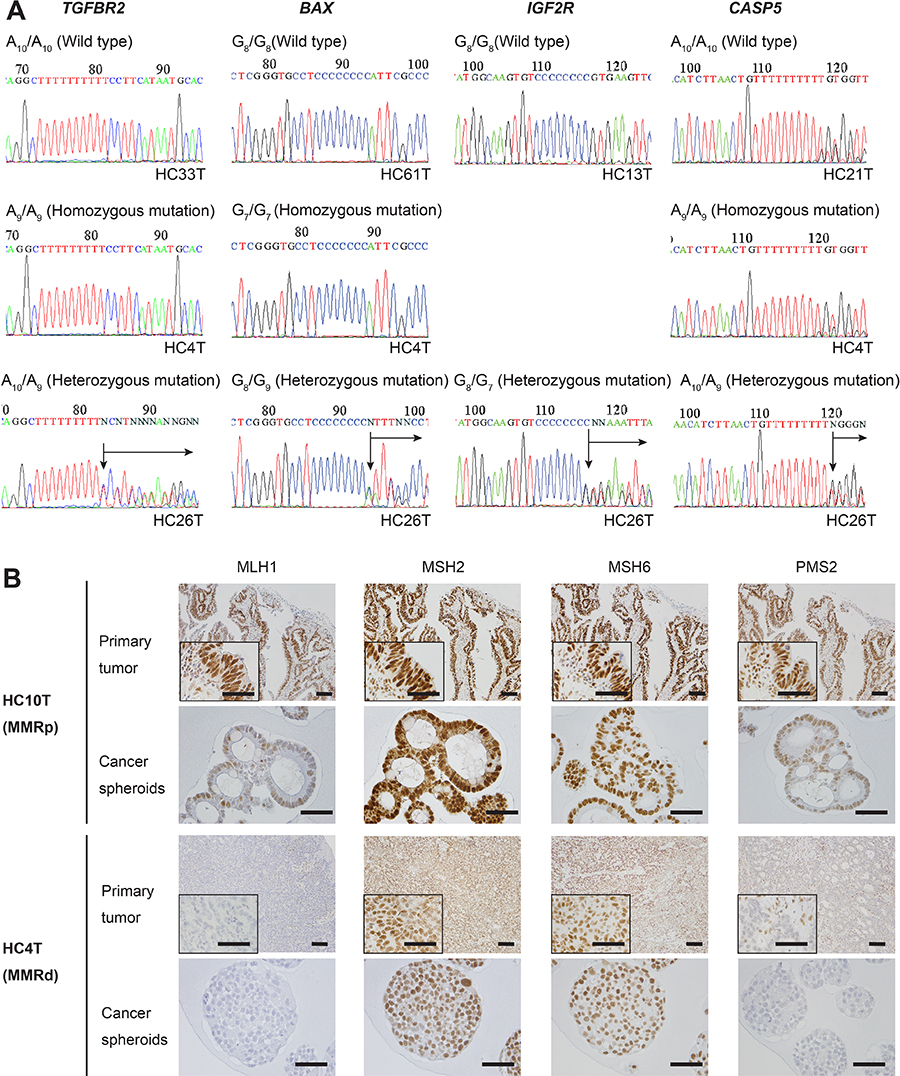
Figure 3: DNA sequencing analysis of some MMR-target coding mononucleotide repeats, and IHC for MMR proteins in primary colorectal cancer tissues or spheroid samples in MSI-H and MSI-L/MSS colorectal cancer cases. (A) DNA sequencing data that cover the mononucleotide repeat regions amplified from the spheroid-derived DNA samples for TGFBR2, BAX, IGF2R, and CASP5. For all markers, the reverse strands of the wild types (top panels), and homozygous (middle) and heterozygous (bottom) mutants are shown. No cases were found with IGF2R homozygous mutation. (B) Immunohistochemistry for mismatch repair (MMR) proteins, MLH1, MSH2, MSH6, and PMS2. The colorectal cancer (CRC) HC10T (top panels) shows a MMR-proficient (MMRp) example of the primary CRC section (Primary tumor), with expression of all MMR proteins. The CRC HC4T (bottom panels) shows a MMR-deficient (MMRd) case. Expression of MLH1 and PMS2 was not detected in the primary tumor tissue. Notably, IHC of spheroid cells derived from the same tumors mirrored the results with the whole tumor sections. Note that some staining of MLH1 and PMS2 in HC4T primary CRC sections is in the stromal cell nuclei.
By this method, we detected frameshift mutations in all seven MSI-H cases (100%) regarding TGFBR2, whereas in six cases (86%) for BAX and CASP5, and in three (43%) for IGF2R. Notably, we did not detect such mutations in any of the MSI-L or MSS spheroids regarding TGFBR2 (104 cases tested) or BAX (49 cases tested) (Figure 4). These results strongly suggest that our on-chip electrophoresis of microsatellites was as accurate as direct sequencing of the key MMR-target coding genes (see below).
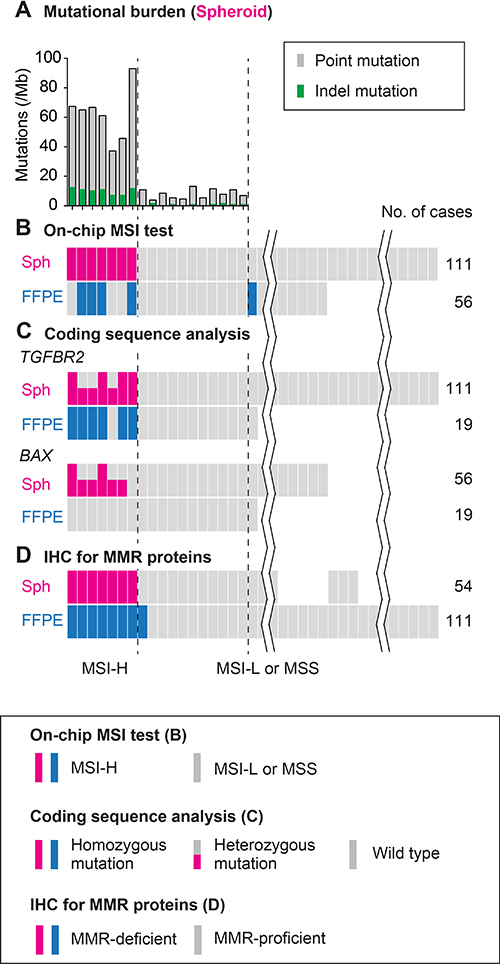
Figure 4: Schematic summary of the analysis results using colorectal cancer spheroids compared with those using FFPE tumors. (A) Mutational burden estimated by exonic sequencing of spheroid DNA for 409 cancer related genes. (B) MSI status judged by on-chip electrophoresis of the Bethesda panel markers. (C) Mutations in the mononucleotide repeats in coding regions of TGFBR2 and BAX determined using spheroid and FFPE tumor DNA. (D) IHC results of the cultured spheroids of tumor-initiating cells and FFPE primary tumors. Color keys are shown in boxes. See text for details.
Immunohistochemistry (IHC) for mismatch repair proteins on FFPE tumor tissues and on cancer spheroids
For clinical diagnosis of MMR-deficient colorectal cancer, it is common to detect loss of MMR proteins in tumor tissue sections by immunohistochemistry (IHC). To investigate how tight the correlation is between the MSI status determined using spheroid DNA and that by MMR protein expression, we next performed immunohistochemistry (IHC) on four MMR proteins, MLH1, MSH2, MSH6, and PMS2 [7] in the primary tumor sections of all 111 cases. Some staining data for the representative cases are shown (Figure 3B). As the result, we detected eight MMR-deficient cases and 103 MMR-proficient ones. Of seven MSI cases, five cases lost both MLH1 and PMS2 protein expression, and were assumed as sporadic cancer [25]. Remaining one case was confirmed for loss of MSH6 expression (HC137T), whereas the other for loss of PMS2 (HC8T), both of which were suspected of Lynch syndrome (Table 2). However we did not pursue further diagnostic tests. Notably, an MSS case with low mutational burden in spheroid DNA genomic analyses appeared as MMR-deficient by IHC (see HC13T below), although this difference of one in 111 cases was too small for statistical significance.
Table 2: MSI-H colon cancer cases assessed by (1) on-chip electrophoresis of five satellite markers, (2) DNA sequencing of four exoninc MMR deficiency target mononucleotide repeats, and (3) IHC for four MMR proteins MLH1, MSH2, MSH6 and PMS2
Case |
On-chip |
Sequencing of MMR deficiency target genesc |
IHC |
|||||
|---|---|---|---|---|---|---|---|---|
MSI statusa |
MSI statusa |
TGFBR2 |
IGF2R |
BAX |
CASP5 |
Absent proteinsd |
Absent proteinsd |
|
HC4T |
H |
L |
A9/A9 |
WT/G7 |
G7/G7 |
A9/A9 |
MLH1, PMS2 |
MLH1, PMS2 |
HC8Te |
H |
H |
WT/A9 |
WT/G9 |
WT/G7 |
A9/A9 |
PMS2f |
PMS2f |
HC26T |
H |
H |
WT/A9 |
WT/G7 |
WT/G9 |
WT/A9 |
MLH1, PMS2 |
MLH1, PMS2 |
HC49T |
H |
H |
A9/A9 |
WT/WT |
G7/G7 |
WT/A9 |
MLH1, PMS2 |
MLH1, PMS2 |
HC44T |
H |
L |
WT/A11 |
WT/WT |
WT/G9 |
WT/A9 |
MLH1, PMS2 |
MLH1, PMS2 |
HC106T |
H |
L |
A8/A9 |
WT/WT |
WT/G9 |
WT/WT |
MLH1, PMS2 |
MLH1, PMS2 |
HC137T |
H |
H |
A9/A9 |
WT/WT |
WT/WT |
A8/A9 |
MSH6 |
MSH6 |
Mutation frequency |
100% |
43% |
86% |
86% |
||||
aH: MSI-High, L: MSI-Low
bSpheroid
cOnly spheroid DNA data were collected.
dAbsent MMR proteins in immunohistochemical staining
eMLH1 V384D mutation (see Discussion, Results and Supplementary Figure 7)
fPMS2 R563X mutation
Colon cancer HC13T was diagnosed as MSS by three genomic analysis methods (on-chip MSI test, mutational burden assessment, and four MMR target mononucleotide repeats) using spheroid DNA derived from two independent clones as well as one FFPE tumor-derived DNA (another DNA sample showed MSI-L) (Supplementary Figure 5). On the other hand, IHC of cancer spheroid cells of two independent spheroid clones demonstrated contradictory results; one MMR-proficient with all four MMR proteins, and the other without MLH1 and PMS2 (Supplementary Figure 6A). Accordingly, we examined 14 sub-legions of the primary tumor for MLH1, MSH6 and PMS2. As the results, IHC for 7 of 13 sub-lesions were evaluated as MMR-deficient because both MLH1 and PMS2 appeared missing (Supplementary Figure 6B, 6C and Supplementary Table 3). Regarding this discrepancy between the DNA and IHC data, it is worth noting that the patient was treated with oxaliplatin before the surgery. It has been reported that platinum-based chemotherapeutics can affect expression of MLH1 and MSH2 in IHC [26], and induce the methylation of MLH1 gene in cancer cells [27]. These results suggested that lack of MLH1 and PMS2 staining in this particular tumor was caused by platinum chemotherapy. Because this took place shortly before the surgery, it is reasonable to speculate that lack of MMR proteins did not accumulate mutations significantly in the whole genome. Accordingly, we concluded that the tumor HC13T was originally MSS, and that the MSI phenotype with IHC was caused by oxaliplatin neoadjuvant therapy. Thus, this particular cancer was not expected to contain enough neoantigens to respond to immune therapies.
It is worth noting that MSI tumor HC8T (Table 2) lacked PMS2 staining and had heterozygous R563X mutation (Supplementary Figure 7). Interestingly, the tumor was immunostained for MLH1, and contained variant mutation V384D [28]. As reported, the MMR phenotype of MLH1 V384D mutation can vary [28]. Here, we found four cases of V384D mutant tumor that ranged from MSI-H (HC8T above) to MSI-L (HC20T; a double cancer with HC8T) or MSS (HC25T and HC34T; Supplementary Figure 7). In IHC of both the primary tumor and spheroid samples, MLH1 immunostaining was absent (–) in HC25T, moderate in HC8T (+) as described above, and positive (++) in HC20T and HC34T. Importantly, immunostaining for PMS2 that forms a complex with MLH1 [8] was positive in all these cases except HC8T that was MSI-high. It is conceivable that MLH1 V384D mutation affected its antigenic epitope detected by the antibody used here and/or its interaction with PMS2.
Altogether, these results of the four methods led us to conclude that seven of 111 cases (6.3%) were diagnosed as MSI-H, although eight were MMR-deficient as described above. As reported earlier, IHC may cause difficulty in diagnosis [29] and spheroid DNA analysis can provide more accurate diagnosis (Figure 4, and Table 2).
Spheroids provide better quality DNA more suitable for MSI diagnosis than FFPE-tumors
In medical research as well as in clinical services, DNA analyses are often performed on tumor DNA extracted from formalin-fixed paraffin-embedded (FFPE) tissues. To evaluate the quality of DNA samples derived from FFPE tumors compared with that from colorectal cancer spheroids, we performed on-chip MSI tests using DNA from both sources for 50 tumors. To this end, we first assessed the chemical purity of DNA samples obtained from spheroids and FFPE tumors by reading the A260/A280 ratio. They were 1.8 and 1.7, respectively, both showing satisfactory purity for further analyses though the difference was statistically significant. We then estimated the DNA chain length by agarose gel electrophoresis. Compared with the spheroid-derived DNA samples, the FFPE tumor DNA showed smears trailing into smaller chain lengths, indicating substantial fragmentation due to chemical damages by formalin exposure (Figure 5A).
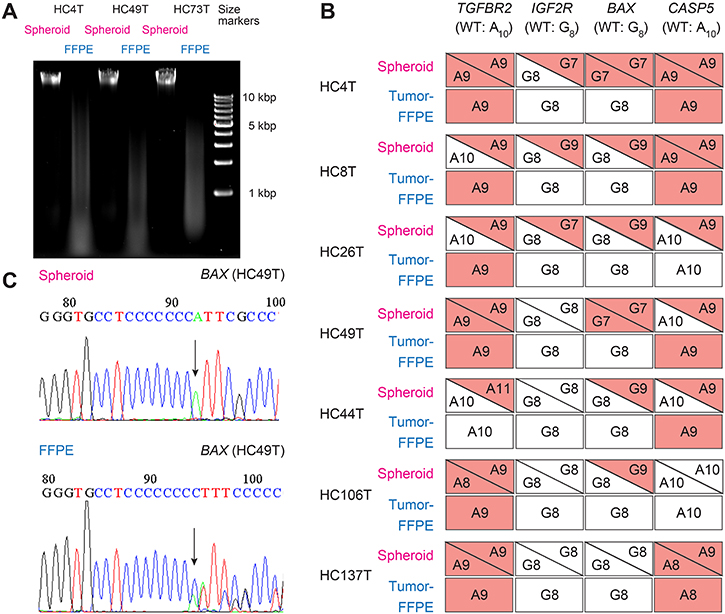
Figure 5: Comparison of cancer spheroid- and FFPE tumor-derived DNA samples in sequence analysis of MMR-target coding mononucleotide repeats. (A) Agarose gel profile comparing the DNA samples from spheroid and FFPE tumors. (B) Mutation profiles of coding mononucleotides in four MMR-target genes for seven MSI cases analyzed with spheroid- and FFPE tissue-derived DNA samples. Red boxes/triangles indicate mutated alleles, whereas white ones show the wild-type. Analysis with spheroid-derived DNA enabled unambiguous identification of both alleles. However, FFPE tissue-derived DNA often gave confusing results. (C, top) An example of homozygous mononucleotide repeat mutation in BAX (G8 → G7), detected using the spheroid-derived DNA. Arrow points 8th G → T (C → A in reverse strand sequenced here). (bottom) With the FFPE tissue-derived DNA, a slightly low peak for the 8th G was detected after seven G peaks (C in reverse strand here). This is likely derived from DNA of the normal (i.e., wild-type) stromal cells rather than cancer epithelial cells.
We next tested the five satellite markers by on-chip electrophoresis. With the spheroid-derived DNA samples of 50 tumors, we diagnosed five cases (10%) as MSI-H, and 45 cases as MSI-L or MSS. With the FFPE-tumor DNA, on the other hand, we detected only three (6.0%) MSI-H cases, misdiagnosing two MSI-H cases as MSI-L, whereas one MSS as MSI-H. Namely, two cases (HC44T and HC4T) were called as MSI-L with FFPE tumor DNA despite that they were MSI-H with the spheroid DNA. By adding the results of two more satellite markers (i.e., BAT40 and MYCL1) in the on-chip electrophoresis, both samples showed more than 30% (i.e., three of seven) unstable markers even with the FFPE tumor DNA. The results of direct sequencing analysis of spheroid DNA for four mononucleotide-repeat markers (i.e., TGFBR2, IGF2R, BAX, and CASP5) confirmed their final diagnosis as MSI-H (Supplementary Figure 8, Figure 5B, and Supplementary Table 3). These additional assessments were performed to resolve discrepant MSI status resulted from the data with spheroid and FFPE DNA samples. Thus, analysis on spheroid-derived DNA allowed us to promptly detect all MSI-H cases according to the Bethesda criteria with five markers (1).
On the other hand, case HC24T was diagnosed as MSI-H with the FFPE tumor DNA by on-chip electrophoresis of the five marker-panel despite it was MSS with the spheroid-derived DNA (Supplementary Figure 9A and 9B, FFPE1). Upon sequencing of spheroid DNA for the four MMR-target mononucleotide repeats, all markers were homozygous wild type (Supplementary Table 3). Interestingly, when we analyzed by on-chip MSI test the FFPE tumor DNA samples extracted from two more separate subregions of the HC24T tumor, both turned out as MSI-low or MSS (Supplementary Figure 9B, FFPE2 and 3). Thus, we diagnosed this particular case as MSS (Supplementary Table 3; see also the IHC results). One of the conceivable causes for this inconsistency regarding HC24T was the poor resolution of the electropherogram with the FFPE tissue-derived DNA as we described above, although the possibility of tumor microheterogeneity could not be excluded thoroughly.
Accordingly, the sensitivity (the MSI-H frequency by on-chip tests out of the cases diagnosed firmly by DNA sequencing and IHC analyses) and specificity (the frequency of the true MSI-H cases out of the cases designated as MSI-H by on-chip tests including false positives) of our on-chip MSI test with spheroid-derived DNA samples were both 100%, whereas those with the FFPE tissue-derived DNA was 60% and 98%, respectively. Thus, these results showed that spheroid-derived DNA samples provided more reliable data than FFPE tumor DNA.
To compare the quality of the DNA samples from the two sources further, we analyzed the MSI-H cases regarding the mononucleotide-repeat sequences of the four target genes that could be affected by MMR deficiency. The frameshift-mutation frequencies in the respective genes in spheroid-derived DNA were 100% for TGFBR2, 43% for IGFR2, and 86% for BAX and CASP5 (Figure 5B). Notably, the frequencies detected with FFPE tissue-derived DNA samples were significantly lower than those with spheroid DNA (Figure 5B). It is likely that the compromised sensitivity of FFPE tumor DNA was caused by the stromal cells in cancer tissues. For example, the colon cancer HC49T had G7/G7 homozygous mutation of BAX in the spheroid-derived DNA (Figure 5C top). With the FFPE tumor DNA, however, peaks representing the wild-type stromal cells dominated, confused the interpretation, and led to misdiagnosis of this case as “wild-type” (Figure 5C bottom).
On the other hand, colon cancer HC106T was diagnosed as MSI-L by on-chip electrophoresis with FFPE tumor DNA. However, the spheroid DNA of the same tumor was diagnosed as MSI-H in seven-marker on-chip analysis because it had multiple peaks shifted from the normal cell DNA for D2S123, BAT40 and MYCL as described above (Supplementary Figure 1). In addition, the IHC results showed lack of MLH1 and PMS2 in both the whole tumor and spheroids (data not shown). Consistently, direct sequencing of four MMR-target mononucleotide repeats showed that TGFBR2 and BAX contained homozygous and heterozygous frameshift mutations, respectively, although their IGF2R and CASP5 were homozygous wild type (Table 2). Mutational burden assessment also showed 61 and 9 total and indel mutations, respectively. Again, the discrepancy of the on-chip electrophoresis data between the DNA samples derived from spheroids and FFPE tumors are likely to be explained by the poor quality of DNA samples from FFPE tumors.
DISCUSSION
The MMR-deficient colorectal cancer syndromes include mainly two types; Lynch syndrome (LS) that is caused by hereditary (i.e., familial) mutations in the MMR protein genes, and sporadic disease without familial background [1]. The latter can be caused either by somatic mutations or epigenetic inactivation of the MMR protein genes [17]. Diagnosis of Lynch syndrome patient could be performed by germline mutation analysis of MMR protein genes or EpCAM using DNA extracted from blood cells [24]. In this study, however, we did not distinguish these two subtypes. This is because both types of patients respond to immune checkpoint therapy very well likely because of the neoantigens generated by frameshift mutations in the coding regions of multiple genes [30]. Accordingly, it is worth the effort to maximize the diagnostic accuracy for identifying possible responders to the immune checkpoint therapy.
Notably, recent recommendations suggest that all colorectal cancer patients should be screened for MMR-deficiency first by IHC which is currently more economical than PCR-based DNA sequencing [6]. As we faced misdiagnosis (Supplementary Figure 6), however, IHC is not without pitfalls [29]. For example, it was reported that IHC of MMR proteins caused considerable errors; 10 of 14 cases assessed by IHC showed discrepant results with that of PCR analysis [29]. Besides, oxaliplatin-induced MLH1 methylation and a specific mutation V384D in the same gene can give confusing results in IHC (Supplementary Figures 6 and 7). It is also worth noting that mutations in the DNA polymerase ε catalytic subunit gene (DPOE1) can cause hypermutational phenotype similar to MSI [18], although we found no such cases among 49 tested.
Here we evaluated multiple diagnostic measures using patient-derived TIC spheroid DNA and IHC, and demonstrated that the accuracy of MSI diagnosis was improved substantially. Because preparation of cancer spheroids from excised tumors has become less labor-intensive and more cost-effective [14], it appears worth the effort considering the very high therapeutic costs for immune checkpoint therapy.
With more advances in technology, mutational burden assessment may become cheaper in a near future [31]. Moreover, the pure and high quality cancer spheroid DNA can make the analysis scheme simpler. On the other hand, analyses of frozen cancer tissues, not to mention FFPE ones, need additional improvements to eliminate noises caused by non-cancer stromal cells. Because tumor mutational burden was distinctively high in MSI-H colorectal cancer when determined with spheroid DNA, it is possible practically to reduce the sequencing depth from the current > 500 repeats down to > 250 or less, as reported in a similar analysis [19, 21], which helps reduce the sequencing cost.
We have demonstrated that heterozygous mutations in the coding mononucleotide-repeats were detected unambiguously with cancer spheroid DNA, which cannot be attained with FFPE-tumor DNA. Although such heterozygous mutations do not cause loss of the gene functions usually, they can generate neoantigens that are targeted by the host immune system, which awaits further studies.
Some diagnostic markers are being implemented to stratify the cancer patients for immune checkpoint therapy. For example, expression of PD-1 ligands on cancer cell surface detected by IHC correlates with good response to the therapy [32]. Likewise, mutations in the 3’-untranslated region of the PD-L1 gene affect its expression level through mRNA stabilization [33]. Accurate diagnosis of MMR-deficient colorectal cancer cases in combination with other functional markers should help maximize the patient benefits.
Here we have evaluated multiple diagnostic measures using spheroid-derived DNA and spheroid IHC. While the final recommendation of the diagnostic method can be made based on the disease stage and socioeconomic conditions of the patient, it is worth culturing and utilizing the TIC spheroids from the excised primary cancer, which helps improve diagnostic accuracy significantly.
MATERIALS AND METHODS
Human samples
A total of 111 human colorectal cancer samples were obtained from patients who underwent resection operations between January 2015 and October 2017 at Kyoto University Hospital (KUHP). The study protocol was approved by the institutional review board of Kyoto University, and written informed consents were obtained from the patients.
Establishment and maintenance of patient-derived epithelial spheroid lines
Patient-derived spheroids of the colorectal cancer and normal colonic epithelium were established and maintained according to previous protocols [13, 14]. Ninety cancer spheroid lines from 89 patients and their genetic mutations were reported previously [14], whereas 21 lines from 21 patients were established thereafter in the present study.
Preparation of spheroid DNA samples
Genomic DNA samples were extracted from normal and cancer epithelial spheroids cultured in 4 wells of 12-well cell-culture plates (~105 cells) according to the previous protocol [14].
Genomic DNA preparation from formalin-fixed paraffin-embedded tissue sections
All cancer tissue sections were evaluated pathologically with H&E-stained slides to select tumor cell-enriched areas by a board-certified pathologist. Normal and cancer tissues were macrodissected with scalpel blades from at least three 10-μm FFPE tissue sections adjoining the H&E-stained section for each primary lesion. Dissected cancer tissue samples contained > 50% cancer cells. Normal colonic epithelium was collected from resected samples and their stem cells were cultured in vitro [14]. Genomic DNA was extracted using QIAamp DNA FFPE tissue kit (Qiagen, Hilden, Germany).
On-chip microsatellite instability test
Microsatellite DNA fragments were amplified from spheroid DNA samples using Multiplex PCR Kit (Qiagen) and primer pairs for the Bethesda panel markers [2, 34] (Supplementary Table 4). Three electrophoretic runs were performed for each sample (BAT25/D2S123, D5S346/D17S250, and BAT26) according to the standard protocol [16] (Supplementary Table 5). Amplified DNA fragments were purified with QIAquick PCR Purification Kit (Qiagen), applied on the DNA LabChip of the Agilent DNA 1000 Kit (Agilent Technologies, Santa Clara, CA, USA), and analyzed with an Agilent 2100 Bioanalyser instrument (Agilent Technologies) according to the previous protocol [16]. The electropherogram of each cancer spheroid line was overlaid with that of the normal epithelial spheroid line derived from the same patient. A marker was diagnosed as unstable if any of the cancer peaks for the marker shifted more than 0.5 seconds. The MSI status of each case was determined as follows: MSI-high (MSI-H), with more than one unstable markers; MSI-low (MSI-L), with one unstable marker; and MSS, with no unstable markers. If only dinucleotide repeat markers as D2S123, D5S346 and/or D17S250 were mutated, a secondary panel of markers with mononucleotide repeats (BAT40 and MYCL) was tested as recommended [1].
Sequence analysis of mononucleotide repeats in cancer-related coding genes
The DNA fragments containing mononucleotide repeats in TGFBR2 (A10), BAX (G8), IGF2R (G8), and CASP5 (A10) were amplified from cancer spheroid DNA samples using JumpStart Taq DNA Polymerase (Sigma-Aldrich, St. Louis, MO, USA) or PrimeSTAR Max DNA Polymerase (Takara Bio, Kusatsu, Japan). Primer sequences and PCR conditions are shown in Supplementary Tables 4 and 5 [35]. Purified PCR fragments were sequenced by Macrogen (Seoul, Republic of Korea).
Next generation sequencing
Next generation sequencing analyses of somatic cancer mutations were performed by Macrogen. In brief, DNA fragments that cover all exons of 409 cancer-related genes spanning a total length of 1.29 Mb (Ion AmpliSeq Comprehensive Cancer Panel; Thermo Fisher) were amplified from DNA samples of 7 MSI-H and 11 MSI-L or MSS cancer spheroid lines, and sequenced with the Ion Proton sequencer (Thermo Fisher).
Estimation of mutational burdens (see Supplementary Figure 2)
The sequencing data were processed using Ion Torrent Suite Software v5.0.4 (Thermo Fisher), and variants against the hg19 human genome reference were called using Torrent Variant Caller v5.0.4 (Thermo Fisher). Possible cancer-specific mutations were selected through the following processes. The polymorphic alleles in 1,200 Japanese individuals were removed from each variant [36]. Only variants in the coding regions with > 20% frequency without polymorphism were scored. Erroneous mutations were eliminated by surveying their coverage tracks on Integrative Genomics Viewer software v2.3 (Broad Institute).
Histological specimens
Formalin-fixed paraffin-embedded (FFPE) tumor specimens were prepared by the standard procedures in Department of Diagnostic Pathology, Kyoto University Hospital. A board-certificated pathologist selected multiple and separate cancer lesions from a single tumor. Specimens of FFPE cancer spheroids were prepared as previously reported [14]. These specimens were sectioned at 4-μm thickness, and stained with H&E or immunostained for MLH-1 (M1, Ventana, Tucson, AZ, USA), MSH2 (G219-1129, Ventana), MSH6 (EPR3945, Abcam, Cambridge, UK), or PMS2 (EPR 3947, Ventana) followed by hematoxylin counterstaining for nuclei.
Agarose gel electrophoresis of genomic DNA
Genomic DNA samples from spheroids and FFPE tissues were electrophoresed on a 0.7% agarose gel. The gel was stained with ethidium bromide, and DNA fragments were visualized using Gel Doc XR+ (BioRad, Hercules, CA, USA) and Image Lab Software 3.0.1 Beta 2 (BioRad).
Statistical analysis
Statistical analyses were conducted using GraphPad Prism 6 (Graph Pad Software, La Jolla, CA, USA). Mann-Whitney test was applied to compare DNA quality and electropherogram peak heights between spheroid- and FFPE-derived samples, and to compare total or indel mutational burden between MSI-H cases and MSI-L or MSS cases.
Abbreviations
mismatch repair: MMR; microsatellite instability: MSI; programmed cell death 1: PD-1; programmed cell death 1 ligand 1: PD-L1; tumor-initiating cells: TIC; immunohistochemistry: IHC; formalin-fixed paraffin-embedded: FFPE; microsatellite instability-high: MSI-H; microsatellite instability-low: MSI-L; microsatellite-stable: MSS; transforming growth factor-beta receptor type 2: TGFBR2; BCL2-associated X protein: BAX; insulin-like growth factor 2 receptor: IGF2R; caspase 5: CAS P5; wild type: WT; Lynch syndrome: LS; DNA polymerase ε catalytic subunit: DPOE1; colorectal cancer: CRC; mismatch repair proficient: MMRp; mismatch repair deficient: MMRd.
Author contributions
Conception and design: T. Yamaura, H. Miyoshi, M.M. Taketo; Development of methodology: T. Yamaura, H. Miyoshi; Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): T. Yamaura, H. Miyoshi, H. Maekawa, T. Morimoro, T. Yamamoto, F. Kakizaki, K. Higasa; Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): T. Yamaura, H. Miyoshi, M.M. Taketo; Writing, review, and/or revision of the manuscript: T. Yamaura, H. Miyoshi, K. Kawada, Y. Sakai, M.M. Taketo; Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): K. Kawada, Y. Sakai, F. Matsuda; Study supervision: T. Yamaura, H. Miyoshi, M.M. Taketo.
ACKNOWLEDGMENTS
We thank the Medical Research Support Center, Graduate School of Medicine, Kyoto University for technical supports. We also thank the members of Division of Gastrointestinal Surgery, Department of Surgery for help in collecting surgical specimens. We are grateful to T Sakurai and H Haga of Department of Clinical Pathology at Kyoto University Hospital for their pathological diagnosis. We are also grateful to M Muto of Department of Clinical Oncology, Kyoto University Hospital for clinical discussions on chemotherapy.
CONFLICTS OF INTEREST
Authors have no potential conflicts of interests to report.
FUNDING
This work was supported by Program for Creating STart-ups from Advanced Research and Technology (START, ST261001TT) from Japan Science and Technology Agency (JST); Practical Research for Innovative Cancer Control (ck0106195h) from Japan Agency for Medical Research and Development (AMED); and Kyoto University Venture Incubation from Kyoto University Office of Society-Academia Collaboration for Innovation (all to M.M.T.).
REFERENCES
1. Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Rüschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004; 96:261–8.
2. Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998; 58:5248–57.
3. Goldstein J, Tran B, Ensor J, Gibbs P, Wong HL, Wong SF, Vilar E, Tie J, Broaddus R, Kopetz S, Desai J, Overman MJ. Multicenter retrospective analysis of metastatic colorectal cancer (CRC) with high-level microsatellite instability (MSI-H). Ann Oncol. 2014; 25:1032–8. https://doi.org/10.1093/annonc/mdu100.
4. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015; 372:2509–20. https://doi.org/10.1056/NEJMoa1500596.
5. Dudley JC, Lin MT, Le DT, Eshleman JR. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin Cancer Res. 2016; 22:813–20. https://doi.org/10.1158/1078-0432.CCR-15-1678.
6. Ottawa (ON): Canadian Agency for Drugs and Technologies in Health; 2016 Aug. DNA Mismatch Repair Deficiency Tumour Testing for Patients With Clorectal Cancer: Recommendations. CADTH Optimal Use Report; No. 5.3d. 2016.
7. Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J Mol Diagn. 2008; 10:293–300. https://doi.org/10.2353/jmoldx.2008.080031.
8. Taketo MM, Edelmann W. Mouse models of colon cancer. Gastroenterology. 2009; 136:780–98. https://doi.org/10.1053/j.gastro.2008.12.049.
9. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–7. https://doi.org/10.1038/nature11252.
10. Duval A, Hamelin R. Mutations at coding repeat sequences in mismatch repair-deficient human cancers: toward a new concept of target genes for instability. Cancer Res. 2002; 62:2447–54.
11. Giardiello FM, Allen JI, Axilbund JE, Boland CR, Burke CA, Burt RW, Church JM, Dominitz JA, Johnson DA, Kaltenbach T, Levin TR, Lieberman DA, Robertson DJ, et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-society Task Force on colorectal cancer. Am J Gastroenterol. 2014; 109:1159–79. https://doi.org/10.1038/ajg.2014.186.
12. Shin HT, Choi YL, Yun JW, Kim NK, Kim SY, Jeon HJ, Nam JY, Lee C, Ryu D, Kim SC, Park K, Lee E, Bae JS, et al. Prevalence and detection of low-allele-fraction variants in clinical cancer samples. Nat Commun. 2017; 8:1377. https://doi.org/10.1038/s41467-017-01470-y.
13. Miyoshi H, Stappenbeck TS. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat Protoc. 2013; 8:2471–82. https://doi.org/10.1038/nprot.2013.153.
14. Miyoshi H, Maekawa H, Kakizaki F, Yamaura T, Kawada K, Sakai Y, Taketo MM. An improved method for culturing patient-derived colorectal cancer spheroids. Oncotarget. 2018; 9:21950–64. https://doi.org/10.18632/oncotarget.25134.
15. Maekawa H, Miyoshi H, Yamaura T, Itatani Y, Kawada K, Sakai Y, Taketo MM. A Chemosensitivity Study of Colorectal Cancer Using Xenografts of Patient-Derived Tumor Initiating Cells. Mol Cancer Ther. 2018; 10:2187–2196. https://doi.org/10.1158/1535-7163.MCT-18-0128.
16. Odenthal M, Barta N, Lohfink D, Drebber U, Schulze F, Dienes HP, Baldus SE. Analysis of microsatellite instability in colorectal carcinoma by microfluidic-based chip electrophoresis. J Clin Pathol. 2009; 62:850–2. https://doi.org/10.1136/jcp.2008.056994.
17. Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008; 135:1079–99. https://doi.org/10.1053/j.gastro.2008.07.076.
18. Kim TM, Laird PW, Park PJ. The landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell. 2013; 155:858–68. https://doi.org/10.1016/j.cell.2013.10.015.
19. Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, Stephens PJ, Daniels GA, Kurzrock R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol Cancer Ther. 2017; 16:2598–608. https://doi.org/10.1158/1535-7163.MCT-17-0386.
20. Johnson DB, Frampton GM, Rioth MJ, Yusko E, Xu Y, Guo X, Ennis RC, Fabrizio D, Chalmers ZR, Greenbowe J, Ali SM, Balasubramanian S, Sun JX, et al. Targeted Next Generation Sequencing Identifies Markers of Response to PD-1 Blockade. Cancer Immunol Res. 2016; 4:959–67. https://doi.org/10.1158/2326-6066.CIR-16-0143.
21. Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, Sun J, Juhn F, Brennan K, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013; 31:1023–31. https://doi.org/10.1038/nbt.2696.
22. Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, Nowak JA, Nishihara R, Qian ZR, Inamura K, Morikawa T, Nosho K, Abril-Rodriguez G, et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discov. 2018; 8:730–49. https://doi.org/10.1158/2159-8290.CD-17-1327.
23. Tiriac H, Belleau P, Engle DD, Plenker D, Deschênes A, Somerville TDD, Froeling FEM, Burkhart RA, Denroche RE, Jang GH, Miyabayashi K, Young CM, Patel H, et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018; 8:1112–29. https://doi.org/10.1158/2159-8290.CD-18-0349.
24. Sinicrope FA. Lynch Syndrome-Associated Colorectal Cancer. N Engl J Med. 2018; 379:764–73. https://doi.org/10.1056/NEJMcp1714533.
25. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010; 138:2073–87.e3. https://doi.org/10.1053/j.gastro.2009.12.064.
26. Samimi G, Fink D, Varki NM, Husain A, Hoskins WJ, Alberts DS, Howell SB. Analysis of MLH1 and MSH2 expression in ovarian cancer before and after platinum drug-based chemotherapy. Clin Cancer Res. 2000; 6:1415–21.
27. Zeller C, Dai W, Steele NL, Siddiq A, Walley AJ, Wilhelm-Benartzi CS, Rizzo S, van der Zee A, Plumb JA, Brown R. Candidate DNA methylation drivers of acquired cisplatin resistance in ovarian cancer identified by methylome and expression profiling. Oncogene. 2012; 31:4567–4576. https://doi.org/10.1038/onc.2011.611.
28. Fan Y, Wang W, Zhu M, Zhou J, Peng J, Xu L, Hua Z, Gao X, Wang Y. Analysis of hMLH1 missense mutations in East Asian patients with suspected hereditary nonpolyposis colorectal cancer. Clin Cancer Res. 2007; 13:7515–21. https://doi.org/10.1158/1078-0432.CCR-07-1028.
29. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, Desai J, Hill A, Axelson M, Moss RA, Goldberg MV, Cao ZA, Ledeine JM, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017; 18:1182–91. https://doi.org/10.1016/S1470-2045(17)30422-9.
30. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, Wong F, Azad NS, Rucki AA, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017; 357:409–13. https://doi.org/10.1126/science.aan6733.
31. Hayden EC. Technology: The $1,000 genome. Nature. 2014; 507:294–5. https://doi.org/10.1038/507294a.
32. Diggs LP, Hsueh EC. Utility of PD-L1 immunohistochemistry assays for predicting PD-1/PD-L1 inhibitor response. Biomark Res. 2017; 5:12. https://doi.org/10.1186/s40364-017-0093-8.
33. Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, Maeda T, Nagata Y, Kitanaka A, Mizuno S, Tanaka H, Chiba K, Ito S, et al. Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature. 2016; 534:402–6. https://doi.org/10.1038/nature18294.
34. Dietmaier W, Wallinger S, Bocker T, Kullmann F, Fishel R, Rüschoff J. Diagnostic microsatellite instability: definition and correlation with mismatch repair protein expression. Cancer Res. 1997; 57:4749–56.
35. Takenoshita S, Tani M, Nagashima M, Hagiwara K, Bennett WP, Yokota J, Harris CC. Mutation analysis of coding sequences of the entire transforming growth factor beta type II receptor gene in sporadic human colon cancer using genomic DNA and intron primers. Oncogene. 1997; 14:1255–8. https://doi.org/10.1038/sj.onc.1200938.
36. Jambaljav B, Tanaka D, Nagashima K, Harashima SI, Harada N, Harada T, Fujiwara Y, Wang Y, Liu Y, Tabara Y, Matsuda F, Koizumi A, Inagaki N. Whole-exome sequencing in a Japanese family with highly aggregated diabetes identifies a candidate susceptibility mutation in ADAMTSL3. Diabetes Res Clin Pract. 2018; 135:143–9. https://doi.org/10.1016/j.diabres.2017.11.012.