
INTRODUCTION
Malignant neoplasm, commonly known as cancer, is a well known life threatening disease and among its diverse classification melanoma is largely considered to be a chemotherapy refractory tumor [1, 2]. The treatment for metastatic melanoma is challenging while in the ample amount of cases, unsuccessful despite advances in ongoing medical research. Therefore, there is a need of detailed mechanistic study of cellular pathways related to available treatments [3–5]. In the past two decades, studies on sphingolipids revealed the important role of its bioactive forms such as ceramide, in the regulation of multiple biological functions especially in apoptosis [6, 7]. Inhibiting cell death by interference on ceramide signaling is a key strategy for tumorigenesis and escape from apoptosis stimuli [8, 9]. Therefore, for effective cancer therapy development, ceramide metabolic pathways have become a major concern. Anti-carcinogenic effect of ceramide in the treatment of malignancy depends on the stimulation of diverse apoptotic pathways.
Cisplatin, an effective platinum-containing anti-neoplastic agent is widely utilized in the treatment of a variety of cancers [10, 11], but its adverse effect due to its ability to generate toxicity in various tissue types is a major concern [12, 13]. While regulating the cellular signaling axis to trigger cell death, cisplatin also targets ceramide pathway to establish its utility in cancer deterioration through apoptosis [14]. There are other reports that ceramide production is regulated through protein kinase C (PKC) signaling pathway. PKCs are serine-threonine kinases, different isotypes of which play a key regulatory role in the proliferation, survival, or apoptosis of various cell types. Amongst different PKC isotypes, PKCδ induces apoptosis in cancer cell and plays a potential tumor suppressor role [15, 16]. It has also been reported that cisplatin mediated ceramide generation in cancer cells leads to tumor regression. Moreover, PKCδ plays a major role in ceramide generation in cancer cells leading to the induction of apoptosis [17, 18]. Furthermore, reports also suggest PKCδ as critical mediator of cisplatin induced kidney cell injury as well as death while inhibition of PKCδ retains chemotherapeutic efficacy of the drug with simultaneous abrogation of the nephrotoxicity [19, 20].
Previous studies from our laboratory reported that specific PKC isotypes were reciprocally regulated and utilized for enhancing the ceramide generation to induce apoptosis in the B16F10 melanoma cells [18]. In continuation with the exploration of the mechanism of endogenous ceramide generation, we investigated the role of cisplatin on the growth of mouse B16F10, human A375 melanoma cells in vitro and murine melanoma tumor in vivo under PKCδ deficient condition. Therefore, for the first time our study highlighted the cisplatin mediated inhibition of cancer cell growth in a PKCδ independent manner. Major focus of our study related to the apoptosis of melanoma cells is to understand the mechanism of ceramide generation by cisplatin in PKCδ deficient cell, while IRF-1 and TNFα emerged as key regulatory molecule.
Interferon regulatory factors (IRF) are transcription factors comprising of a large number of isoforms, among which IRF-1 and IRF-8 (or ICSBP) are associated with a vast range of host responses to infection and tumor growth [21–23]. On the other hand, TNFα is a pleiotropic cytokine that regulates a broad range of biological activities including cell differentiation, proliferation and death as well as inflammation and tissue development [24, 25]. Moreover, previous reports demonstrated that the expression of IRF-1, also known as interferon stimulated-gene factor 2 (ISGF-2), is synergistically induced by TNFα and IFNγ [26]. However, key enzymes involved in ceramide signaling pathway also include SphK1 and SphK2, which have distinct roles in sensitivity to cisplatin and other drugs modulation [27–29]. Relating these regulations, our study is majorly focused on the role of cisplatin induced apoptosis through PKCδ independent pathway involving different transcription factors and enzymes. Silencing of PKCδ retains the effect of cisplatin in hypoxic conditions, suggesting a novel regulation in hypoxia, which is an important selective force in the clonal evolution of tumors [30].
With such objectives in mind, the present work has highlighted the important cellular signaling events that sensitize PKCδ deficient melanoma cells towards proliferation inhibition and apoptosis by a de novo pathway. This pathway is also associated with increased generation of pro-inflammatory cytokine TNFα which may provide a useful therapeutic strategy to enhance the ability of cisplatin to eradicate tumors with lesser adverse effects.
RESULTS
Cisplatin inhibits cell cycle progression and induces apoptosis in PKCδ silenced B16F10 cells via ceramide generation
Cisplatin, a well established chemotherapeutic agent, is involved in apoptosis of cancer cells and abrogate malignancy [10]. Cisplatin is also associated with high nephrotoxicity. Therefore, the mechanism of its action is the major area of concern [19]. It is established that ceramide is one of the major key players of cisplatin induced apoptosis, where PKCδ is a well-known modulator of cisplatin induced ceramide generation [14, 18]. However, recent studies have also depicted the involvement of TNFα in cisplatin induced apoptosis process [25]. Therefore, we were interested to investigate whether cisplatin could induce apoptosis of their target cells in a PKCδ independent manner. Accordingly, we silenced PKCδ in B16F10 cells using specific siRNA (Figure 1A and 1B) and the effect of cisplatin on cell cycle progression was studied. Interestingly, cisplatin at 50μM concentration showed a significant increase in the number of cells in sub G0/G1 phase and a concomitant decrease in the number of cells in S and G2/M phase, indicating that cisplatin halted G1-S transition resulting in cell cycle arrest and also gave rise to the sub G0/G1 cells from control to drug treatment, according to the morphological analysis these were apoptotic cells (Figure 1C). Cell proliferation analysis using (3H) – Thymidine incorporation assay revealed a significant decrease in the proliferation of cisplatin treated cells (Figure 1D). In order to check the translocation of phosphatidylserine (PS) externalization from inner cell membrane to outer cell membrane, a characteristic feature of cells undergoing apoptosis, cells were subjected to flow cytometric analysis after staining with Annexin-V-FITC and PI. The percentage of apoptosis in cisplatin treated cells was found to be 37.95% as compared to a very low percentage of apoptotic cells in the untreated PKCδ deficient B16F10 melanoma cells (Figure 1E). On analyzing different caspase activities upon cisplatin mediated apoptosis, caspase-3 and caspase-8 activities were found to be elevated significantly, which confirms their role in cisplatin mediated apoptosis (Figure 1F). Moreover, we wanted to know whether PKCδ deficient melanoma cells could generate ceramide upon cisplatin treatment. We observed that cisplatin significantly increased the level of ceramide (Figure 1G). In order to determine the pathway of ceramide generation, PKCδ deficient B16F10 melanoma cells were treated with FB1 (10μM) or imipramine (10μM), the well-known inhibitors of de novo and ASMase pathway respectively following cisplatin treatment. It was observed that in the presence of imipramine, cisplatin failed to induce ceramide generation (Figure 1G). Furthermore, the mRNA expression of ASMase was upregulated by cisplatin treatment, which was also reduced by imipramine treatment (Figure 1H). Interestingly the six isoforms of ceramide synthase, the key enzymes of the de novo synthesis pathway, showed no effect on the cisplatin mediated ceramide generation (Figure 1H). Taken together, our results confirmed that the ceramide which is produced by sphingomyelinase pathway is involved in cisplatin mediated apoptosis of PKCδ deficient B16F10 melanoma cells.
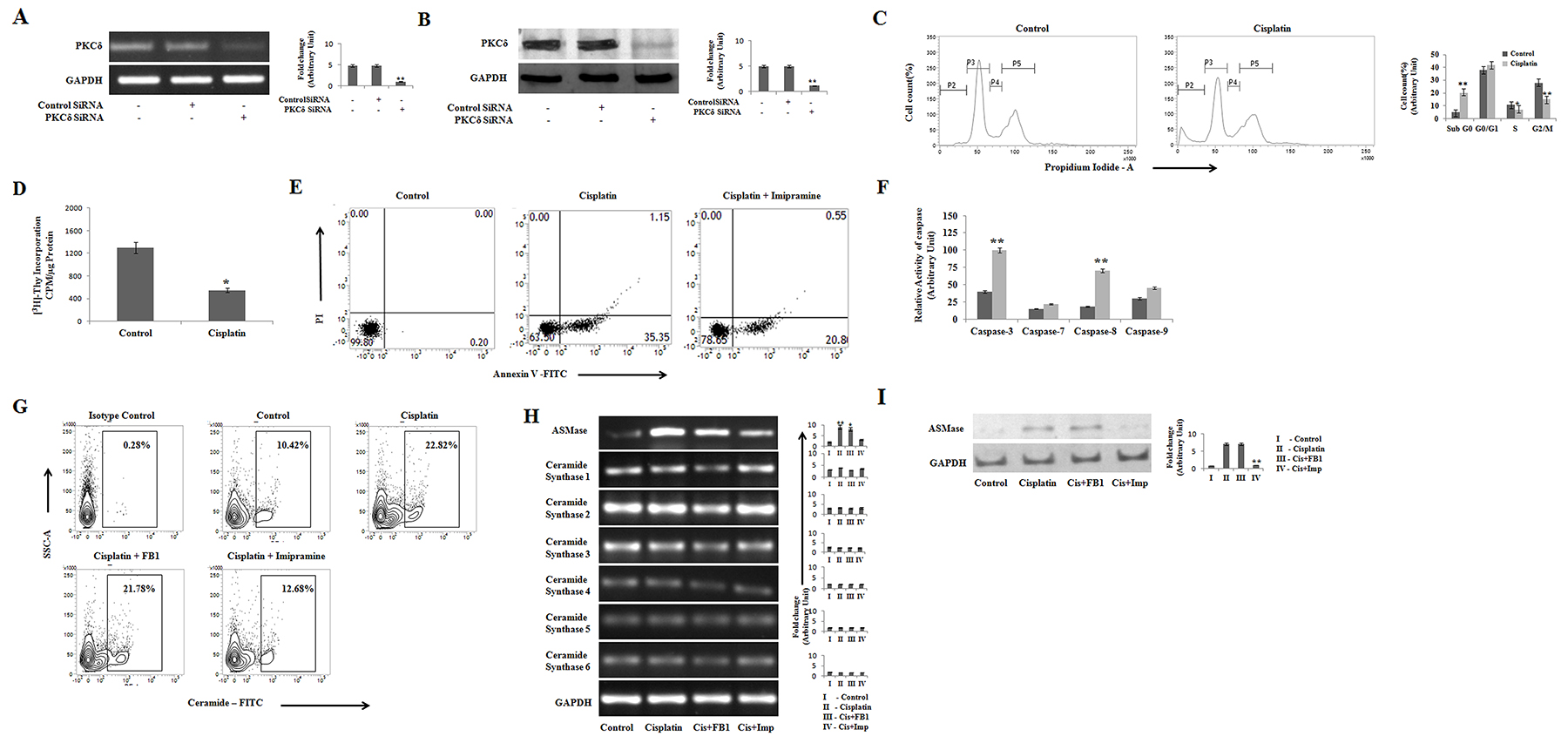
Figure 1: Cisplatin inhibits cell cycle progression and induces apoptosis in PKCδ silenced B16F10 cells via ceramide generation. (A) 2 × 106 B16F10 cells were transfected with PKCδ specific siRNA or control siRNA as mentioned in Materials and Methods. The transfected cells were collected in TRIZOL for mRNA expression of PKCδ by semi quantitative RT-PCR. GAPDH was used as a reference. Data are from one of the three representative experiments. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (B) In a separate experiment, similarly B16F10 cells were transfected with PKCδ specific siRNA or control siRNA as mentioned in Materials and Methods. The expression of PKCδ was analyzed in whole cell lysates by western blot analysis. GAPDH was used as a reference. Data are from one of three representative experiments. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (C) B16F10 cells transfected PKCδ specific siRNA with as mentioned in materials and methods were treated with cisplatin (50μM), harvested for 24 hrs and stained with propidium iodide to measure the DNA content by flow cytometry. (D) Cisplatin treated B16F10 cells transfected PKCδ specific siRNA were measured for incorporation of (3H) - thymidine to determine cell proliferation. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (E) PKCδ silenced B16F10 melanoma cells were treated with cisplatin (50μM) and cisplatin along with imipramine (10μM) for 12 hrs. The treated cells were collected and stained with Annexin-V FITC and PI and subsequently analyzed by flow cytometry. Data are from one of the three representative experiments. (F) B16F10 cells were transfected with PKCδ siRNA followed by cisplatin treatment. Caspase-3, 7, 8, 9 activities were determined from cell lysates using the caspase assay kits according to the manufacturer’s instructions. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (G) PKCδ silenced B16F10 melanoma cells were treated with Cisplatin (50μM), cisplatin and FB1 (10μM), cisplatin and imipramine (10μM) for 12 hrs. The treated cells were subsequently stained with anti-mouse ceramide-FITC, isotype-matched control mouse antibody. Ceramide expression was analysed by flow cytometry. Data are from one of three representative experiments. (H) and (I) Similarly PKCδ specific siRNA transfected B16F10 melanoma cells were treated with cisplatin (50μM), cisplatin and FB1 (10μM), cisplatin and imipramine (10μM) for 12 hrs. The treated cells were collected in TRIZOL for mRNA expression analyses of ceramide synthase (1 – 6) and ASMase by semi quantitative RT-PCR. In a separate experiment, the expression of ASMase was analyzed in whole cell lysates by western blot analysis. (I) GAPDH was used as a reference. Data are from one of three representative experiments. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated.
Cisplatin induces apoptosis in PKCδ silenced B16F10 cells by TNFα mediated pathway
Cytokines released in response to inflammation and immunity can function to inhibit cancer development and progression [31]. Cytokines with various biological effects act as autocrine or paracrine factors and they are capable in modulation of tumor growth [32]. On the other hand, malignant cells can act in response to a few cytokines, which in turn support growth, knock down apoptosis, and assist invasion via metastasis. A more detailed understanding of cytokine-cancer cell interactions provides new opportunities for improving cancer immunotherapy [33]. Therefore, we investigated the status of different cytokines in cisplatin mediated apoptosis of PKCδ deficient B16F10 cells. It was observed that cisplatin in PKCδ deficient B16F10 melanoma cells were unable to induce the expression of IL-12 and IFN-γ (Figure 2A), whereas it increased the expression of TNFα. The expression of anti-inflammatory cytokines, including TGF-β and IL-10 remained unaltered. In all the cases, C2 ceramide (20μM) at 12 h was taken as positive control and the regulatory effect of ceramide on various cytokines was further confirmed by ELISA (Figure 2B).
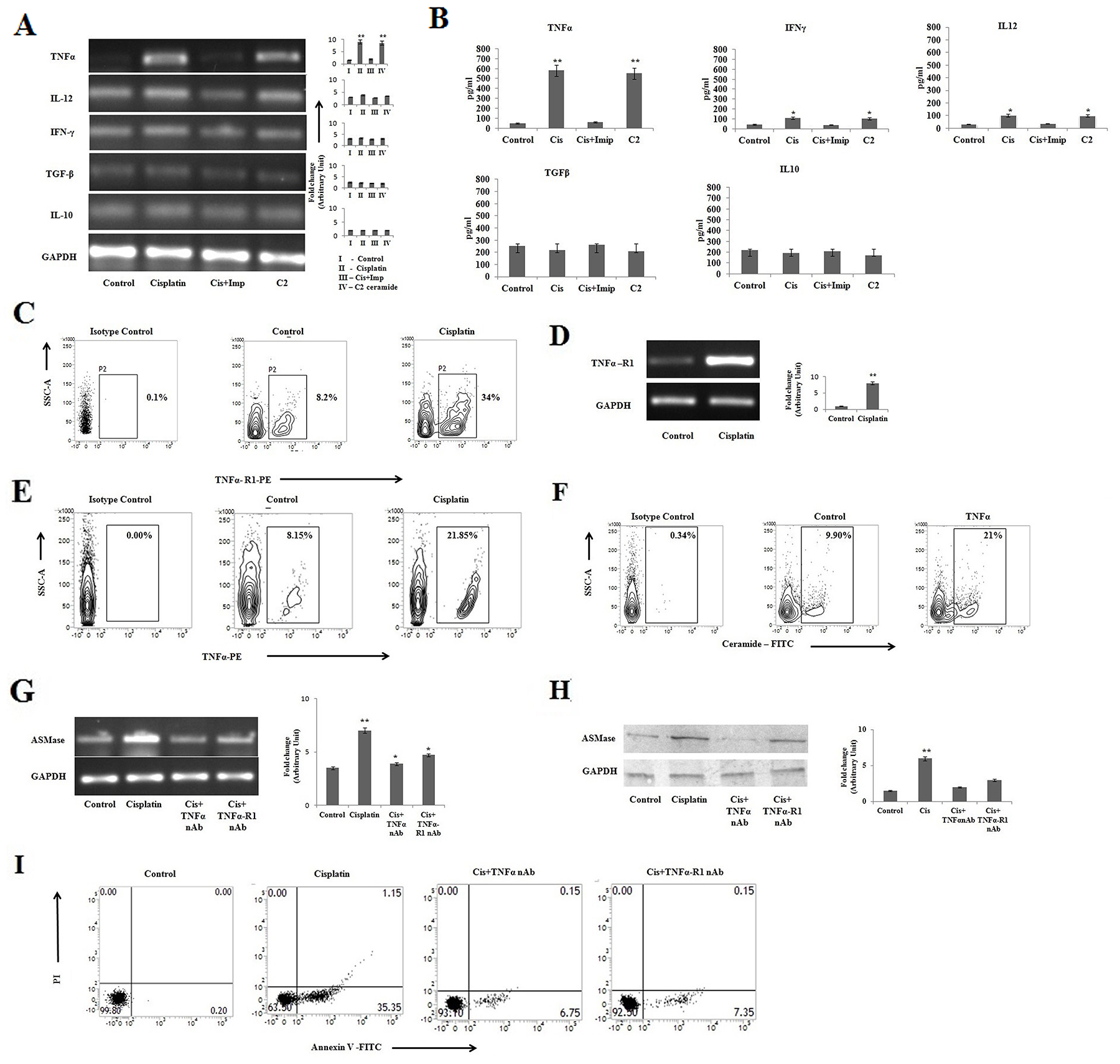
Figure 2: Cisplatin induces apoptosis in PKCδ silenced B16F10 cells by TNFα mediated pathway. (A) and (B) B16F10 cells were transfected with PKCδ siRNA followed by cisplatin, cisplatin along with impiramine, and C2 ceramide treatment. The treated cells were collected in TRIZOL for mRNA expression analyses of TNF-α, IL-12, IFN-γ, IL-10 and TGF-β by semi quantitative RT-PCR. The cell supernatants were collected for estimation of TNF-α, IL-12, IFN-γ, IL-10 and TGF-β by ELISA. GAPDH was used as a reference. Data are from one of three representative experiments. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (C) PKCδ silenced B16F10 melanoma cells were treated with Cisplatin followed by fixation and staining for FITC-conjugated TNF-R1 expression and analyzed by flow cytometry. Data are from one of three representative experiments. (D) In a separate experiment, similarly B16F10 cells transfected with PKCδ specific siRNA were treated with cisplatin and collected in TRIZOL for mRNA expression analyses of TNF-R1 by semiquantitative RT-PCR. GAPDH was used as a reference. Data are from one of the three representative experiments. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (E) PKCδ silenced B16F10 melanoma cells were treated with cisplatin and then the cells were subsequently stained for intracellular cytokine staining with anti-mouse TNFα-PE, isotype-matched control mouse antibody. TNFα expression was analysed by flow cytometry. Data are from one of the three representative experiments. (F) PKCδ silenced B16F10 melanoma cells were treated with exogenous TNFα and then the cells were subsequently stained with anti-mouse ceramide-FITC, isotype-matched control mouse antibody. Ceramide expression was analysed by flow cytometry. Data are from one of three representative experiments. (G) and (H) Similarly PKCδ specific siRNA transfected B16F10 melanoma cells were treated with cisplatin, cisplatin along with TNFα or TNFα-R1 neutralizing antibody. The treated cells were collected in TRIZOL for mRNA expression of ASMase by semi quantitative RT-PCR. GAPDH was used as a reference. Data are from one of the three representative experiments. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (I) In a separate experiment PKCδ specific siRNA transfected B16F10 melanoma cells were treated with cisplatin, cisplatin and TNFα neutralizing antibody together and cisplatin along with TNFα-R1 neutralizing antibody. The treated cells were collected and stained with Annexin-V FITC and PI and subsequently analyzed by flow cytometry. Data are from one of the three representative experiments.
Cisplatin treatment also increased the expression of TNFα receptor which was confirmed by FACS and PCR analyses (Figure 2C and 2D). Moreover, cisplatin treatment increased the production of intracellular TNFα in PKCδ deficient B16F10 cells compared to the untreated cells (Figure 2E). The effect of exogenous recombinant TNFα (50ng/ml) on ceramide generation (Figure 2F) and the effect of TNFα as well as TNFα-R1 neutralizing antibody (10 μg/ml) on ASMase expression in relation to mRNA (Figure 2G), protein level (Figure 2H) and apoptosis (Figure 2I) further confirmed that cisplatin mediated ceramide generation is TNFα and ASMase dependent.
Cisplatin induces IRF-1 dependent TNFα activation for the initiation of apoptosis in PKCδ silenced B16F10 melanoma cells
The cytokine responsiveness depends on interferon regulatory factors (IRFs) expression [26], which in turn can be induced by cisplatin [34]. IRFs can also control tumor progression by regulating the interactions between cancer and immune cells within the tumor microenvironment [35]. Therefore, we examined the effect of cisplatin on the expression of these IRFs in PKCδ deficient B16F10 cells. It was observed that cisplatin differentially modulated the expression of eight IRFs in PKCδ deficient B16F10 (Figure 3A). It was interesting to observe that among the eight IRFs, only the expression of IRF1 was significantly increased compared to the other IRFs upon cisplatin treatment. To further confirm the involvement of cisplatin in the regulation of IRF1, we performed western blot analysis which revealed the similar pattern of expression corroborating the previous findings (Figure 3B). Our results indicated that silencing IRF1 expression abrogated cisplatin mediated increase in IRF1 (Figure 3C) along with TNFα expression (Figure 3D) and protein level (Figure 3E) and ceramide generation (Figure 3H).
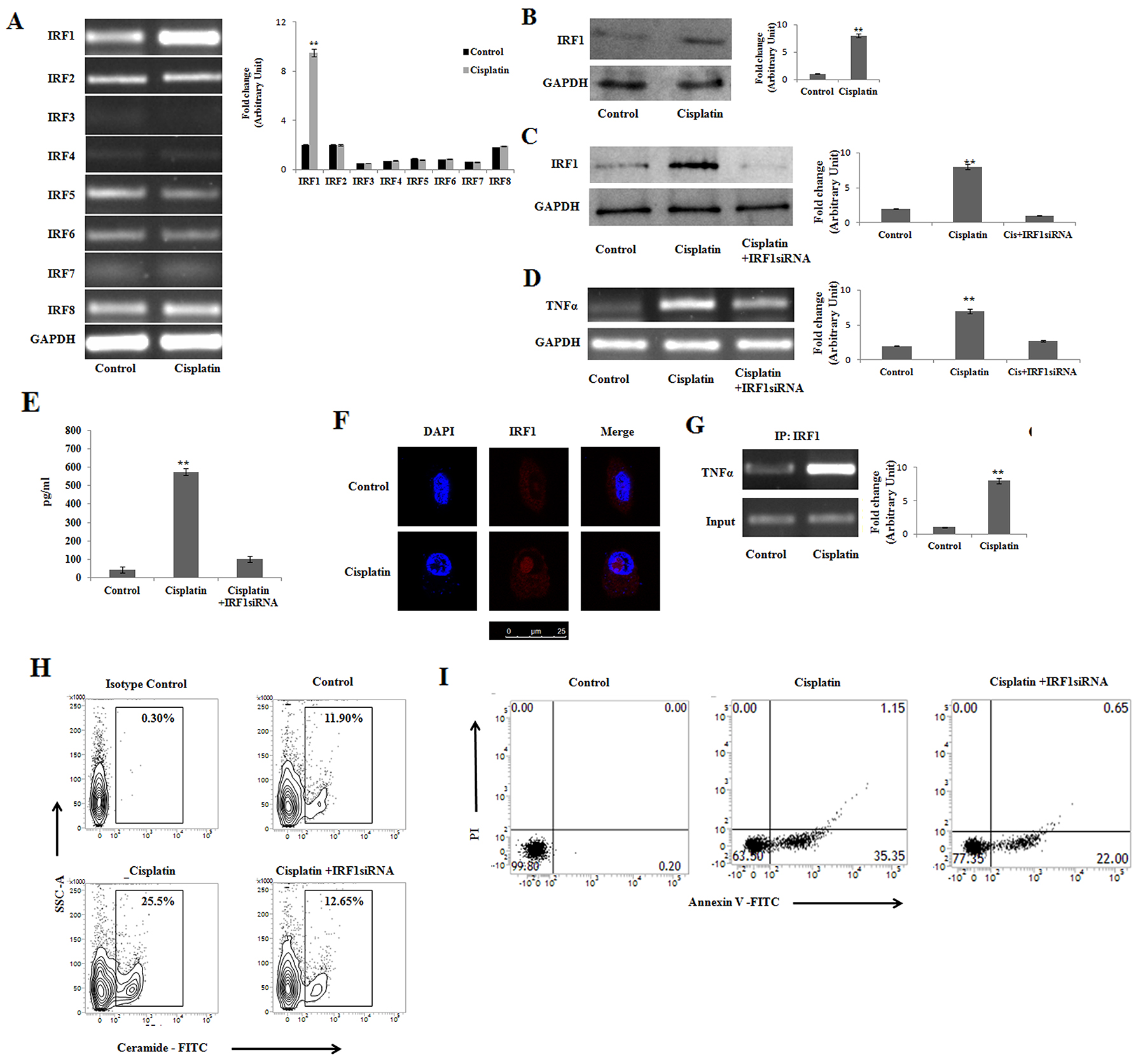
Figure 3: Cisplatin induces IRF-1 dependent TNFα activation for initiation of apoptosis in PKCδ silenced B16F10 melanoma cells. (A) and (B) B16F10 cells were transfected with PKCδ siRNA followed by Cisplatin treatment. The treated cells were collected in TRIZOL for mRNA extraction and semi quantitative RT-PCR analyses for IRF 1- 8 were done. The expression IRF 1 was analyzed in whole cell lysates by Western blotting. GAPDH was used as a reference. Data are from one of three representative experiments. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (C) PKCδ silenced and cisplatin treated B16F10 cells were transfected with IRF1 specific siRNA mentioned in materials and methods and harvested for 24 hrs. The expression with IRF 1 was analysed in whole cell lysates by western blot analysis. GAPDH was used as a reference. Data are from one of the three representative experiments. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (D) and (E) In a separate experiment, similarly PKCδ silenced and cisplatin treated B16F10 cells were transfected with IRF1 specific siRNA as mentioned in Materials and Methods. The treated cells were collected in TRIZOL for TNFα mRNA expression by semiquantitative RT-PCR. GAPDH was used as a reference. The cell supernatants were collected for the estimation of TNFα by ELISA. Data are from one of the three representative experiments. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (F) PKCδ silenced and cisplatin treated B16F10 cells were stained with PE- IRF-1 and analyzed by confocal microscopy. IRF-1 sub cellular localization was studied by confocal microscopy using the anti mouse IRF-1 monoclonal antibody and DAPI for nuclear staining. Scale bar 25 μm. Data are from one of the three representative experiments. (G) B16F10 cells were transfected with PKCδ siRNA followed by Cisplatin treatment. In treated cells IRF1 binding at TNFα promoter (IP: IRF1), was analyzed by ChIP assay as described in materials and methods. Data are from one of the three representative experiments. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (H) PKCδ silenced and cisplatin treated B16F10 cells were transfected with IRF1 specific siRNA mentioned in materials and methods and harvested for 24 hrs. Then the cells were subsequently stained with anti-mouse ceramide-FITC, isotype-matched control mouse antibody. Ceramide expression was analysed by flow cytometry. Data are from one of the three representative experiments. (I) In a separate experiment, similarly PKCδ silenced and cisplatin treated B16F10 cells were transfected with IRF1 specific siRNA as mentioned in Materials and Methods. Cells were collected and stained with Annexin-V FITC and PI and subsequently analyzed by flow cytometry. Data are from one of the three representative experiments.
In order to determine the role of IRF1 in cisplatin induced variations in expression, an in silico analysis was performed to evaluate the possible enrichment of binding sites for transcription factors in the putative promoter regions of the gene subgroups that were differentially regulated by cisplatin. For that we used the free web tool oPOSSUM. The result showed that cisplatin can increase IRF1 expression (data not shown). To further confirm the involvement of IRF1 in response to cisplatin, the localization of IRF1 was studied by confocal microscopy (Figure 3F). Our result showed that upon cisplatin treatment IRF1 nuclear translocation was increased in comparison with the control set. Moreover, a prominent binding of IRF1 to the TNFα promoter was observed in PKCδ deficient B16F10 cells (Figure 3G). Further, FACS analysis of Annexin-V assay using IRF1 siRNA confirmed the role of IRF1 in cisplatin mediated apoptosis (Figure 3I). Thus, our finding suggests that IRF1 regulates TNFα mediated ceramide generation in PKCδ deficient B16F10 cells.
cPLA2 modulates cisplatin mediated apoptosis in PKCδ silenced B16F10 melanoma cells
TNFα amplifies ceramide generation by activating two different intracellular signaling cascades involving ASMase (Figure 2F) as well as cytosolic phospholipase A2 (cPLA2) in PKCδ deficient B16F10 melanoma cells. The activity of cPLA2 is dependent on some proinflamatory cytokines in tumors [36, 37]. As cPLA2 is necessary for TNFα induced ceramide generation [38, 39], we investigated whether cisplatin treatment can regulate the cPLA2 expression. On analyzing cPLA2 enzyme activity upon cisplatin treatment (Figure 4A), cPLA2 activity was found to be elevated significantly. Similar observation was obtained in western blot analysis (Figure 4B). However, upon treatment with TNFα neutralizing (10 μg/ml) antibody, the cPLA2 activity was found to be decreased as confirmed by activity assay (Figure 4C), western blot as well as mRNA expression analysis (Figure 4D).
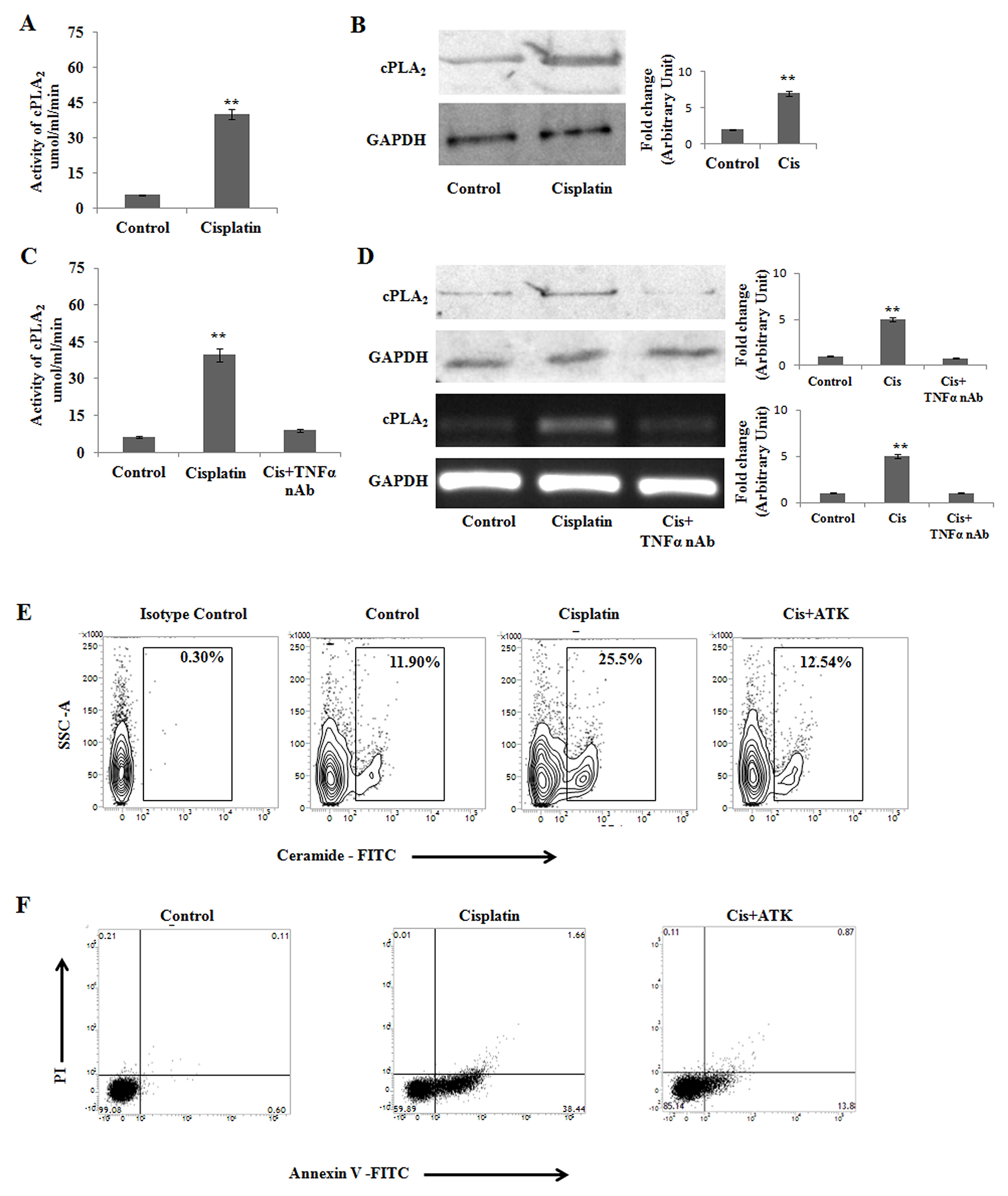
Figure 4: cPLA2 modulates cisplatin mediated apoptosis in PKCδ silenced B16F10 melanoma cells. (A) and (B) B16F10 cells were transfected with PKCδ siRNA followed by Cisplatin treatment. cPLA2 activity was determined from cell lysates using the cytosolic phospholipase A2 assay kit according to the manufacturer’s instructions. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. The expression cPLA2 was analysed in whole cell lysates by western blot analysis. GAPDH was used as a reference. Data are from one of three representative experiments. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (C) PKCδ specific siRNA transfected B16F10 melanoma cells were treated with cisplatin and TNFα neutralizing antibody. cPLA2 activity was determined from cell lysates using the cytosolic phospholipase A2 assay kit according to the manufacturer’s instructions. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (D) Similarly PKCδ specific siRNA transfected B16F10 melanoma cells were treated with cisplatin and TNFα neutralizing antibody. The expression cPLA2 was analysed in whole cell lysates by western blotting. In a separate experiment, the treated cells were collected in TRIZOL for mRNA expression of cPLA2 by semi quantitative RT-PCR. GAPDH was used as a reference. Data are from one of three representative experiments. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (E) and (F) PKCδ silenced and cisplatin treated B16F10 cells were incubated in the presence or absence of the cPLA2 inhibitor ATK. Cells were collected and subsequently stained with anti-mouse ceramide-FITC, isotype-matched control mouse antibody. Ceramide expression was analysed by flow cytometry. In a different experiment, as mentioned in Materials and Methods, cells were stained with Annexin-V FITC and PI and subsequently analyzed by flow cytometry. Data are from one of the three representative experiments.
Moreover, when 10μM arachidonyl trifluoromethyl ketone (ATK; a cPLA2 inhibitor) is treated in PKCδ deficient B16F10 melanoma, it failed to augment ceramide generation (Figure 4E) and apoptosis (Figure 4F) in cisplatin treated cells. Therefore, our results clearly suggest that cPLA2 and ASMase mediated ceramide generation plays a major role in PKCδ deficient melanoma cells to induce apoptosis.
Reciprocal regulation of Sphingosine kinase 1 and Sphingosine kinase 2 on cisplatin treated PKCδ silenced B16F10 melanoma cells
Sphingosine kinase 1 and Sphingosine kinase 2 are two important mediators in ceramide induced apoptosis, where the fine tuning of both the enzymes decides the fate of cell [27, 28]. Activators of Sphk2 act as the promising regulator of apoptosis [40]. Sphk1 and Sphk2 maintained a reciprocal regulation in PKCδ silenced B16F10 melanoma cells in a time dependent manner (Figure 5A), which was found to be correlated with the activity assay of sphingosine kinases (Figure 5B). In the initial hrs of cisplatin treatment Sphk1 was found to be upregulated whereas the expression of Sphk2 was downregulated. In contrary, these expressions were reversed at later time points, indicating the reciprocal regulation of both the enzymes. To further confirm the role of Sphk1 and Sphk2 in the regulation of apoptosis, we performed FACS analysis using their respective siRNAs. The result showed that Sphk2 is majorly responsible for apoptosis in comparison with Sphk1 (Figure 5C). Therefore, our findings indicate that Sphk1 inhibition and Sphk2 activation are important for cisplatin mediated apoptosis in PKCδ deficient B16F10 melanoma cell.
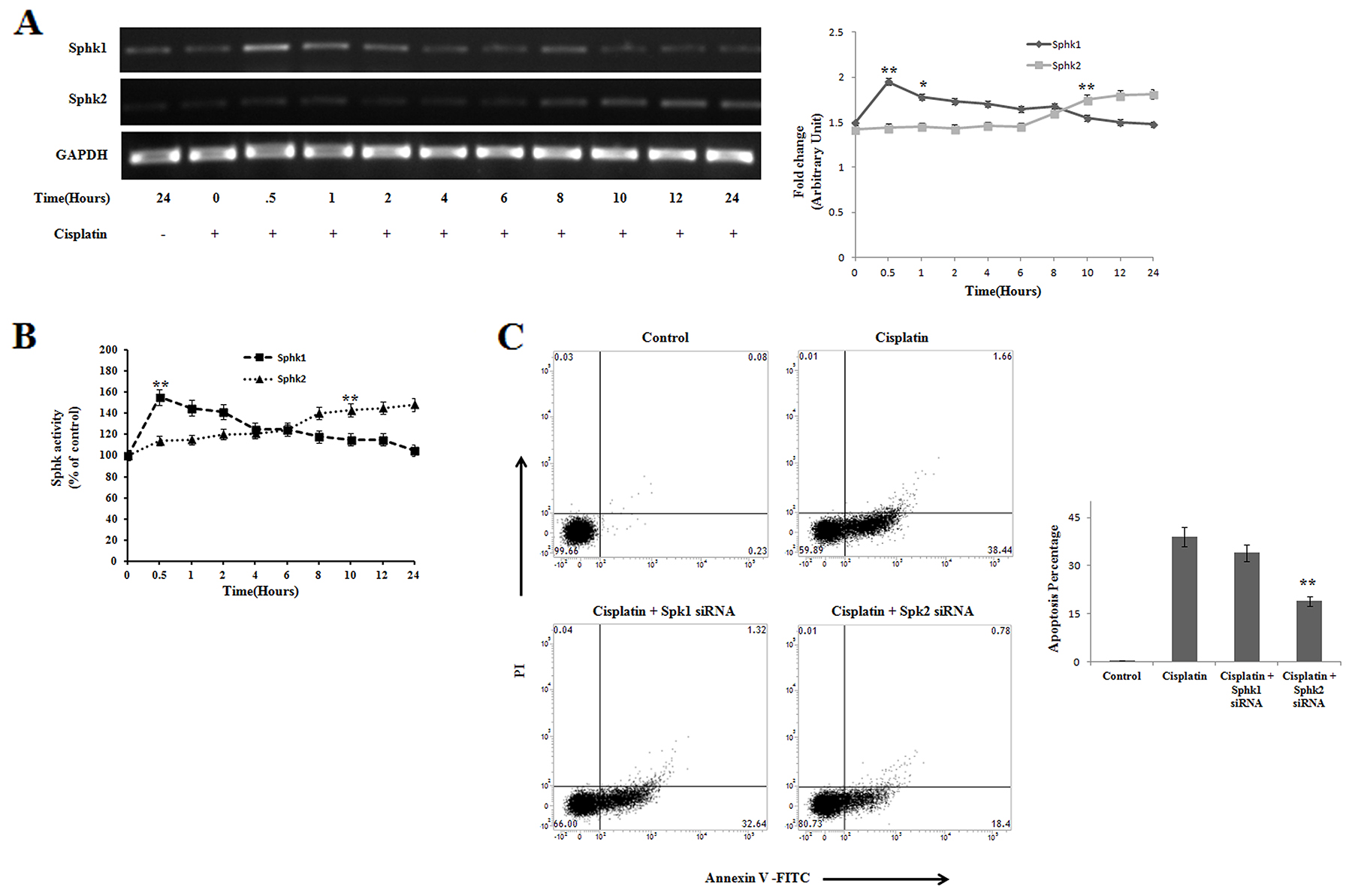
Figure 5: Reciprocal regulation of Sphingosine kinase 1 and Sphingosine kinase 2 on cisplatin treated PKCδ silenced B16F10 melanoma cells. (A) B16F10 cells were transfected with PKCδ siRNA followed by cisplatin treatment in a time dependent manner. At different time points the treated cells were collected in TRIZOL for Sphk1 and Sphk2 mRNA expression by semi quantitative RT-PCR analysis. GAPDH was used as a reference. Data are from one of the three representative experiments. At different time points, data were represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (B) B16F10 cells were transfected with PKCδ siRNA followed by cisplatin treatment. Activities of sphingosine kinase 1 and sphingosine kinase 2 were determined from cell lysates following the protocol described in Materials and Methods. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (C) In a separate experiment, similarly PKCδ silenced cisplatin treated B16F10 cells were transfected with Sphk1 and Sphk2 specific siRNAs as mentioned in Materials and Methods. Cells were collected stained with Annexin-V FITC and PI and subsequently analyzed by flow cytometry. Data are from one of the three representative experiments.
Cisplatin continues to induce apoptosis by ceramide generation during hypoxic condition in PKCδ silenced B16F10 cells
Major tumor environmental regulations are prominent in tumor core, where the extent of hypoxia is more powerful than the periphery. So, generation of hypoxic conditions in vitro can successfully mimic the tumor core regulations [41, 42]. To evaluate the effect of cisplatin in PKCδ deficient conditions and to correlate it with tumor hypoxia, PKCδ deficient B16F10 melanoma cells were incubated in hypoxia chamber, where oxygen tension was maintained at 90% lower than normoxic environment. Exposure of cisplatin treated PKCδ deficient B16F10 melanoma cells to hypoxia continued to generate ceramide (Figure 6A) and induced apoptosis (Figure 6B). Hence, these results clearly demonstrated that cisplatin treatment has a great potential in melanoma therapy even in PKCδ deficient conditions.
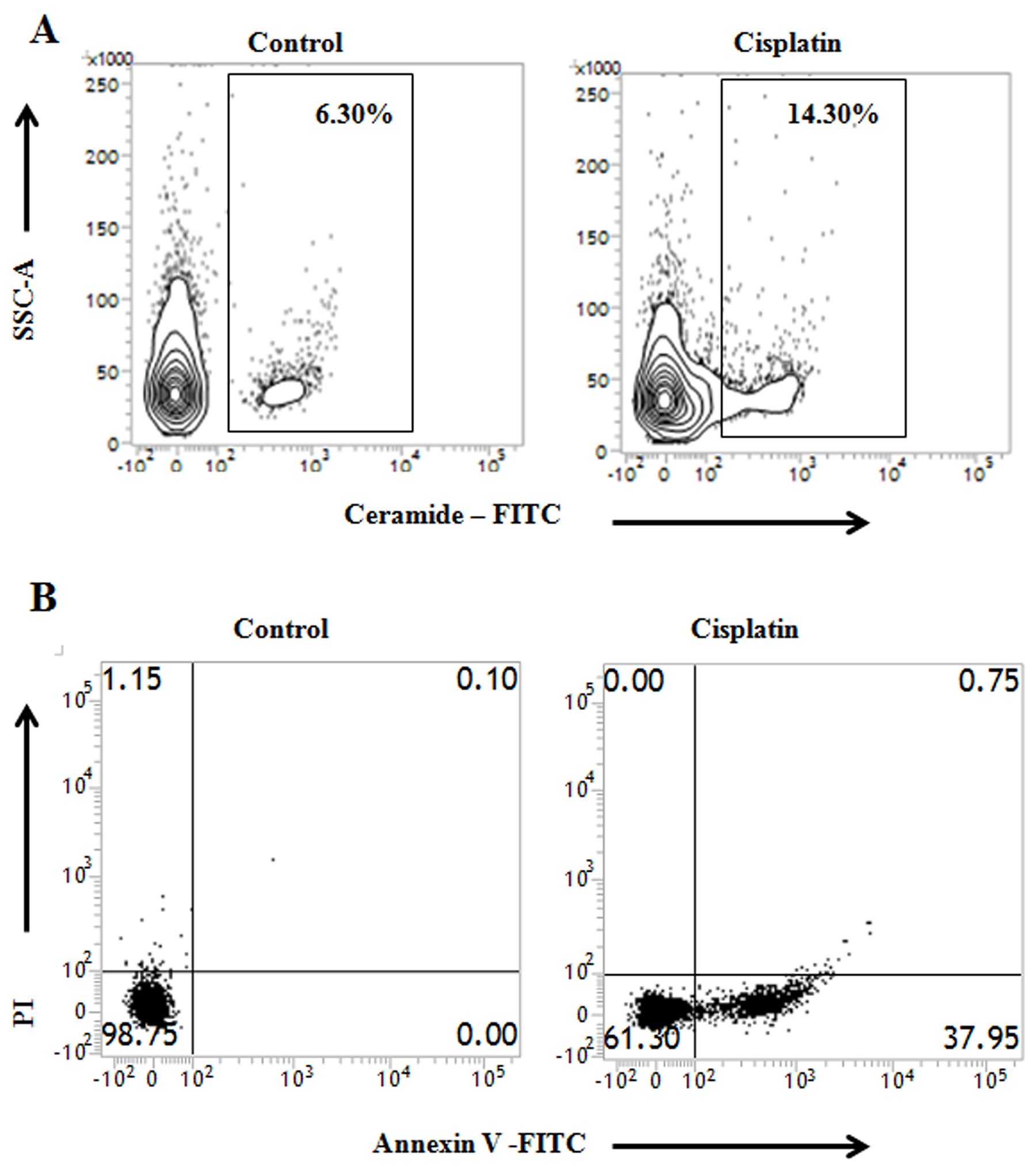
Figure 6: Cisplatin continues to induce apoptosis by ceramide generation during hypoxic condition in PKCδ silenced B16F10 Cells. (A) and (B) B16F10 cells transfected with PKCδ siRNA followed by cisplatin treatment were cultured in hypoxic chamber as described in Materials and Methods. Cells were collected and subsequently stained with anti-mouse ceramide-FITC, isotype-matched control mouse antibody. Ceramide expression was measured by flow cytometry analysis. In a different experiment as mentioned in Materials and Methods, cells were stained with Annexin-V FITC and PI and subsequently analyzed by flow cytometry. Data are from one of the three representative experiments. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated.
Cisplatin treatment in PKCδ silenced A375 human melanoma cells induces ceramide mediated apoptosis through IRF1-TNFα axis
Our previous results suggest a positive regulatory role of IRF1-TNFα interactions in ceramide generation leading to apoptosis in PKCδ silenced B16F10 mouse melanoma cells. Therefore, we intended to validate the role of TNFα signaling on ceramide generation pathway in PKCδ silenced A375 human melanoma cells. In the present study, after successfully silencing PKCδ in A375 cells using specific siRNA (Figure 7A and 7B) the effect of cisplatin on apoptosis was studied. We observed a marked decrease in the ceramide mediated apoptosis in cisplatin (50μM, 24h) treated PKCδ silenced A375 cells when imipramine (10μM), ATK (10μM) or IRF1 siRNA were used in the experiment (Figure 7C and Figure 7D). The treatment of exogenous recombinant TNFα (50ng/ml) also increased ceramide generation in PKCδ silenced A375 melanoma cells (Figure 7C). Interestingly, Sphk2 siRNA, TNFα or TNFα-R1 neutralizing antibody (10 μg/ml) treated cells showed higher percentage of apoptosis upon cisplatin treatment in comparison with the untreated cells (Figure 7D). On the other hand, under hypoxic condition, cisplatin was able to induce apoptosis (Figure 7F) by ceramide generation (Figure 7E) in A375 melanoma cells under PKCδ silenced condition. Hence our data suggests that the TNFα mediated signaling is also operational to increase apoptosis by ceramide generation in PKCδ silenced human melanoma cells.
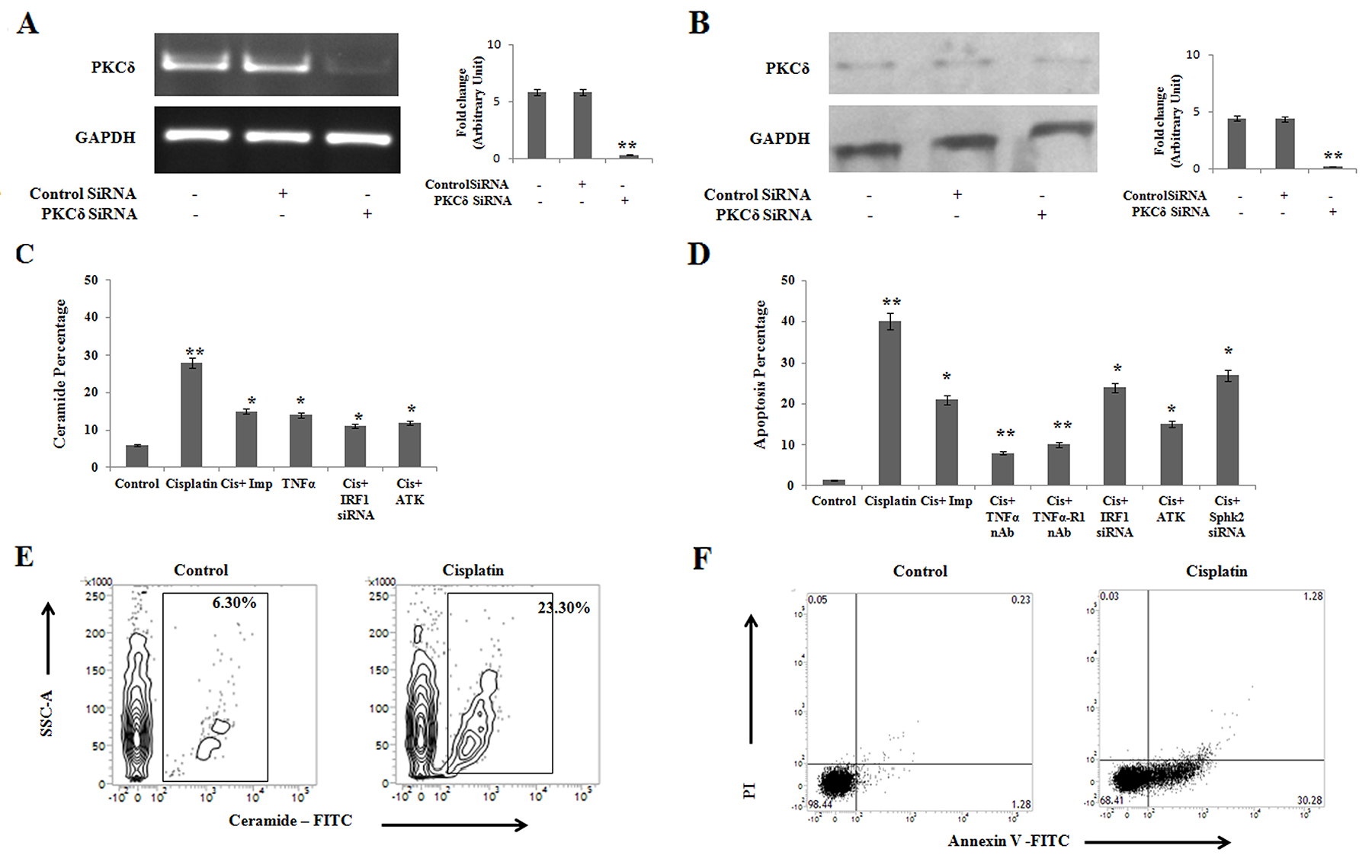
Figure 7: Cisplatin treatment in PKCδ silenced A375 human melanoma cells induces ceramide mediated apoptosis through IRF1-TNFα axis. (A) 2 × 106 A375 cells were transfected with PKCδ specific siRNA or control siRNA as mentioned in Materials and Methods and the transfected cells were collected in TRIZOL for mRNA expression of PKCδ by semi quantitative RT-PCR. GAPDH was used as a reference. Data are from one of the three representative experiments. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (B) In a separate experiment, similarly A375 cells were either transfected with PKCδ specific siRNA or control siRNA as mentioned in Materials and Methods. The expression of PKCδ was analyzed in whole cell lysates by western blot analysis. GAPDH was used as a reference. Data are from one of the three representative experiments. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (C) PKCδ silenced A375 cells were either treated with cisplatin alone or in combination with imipramine, ATK or transfected with IRF1 specific siRNAs. Cells were also treated with exogenous TNFα in absence of cisplatin. Treated cells were subsequently stained with anti-mouse ceramide-FITC, isotype-matched control mouse antibody. Ceramide expression was analysed by flow cytometry. Data are from one of the three representative experiments. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (D) PKCδ silenced A375 cells were either treated with cisplatin alone or in combination with imipramine, ATK, TNFα/TNFα-R1 neutralizing antibody or transfected with IRF1/Sphk2 specific siRNAs as mentioned in Materials and Methods. Cells were collected, stained with Annexin-V FITC and PI and subsequently analyzed by flow cytometry. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (E) and (F) A375 cells, transfected with PKCδ siRNA followed by cisplatin treatment were cultured in hypoxic chamber as described in Materials and Methods. Cells were collected and subsequently stained with anti-mouse ceramide-FITC, isotype-matched control mouse antibody. Ceramide expression was analysed by flow cytometry. In a different experiment cells were stained with Annexin-V FITC and PI and subsequently analyzed by flow cytometry. Data are from one of the three representative experiments.
Effect of cisplatin treatment in PKCδ inhibited melanoma tumor growth in vivo
To evaluate the effect of cisplatin treatment in vivo in PKCδ inhibited B16F10 melanoma tumor growth, B16F10 murine models were developed. We investigated whether the lentiviral expression of PKCδ shRNA would reduce the toxicity of cisplatin treatment in C57BL/6 mice. Tumor-bearing normal mice, lentivirally silenced with control shRNA or PKCδ shRNA mediated mice were treated with PBS or cisplatin (10 mg/kg bw) for 7 days after day 3 of tumor cell inoculation. Animals were sacrificed when the tumors reached a palpable size at day 21. Similar responses were observed in the control shRNA treated mice as well as in mice with no shRNA treatment in all the experiments (data of control shRNA treated mice not shown). As shown in (Figure 8A), cisplatin significantly reduced PKCδ inhibited B16F10 tumor growth in comparison with untreated tumor. Moreover, the cisplatin treatment in PKCδ inhibited B16F10 tumor bearing mice did not reduce the body weight, suggesting that PKCδ inhibition has no significant toxicity (Figure 8B). Interestingly, we observed a reduction in the cisplatin mediated nephrotoxicity in kidney sections under PKCδ inhibited mice in comparison with uninhibited groups (Figure 8C). Control sets revealed normal glomeruli in the kidney section whereas cisplatin treatment revealed distorted glomerular kidney along with severe tubular damages. It is noteworthy that the cisplatin treatment in PKCδ silenced group was able in restoration of the normal glomerular kidney. Furthermore, the tumor samples of mice bearing B16F10 cells were isolated and experiments were conducted to determine whether cisplatin was able to generate ceramide in vivo. However, samples from mice treated with cisplatin for both PKCδ inhibited and uninhibited condition showed higher percentage of ceramide generation (Figure 8D). Hence, these results demonstrated that the cisplatin treatment under PKCδ inhibited condition could reduce ceramide mediated tumor growth with lesser toxic effects.
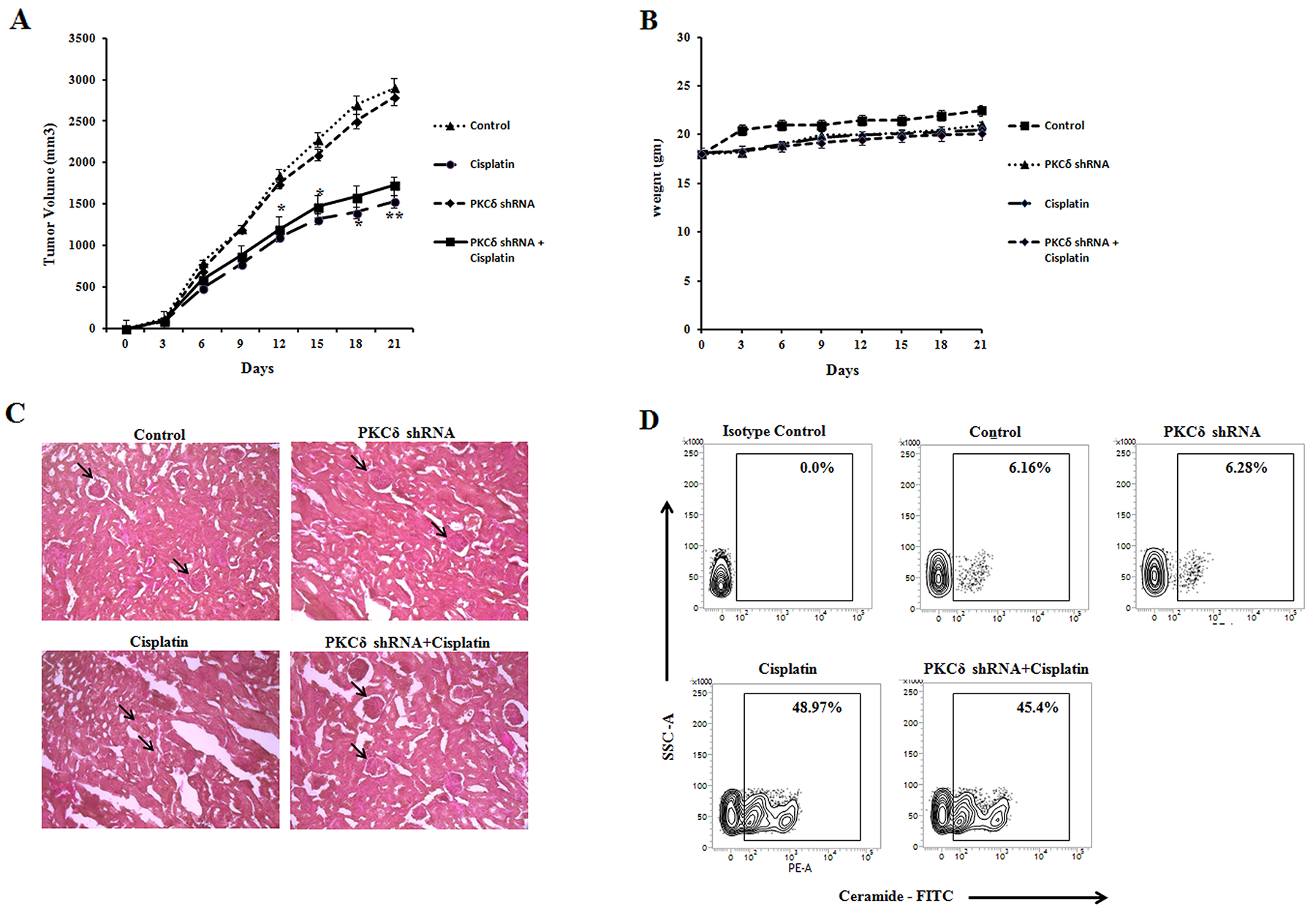
Figure 8: Effect of cisplatin treatment in PKCδ inhibited melanoma tumor growth in vivo. (A) and (B) 3 × 105 B16F10 melanoma cells were injected subcutaneously into the right flank of control and lentivirally PKCδ silenced female C57BL/6 mice and were left untreated or treated with cisplatin. The tumor volumes were monitored on every two days and the final tumor volume was determined at 21days post injection of tumor cells. The data shown here are representative of the three independent experiments. n = 4/ mice group. Bar diagram is represented as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (C) Representative kidney sections with or without PKCδ inhibited cisplatin treated mice groups were stained with hematoxylin and eosin (H&E). Arrows indicate conditions of glomerulus. Original magnification is 200X. (D) After enzymatic digestion of tumor, single cells were isolated and subsequently stained with anti-mouse ceramide-FITC, isotype-matched control mouse antibody. Ceramide expression was analysed by flow cytometry. Data are from one of the three representative experiments.
DISCUSSION
The complete cure of metastatic melanoma is still a challenge whereas surgery and radiation therapy can only play a role in the palliation of symptoms [2–5]. One of the major reasons for the difficulty in metastatic melanoma treatment is its heterogeneity [43]. Henceforth detailed investigation of signal transduction and the importance of apoptosis as the crucial player of tissue homeostasis maintenance, have gained more attention. Our study deals with this challenge revealing new details about the mode of action of cisplatin.
From decades cisplatin has been consumed as a traditional chemotherapeutic medicine for its antitumor activity and still remains as a significant interest [10]. As chemotherapeutic approaches may have a palliative benefit, the effects of which in the modulation of natural cellular components during cancer regression are major topics of discussion. In relation with this, ceramide, the central molecule in sphingolipids metabolism can function as a tumor-suppressor lipid inducing antiproliferative and apoptotic responses in various cancer cells [6–9]. Conversely, S1P induced responses render S1P as a tumor-promoting lipid [44]. These discoveries are paving the way for the advancement of anticancer therapies. Ceramide mediated signaling is involved in diverse functioning. In Leishmania infection ceramide plays a negative role for host by creating a favorable environment for the parasite entry [45, 46], whereas in case of cancer, ceramide induces programmed cell death which in turn favors host survival. The modulations of ceramide signaling as an effective therapy have been reported against various types of cancer [8, 9]. Previous study from our laboratory reported that PKCδ over expression results in ceramide generation, which leads to cancer cell apoptosis [18]. Even exogenous ceramide administrations show anti-proliferative effects and has been associated with induction of apoptosis in many cancers [47]. Endogenous ceramide is produced in the cell via two distinct pathways i.e., de novo pathway and the salvage pathway depending on various stimuli. The salvage pathway of ceramide production needs sphingomyelinase activity for the generation of ceramide from sphingomyelin which is known as one of the key pathway for the regulation of apoptosis [7]. The salvage pathway of ceramide production is specifically regulated by some PKC isotype mediated sphingomyelinase activity. Report suggested that anticancer drug etoposide is capable of inducing PKCδ mediated ceramide generation by both de novo and neutral sphingomyelinase (nSMase) pathway in prostate cancer cell [48]. PKCs are serine/threonine kinases which play vivid roles in cell proliferation, apoptosis, differentiation and angiogenesis [16]. Previous reports suggested that cisplatin via PKCδ causes regression of tumor, but at the same time PKCδ is found to be the major reason behind cisplatin induced nephrotoxicity [19, 20]. In order to find out the effective therapy against melanoma, we investigated whether cisplatin is able to overcome protein kinase C dependency during generation of ceramide, which has a broad spectrum of antineoplastic actions with lesser adverse effects and a noticeable efficacy in a wide range of tumors. Initial experiments of our studies surprisingly revealed that cisplatin can induce ceramide generations in PKCδ independent manner via a novel pathway which involves increased generation of TNFα.
For the first time, we examined the effect of cisplatin treatment on proliferation of melanoma cells in PKC silenced conditions and interestingly, it was effective to reduce the cancer progression significantly. On the other hand, cisplatin also upregulated the ceramide generation via sphingomyalinase pathway by increasing ASMase expression, but the de novo pathway remained unaltered. Further studies revealed that increased ASMase expression is dependent on TNFα and TNFαR1 expression on cisplatin treatment in PKCδ silenced melanoma cells. When exogenous TNFα was administered, it continued to increase ceramide expression whereas its neutralization reduced the ASMase activity. Previous studies support our result that TNFα is mainly expressed by activated immune cells especially macrophages and T cells, but can also be produced by tumor cells and capable of exhibiting anticarcinogenic effect in various subtypes of cancer [49, 50].
Damages in DNA due to drugs, such as etoposide and doxorubicin result in the upregulation of the transcription factors like IRF-1, IRF-3 and IRF-7 [51–54]. IRF-1 was reported to play a significant role in modulation of the immune response, cell proliferation, differentiation and inflammation. Subsequently, it has been shown that IRF1 and IRF3 act as tumor suppressor genes by preventing oncogene mediated malignancy. While investigating the probable mediators of cisplatin induced TNFα elevation, IRF1 was diagnosed as a key transcription factor. IRF1 was found to be translocated to nucleus on cisplatin treatment and by binding to TNFα promoter it augmented TNFα production leading to enhanced ceramide generations. Apart from ASMase, cPLA2 was also found to be upregulated by TNFα and continued to magnify ceramide generation and corresponding apoptosis even in PKCδ silenced condition following cisplatin treatment. Earlier report suggested that PLA2 enzymes comprise a large and growing number of signal transduction enzymes [55] and these PLA2 isoforms can be induced by the inflammatory mediators like IL-1β and TNFα [56]. In addition, another two key enzymes of sphingolipid biosynthesis, Sphk1 and Sphk2 were found to get reciprocally regulated in a time dependent manner during cisplatin treatment. The role of two isoforms of sphingosine kinase (Sphk) is to catalyze the formation of sphingosine 1-phosphate, the key sphingolipid mediator [27]. Sphk1 stimulates cell growth and survival of cells whereas Sphk2 enhances apoptosis along with suppressed cellular proliferation [29, 40]. Our finding confirmed that Sphk2 causes apoptosis in PKCδ deficient B16F10 cells.
In advanced stage of cancer, hypoxic microenvironment plays a critical role in chemoresistance and it is the major mediator for various regulations in cancer [30, 57–59]. Hypoxia, which is majorly present in solid tumor, is generated due to the proliferation of tumor cells and outpaces the blood vessel formation in the tumor mass. To explore the effect of hypoxia during cisplatin induced apoptosis, we mimicked in vivo hypoxic tumor core conditions using hypoxic chamber. The results clearly indicated that cisplatin induced ceramide generation followed by apoptosis in PKCδ silenced conditions. Moreover, we have performed the key experiments in human A375 melanoma cells to increase the acceptability of our study. It was interesting to observe that the study on human melanoma cells revealed similar responses to the results obtained from the study carried out on B16F10 cells. Further, cisplatin administered PKCδ silenced murine melanoma model showed increased ceramide generation and simultaneous inhibition of tumor growth with reduced toxicity as revealed from the kidney sections. This finding validated our study in in vivo system. However, detailed mechanistic studies are required for better understanding of our findings in future.
Taken together, our data decipher a novel mechanism exploited by cisplatin in PKCδ deficient conditions. It highlighted the involvement of bioactive sphingolipids ceramide and its impact to facilitate the host immune surveillance, restricting tumor growth through pro-inflammatory cytokine TNFα generation (Figure 9). These mechanistic details may open up a new insight of chemotherapy with lesser adverse effects exploiting natural cellular components like ceramide.
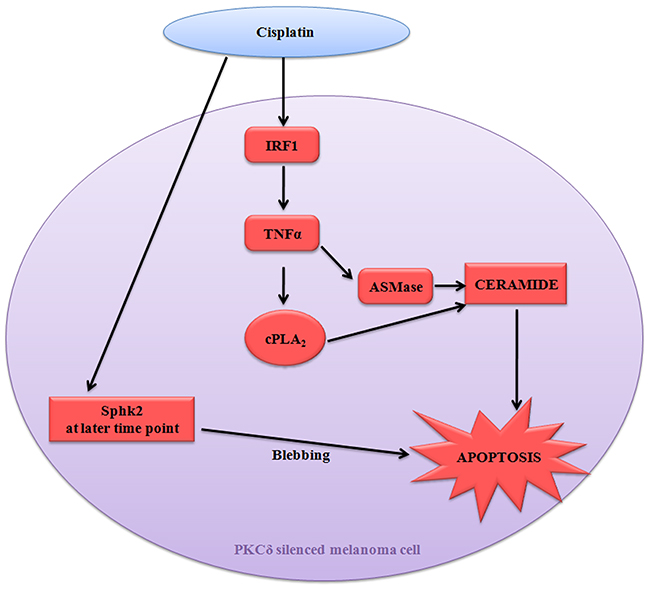
Figure 9: Diagrammatic representation of cisplatin mediated apoptosis of B16F10 melanoma cells in PKCδ independent manner.
MATERIALS AND METHODS
Reagents and chemicals
DMEM medium, penicillin, streptomycin, L-glutamine, Cisplatin, AKT, Fumonisin B-1 (FB-1), Imipramine and Trizol were purchased from Sigma (St Louis, MO, USA). Fetal calf serum was from Gibco BRL (Grand Island, NY, USA). Deoxynucleoside triphosphates, RevertAid M-MuLV Reverse Transcriptase, oligo-dT, RNase inhibitor and other chemicals for cDNA synthesis were purchased from Fermentas (Ontario, Canada). Recombinant mouse TNFα was obtained from R&D (Minneapolis, Canada). Mouse TNFα, TNFα-R1 neutralizing antibody was from CST (Danvers, USA). Anti- cPLA2, PKCδ, Ceramide, IRF1, TNFα, TNFα-R1, GAPDH antibodies were purchased from Santa Cruz Biotechnology (San Jose, CA, USA). Mouse specific IRF1 small-interfering RNA (siRNA), human specific PKCδ, IRF1, Sphk2 siRNA and control siRNAs were obtained from Santa Cruz Biotechnology.
Cell culture and transfection
B16F10 murine melanoma cells and A375 human melanoma cells were obtained from the National Centre for Cell Sciences, (Pune, India) and were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS), 120 mg/ml penicillin, 200 mg/ml streptomycin and 2 mM L-glutamine. The cells were cultured in a humidified 5% CO2 incubator at 37°C. Cells were regularly cultured to maintain exponential growth of cells. Twenty-four hrs before transfection, cells were diluted in fresh medium without antibiotics and transferred to 24-well plates. Transient transfection of PKCδ siRNA was performed using a Lipofectamine PLUS reagent (Invitrogen, CA, USA) according to the manufacturer’s recommendations. The efficiency of the transfection was monitored every 12 hrs. At 36 hrs, PKCδ expression was totally inhibited. Specific silencing was confirmed by at least three independent experiments. The cells were also treated with transfection reagent alone or with the non silencing scrambled PKCδ siRNA.
Preparation of cell lysates
The adherent melanoma cell population was scraped and centrifuged at 2000 g for 10 mins at 4°C. The cells were then resuspended in ice-cold extraction buffer containing 50 mM Tris-HCl (pH-7.5), 50mM EGTA, protease inhibitor cocktail, and 50mM β-mercaptoethanol. Anti-protease mixture consisted of 0.33 mM leupeptin, 0.2 mM phenyl methyl sulfonyl fluoride (PMSF), and 0.35 mM of aprotinin. The cell containing suspension was sonicated at 4°C and centrifuged at 8000g for 15 mins at 4°C. The supernatant was used for experiments after determination of protein content.
Gel electrophoresis and immunoblotting
Whole cell lysates were prepared in lysis buffer (10mM Tris; pH 7.5, 4.5 mM EGTA, 0.2 mM PMSF, 1.0 mM sodium orthovanadate, 4.8 Trypsin inhibitor units of aprotinin per ml and 0.33 mM leupeptin). The supernatants were assayed for protein estimation by Folin Lowry method. The protein samples in equal amounts for a single set of experiment were analyzed by SDS- PAGE together with protein molecular weight markers (Bio-Rad) and transferred to nitro-cellulose membrane [60]. The blots were washed by Tris Buffered saline (TBS) pH 7.5 and blocked with 3% BSA (MERCK) in TBS. The blots were then probed with the primary antibody (1:500) for 4 hrs. The blots were then washed three times with TBS containing polysorbate Tween-20 (TBST) and TBS sequentially, and incubated with secondary antibody (1:1000) covalently linked to Alkaline Phosphatase for 1h. Nitro blue Tetrazolium- Bromo Chloro Indolyl Phosphate (NBT-BCIP) was used as substrate for color development (NBT-7.5 mg dissolved in 175 μl Dimethyl formamide and 75μl double distilled water. BCIP- 3.75 mg dissolved in 250μl Dimethyl Formamide).
Densitometry analysis
Immunoblots were analyzed using a model GS-700 Imaging Densitometer and Molecular Analyst (version 1.5; Bio-Rad Laboratories).
Measurement of cytokines by SANDWICH ELISA
The level of mouse IL-12, IFN-γ, TNF-α, IL-10 and TGF-β in the conditioned medium of melanoma cell was measured using the sandwich Enzyme-Linked Immuno-Sorbent Assay (ELISA) kit (Quantikine M; R and D systems, Minneapolis, MN, USA). The assay was performed as per the detailed instructions of the manufacturer.
Isolation of RNA and RT-PCR
RNA was isolated according to the standard protocol [61]. Briefly total RNA extracted using TRIZOLTM reagent (SIGMA). Isolated total RNA was then reverse transcribed using Revert Aid TM M-MuLV Reverse Transcriptase (Fermentas). PCR amplification of the cDNA was conducted in a reaction volume of 20 μl using a Perkin Elmer Gen Amp PCR system 2400 and 0.5 unit of Taq polymerase set for 35 cycles. PCR amplified product was subsequently size fractioned on 1.5% agarose gel, stained with Ethidium Bromide and visualized under UV-light, the mRNA expression were compared, and normalized to GAPDH. The primer sequences are listed in Table 1.
Table 1: The primer sequences
Gene Name |
Primer Sequence |
|---|---|
Mouse IFN-γ |
Forward 5′-GGATATCTGGAGGAACTGGC-3′ |
Mouse IL-10 |
Forward 5′-CGGGAAGACAATAACTG-3′ |
Mouse TGF-β |
Forward 5′-GGATACCAACTATTGCTTCAGCTCC-3′ |
Mouse IL-12p40 |
Forward 5′-CAACATCAAGAG CAGTAGCAG-3′ |
Mouse TNF-α |
Forward 5′-GGCAGGTCTACTTT GGAGTCATTGC-3′ |
Mouse GAPDH |
Forward 5′-CAAGGCTG TGGGCAAGGTCA-3′ |
Mouse Ceramide Synthase 1 |
Forward 5′-TGCCATCGTTTTTGCGACCA-3′ |
Mouse Ceramide Synthase 2 |
Forward 5′-AAGCAGGTGGAGGTAGACCTTT-3′ |
Mouse Ceramide Synthase 3 |
Forward 5′-GGGTCAGTTCGTCAGTTGTTGT-3′ |
Mouse Ceramide Synthase 4 |
Forward 5′-AAAGCAGGGCCCAGTTTCAA-3′ |
Mouse Ceramide Synthase 5 |
Forward 5′-GCAATGGTGCCAACTGCAT-3′ |
Mouse Ceramide Synthase 6 |
Forward 5′-TTTGGCTTCCGCACAATGTCA-3′ |
Mouse ASMase |
Forward 5′-CTCCGCCTCATCTCTCTCA-3′ |
Mouse PKCδ |
Forward 5′-ACAAATGCAGGCAATGCAACG-3′ |
Mouse IRF-1 |
Forward 5′- CAGAGGAAAGAGAGAAAGTCC-3′ |
Mouse IRF-2 |
Forward 5′- CAGTTGAGTCATCTTTGGGGC-3′ |
Mouse IRF-3 |
Forward 5′- TACGTGAGGCATGTGCTGA-3′ |
Mouse IRF-4 |
Forward 5′- GTGACTGTGCCCTGGCTTAT-3′ |
Mouse IRF-5 |
Forward 5′- AATACCCCACCACCTTTTGA-3′ |
Mouse IRF-6 |
Forward 5′- GATGTACGATGGCACCAAGG-3′ |
Mouse IRF-7 |
Forward 5′- TGCAGAAGGTGGTGGGACA-3′ |
Mouse IRF-8 |
Forward 5′-AACTGTGCTCTGGGCTCATC-3′ |
Mouse Sphk1 |
Forward 5′-TTTGGAGGTTGCTGACGAGGTA-3′ |
Mouse Sphk2 |
Forward 5′-TTCTCGATGGTATGTGGGAGGA-3′ |
Mouse cPLA2 |
Forward 5′-TGTGTACAATCTTTGTGTTGTTTCA-3′ |
Mouse TNFR1 |
Forward 5′-GGGCACCTTTACGGCTTCC3-3′ |
Human PKCδ |
Forward 5′-AAAGGCAGCTTCGGGAAGGT-3′ |
Human GAPDH |
Forward 5′-GTTCGACAGTCAGCCGCATC-3′ |
Preparation of small interfering RNA
PKCδ (Sense 5′-AAC CTC ACT ACG CAT AGA CTG CCT GTC TC-3′), (Antisense 5′-AAC CTC ACT ACG CAT AGA CTG CCT GTC TC-3′), Sphk1 (Sense 5′-AAG AGC TGC AGA GCC TTG CCC CCT GTC TC-3′), (Antisense 5′-AAG GGC AAG GCT CTG CAG CTC CCT GTC TC-3′), Sphk2 (Sense 5′- AAG TCG CTG TAT GTG TAG GGC CCT GTC TC-3′), (Antisense 5′ AAG CCC TAC ACA TAC AGC GAC CCT GTC TC -3′) specific small interfering RNA (siRNA) were prepared using the Silencer siRNA Construction kit (Ambion) according to manufacturer’s protocol. A nonspecific scrambled siRNA (control siRNA) was generated with same GC content for control.
cPLA2 activity assay
cPLA2 activity assay was conducted using a cytosolic phospholipase A2 assay kit (Abcam, Cambridge, United Kingdom) according to the manufacturer’s instructions.
Caspase activity assay
Caspase activity assay was conducted using caspase assay kits for caspase-3, 7, 8, 9 (Abcam, Cambridge, United Kingdom) according to the manufacturer’s protocol.
Sphingosine kinase activity assay
Sphingosine kinase activity was performed according to the standard protocol as described elsewhere [62]. Briefly, Sphingosine kinase activity was measured from cell lysates in the presence of 20 μmol/L sphingosine, 1 mmol/L ATP, 10 mmol/L MgCl2, and 10 μCi/mL [3-3H] D-erythro-sphingosine (PerkinElmer, San Diego, USA). Sphk1 activity was determined in the presence of 0.25% Triton X-100, which inhibits Sphk2 activity. Sphk2 activity was determined with sphingosine added as a complex with 4 mg/mL BSA and 1 mol/L KCl, conditions in which Sphk2 activity is optimal and activity of Sphk1 is strongly inhibited. The reaction was performed at 37°C for 30 min and stopped on ice.
Hypoxic treatment
To mimic hypoxic condition, melanoma cells were cultured in a humidified incubator at 37°C in an atmosphere containing 5% O2, 85% N2 and 10% CO2 with the help of a N2 and a CO2 gas mixture in the Stem cell technology hypoxia chamber (Canada).
Chromatin immuno-precipitation (ChIP) assay
CHIP assays were conducted using the CHIP Assay kit following the manufacturers Protocol (Millipore, Billerica, MA) as described elsewhere [63]. With the extracted DNA, PCR was conducted using TNFα promoter specific primers, specifically IRF1 binding sites. PCR amplified product was resolved on 2% agarose gel, stained with ethidium bromide and visualized under UV-light. The primers from TNFα promoter used for PCR were: TNF - 533, 5’- CCT CCA AGA ACT CAA ACA GGG GGC TTT CCC-3’; TNF + 45, 5’-CTC CTG GCT AGT CCC TTG CTG TCC TCG CTG-3’.
Flow cytometry analysis
Melanoma cells in different experimental conditions were harvested and washed twice in ice-cold fluorescence-activated cell sorter (FACS) buffer (PBS containing 10% (w/v) BSA and 0.1% (w/v) sodium azide). Next, cells were collected by centrifugation (2000 rpm for 5 min), and exposed to FITC-conjugated anti-mouse ceramide antibody, anti-mouse TNFα receptor antibody for 1–2 h at 4°C in the dark. For intracellular cytokine staining, brefeldin A (10 mg/ml) treated melanoma cells were permeabilized (0.1% saponin) and stained with anti-mouse TNFα-PE antibodiy. After incubation, cells were washed in FACS buffer, and the percentage of cells expressing the targets were determined by comparison of fluorescence emission intensities collected using FACS Verse (Beckton Dickinson, NJ, USA).
Detection of cell proliferation
Melanoma cells in different condition were harvested and washed twice with PBS. Intracellular DNA was labeled with 1 ml of cold propidium iodide solution containing 0.1% Triton X- 100, 0.1 mM EDTA, 0.05 mg/ml RNase A, 50 ug/ml propidium iodide in PBS, and further incubated on ice for 30 min in the dark. Cytometric analyses were performed using a flow cytometer (FACS Verse, Beckton Dickinson, NJ, USA) and Cell Quest software.
Alternatively, (3H)-thymidine in corporation was determined. Cells were seeded in 96-well multi-plates. Each well was then pulsed with 1m Ci(3H) –thymidine (specific activity 20Ci/mmol). Sixteen hrs later, cells were washed twice with ice-cold PBS, treated with 5% trichloroaceticacid for 30 min at 4°C and solubilized with 0.5N NaOH. (3H)-Thymidine incorporation was determined on liquid scintillation counter (Wallac1409DSA, Finland). Data points for all assays were obtained in triplicate, and expressed as cpm/ug protein. Background radioactivity from cell-free wells was determined and subtracted from all data points
In vitro tumor apoptosis assay
In vitro apoptosis of tumor cells was determined using the apoptosis detection kit (BD Pharmingen) as per the manufacturer’s protocol. In brief, treated melanoma cells were incubated for 37°C in a humidified atmosphere with 5% CO2. After 24 hrs cells were washed with cold PBS and suspended in binding buffer and incubated with Annexin V-FITC and propidium iodide for 15 min at room temperature in the dark. Apoptotic cells were monitored by flow cytometric analysis.
Confocal microscopy
For IRF1 translocation study, cells on cover slips were fixed with 3% paraformaldehyde for 15 min in ice. Then cells were incubated in 0.5% Triton-X in PBS (25 min, room temperature) then washed twice with PBS. Slides were blocked with 1% BSA for 1 h at room temperature, and the rabbit polyclonal IRF1 antibody (final concentration 1:200) was added to the solution and incubated for 4 h. Slides were rinsed twice and washed three times with PBS for 5 min. The secondary antibody goat anti-rabbit IgG–PE conjugate (1:500) was incubated with the cells for 1 h at room temperature, followed by washes with PBS. The nuclei were stained with DAPI present in Vectashield, which was used for mounting. Finally, the slides were visualized under a Leica TCS SP8 Confocal platform.
Production of lentiviral particles for PKCδ and control shRNA
PKCδ shRNA (GenBank accession no. NM_011103) in pGIPZ lentivirus vector and control shRNA in pGIPZ lentivirus vector were purchased from Dharmacon (Lafayette, CO, USA). These shRNA were used for lentiviral production using Trans-Lentiviral packaging system (Dharmacon, USA) in HEK293T cell line following the manufacturer’s protocol.
Animal
Female 6-8 weeks old C57BL/6 mice were obtained from National Center for Laboratory Animal Sciences, Hyderabad, India. Mice were maintained under strict regulation with proper hygiene and every experiment was conducted in compliance with the regulations of Institutional Animal Ethics Committee.
Tumor induction and treatment
Mice were injected (i.v) with 5 × 106 transduction units (TU) of lentivirus-expressing PKCδ shRNA and control shRNA before day 3 of tumor induction. The mice were injected subcutaneously with B16F10 cells (3 × 105 cells) in 100μl PBS into the right flank of each mouse. Mice were divided into 6 groups (n = 4). The control group received vehicle (PBS), while treatment group received intradermal (i.d.) injection of cisplatin (10 mg/kg bw) for 7 days after day 3 of tumor cell implantation. Tumor size was measured bi-dimensionally with calipers every 2 days, and tumor volume was estimated using the formula: (a × b2)/2, where a is the largest diameter and b is its perpendicular. Mice were euthanized after day 21 and excised surgically.
Histological studies
Kidney tissues of normal mice and treated mice were fixed by immersion in 10% neutral buffered formalin for 24 h at room temperature and then embedded in paraffin. 5μm thin tissue sections were then stained with hematoxylin-eosin (HE) and pathophysiological changes were observed under bright field microscope (Leica 269Microsystem DN1000; camera: DFC450 C).
Statistical analysis
The data, represented as mean ± standard deviation (SD), is from one experiment, which was performed at least three times. Student’s t test was employed to assess the significance of the differences between the mean values of control and experimental groups. A P value of less than 0.05 was considered significant and less than 0.001 was considered highly significant.
Abbreviations
PKCδ, protein kinase Cδ; IRF, interferon regulatory factor; TNFα, tumor necrosis factor alpha; ASMase, acid sphingomyelinase; cPLA2, cytosolic phospholipase A2; Sphk, sphingosine kinase; FB1, fumonisin B1; IL, interleukin; AKT, arachidonyl trifluoromethyl ketone; IFN-γ, interferon gamma; TGF-β, transforming growth factor beta; S1P, sphingosine-1-phosphate.
Author contributions
SG designed and performed the experiments, analyzed the data and wrote the paper; JJJ and KH performed the experiments and analyzed the data; SB, BPC, SKJ and SBM performed experiments; AS, AB and RNB helped in performing some experiments; SM designed the project, analyzed the data and wrote the paper.
ACKNOWLEDGMENTS
We are grateful to the Director, Bose Institute (Kolkata, India) for his continuous encouragement. We thank Mr. Prabal Gupta for his technical expertise and the Central Instrument Facility for their assistances. SG gratefully acknowledges CSIR, New Delhi for fellowship.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
FUNDING
The work was supported by the Council of Scientific and Industrial Research (CSIR), India (Grant No: 09/015(0475)2015-EMR-1).
REFERENCES
1. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011; 147: 275-92.
2. Bhatia S, Tykodi SS, Thompson JA. Treatment of metastatic melanoma: an overview. Oncology (Williston Park). 2009; 23: 488-96.
3. Eton O, Legha SS, Moon TE, Buzaid AC, Papadopoulos NE, Plager C, Burgess AM, Bedikian AY, Ring S, Dong Q, Glassman AB, Balch CM, Benjamin RS. Prognostic factors for survival of patients treated systemically for disseminated melanoma. J Clin Oncol. 1998; 16: 1103-11.
4. Wong JH, Skinner KA, Kim KA, Foshag LJ, Morton DL. The role of surgery in the treatment of nonregionally recurrent melanoma. Surgery. 1993; 113: 389-94.
5. Lui P, Cashin R, Machado M, Hemels M, Corey-Lisle PK, Einarson TR. Treatments for metastatic melanoma: synthesis of evidence from randomized trials. Cancer Treat Rev. 2007; 33: 665-80.
6. Coderch L, López O, de la Maza A, Parra JL. Ceramides and skin function. Am J Clin Dermatol. 2003; 4: 107-29.
7. Morad SA, Cabot MC. Ceramide-orchestrated signalling in cancer cells. Nat Rev Cancer. 2013; 13: 51-65.
8. Dbaibo GS, Hannun YA. Signal transduction and the regulation of apoptosis: roles of ceramide. Apoptosis. 1998; 3: 317-34.
9. Pettus BJ, Chalfant CE, Hannun YA. Ceramide in apoptosis: an overview and current perspectives. Biochim Biophys Acta. 2002; 1585: 114-25.
10. Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014; 740: 364-78.
11. Florea AM, Büsselberg D. Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects. Cancers (Basel). 2011; 3: 1351-71.
12. Eljack ND, Ma HY, Drucker J, Shen C, Hambley TW, New EJ, Friedrich T, Clarke RJ. Mechanisms of cell uptake and toxicity of the anticancer drug cisplatin. Metallomics. 2014; 6: 2126-33.
13. Dakup PP, Porter KI, Little AA, Gajula RP, Zhang H, Skornyakov E, Kemp MG, Van Dongen HPA, Gaddameedhi S. The circadian clock regulates cisplatin-induced toxicity and tumor regression in melanoma mouse and human models. Oncotarget. 2018; 9:14524-14538. https://doi.org/10.18632/oncotarget.24539.
14. Noda S, Yoshimura S, Sawada M, Naganawa T, Iwama T, Nakashima S, Sakai N. Role of ceramide during cisplatin-induced apoptosis in C6 glioma cells. J Neurooncol. 2001; 52: 11-21.
15. Brodie C, Blumberg PM. Regulation of cell apoptosis by protein kinase c delta. Apoptosis. 2003; 8: 19-27.
16. Yoshida K. PKCdelta signaling: mechanisms of DNA damage response and apoptosis. Cell Signal. 2007; 19: 892-901.
17. Zeidan YH, Hannun YA. Activation of acid sphingomyelinase by protein kinase Cdelta-mediated phosphorylation. J Biol Chem. 2007; 282: 11549-61.
18. Halder K, Banerjee S, Bose A, Majumder S, Majumdar S. Overexpressed PKCδ downregulates the expression of PKCα in B16F10 melanoma: induction of apoptosis by PKCδ via ceramide generation. PLoS One. 2014; 9: e91656.
19. Pabla N, Dong G, Jiang M, Huang S, Kumar MV, Messing RO, Dong Z. Inhibition of PKCδ reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J Clin Invest. 2011; 121: 2709-22.
20. Pabla N, Dong Z. Curtailing side effects in chemotherapy: a tale of PKCδ in cisplatin treatment. Oncotarget. 2012; 3:107-111. https://doi.org/10.18632/oncotarget.439.
21. Zhang J, Qian X, Ning H, Yang J, Xiong H, Liu J. Activation of IL-27 p28 gene transcription by interferon regulatory factor 8 in cooperation with interferon regulatory factor 1. J Biol Chem. 2010; 285: 21269-81.
22. Yanai H, Negishi H, Taniguchi T. The IRF family of transcription factors: inception, impact and implications in oncogenesis. Oncoimmunology. 2012; 1: 1376-1386.
23. Chen FF, Jiang G, Xu K, Zheng JN. Function and mechanism by which interferon regulatory factor-1 inhibits oncogenesis. Oncol Lett. 2013; 5: 417-423.
24. Waters JP, Pober JS, Bradley JR. Tumour necrosis factor and cancer. J Pathol. 2013; 230: 241-8.
25. Shamimi-Noori S, Yeow WS, Ziauddin MF, Xin H, Tran TL, Xie J, Loehfelm A, Patel P, Yang J, Schrump DS, Fang BL, Nguyen DM. Cisplatin enhances the antitumor effect of tumor necrosis factor-related apoptosis-inducing ligand gene therapy via recruitment of the mitochondria-dependent death signaling pathway. Cancer Gene Ther. 2008; 15: 356-70.
26. Murtas D, Maric D, De Giorgi V, Reinboth J, Worschech A, Fetsch P, Filie A, Ascierto ML, Bedognetti D, Liu Q, Uccellini L, Chouchane L, Wang E, et al. IRF-1 responsiveness to IFN-γ predicts different cancer immune phenotypes. British Journal of Cancer. 2013; 109: 76-82.
27. Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R, Collier C, Zhang M, Satin LS, Merrill AH Jr, Milstien S, Spiegel S. SphK1 and SphK2, Sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J Biol Chem. 2005; 280: 37118-29.
28. Bektas M, Jolly PS, Müller C, Eberle J, Spiegel S, Geilen CC. Sphingosine kinase activity counteracts ceramide-mediated cell death in human melanoma cells role of Bcl-2 expression. Oncogene. 2005; 24: 178-87.
29. Min J, Mesika A, Sivaguru M, Van Veldhoven PP, Alexander H, Futerman AH, Alexander S. (Dihydro)ceramide synthase 1 regulated sensitivity to cisplatin is associated with the activation of p38 mitogen-activated protein kinase and is abrogated by sphingosine kinase 1. Mol Cancer Res. 2007; 5: 801-12.
30. Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia. 2015; 3: 83-92.
31. Elias EG, Hasskamp JH, Sharma BK. Cytokines and growth factors expressed by human cutaneous melanoma. Cancers (Basel). 2010; 2: 794-808.
32. Lázár-Molnár E, Hegyesi H, Tóth S, Falus A. Autocrine and paracrine regulation by cytokines and growth factors in melanoma. Cytokine. 2000; 12: 547-54.
33. Lee S, Margolin K. Cytokines in cancer immunotherapy. Cancers. 2011; 3: 3856-3893.
34. Pavan S, Olivero M, Corà D, Di Renzo MF. IRF-1 expression is induced by cisplatin in ovarian cancer cells and limits drug effectiveness. Eur J Cancer. 2013; 49: 964-73.
35. Mattei F, Schiavoni G, Sestili P, Spadaro F, Fragale A, Sistigu A, Lucarini V, Spada M, Sanchez M, Scala S, Battistini A, Belardelli F, Gabriele L. IRF-8 controls melanoma progression by regulating the cross talk between cancer and immune cells within the tumor microenvironment. Neoplasia. 2012; 14: 1223-35.
36. Mutch DG, Powell CB, Kao MS, Collins JL. Resistance to cytolysis by tumor necrosis factor alpha in malignant gynecological cell lines is associated with the expression of protein(s) that prevent the activation of phospholipase A2 by tumor necrosis factor alpha. Cancer Res. 1992; 52: 866-72.
37. Hollenbach PW, Zilli DL, Laster SM. Inhibitors of transcription and translation act synergistically with tumor necrosis factor to cause the activation of phospholipase A2. J Biol Chem. 1992; 267: 39-42.
38. Voelkel-Johnson C, Thorne TE, Laster SM. Susceptibility to TNF in the presence of inhibitors of transcription or translation is dependent on the activity of cytosolic phospholipase A2 in human melanoma tumor cells. J Immunol. 1996; 156: 201-7.
39. Hoeck WG, Ramesha CS, Chang DJ, Fan N, Heller RA. Cytoplasmic phospholipase A2 activity and gene expression are stimulated by tumor necrosis factor: dexamethasone blocks the induced synthesis. Proc Natl Acad Sci U S A. 1993; 90: 4475-9.
40. Don AS, Martinez-Lamenca C, Webb WR, Proia RL, Roberts E, Rosen H. Essential requirement for sphingosine kinase 2 in a sphingolipid apoptosis pathway activated by FTY720 analogues. J Biol Chem. 2007; 282: 15833-42.
41. Cosse JP, Michiels C. Tumour hypoxia affects the responsiveness of cancer cells to chemotherapy and promotes cancer progression. Anticancer Agents Med Chem. 2008; 8: 790-7.
42. Kunz M, Ibrahim SM. Molecular responses to hypoxia in tumor cells. Mol Cancer. 2003; 2: 23.
43. Grzywa TM, Paskal W, Włodarski PK. Intratumor and intertumor heterogeneity in melanoma. Translational Oncology. 2017; 10: 956-975.
44. Tabasinezhad M, Samadi N, Ghanbari P, Mohseni M, Saei AA, Sharifi S, Saeedi N, Pourhassan A. Sphingosin 1-phosphate contributes in tumor progression. J Cancer Res Ther. 2013; 9: 556-63.
45. Dey R, Majumder N, Bhattacharjee S, Majumdar SB, Banerjee R, Ganguly S, Das P, Majumdar S. Leishmania donovani-induced ceramide as the key mediator of Akt dephosphorylation in murine macrophages: role of protein kinase Czeta and phosphatase. Infect Immun. 2007; 75: 2136-42.
46. Majumder S, Dey R, Bhattacharjee S, Rub A, Gupta G, Bhattacharyya Majumdar S, Saha B, Majumdar S. Leishmania-induced biphasic ceramide generation in macrophages is crucial for uptake and survival of the parasite. J Infect Dis. 2012; 205: 1607-16.
47. Zhu W, Wang X, Zhou Y, Wang H. C2-ceramide induces cell death and protective autophagy in head and neck squamous cell carcinoma cells. International Journal of Molecular Sciences. 2014; 15: 3336-3355.
48. Sumitomo M, Ohba M, Asakuma J, Asano T, Kuroki T, Asano T, Hayakawa M. Protein kinase Cδ amplifies ceramide formation via mitochondrial signaling in prostate cancer cells. The Journal of Clinical Investigation. 2002; 109: 827-836.
49. Wang X, Lin Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol Sin. 2008; 29: 1275-1288.
50. Lu L, Shi W, Deshmukh RR, Long J, Cheng X, Ji W, Zeng G, Chen X, Zhang Y, Dou QP. Tumor necrosis factor-α sensitizes breast cancer cells to natural products with proteasome-inhibitory activity leading to apoptosis. PLoS One. 2014; 9: e113783.
51. Tanaka N, Ishihara M, Lamphier MS, Nozawa H, Matsuyama T, Mak TW, Aizawa S, Tokino T, Oren M, Taniguchi T. Cooperation of the tumour suppressors IRF-1 and p53 in response to DNA damage. Nature. 1996; 382: 816-8.
52. Kim T, Kim TY, Song YH, Min IM, Yim J, Kim TK. Activation of interferon regulatory factor 3 in response to DNA-damaging agents. J Biol Chem. 1999; 274: 30686-9.
53. Kim TK, Kim T, Kim TY, Lee WG, Yim J. Chemotherapeutic DNA-damaging drugs activate interferon regulatory factor-7 by the mitogen-activated protein kinase kinase-4-cJun NH2-terminal kinase pathway. Cancer Res. 2000; 60: 1153-6.
54. Savitsky D, Tamura T, Yanai H, Taniguchi T. Regulation of immunity and oncogenesis by the IRF transcription factor family. Cancer Immunol Immunother. 2010; 59: 489-510.
55. Bonventre JV. Phospholipase A2 and signal transduction. J Am Soc Nephrol. 1992; 3: 128-50.
56. Anthonsen MW, Solhaug A, Johansen B. Functional coupling between secretory and cytosolic phospholipase A2 modulates tumor necrosis factor-alpha- and interleukin-1beta-induced NF-kappa B activation. J Biol Chem. 2001; 276: 30527-36.
57. Banerjee S, Halder K, Ghosh S, Bose A, Majumdar S. The combination of a novel immunomodulator with a regulatory T cell suppressing antibody (DTA-1) regress advanced stage B16F10 solid tumor by repolarizing tumor associated macrophages in situ. Oncoimmunology. 2015; 4: e995559.
58. Halder K, Banerjee S, Ghosh S, Bose A, Das S, Chowdhury BP, Majumdar S. Mycobacterium indicus pranii (Mw) inhibits invasion by reducing matrix metalloproteinase (MMP-9) via AKT/ERK-1/2 and PKCα signaling: a potential candidate in melanoma cancer therapy. Cancer Biology & Therapy. 2017; 18: 850-862.
59. Ghosh T, Barik S, Bhuniya A, Dhar J, Dasgupta S, Ghosh S, Sarkar M, Guha I, Sarkar K, Chakrabarti P, Saha B, Storkus WJ, Baral R, Bose A. Tumor-associated mesenchymal stem cells inhibit naïve T cell expansion by blocking cysteine export from dendritic cells. Int J Cancer. 2016; 139: 2068-81.
60. Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979; 76: 4350-4.
61. Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987; 162: 156-9.
62. Blondeau N, Lai Y, Tyndall S, Popolo M, Topalkara K, Pru JK, Zhang L, Kim H, Liao JK, Ding K, Waeber C. Distribution of sphingosine kinase activity and mRNA in rodent brain. J Neurochem. 2007; 103: 509-17.
63. Banerjee S, Halder K, Bose A, Bhattacharya P, Gupta G, Karmahapatra S, Das S, Chaudhuri S, Bhattacharyya Majumdar S, Majumdar S. TLR signaling-mediated differential histone modification at IL-10 and IL-12 promoter region leads to functional impairments in tumor-associated macrophages. Carcinogenesis. 2011; 32: 1789-97.