
INTRODUCTION
Recent reviews of the expression of radioresistance in cancer of the pancreas [1], gliomas [2], head and neck cancers [3], prostate cancer [4], urinary bladder cancer [5] and cancers of other organs [6, 7] emphasize the clinical importance of the state of radioresistance and the complexity of its molecular mechanisms. Radioresistance may be an inherent quality of a tumor or may emerge in the course of cancer management.
Spontaneous or medically induced hypothyroidism may beneficially alter the clinical behavior of a number of cancers, e.g., breast cancer [8], glioblastoma [9], head-and-neck tumors [10] and renal cell carcinoma [11]. Induction of the clinical state of euthyroid hypothyroxinemia—in which euthyroidism is maintained by administered 3,5,3′-triiodo-L-thyronine (T3) in the absence of host L-thyroxine (T4)—has also been shown to slow the course of end-stage carcinomas of various origins [12]. T4 in physiological concentrations in vitro has been shown to cause proliferation of a wide variety human cancer cells [13–16], whereas T3 in physiological concentrations does not appear to promote cancer cell proliferation in vitro [14]. Reports that T3 may stimulate tumor cell proliferation in vitro have relied upon high concentrations of the hormone [17, 18]. Clinically, circulating T3 may be quite low in cancer patients subject to the nonthyroidal illness syndrome [19].
The proliferative effects of T4 on cancer cells are initiated at a cell surface receptor for thyroid hormone on the extracellular domain of integrin αvβ3 [20, 21]. T4 is also anti-apoptotic via the receptor on αvβ3 [22] and supports tumor-relevant angiogenesis via this receptor [23]. The tumor-support actions of T4 at integrin αvβ3 on cancer cells are blocked by a derivative of T4, tetraiodothyroacetic acid (tetrac), either unmodified or reformulated as a nanoparticulate agent (Nano-diamino-tetrac (NDAT) or Nanotetrac) [20, 21], and these compounds by their anti-proliferative and pro-apoptotic actions [22] also reduce tumor xenograft size [24–29].
Importantly, tetrac has also been shown to radiosensitize glioma cells [30, 31]. Studies in vitro disclosed that this thyroid hormone derivative decreased repair of double-strand DNA breaks (DSBs) induced by radiation [31] and that tetrac itself may cause DSBs. Unmodified tetrac and Nanotetrac that act exclusively at integrin αvβ3 block the binding of T4 to the integrin and prevent the proliferative and anti-apoptotic actions of T4 initiated at this site [20, 21]. The observation that tetrac compounds acting exclusively at the integrin are capable of radiosensitizing tumor cells suggests that T4, acting at exactly the same site and displaced by tetrac, contributes to the phenomenon of radioresistance. We have shown that exposure of human cancer cells in vitro to X-irradiation rapidly activates αvβ3 [32] and that unmodified tetrac blocks this profound change in the physical state of the integrin. The silencing mediator for retinoid and thyroid hormone receptor (SMRT) is a nuclear corepressor protein that appears to be important to repair of DSBs that are radiation-induced [33], possibly implicating thyroid hormone in the promotion of this mechanism of radioresistance. The integrin has been implicated in regulation of radioresistance/radiosensitivity [34, 35].
Tumor irradiation in vitro has recently been shown to activate cancer cell integrin αvβ3 [32]. Activation of the extracellular domain of the integrin is an extended state and is induced within minutes. It appears to be associated with radioresistance, and this response to radiation is blocked by tetrac. It is thought that the change in physical state of the integrin contributes to radioresistance, perhaps by inducing more complex intercellular interactions and decreasing cell division. The radiosensitizing properties of tetrac may include this action on the state of integrin αvβ3.
What is clear from the actions of tetrac formulations initiated at integrin αvβ3 is that iodothyronine receptor site on the integrin controls radiosensitivity-radioresistance, and that tetrac molecules acting at this site control a substantial number of signaling pathways that contribute to the radiation sensitivity status of the tumor cells expressing the integrin. We will in brief review here a) the actions of tetrac—and, in some cases, thyroid hormone—on cancer cell radiosensitivity and b) the effects of Nanotetrac on the signaling pathways relevant to the radiation sensitivity state of tumor cells.
MECHANISMS THAT MAY CONTRIBUTE TO DEVELOPMENT OF RADIORESISTANCE
Tetrac and tumor radiosensitivity
As noted above, tetrac has been shown to radiosensitize murine glioma GL261 cells [30] and human glioblastoma U87MG cells [31], as well as to restore radiosensitivity to resistant human basal cell carcinoma (TE.354.T) cells [36]. In the studies of U87MG cells, exposure of cells in vitro for 1 hour prior to radiation resulted in a more than 70% reduction in repair of DSBs induced by X-irradiation. The molecular basis of this action of unmodified tetrac on DNA repair is not established. The anticancer actions of unmodified tetrac and Nanotetrac are initiated exclusively at plasma membrane integrin αvβ3, but many effects of the agent are expressed downstream in specific gene expression or altered activities of nuclear co-repressor and co-activator proteins [21, 23]. The integrin is expressed generously by cancer cells but not by normal cells; hence, tetrac is unlikely to make nonmalignant cells more radiosensitive.
Signaling pathways involved in regulation of radiosensitivity and known effects of thyroid hormone analogues on these pathways
AKT
The phosphatidylinositol 3-kinase (PI-3K)/AKT (protein kinase B)/mTOR signal transduction pathway is frequently disordered in cancer cells and its excessive activity contributes to tumor cell proliferation and radioresistance [37, 38]. The latter is a function of enhanced repair of DSBs that are induced by radiation [39]. Although it has been stated that it is unclear how AKT may be stimulated in cancer cells in the setting of radiation so that radioresistance is generated [2], we would point out that physiological levels of thyroid hormone as T4 activate the AKT pathway via the cell surface thyroid hormone receptor on integrin αvβ3 [20, 21]. We propose that endogenous T4 in the intact organism—preclinically in animal models or in the clinical setting—is a mechanism of support in tumor cells for AKT-dependent DSB repair.
STAT3
Among its multiple contributions to intracellular signaling in nonmalignant cells [40] and cancer cells, signal transducer and activator of transcription 3 (STAT3) is known when activated to promote radioresistance in a variety of tumors [7, 41], including gliomas and lung cancer. We showed that T4 nongenomically activates STAT3, where activation is defined as specific tyrosine phosphorylation of the protein and subsequent nuclear translocation of the STAT [42]. T4 at physiological free concentrations also was found nongenomically to potentiate induction by epidermal growth factor (EGF) of c-Fos expression, and c-Fos has subsequently been found to promote cancer cell radioresistance in glioma cells [43]. Thus, one or more STAT3-dependent mechanisms exist by which thyroid hormone as T4 may contribute to tumor radioresistance.
Wnt/β-catenin
A third signaling pathway that has been implicated in the induction of the radioresistant state in tumor cells is that involving Wnt/β-catenin. The pathway is involved in stem cell biology and is also an index of tumor cell aggressiveness [44–46]. In several models that enabled study of radioresistance development in glioblastoma cells, frankly increased levels of β-catenin correlated with radioresistance [47]. Nuclear uptake of β-catenin occurs in radiated glioblastoma cells and accompanies tumor aggressiveness that is reversed by inhibition of this signaling pathway [48]. T4 promotes β-catenin-dependent proliferation of colorectal cancer cells [49]. Inhibition of nongenomic T4-initiated actions at integrin αvβ3 with the T4 analogue, tetrac, increases tumor cell nucleus accumulation of Cby1, a protein inhibitor of nuclear actions of β-catenin and also downregulates nuclear accumulation of the catenin [50]. Such results are consistent with the function of this pathway as another mediator of the actions of T4 to induce the radioresistant state.
Epithelial-mesenchymal transition (EMT) in conjunction with radioresistance
Radiation exposure of cancer cells may support cell aggressiveness via the EMT process [6]. For example, in an esophageal cancer cell line in which radioresistance was induced, the development of radioresistance was coupled with AKT-induced EMT [51]. Physiological levels of T4 act at integrin αvβ3 to promote EMT in ovarian cancer cells [52], and this may be an ancillary mechanism by which the hormone contributes to radioresistance, as well as to cancer cell invasiveness.
Hypoxia
Cancer cells are significantly more radioresistant under hypoxic conditions [2, 6]. The resistance is at least in part due to generation of hypoxia-inducible factor 1α (HIF-1α) [2, 6] that stabilizes cells subjected to the hypoxic state. The molecular basis of the generation of radioresistance by HIF-1α is not yet clear. A number of studies have shown that thyroid hormone can stimulate transcription of HIF-1α [53–55]. However, supraphysiological amounts of T3 appear to be required at the hormone receptor on integrin αvβ3 in order to stimulate HIF-1α gene expression [53] and this may also be the case at the nuclear thyroid hormone receptor (TR) [54]. Further, hypoxic conditions may activate deiodinase 3 (DIO3, D3) and lead to deiodination and inactivation of T3 [56]. T4 does not affect HIF-1α transcription at integrin αvβ3 [53]. We can conclude that it is unclear whether actions of thyroid hormone on HIF-1α can be relevant to pathogenesis of radioresistance.
microRNAs
A substantial panel of miRNAs has been identified that modulates radiosensitivity of cancer cells [2–4]. Relatively little information is available on thyroid hormone-specific miRNA abundance and cancer and radiosensitivity. Actions of thyroid hormone on miRNA-21 abundance and activity have been studied in basal cell carcinoma cells [57], and this miRNA is known to be associated with radioresistance and cell proliferation in glioma cells [2]. miRNA-21 is downregulated by T3 in basal cells [57], a potentially desirable effect that is interestingly attenuated by concomitant increase in D3 generated by the hormone. Recent clinical experience with euthyroid hypothyroxinemia [12] indicates that T3 does not stimulate growth of a variety of solid tumors, including glioblastoma. In this setting, the action of T3 on D3 is insufficient to block the tumor response to triiodothyronine. Additional information is needed on the possible actions of thyroid hormones on amounts of tumor-specific miRNAs and radiosensitivity.
In addition to the factors discussed in this review that are relevant to tumor cell radiosensitivity, the Hedgehog pathway, Notch, P53 and other cell mechanisms may condition the state of radioresistance [1, 2, 6, 58]. Certain of these factors may also be controlled by thyroid hormone via its receptor on integrin αvβ3, e.g., P53, whose activation/phosphorylation is inhibited by T4 [22], supporting anti-apoptosis in cells exposed to thyroid hormone. But interpreting changes in P53 behavior and apoptosis across cell lines [59] and in response to radiation [60, 61] has been shown to be complex.
IMPLICATION OF INVOLVEMENT OF THYROID HORMONE AND INTEGRIN αVβ3 IN RADIOSENSITIVITY STATE ON CANCER CELLS
Circulating T4 levels during periods of radiotherapy of cancer in euthyroid patients
Evidence discussed above supports the possibility that the thyroid hormone analogue receptor on integrin αvβ3 supports the expression of radioresistance by cancer cells. From these reports, it is the action of T4, rather than that of T3, that appears to be particularly relevant to the radiosensitivity state of the tumor cell (Figure 1). This possibility deserves additional preclinical investigation that compares tumor cell or xenograft sensitivity to X-irradiation in the presence and absence of T4 and T3, evaluating these forms of thyroid hormone individually.
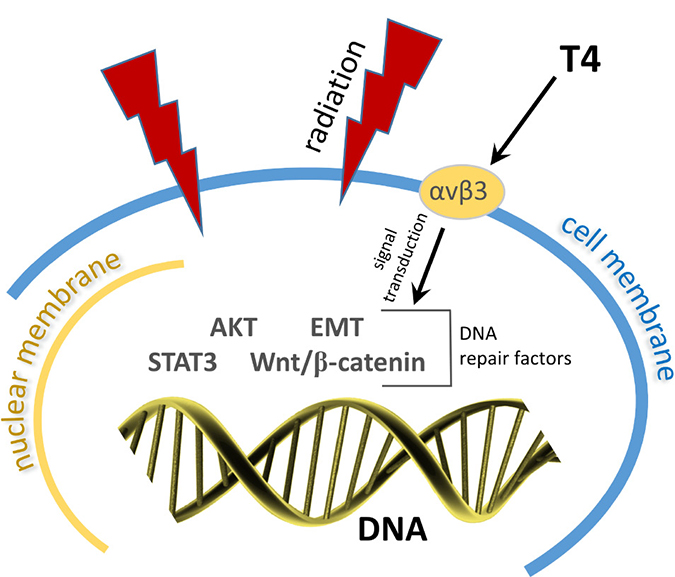
Figure 1: Radiation induces double-strand DNA breaks. By several signaling pathways T4 acts at the tumor cell plasma membrane to activate multiple DNA repair factors (AKT, EMT, STAT3, Wnt/β-catenin, and possibly others).
There may be existing clinical settings in which a possible contribution of T4 to radioresistance may be assessed. Patients with advanced cancers who are managed palliatively with euthyroid hypothyroxinemia are a clinical population sometimes subjected to radiation for relief of pain, and it may be useful to evaluate the effectiveness of such radiation in term of analgesia, but also in terms of tumor size response to radiotherapy when a history of radioresistance is present. In addition, in cancer patients with hypothyroidism who may be candidates for radiotherapy, thyroid hormone replacement may be achieved with T3, and studies are needed that are designed to assess the possibility that radiosensitivity—when radiation therapy is indicated—can be conditioned by T4. Thus, it may be useful to determine whether differences in radiosensitivity in tumor patients can be in part conditioned by whether circulating endogenous T4 levels are in the upper or lower quartile of the normal range.
Finally, tyrosine kinase inhibitor (TKI) anticancer drugs may induce thyroiditis that leads to usually transient hyperthyroidism and then to hypothyroidism [62, 63]. We would propose that radiotherapy should be avoided during a period of hyperthyroidism that is caused by TKI therapy.
CONCLUSIONS
In this review, we have examined selected host factors that at the molecular level mediate the development of radioresistance in cancer cells. We have shown that thyroid hormone, particularly as T4, can activate these factors and, therefore, that T4 may support the emergence of radioresistance. We have preliminary in vitro evidence that T4 may reduce the effectiveness of X-irradiation in tumor cells and we recommend that additional preclinical evidence be sought that tests this possibility. Tetrac, an anti-T4 agent that acts at integrin αvβ3, has been shown to restore radiosensitivity in tumor cells. There are existing clinical settings in which the tumor radiosensitivity in the presence of T4 may be compared with that in the presence of T3 to test the possibility that T4 is a clinically relevant inducer of radioresistance.
CONFLICTS OF INTEREST
None.
FUNDING
This work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
REFERENCES
1. Seshacharyulu P, Baine MJ, Souchek JJ, Menning M, Kaur S, Yan Y, Ouellette MM, Jain M, Lin C, Batra SK. Biological determinants of radioresistance and their remediation in pancreatic cancer. Biochim Biophys Acta. 2017; 1868:69–92.
2. Han X, Xue X, Zhou H, Zhang G. A molecular view of the radioresistance of gliomas. Oncotarget. 2017; 8:100931–100941. https://doi.org/10.18632/oncotarget.21753.
3. Ahmad P, Sana J, Slavik M, Slampa P, Smilek P, Slaby O. MicroRNAs involvement in radioresistance of head and neck cancer. Dis Markers. 2017; 2017:8245345.
4. Ni J, Bucci J, Chang L, Malouf D, Graham P, Li Y. Targeting microRNAs in prostate cancer radiotherapy. Theranostics. 2017; 7:3243–3259.
5. Mari A, D’Andrea D, Abufaraj M, Foerster B, Kimura S, Shariat SF. Genetic determinants for chemo- and radiotherapy resistance in bladder cancer. Transl Androl Urol. 2017; 6:1081–1089.
6. Chen GZ, Zhu HC, Dai WS, Zeng XN, Luo JH, Sun XC. The mechanisms of radioresistance in esophageal squamous cell carcinoma and current strategies in radiosensitivity. J Thorac Dis. 2017; 9:849–859.
7. Harada D, Takigawa N, Kiura K. The role of STAT3 in non-small cell lung cancer. Cancers (Basel). 2014; 6:708–722.
8. Cristofanilli M, Yamamura Y, Kau SW, Bevers T, Strom S, Patangan M, Hsu L, Krishnamurthy S, Theriault RL, Hortobagyi GN. Thyroid hormone and breast carcinoma. Primary hypothyroidism is associated with a reduced incidence of primary breast carcinoma. Cancer. 2005; 103:1122–1128.
9. Hercbergs AA, Goyal LK, Suh JH, Lee S, Reddy CA, Cohen BH, Stevens GH, Reddy SK, Peereboom DM, Elson PJ, Gupta MK, Barnett GH. Propylthiouracil-induced chemical hypothyroidism with high-dose tamoxifen prolongs survival in recurrent high grade glioma: a phase I/II study. Anticancer Res. 2003; 23:617–626.
10. Nelson M, Hercbergs A, Rybicki L, Strome M. Association between development of hypothyroidism and improved survival in patients with head and neck cancer. Arch Otolaryngol Head Neck Surg. 2006; 132:1041–1046.
11. Schmidinger M, Vogl UM, Bojic M, Lamm W, Heinzl H, Haitel A, Clodi M, Kramer G, Zielinski CC. Hypothyroidism in patients with renal cell carcinoma: blessing or curse? Cancer. 2011; 117:534–544.
12. Hercbergs A, Johnson RE, Ashur-Fabian O, Garfield DH, Davis PJ. Medically induced euthyroid hypothyroxinemia may extend survival in compassionate need cancer patients: an observational study. Oncologist. 2015; 20:72–76.
13. Cohen K, Ellis M, Khoury S, Davis PJ, Hercbergs A, Ashur-Fabian O. Thyroid hormone is a MAPK-dependent growth factor for human myeloma cells acting via αvβ3 integrin. Mol Cancer Res. 2011; 9:1385–1394.
14. Davis FB, Tang HY, Shih A, Keating T, Lansing L, Hercbergs A, Fenstermaker RA, Mousa A, Mousa SA, Davis PJ, Lin HY. Acting via a cell surface receptor, thyroid hormone is a growth factor for glioma cells. Cancer Res. 2006; 66:7270–7275.
15. Meng R, Tang HY, Westfall J, London D, Cao JH, Mousa SA, Luidens M, Hercbergs A, Davis FB, Davis PJ, Lin HY. Crosstalk between integrin αvβ3 and estrogen receptor-α is involved in thyroid hormone-induced proliferation in human lung carcinoma cells. PLoS One. 2011; 6:e27547.
16. Shinderman-Maman E, Cohen K, Weingarten C, Nabriski D, Twito O, Baraf L, Hercbergs A, Davis PJ, Werner H, Ellis M, Ashur-Fabian O. The thyroid hormone-αvβ3 integrin axis in ovarian cancer: regulation of gene transcription and MAPK-dependent proliferation. Oncogene. 2016; 35:1977–1987.
17. Hall LC, Salazar EP, Kane SR, Liu N. Effects of thyroid hormones on human breast cancer cell proliferation. J Steroid Biochem Mol Biol. 2008; 109:57–66.
18. Perri A, Catalano S, Bonofiglio D, Vizza D, Rovito D, Qi H, Aquila S, Panza S, Rizza P, Lanzino M, Ando S. T3 enhances thyroid cancer cell proliferation through TRβ1/Oct-1-mediated cyclin D1 activation. Mol Cell Endocrinol. 2014; 382:205–217.
19. Hercbergs A, Mousa SA, Davis PJ. Nonthyroidal illness syndrome and thyroid hormone actions at integrin αvβ3. J Clin Endocrinol Metab. 2018; 103:1291–1295.
20. Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010; 31:139–170.
21. Davis PJ, Goglia F, Leonard JL. Nongenomic actions of thyroid hormone. Nat Rev Endocrinol. 2016; 12:111–121.
22. Lin HY, Glinsky GV, Mousa SA, Davis PJ. Thyroid hormone and anti-apoptosis in tumor cells. Oncotarget. 2015; 6:14735–14743. https://doi.org/10.18632/oncotarget.4023.
23. Davis PJ, Sudha T, Lin HY, Mousa SA. Thyroid hormone, hormone analogs, and angiogenesis. Compr Physiol. 2015; 6:353–362.
24. Sudha T, Bharali DJ, Yalcin M, Darwish NH, Debreli Coskun M, Keating KA, Lin HY, Davis PJ, Mousa SA. Targeted delivery of paclitaxel and doxorubicin to cancer xenografts via the nanoparticle of nano-diamino-tetrac. Int J Nanomedicine. 2017; 12:1305–1315.
25. Yalcin M, Lin HY, Sudha T, Bharali DJ, Meng R, Tang HY, Davis FB, Stain SC, Davis PJ, Mousa SA. Response of human pancreatic cancer cell xenografts to tetraiodothyroacetic acid nanoparticles. Horm Cancer. 2013; 4:176–185.
26. Mousa SA, Yalcin M, Bharali DJ, Meng R, Tang HY, Lin HY, Davis FB, Davis PJ. Tetraiodothyroacetic acid and its nanoformulation inhibit thyroid hormone stimulation of non-small cell lung cancer cells in vitro and its growth in xenografts. Lung Cancer. 2012; 76:39–45.
27. Yalcin M, Dyskin E, Lansing L, Bharali DJ, Mousa SS, Bridoux A, Hercbergs AH, Lin HY, Davis FB, Glinsky GV, Glinskii A, Ma J, Davis PJ, et al. Tetraiodothyroacetic acid (tetrac) and nanoparticulate tetrac arrest growth of medullary carcinoma of the thyroid. J Clin Endocrinol Metab. 2010; 95:1972–1980.
28. Davis PJ, Glinsky GV, Lin HY, Leith JT, Hercbergs A, Tang HY, Ashur-Fabian O, Incerpi S, Mousa SA. Cancer cell gene expression modulated from plasma membrane integrin αvβ3 by thyroid hormone and nanoparticulate tetrac. Front Endocrinol (Lausanne). 2014; 5:240.
29. Glinskii AB, Glinsky GV, Lin HY, Tang HY, Sun M, Davis FB, Luidens MK, Mousa SA, Hercbergs AH, Davis PJ. Modification of survival pathway gene expression in human breast cancer cells by tetraiodothyroacetic acid (tetrac). Cell Cycle. 2009; 8:3562–3570.
30. Hercbergs A, Davis PJ, Davis FB, Ciesielski MJ, Leith JT. Radiosensitization of GL261 glioma cells by tetraiodothyroacetic acid (tetrac). Cell Cycle. 2009; 8:2586–2591.
31. Hercbergs AH, Lin HY, Davis FB, Davis PJ, Leith JT. Radiosensitization and production of DNA double-strand breaks in U87MG brain tumor cells induced by tetraiodothyroacetic acid (tetrac). Cell Cycle. 2011; 10:352–357.
32. Leith JT, Hercbergs A, Kenney S, Mousa SA, Davis PJ. Activation of tumor cell integrin αvβ3 by radiation and reversal of activation by chemically modified tetraiodothyroacetic acid (tetrac). Endocr Res. 2018; 43:1–5.
33. Yu J, Palmer C, Alenghat T, Li Y, Kao G, Lazar MA. The corepressor silencing mediator for retinoid and thyroid hormone receptor facilitates cellular recovery from DNA double-strand breaks. Cancer Res. 2006; 66:9316–9322.
34. Monferran S, Skuli N, Delmas C, Favre G, Bonnet J, Cohen-Jonathan-Moyal E, Toulas C. αvβ3 and αvβ5 integrins control glioma cell response to ionising radiation through ILK and RhoB. Int J Cancer. 2008; 123:357–364.
35. Ou J, Luan W, Deng J, Sa R, Liang H. αV integrin induces multicellular radioresistance in human nasopharyngeal carcinoma via activating SAPK/JNK pathway. PLoS One. 2012; 7:e38737.
36. Leith JT, Davis PJ, Mousa SA, Hercbergs AA. In vitro effects of tetraiodothyroacetic acid combined with X-irradiation on basal cell carcinoma cells. Cell Cycle. 2017; 16:367–373.
37. Mehta M, Khan A, Danish S, Haffty BG, Sabaawy HE. Radiosensitization of primary human glioblastoma stem-like cells with low-dose AKT inhibition. Mol Cancer Ther. 2015; 14:1171–1180.
38. Kwiatkowska A, Symons M. Signaling determinants of glioma cell invasion. Adv Exp Med Biol. 2013; 986:121–141.
39. Golding SE, Morgan RN, Adams BR, Hawkins AJ, Povirk LF, Valerie K. Pro-survival AKT and ERK signaling from EGFR and mutant EGFRvIII enhances DNA double-strand break repair in human glioma cells. Cancer Biol Ther. 2009; 8:730–738.
40. de la Iglesia N, Puram SV, Bonni A. STAT3 regulation of glioblastoma pathogenesis. Curr Mol Med. 2009; 9:580–590.
41. Luwor RB, Stylli SS, Kaye AH. The role of Stat3 in glioblastoma multiforme. J Clin Neurosci. 2013; 20:907–911.
42. Lin HY, Shih A, Davis FB, Davis PJ. Thyroid hormone promotes the phosphorylation of STAT3 and potentiates the action of epidermal growth factor in cultured cells. Biochem J. 1999; 338:427–432.
43. Liu ZG, Jiang G, Tang J, Wang H, Feng G, Chen F, Tu Z, Liu G, Zhao Y, Peng MJ, He ZW, Chen XY, Lindsay H, et al. c-Fos over-expression promotes radioresistance and predicts poor prognosis in malignant glioma. Oncotarget. 2016; 7:65946–65956. https://doi.org/10.18632/oncotarget.11779.
44. Liu XR, Wang L, Zhao SF, Ji XM, Luo YM, Ling F. beta-Catenin overexpression in malignant glioma and its role in proliferation and apoptosis in glioblastma cells. Med Oncol. 2011; 28:608–614.
45. Yamamoto H, Oue N, Sato A, Hasegawa Y, Yamamoto H, Matsubara A, Yasui W, Kikuchi A. Wnt5a signaling is involved in the aggressiveness of prostate cancer and expression of metalloproteinase. Oncogene. 2010; 29:2036–2046.
46. Anson M, Crain-Denoyelle AM, Baud V, Chereau F, Gougelet A, Terris B, Yamagoe S, Colnot S, Viguier M, Perret C, Couty JP. Oncogenic beta-catenin triggers an inflammatory response that determines the aggressiveness of hepatocellular carcinoma in mice. J Clin Invest. 2012; 122:586–599.
47. Kim Y, Kim KH, Lee J, Lee YA, Kim M, Lee SJ, Park K, Yang H, Jin J, Joo KM, Lee J, Nam DH. Wnt activation is implicated in glioblastoma radioresistance. Lab Invest. 2012; 92:466–473.
48. Dong Z, Zhou L, Han N, Zhang M, Lyu X. Wnt/beta-catenin pathway involvement in ionizing radiation-induced invasion of U87 glioblastoma cells. Strahlenther Onkol. 2015; 191:672–680.
49. Lee YS, Chin YT, Shih YJ, Nana AW, Chen YR, Wu HC, Yang YS, Lin HY, Davis PJ. Thyroid hormone promotes β-catenin activation and cell proliferation in colorectal cancer. Horm Cancer. 2018; 9:156–165.
50. Nana AW, Chin YT, Lin CY, Ho Y, Bennett JA, Shih YJ, Chen YR, Changou CA, Pedersen JZ, Incerpi S, Liu LF, Whang-Peng J, Fu E, et al. Tetrac downregulates beta-catenin and HMGA2 to promote the effect of resveratrol in colon cancer. Endocr Relat Cancer. 2018; 25:279–293.
51. He E, Pan F, Li G, Li J. Fractionated ionizing radiation promotes epithelial-mesenchymal transition in human esophageal cancer cells through PTEN deficiency-mediated Akt activation. PLoS One. 2015; 10:e0126149.
52. Weingarten C, Jenudi Y, Tshuva RY, Moskovich D, Alfandari A, Hercbergs A, Davis PJ, Ellis M, Ashur-Fabian O. The interplay between epithelial-mesenchymal transition (EMT) and the thyroid hormones-αvβ3 axis in ovarian cancer. Horm Cancer. 2018; 9:22–32.
53. Lin HY, Sun M, Tang HY, Lin C, Luidens MK, Mousa SA, Incerpi S, Drusano GL, Davis FB, Davis PJ. L-Thyroxine vs. 3,5,3'-triiodo-L-thyronine and cell proliferation: activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Am J Physiol Cell Physiol. 2009; 296:C980–991.
54. Moretto FC, De Sibio MT, Luvizon AC, Olimpio RM, de Oliveira M, Alves CA, Conde SJ, Nogueira CR. Triiodothyronine (T3) induces HIF1A and TGFA expression in MCF7 cells by activating PI3K. Life Sci. 2016; 154:52–57.
55. Otto T, Fandrey J. Thyroid hormone induces hypoxia-inducible factor 1α gene expression through thyroid hormone receptor β/retinoid x receptor α-dependent activation of hepatic leukemia factor. Endocrinology. 2008; 149:2241–2250.
56. Simonides WS, Mulcahey MA, Redout EM, Muller A, Zuidwijk MJ, Visser TJ, Wassen FW, Crescenzi A, da-Silva WS, Harney J, Engel FB, Obregon MJ, Larsen PR, et al. Hypoxia-inducible factor induces local thyroid hormone inactivation during hypoxic-ischemic disease in rats. J Clin Invest. 2008; 118:975–983.
57. Di Girolamo D, Ambrosio R, De Stefano MA, Mancino G, Porcelli T, Luongo C, Di Cicco E, Scalia G, Vecchio LD, Colao A, Dlugosz AA, Missero C, Salvatore D, et al. Reciprocal interplay between thyroid hormone and microRNA-21 regulates hedgehog pathway-driven skin tumorigenesis. J Clin Invest. 2016; 126:2308–2320.
58. Wang H, Mu XY, He H, Zhang XD. Cancer radiosensitizers. Trends Pharmacol Sci. 2018; 39:24–48.
59. Stewart-Ornstein J, Lahav G. p53 dynamics in response to DNA damage vary across cell lines and are shaped by efficiency of DNA repair and activity of the kinase ATM. Sci Signal. 2017; 10.
60. Balcer-Kubiczek EK. Apoptosis in radiation therapy: a double-edged sword. Exp Oncol. 2012; 34:277–285.
61. Lu C, El-Deiry WS. Targeting p53 for enhanced radio- and chemo-sensitivity. Apoptosis. 2009; 14:597–606.
62. Hamnvik OP, Larsen PR, Marqusee E. Thyroid dysfunction from antineoplastic agents. J Natl Cancer Inst. 2011; 103:1572–1587.
63. Ahmadieh H, Salti I. Tyrosine kinase inhibitors induced thyroid dysfunction: a review of its incidence, pathophysiology, clinical relevance, and treatment. Biomed Res Int. 2013; 2013:725410.