
INTRODUCTION
Whilst the majority of melanoma patients are cured by surgical resection, approximately 30% of patients will present with systemic recurrence at some point during their lifetime [1], sometimes 10 or more years after diagnosis of primary melanoma [2–7]. Numerous factors such as clinical staging as well as host and tumor-dependent variables have been associated with recurrence [1, 7, 8]. Such characteristics however are not infallible and do not account for all patients that present with recurrence [4]. The risk of melanoma recurrence therefore is never entirely eliminated and the ability to correctly identify a subset of patients at risk of recurrence from a patient population with an expected favorable outcome is therefore challenging.
Timely detection of recurrence is of significant importance for improved patient outcomes given that surgical removal of solitary or a small number of isolated metastases most often improves overall survival [9–11]. Moreover, it has recently been shown that metastatic melanoma patients are more likely to achieve a long lasting response to systemic therapy when it is administered to patients with a low burden of disease [12, 13]. Currently, sentinel lymph node biopsies (SLNB), physical examination and radiological imaging methods such as 18F-FDG PET/CT are employed to stage primary melanomas and detect metastatic disease. Whilst 18F-FDG PET/CT provides valuable information on the location and metabolic activity of metastatic disease through real-time whole-body imaging [14], such imaging is not routinely used in the follow-up of early stage patients (American Joint Committee on Cancer (AJCC) stages 0-IIA) but rather for staging symptomatic patients or monitoring high-risk patients [15]. SLNBs are occasionally recommended for staging purposes for those with ≥1 mm thick melanomas [16]. However, they are relatively invasive and prohibitively costly for ongoing routine monitoring [17, 18]. Consequently, physical examination (including lymph node palpation) and radiological assessments are the primary surveillance strategies for patients having undergone primary tumor resection [15]. There is however, little consensus on the frequency of clinical assessment; the National Comprehensive Cancer Network (NCCN) guidelines suggest physical examinations be conducted every 3 to 12 months for the first 5 years and then annually thereafter for AJCC stage IA-IIA, and every 3 to 6 months for 2 years, then every 3 to 12 months for 3 years followed by annual checks and optional imaging for stages IIB to IV [15]. Importantly, physical examinations are usually inefficient at detecting distant metastases (disease that has spread to other sites such as lung, liver, bone or brain) [17], and thus melanoma recurrence is often detected at a late stage. Thus, there remains a need to improve follow-up for early detection of melanoma recurrence, which could lead to timely interventions and earlier treatment options that may improve patient outcomes.
CtDNA is an easily accessible marker that may be suitable for routine monitoring of disease recurrence [18–22]. The exact mechanism of ctDNA production remains elusive but it is thought to be released from cells predominantly by apoptosis and possibly through active secretion [23, 24]. Recent studies show that ctDNA levels exhibit a close correlation with disease status in a number of different cancers including gastric [25], colorectal [26], colon [27] and breast [19, 28], although, most studies have been conducted in the metastatic setting where ctDNA is likely to be relatively abundant. A few studies have demonstrated a potential utility for detecting ctDNA post-surgery in early stage cancers [29]. The presence of ctDNA was found to correlate with minimal residual disease in breast [28] and colon [27] cancers, and provide an early measure of clinically detectable recurrence in early stage lung cancer [30].
While there is now growing evidence that ctDNA has the potential to detect disease recurrence earlier or at the same time as conventional methods [27, 28, 30–32], the use of ctDNA for monitoring of residual disease and recurrence in clinically disease-free patients remains limited [27, 30, 31]. Thus, it remains unclear whether ctDNA is detectable at early stages and/or can provide early evidence of disease progression.
In this study, we aimed to determine whether in melanoma, ctDNA could indicate the presence of disease recurrence and/or of metastatic disease, earlier than conventional methods. To do this, we analyzed BRAF, NRAS and TERT promoter mutant ctDNA using serial blood samples from melanoma patients with and without confirmed disease recurrence.
RESULTS
Patient samples and cohort summary
From the original cohort of 139 patients, we selected 3 patients who developed metastatic disease and 27 patients with no evidence of disease recurrence at last follow-up or at the time of unrelated death for retrospective analysis (Figures 1 and 2). The median age was 63.7 (range 28 to 92 years). The median time from primary melanoma resection to enrolment was 14.7 months (range 0 to 134.6 months) and the median follow-up was 22 months (range 5.9 to 48.8 months).
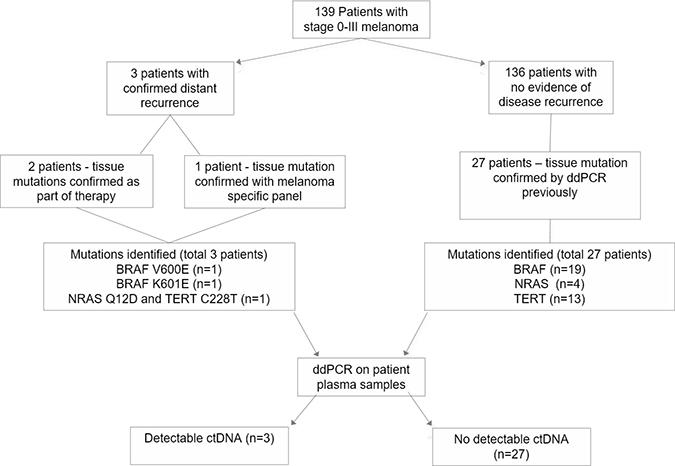
Figure 1: Overview of patient enrolment, sample collection and analysis. CtDNA was detectable in all three patients with disease recurrence but not in patients with no evidence of disease recurrence.
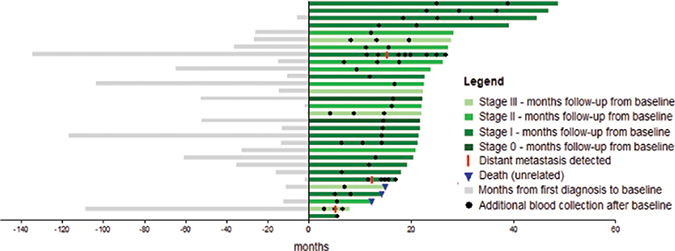
Figure 2: Swimmers plot in months from diagnosis of first primary melanoma to last follow-up. A total of 30 patients were assessed in the study. Bar colors indicate the melanoma stage at enrolment. All patients had a blood sample drawn at enrolment (zero months) and at the time points indicated (black dots). Three patients experienced recurrence during study follow up (red line) and another three cases died from reasons unrelated to melanoma (blue triangle).
Nineteen patients harbored a BRAF mutation, V600E (n = 8), V600K (n = 10) or K601E (n = 1); 4 patients had NRAS mutations, Q61K (n = 3), Q12D (n = 1); 13 had TERT promoter mutations, C228T (n = 8), C250T (n = 5) and 10 patients had more than one mutation. Of the patients that presented with disease recurrence, patient 2 harbored the BRAF K601E mutation, patient 6 harbored NRAS Q12D and TERT C228T mutations and patient 23 harbored the BRAF V600E mutation. Patient characteristics and their mutation status are described in Table 1.
Table 1: Patient and primary tumor characteristics
Patient number |
Sex |
Age at diagnosis |
AJCC stage |
Classification |
Breslow (mm) |
Ulceration |
Mitoses (rate per sq.mm) |
Regression |
Mutation* |
|---|---|---|---|---|---|---|---|---|---|
1 |
F |
38 |
I |
Unclassified |
0.35 |
- |
- |
Yes |
BRAF V600E |
2 |
M |
50 |
III |
Nodular |
8.3 |
Incipient |
4 |
No |
BRAF K601E |
3 |
M |
90 |
II |
SSM |
3.7 |
Yes |
10 |
No |
TERT C228T |
4 |
M |
83 |
I |
SSM |
1 |
No |
1 |
Slight |
BRAF V600E, NRAS Q61K, TERT C228T |
5 |
M |
93 |
III |
- |
- |
- |
- |
- |
TERT C228T |
6 |
M |
81 |
I |
Unclassified |
1.1 |
No |
No |
No |
NRAS Q12D, TERT C228T |
7 |
M |
75 |
I |
CBD |
1.2 |
No |
No |
Yes |
NRAS Q61K, TERT C250T |
8 |
F |
28 |
I |
SSM |
0.38 |
No |
<1 |
No |
BRAF V600E |
9 |
M |
46 |
I |
SSM |
0.45 |
No |
No |
Yes |
BRAF V600K |
10 |
M |
89 |
II |
Nodular |
5.5 |
No |
6 |
- |
BRAF V600K, TERT C250T |
11 |
F |
92 |
I |
SSM |
0.3 |
No |
No |
No |
BRAF V600K |
12 |
F |
45 |
I |
Unclassified |
0.3 |
- |
- |
No |
BRAF V600K |
13 |
M |
38 |
I |
SSM |
0.3 |
No |
No |
No |
BRAF V600E |
14 |
M |
41 |
0 |
- |
- |
- |
- |
- |
TERT C228T |
15 |
M |
73 |
III |
- |
- |
- |
- |
- |
BRAF V600K, TERT C250T |
16 |
M |
58 |
II |
SSM |
7 |
No |
No |
No |
NRAS Q61K |
17 |
M |
56 |
0 |
- |
- |
- |
- |
- |
BRAF V600E |
18 |
M |
58 |
III |
- |
- |
- |
- |
- |
NRAS Q61K, TERT C228T |
29 |
M |
44 |
II |
SSM |
1 |
Yes |
Active |
No |
TERT C250T |
20 |
F |
43 |
I |
Unclassified |
0.5 |
No |
No |
No |
BRAF V600K |
21 |
M |
67 |
II |
Nodular |
25 |
Yes |
5 |
No |
BRAF V600E, TERT C228T |
22 |
M |
74 |
II |
Nodular |
2 |
Yes |
10 |
Yes |
BRAF V600E, TERT C250T |
23 |
M |
62 |
I |
Nodular |
1.82 |
- |
20 |
- |
BRAF V600E |
24 |
M |
75 |
II |
Lentigo |
2.1 |
No |
8 |
Yes |
BRAF V600K |
25 |
M |
90 |
III |
- |
- |
- |
- |
- |
BRAF V600K |
26 |
M |
88 |
II |
SSM |
0.4 |
- |
- |
Yes |
TERT C228T |
27 |
M |
69 |
I |
SSM |
0.4 |
No |
No |
No |
BRAF V600K |
28 |
M |
58 |
I |
SSM |
0.45 |
No |
No |
Yes |
BRAF V600K |
29 |
F |
68 |
I |
Unclassified |
0.3 |
- |
- |
Yes |
BRAF V600E |
30 |
M |
40 |
I |
Unclassified |
0.45 |
- |
- |
Yes |
BRAF V600E |
Abbreviations: SSM = Superficial spreading melanoma; CBD = cannot be determined; – = unavailable. *Assessed by ddPCR
Patients 2, 6 and 23 presented with disease recurrence and had their tumors assessed for mutations by NGS sequencing in addition to ddPCR analysis
Patients indicated in bold are those that suffered recurrence.
Quantification of mutant specific ctDNA from plasma
Overall, cfDNA was isolated from 73 plasma samples taken from 30 patients. The number of mutant ctDNA copies per mL of plasma was quantified by ddPCR. CtDNA was measurable in all 3 patients (Figure 3) that presented with disease recurrence and in none of those without recurrence at last follow-up.
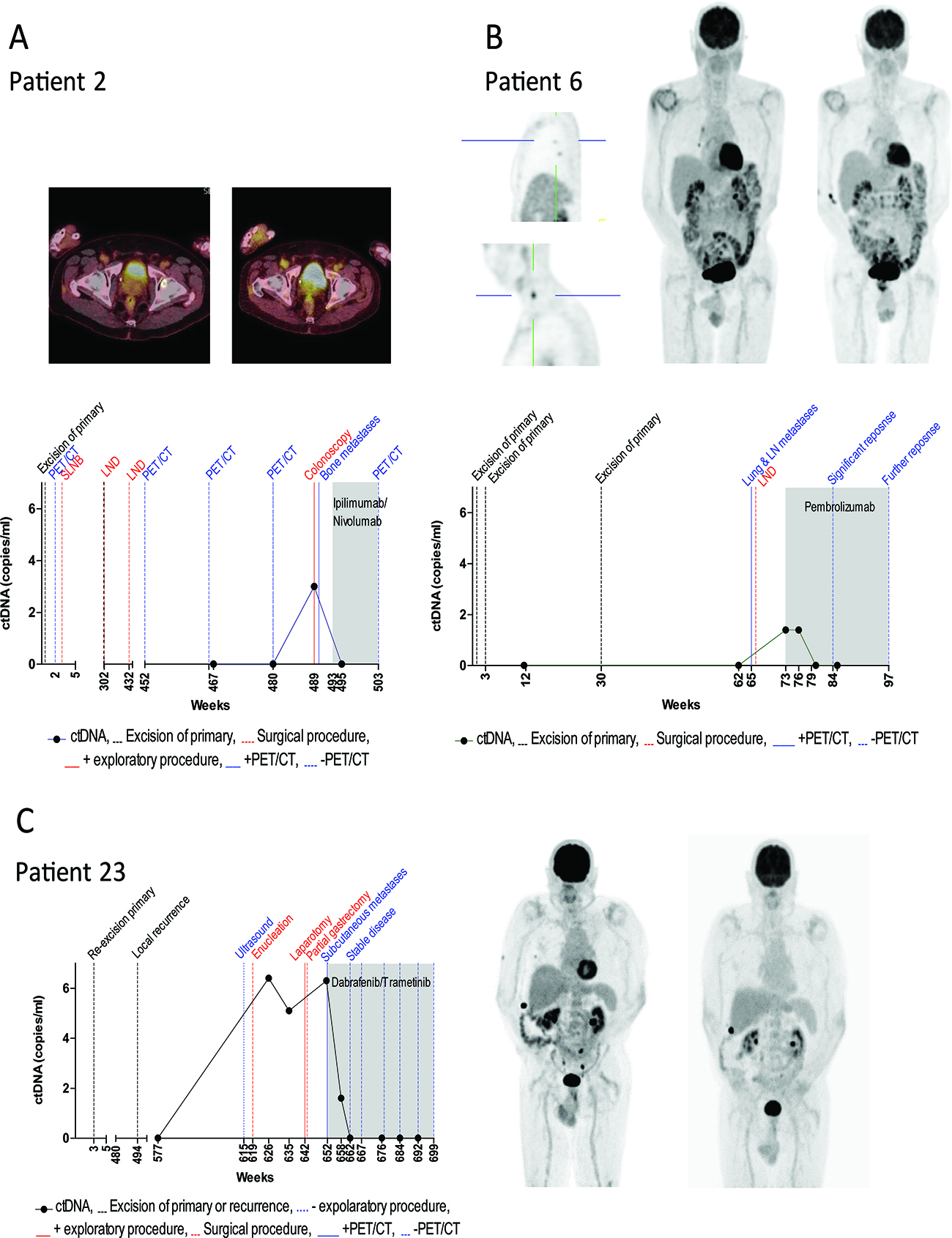
Figure 3: Monitoring ctDNA levels during clinical disease course. The levels of plasma ctDNA for three melanoma patients presenting with disease recurrence is represented in relation to the patients’ clinical history in weeks from first primary diagnosis. CtDNA concentrations were defined by (A) BRAF K601E (patient 2), (B) NRAS Q12D (patient 6) and (C) BRAF V600E (patient 23) mutant copies. The dotted black line indicates the time of resection of primary melanoma or local recurrence. Positive and negative radiological outcomes are represented by a solid and dashed blue line, respectively. Surgical procedures are represented by dashed red lines. Solid and dotted red lines represent positive and negative exploratory procedures respectively. The shaded area indicates the period during which systemic therapy was administered.
Patient 2
Curative resection was performed for a primary nodular melanoma located on the neck with a Breslow measurement of 8.35mm. PET/CT and SLNB conducted immediately after resection revealed no regional or distant metastases. The patient was disease free for approximately five years, then presented with two positive lymph node (LN) metastases at 302 weeks and with one positive LN at 432 weeks after primary diagnosis. All positive LNs were resected at time of identification. Approximately 36 weeks after the last resection the patient was recruited into this study at which time ctDNA was not detected (Figure 3A). 13 weeks later (48 weeks post lymph node dissection (LND)), PET/CT revealed no evidence of metastases and ctDNA remained negative. Following an investigative colonoscopy one-year post LND and 9 weeks post negative PET/CT, a subsequent biopsy revealed stage IV melanoma with bowel metastasis at which time ctDNA was detected at 3 copies/mL of plasma. A subsequent PET/CT also revealed bone metastases. The patient commenced full dose combination Ipilimumab/Nivolumab therapy and within three weeks, ctDNA was undetectable. At last follow-up (week 503), the patient had no evidence of metastatic disease on PET/CT.
Patient 6
Patient 6 underwent curative resection for a 1.1 mm primary melanoma on the right anterior scalp, with a mitotic rate of 2/sq. mm but no ulceration, regression, perineural or lymphovascular invasion. Re-excision was performed at week 3. At week 12 the patient was recruited into the study and ctDNA was not detected (Figure 3B). Approximately 30 weeks following initial diagnosis, a second primary invasive melanoma on the scalp measuring 3.1 mm with vascular involvement was excised with a wide margin. At week 62, ctDNA was not detected, but clinical and radiological evaluation three weeks later revealed a level III cervical lymph node (which was confirmed as metastatic melanoma on pathology) and five FDG avid nodules in the right lung (largest measuring 6 mm). No blood was collected at this time. Prior to commencing treatment of pembrolizumab almost eight weeks later, NRAS Q12D mutant ctDNA was detected at 1.4 copies/mL of blood. Within 3 weeks of commencing treatment, ctDNA was undetectable. Following PET/CT at week 84 post initial diagnosis, a significant but incomplete improvement was observed when compared to the previous PET/CT scan and ctDNA was not detected. The patient subsequently showed complete response on PET/CT and eighteen months post treatment cessation, remains disease free. The TERT promoter mutation found in the tumor, was not detected in plasma at any time point in this patient.
Patient 23
In 2004, the patient underwent resective surgery for a primary nodular melanoma on the left thigh. Histology reported the melanoma contained an unusual polypoidal appearance however demonstrated invasion of the reticular dermis (Clark level 4) and a maximum thickness of 1.82 mm. The lesion extended to within 1mm of a lateral resection margin and hence further wide local excision was recommended. Re-excision was completed within 3 weeks with no further residual tumor identified. There was at least 10 mm of skin and subcutaneous fat at either edge of the scar. Nodules of metastatic melanoma were identified and excised from the dermis and subcutis within and adjacent to the scar site 494 weeks after the primary melanoma was identified. Excision was complete with clearance of 0.5 mm from the deep and side margins. At week 577 the patient was recruited into the study and ctDNA was not detected (Figure 3C). Abdominal ultrasound at week 615 showed no evidence of metastatic disease. Approximately 42 weeks later at week 619, the patient presented with a red, painful eye and ocular melanoma was found. The patient elected to undergo enucleation. We did not have a blood sample to test ctDNA at this time point. Histological and molecular analysis of the tumor indicated the presence of a BRAF V600E mutation. BRAF V600E mutant ctDNA was detected at 6.4 copies/mL of plasma at week 626, and again at week 635 at 5.1 copies/mL of plasma. Having refused PET/CT scans due to claustrophobia, the patient underwent a gastroscopy for anemia at week 642, which revealed a 4 cm gastric metastasis and 2 ileum metastases (10 cm apart). All metastases were surgically removed following confirmation of metastatic melanoma on biopsy. Histological and molecular analysis of the gastric tumor also indicated the presence of a BRAF V600E mutation. No blood samples were collected at this time. At week 652 PET/CT revealed progressive disease with bone metastases. At this time, ctDNA was detected at 6.3 copies per mL of plasma and combination therapy of dabrafenib and trametinib was commenced. CtDNA levels declined to 1.6 copies/ml at 6 weeks and were undetectable at 10 weeks post treatment commencement. At week 667 (14 weeks post treatment initiation), PET/CT revealed complete response which has persisted and ctDNA levels have remained undetectable.
DISCUSSION
This report describes the results of a retrospective assessment of mutant specific ctDNA for patients with cutaneous melanoma after removal of a primary lesion (n = 30). In three patients with disease recurrence, ctDNA was detectable at the time of clinically confirmed disease recurrence. In one case, plasma ctDNA detection preceded clinical identification of recurrence by an interval of 4 months. By contrast, ctDNA was not detected at any time point in patients with no clinical evidence of disease recurrence.
Although there was variation in the timing of plasma collections, which did not necessarily coincide with functional imaging, three plasma samples were available for each recurring patient before the initiation of systemic therapy. In this study, no mutant specific ctDNA was detected in any patient prior to detection of metastatic disease, however ctDNA was later detected in all three patients that presented with disease recurrence and subsequently diagnosed with stage IV metastatic melanoma. This is in contrast with previous studies in stage II and III melanoma [33], uveal melanoma (Beasley), breast [28] and colon [27] cancers, which showed that ctDNA was detectable in patients with early stage disease.
In patient 23, BRAF V600E mutant ctDNA was detectable 4 months prior to metastatic disease being clinically evident. Notably, this patient was not undergoing routine PET/CT scans due to severe claustrophobia, and metastatic disease was discovered during a gastroscopy performed to evaluate asymptomatic anemia. Importantly however, this suggests that ctDNA may serve as an alternative option to those not wishing to undertake routine PET/CT scans.
Although we detected NRAS Q12D mutant ctDNA at 1.4 copies per mL of plasma at one time point in patient 6, we did not detect TERT C228T mutant ctDNA at any time point in this patient, although it was detected in the tumor. This may be explained by the fact that TERT promoter mutations demonstrate lower copy numbers relative to other driver mutations [34] (Calapre et al., manuscript submitted). Moreover, NRAS mutant ctDNA was not detected in the plasma sample collected three weeks prior to PET/CT which identified pulmonary metastases, nor was it found in the blood sample collected after initiation of systemic therapy where the PET/CT scan revealed a significant but incomplete response. This highlights a limitation in the sensitivity of our ctDNA detection approach which may be improved with increasing the amount of plasma from which cfDNA is extracted, as well as by introducing pre-amplification of cfDNA prior to ctDNA quantification. The use of next generation sequencing custom panels using unique molecular identifiers may also improve sensitivity as well as enable monitoring of a large number of mutations. Previously, we have shown a correlation between ctDNA and quantitative metabolic tumour burden (MTB) derived from PET/CT in treatment naïve patients [35]. In addition, future studies may further benefit from a correlation between ctDNA and MTB in patients undergoing treatment as this will further indicate the limit at which ctDNA can be detected in patients presenting with distant metastasis(es).
Of importance is the evidence of distant metastatic disease in patient 2 almost 9 years after the primary melanoma was resected and more than 3 years after stage III disease was initially diagnosed which supports the evidence that recurrences have been observed many years after primary resection [2, 3, 6]. During this time, 2 PET/CT scans were performed, and it was ultimately the colonoscopy which detected metastatic disease and prompted the third PET/CT scan which highlights the need for improvements in routine monitoring.
Whilst stage III melanoma patients have a higher risk of recurrence than stage I [36, 37], in our cohort two of the patients with stage 1 disease and only 1 of the patients with stage III disease at time of diagnosis of the primary, developed metastatic disease. Furthermore tumor thickness has been considered to be an important prognostic risk factor for recurrence [38], however here we report recurrence from primary tumors of 1.1 mm, 1.83 mm and 8.3 mm thickness. Previous studies that have compared time to recurrence with clinical staging as well as host and tumor-dependent variables, have shown that older age, gender (male), thin and non-ulcerated melanomas are each independently associated with a late recurrence (>8 years from time of diagnosis) [7, 8]. In our study, we report distant recurrence in one patient with a 1.1 mm thick non-ulcerated primary 63 weeks after initial diagnosis (Figure 3B). Conversely, Faries et al. reported tumors less than 1.23 mm and the lack of ulceration to each be independently associated with a late recurrence which they defined as >10 years from initial diagnosis [7]. In one patient with a 1.83mm thick primary, we report a local recurrence more than 9 years and metastatic disease more than 12 years after initial diagnosis (Figure 3C), however Faries et al. reported thicker tumors (2.93 mm) to be independently associated with an early recurrence (within 3 years of diagnosis) [7]. Whilst we only report 3 recurrences, the heterogeneity between patient outcomes and tumor-dependent variables highlight the challenges associated with correctly identifying a subset of patients at risk of recurrence from a patient population with an expected favorable outcome.
The major limitations of this study are the relatively short follow up time and the low number of patients analyzed, hence, the low number of observed recurrences. Nevertheless, we provide a proof of concept indicating that monitoring ctDNA during follow-up of melanoma patients after surgery may provide a potentially useful clinical test. Our experience confirms that ctDNA surveillance provides a supplementary method of monitoring patients without the additional risk of radiation exposure, invasiveness, and high cost of radiological scan, and that ctDNA tests indicate disease recurrence at least at the time of clinical confirmation. The detection of disease recurrence earlier may provide an increased window of opportunity for intervention that may positively impact on the patient’s outcomes. Furthermore, ctDNA testing may offer an alternative option for routine monitoring for patients who are unable, for whatever reason, to undergo functional imaging. Future prospective studies are needed to demonstrate the utility of ctDNA for routine monitoring of early stage melanoma patients.
In conclusion, this study has provided proof-of-concept evidence for the prospective and frequent monitoring of ctDNA in melanoma patients. However, prospective studies and improved assay sensitivity are needed before it can be used for monitoring patients after resection of their primary for early detection of metastatic disease.
MATERIALS AND METHODS
Patient and sample collection
Patient enrolment and study overview are presented in Figure 1. We analyzed a group of patients that were prospectively accrued into the “Blood Based Biomarkers: Prognostic Tools for Melanoma Recurrence” project between July 2013 and February 2017. The study was approved by the Human Research Ethics Committee of Edith Cowan University under reference numbers 11543 and 13313 and Sir Charles Gairdner Hospital (no.2013-246). A cohort of 139 melanoma patients (stage 0–III), diagnosed and resected in the previous 10 years were eligible for enrolment. A blood sample was collected at enrolment and then again between 3–25 months thereafter. Overall, three cases experienced distant disease recurrence during the sample collection period. The BRAF mutation status of these three tumors was determined previously [38] and by Next Generation Sequencing (NGS) using a custom targeted sequencing panel for a third case which was found to be BRAF wild-type. A subset of patients (n = 27) with no clinical evidence of disease recurrence at last follow-up was selected for ctDNA assessment. The main criteria for their selection was based on the availability of primary tumor mutational data determined as part of a previous study [39] in which they were found to be positive for at least one of the common melanoma mutations (BRAF, NRAS, TERT).
Isolation and quantification of circulating tumor DNA from plasma
Blood samples were collected into EDTA vacutainer tubes and stored at 4° C. Plasma was separated within 24 hours by centrifugation at 1600 g for 10 minutes, followed by a second centrifugation at 2000 g for 10 minutes, then stored at −80° C until extraction. Cell free DNA (cfDNA) was isolated from 5 mL of plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen) as per the manufacturer’s instructions, eluted in 40 μl AVE buffer (Qiagen) and stored at –80° C until ctDNA quantification. CtDNA was quantified by ddPCR targeting the specific mutation found in the patient’s tumor. BRAF mutations were detected using in-house primer/probe sets [40], NRAS mutations were tested using PrimePCR assays (Bio-Rad) and TERT promoter mutations were detected using in-house primer/probe sets [41]. Amplifications were carried out in a 20 μL reaction mixture containing ddPCR Supermix (Bio-Rad), 900 nM primers, 250 nM of each probe and 5 ul (for TERT assay) or 8 μL (for BRAF/NRAS assays) cfDNA. Droplets were generated using the Automatic Droplet generator QX200 AutoDG (Bio-Rad). Droplets were analyzed through a QX200 droplet reader (Bio-Rad) and data was analyzed using QuantaSoft analysis software (Bio-Rad). For quantification, a minimum of 10,000 acceptable droplets was required per 20 uL reaction. To ensure the accuracy of results, each sample was tested minimally in duplicate with each run including a non-template control and gDNA from cell lines containing the relevant mutation. Samples were deemed positive according to criteria previously described [40, 41].
In addition, given that 82% of benign melanocytic nevi carry BRAF mutations [42], we tested a cohort of healthy controls with ≥50 nevi for BRAF V600E and BRAF V600K mutant specific cfDNA (Supplementary Table 1). None of the samples were found positive, suggesting that BRAF V600E/K cfDNA is specific to tumor derived DNA and/or nevi do not release sufficient mutated DNA to be detectable in plasma using these assays.
Author contributions
All authors contributed to the work presented in this study and reviewed the final manuscript. ACM designed the study, performed experiments, analyzed data and wrote the manuscript. MRP and AR performed experiments and analyzed data. RR, LC, MAK, MM and TMM recruited and clinically assessed patients and provided expert advice. ZA assessed radiological imaging and provided expert advice. ESG and MZ designed experiments, analyzed data and supervised the study and writing of the manuscript.
ACKNOWLEDGMENTS
The authors thank all the participants for their assistance with the study. We would also like to thank Dr Benhur Amanuel and Dr Tindaro Giardina for molecular pathology service and advice.
CONFLICTS OF INTEREST
ESG has received travel and accommodation sponsorship from Bio-Rad Laboratories.
GRANT SUPPORT
ACM has received financial support through an “Australian Government Research Training Program” scholarship and the Edith Cowan University “Inspiring Minds” scholarship. ESG is supported by a fellowship from the Cancer Research Trust. This study was funded by a Western Australia Cancer Council grant (1100249) to ESG and National Health and Medical Research (NHMRC) grants (1046711 and 1119791) and ECU Perpetual and DoH WA grants to MZ, MM, ESG, MK, RP and LC. The funding bodies had no role in the design of the study, collection, analysis and interpretation of the data, nor in writing the manuscript.
REFERENCES
1. Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, Buzaid AC, Cochran AJ, Coit DG, Ding S, Eggermont AM, Flaherty KT, Gimotty PA, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009; 27:6199–206.
2. Crowley NJ, Seigler HF. Late recurrence of malignant melanoma. Analysis of 168 patients. Ann Surg. 1990; 212:173–77. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1358053/pdf/annsurg00162-0061.pdf.
3. Tsao H, Cosimi AB, Sober AJ. Ultra-late recurrence (15 years or longer) of cutaneous melanoma. Cancer. 1997; 79:2361–70.
4. Kalady MF, White RR, Johnson JL, Tyler DS, Seigler HF. Thin melanomas: predictive lethal characteristics from a 30-year clinical experience. Ann Surg. 2003; 238:528–35.
5. Dong XD, Tyler D, Johnson JL, DeMatos P, Seigler HF. Analysis of prognosis and disease progression after local recurrence of melanoma. Cancer. 2000; 88:1063–71.
6. Hohnheiser AM, Gefeller O, Göhl J, Schuler G, Hohenberger W, Merkel S. Malignant melanoma of the skin: long-term follow-up and time to first recurrence. World J Surg. 2011; 35:580–89.
7. Faries MB, Steen S, Ye X, Sim M, Morton DL. Late recurrence in melanoma: clinical implications of lost dormancy. J Am Coll Surg. 2013; 217:27–34.
8. Brauer JA, Wriston CC, Troxel AB, Elenitsas R, Shin DB, Guerry D, Ming ME. Characteristics associated with early and late melanoma metastases. Cancer. 2010; 116:415–23.
9. Petersen RP, Hanish SI, Haney JC, Miller CC 3rd, Burfeind WR Jr, Tyler DS, Seigler HF, Wolfe W, D'Amico TA, Harpole DH Jr. Improved survival with pulmonary metastasectomy: an analysis of 1720 patients with pulmonary metastatic melanoma. J Thorac Cardiovasc Surg. 2007; 133:104-10. https://doi.org/10.1016/j.jtcvs.2006.08.065.
10. Chua TC, Saxena A, Morris DL. Surgical metastasectomy in AJCC stage IV M1c melanoma patients with gastrointestinal and liver metastases. Ann Acad Med Singapore. 2010; 39:634–39. https://www.ncbi.nlm.nih.gov/pubmed/20838706.
11. Wasif N, Bagaria SP, Ray P, Morton DL. Does metastasectomy improve survival in patients with Stage IV melanoma? A cancer registry analysis of outcomes. J Surg Oncol. 2011; 104:111–15.
12. Schadendorf D, Long GV, Stroiakovski D, Karaszewska B, Hauschild A, Levchenko E, Chiarion-Sileni V, Schachter J, Garbe C, Dutriaux C, Gogas H, Mandalà M, Haanen JB, et al. Three-year pooled analysis of factors associated with clinical outcomes across dabrafenib and trametinib combination therapy phase 3 randomised trials. Eur J Cancer. 2017; 82:45–55.
13. Luke JJ, Flaherty KT, Ribas A, Long GV. Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat Rev Clin Oncol. 2017; 14:463–82.
14. Gellén E, Sántha O, Janka E, Juhász I, Péter Z, Erdei I, Lukács R, Fedinecz N, Galuska L, Remenyik É, Emri G. Diagnostic accuracy of (18)F-FDG-PET/CT in early and late stages of high-risk cutaneous malignant melanoma. J Eur Acad Dermatol Venereol. 2015; 29:1938–44.
15. Trotter SC, Sroa N, Winkelmann RR, Olencki T, Bechtel M. A global review of melanoma follow-up guielines. J Clin Aesthet Dermatol. 2013; 6:18–26.
16. Coit DG, Andtbacka R, Anker CJ, Bichakjian CK, Carson WE 3rd, Daud A, Dimaio D, Fleming MD, Guild V, Halpern AC, Hodi FS Jr, Kelley MC, Khushalani NI, et al, and National Comprehensive Cancer Network (NCCN). Melanoma, version 2.2013: featured updates to the NCCN guidelines. J Natl Compr Canc Netw. 2013; 11:395–407.
17. Danielsen M, Højgaard L, Kjær A, Fischer BM. Positron emission tomography in the follow-up of cutaneous malignant melanoma patients: a systematic review. Am J Nucl Med Mol Imaging. 2013; 4:17–28.
18. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014; 6:224ra24. https://doi.org/10.1126/scitranslmed.3007094.
19. Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B, Rajan S, Humphray S, Becq J, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013; 368:1199–209.
20. Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS, Vogelstein B. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012; 486:537–40. https://www.nature.com/articles/nature11219#supplementary-information.
21. Heitzer E, Ulz P, Geigl JB. Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem. 2015; 61:112–23.
22. Gray ES, Rizos H, Reid AL, Boyd SC, Pereira MR, Lo J, Tembe V, Freeman J, Lee JH, Scolyer RA, Siew K, Lomma C, Cooper A, et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget. 2015; 6:42008–42018. https://doi.org/10.18632/oncotarget.5788.
23. Stroun M, Lyautey J, Lederrey C, Olson-Sand A, Anker P. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin Chim Acta. 2001; 313:139–42.
24. Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch RD, Knippers R. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001; 61:1659–65.
25. Hamakawa T, Kukita Y, Kurokawa Y, Miyazaki Y, Takahashi T, Yamasaki M, Miyata H, Nakajima K, Taniguchi K, Takiguchi S, Mori M, Doki Y, Kato K. Monitoring gastric cancer progression with circulating tumour DNA. Br J Cancer. 2015; 112:352–56. https://www.nature.com/articles/bjc2014609#supplementary-information.
26. Reinert T, Schøler LV, Thomsen R, Tobiasen H, Vang S, Nordentoft I, Lamy P, Kannerup AS, Mortensen FV, Stribolt K, Hamilton-Dutoit S, Nielsen HJ, Laurberg S, et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut. 2016; 65:625–34.
27. Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, Silliman N, Tacey M, Wong HL, Christie M, Kosmider S, Skinner I, Wong R, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. 2016; 8:346ra92. https://doi.org/10.1126/scitranslmed.aaf6219.
28. Beaver JA, Jelovac D, Balukrishna S, Cochran R, Croessmann S, Zabransky DJ, Wong HY, Toro PV, Cidado J, Blair BG, Chu D, Burns T, Higgins MJ, et al. Detection of cancer DNA in plasma of patients with early-stage breast cancer. Clin Cancer Res. 2014; 20:2643–50.
29. Phallen J, Sausen M, Adleff V, Leal A, Hruban C, White J, Anagnostou V, Fiksel J, Cristiano S, Papp E, Speir S, Reinert T, Orntoft MW, et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci Transl Med. 2017; 9.
30. Sozzi G, Conte D, Mariani L, Lo Vullo S, Roz L, Lombardo C, Pierotti MA, Tavecchio L. Analysis of circulating tumor DNA in plasma at diagnosis and during follow-up of lung cancer patients. Cancer Res. 2001; 61:4675–78.
31. Oshiro C, Kagara N, Naoi Y, Shimoda M, Shimomura A, Maruyama N, Shimazu K, Kim SJ, Noguchi S. PIK3CA mutations in serum DNA are predictive of recurrence in primary breast cancer patients. Breast Cancer Res Treat. 2015; 150:299–307.
32. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA Jr. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008; 14:985–90. https://www.nature.com/articles/nm.1789#supplementary-information.
33. Lee RJ, Gremel G, Marshall A, Myers KA, Fisher N, Dunn JA, Dhomen N, Corrie PG, Middleton MR, Lorigan P, Marais R. Circulating tumor DNA predicts survival in patients with resected high-risk stage II/III melanoma. Ann Oncol. 2018; 29:490–496.
34. Wong SQ, Raleigh JM, Callahan J, Vergara IA, Ftouni S, Hatzimihalis A, Colebatch AJ, Li J, Semple T, Doig K, Mintoff C, Sinha D, Yeh P, et al. Circulating tumor DNA analysis and functional imaging provide complementary approaches for comprehensive disease monitoring in metastatic melanoma. JCO Precision Oncology 2017. 1:1, 1–14.
35. McEvoy AC, Warburton L, Al-Ogaili Z, Celliers L, Calapre L, Pereira MR, Khattak MA, Meniawy TM, Millward M, Ziman M, Gray ES. Correlation between circulating tumour DNA and metabolic tumour burden in metastatic melanoma patients. BMC Cancer. 2018; 18:726.
36. Romano E, Scordo M, Dusza SW, Coit DG, Chapman PB. Site and timing of first relapse in stage III melanoma patients: implications for follow-up guidelines. J Clin Oncol. 2010; 28:3042–47.
37. Leiter U, Buettner PG, Eigentler TK, Bröcker EB, Voit C, Gollnick H, Marsch W, Wollina U, Meier F, Garbe C. Hazard rates for recurrent and secondary cutaneous melanoma: an analysis of 33,384 patients in the German Central Malignant Melanoma Registry. J Am Acad Dermatol. 2012; 66:37–45.
38. Lyth J, Falk M, Maroti M, Eriksson H, Ingvar C. Prognostic risk factors of first recurrence in patients with primary stages I-II cutaneous malignant melanoma - from the population-based Swedish melanoma register. J Eur Acad Dermatol Venereol. 2017; 31:1468–74.
39. McEvoy AC, Wood BA, Ardakani NM, Pereira MR, Pearce R, Cowell L, Robinson C, Grieu-Iacopetta F, Spicer AJ, Amanuel B, Ziman M, Gray ES. Droplet digital PCR for mutation detection in foramalin fixed parafin embedded melanoma tissues: a comparison with Sanger sequencing and pyrosequencing. J Mol Diagn. 2018; 20:240–52.
40. Reid AL, Freeman JB, Millward M, Ziman M, Gray ES. Detection of BRAF-V600E and V600K in melanoma circulating tumour cells by droplet digital PCR. Clin Biochem. 2015; 48:999–1002.
41. McEvoy AC, Calapre L, Pereira MR, Giardina T, Robinson C, Khattak MA, Meniawy TM, Pritchard AL, Hayward NK, Amanuel B, Millward M, Ziman M, Gray ES. Sensitive droplet digital PCR method for detection of TERT promoter mutations in cell free DNA from patients with metastatic melanoma. Oncotarget. 2017; 8:78890–78900. https://doi.org/10.18632/oncotarget.20354.
42. Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, Moses TY, Hostetter G, Wagner U, Kakareka J, Salem G, Pohida T, Heenan P, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003; 33:19–20.