
INTRODUCTION
Grade IV glioblastomas (GBM) demonstrate dismal prognoses due to their aggressive nature and resistance to standard of care therapies. Despite multi-modality treatment with maximal safe resection, chemotherapy and radiation, the median overall survival for these patients is only 12-15 months [1, 2]. In 2008, the IDH1/2 mutation was discovered in gliomas [3] and has been associated with improved prognoses in gliomas independent of tumor grade (Grade II-IV) or histologic subtype (astrocytoma, oligodendroglioma, oligoastrocytoma) [4]. The World Health Organization (WHO) revised the low(er) grade glioma (LGG) classification system in 2016 to include isocitrate dehydrogenase (IDH1/2) mutations and 1p/19q co-deletion status in addition to glioma grade and histology [5]. As a result, IDH1/2 mutation status is routinely being utilized in the clinic to help predict tumor prognosis and guide management decisions for glioma patients [6]. While IDH1/2 mutations most commonly occur in Grade II and III gliomas, they are also present in approximately 5% of GBM that have progressed from lower grade gliomas, known as secondary GBM [3, 7]. Regardless of the grade, IDH1/2 mutations are a favorable prognostic factor amongst all gliomas. In fact, secondary GBM patients harboring an IDH1/2 mutation (median survival of 2.1 years) often have improved survival compared to LGG without the mutation, referred to as IDH1/2 wild-type (IDH1/2 WT) (median survival of 1.7 years) [4, 8, 9].
The IDH1/2 enzyme catalyzes the oxidative decarboxylation of isocitrate to form α-ketoglutarate (α-KG), an important reaction for glutamine metabolism, lipogenesis, and regulation of cellular redox status in the cell [10]. IDH1 and IDH2 proteins are encoded by separate genes that share approximately 70% sequence homology in humans [10]. The heterozygous R132H mutation in IDH1 accounts for 95% of IDH1/2 mutations in gliomas [11], however heterozygous mutations also occur in the analogous amino acid of IDH2 (R172), including R172G, R172K, and R172M [4, 12]. Both the R132 and R172 residues are located at the active site of IDH1/2 and form hydrogen bonds with the isocitrate substrate, therefore decreasing its binding affinity. As a result, IDH1 and IDH2 mutations compromise α-KG production, but at the same time result in neomorphic enzymatic activity that reduces α-KG to 2-hydroxyglutarate (2-HG). The mechanism by which aberrant 2-HG causes oncogenesis is poorly understood. It has been well-established that 2-HG inhibits dioxygeases, including DNA demethylases, that result in a global hypermethylated phenotype and gene silencing, known as glioma CpG-island hypermethylator phenotype (G-CIMP) [13]. The specific gene expression changes associated with this glioma hypermethylated phenotype in IDH1/2 mutant gliomas are yet to be elucidated. In this study, we performed molecular profiling of IDH1/2 WT versus mutant low(er) grade II and III gliomas to identify key molecular pathways that may contribute to differences in survival outcomes. To this end, by employing an mRNA and proteomics profiling approach, we identified transgelin-2 (TAGLN2) to be differentially expressed between IDH1/2 WT and mutant gliomas.
TAGLN2 is an actin-polymerizing protein important for regulation of the actin cytoskeleton during normal physiologic processes, including cell proliferation, differentiation, migration, and immunologic synapse formation (reviewed in [14–16]). TAGLN2 and transgelin-3 (TAGLN3) are both homologues of transgelin-1 (TAGLN1), a marker of smooth muscle cell differentiation, that appears to be widely expressed in tissues with similar biological functions as its homologues [15]. TAGLN2 is highly expressed in epithelial cells, while TAGLN3 is neuron-specific [14].
TAGLN2 has also been shown to be dysregulated in multiple cancer types, such as colorectal [15, 17, 18], breast [19, 20], cervical [21], lung [22, 23], hepatocellular [24, 25], pancreatic [26, 27], head and neck [28], meningiomas [29] and more recently gliomas [30]. While the role of TAGLN2 in cancers has been controversial, multiple studies have shown that TAGLN2 contributes to invasion, cell proliferation, metastases, treatment resistance and poor prognosis among several cancer types, however little is known about its role in glioma biology. Han et al. [30] recently reported that silencing of TAGLN2 decreases proliferation and invasion in gliomas and that TAGLN2 may be a prognostic biomarker in gliomas. Thus, it remains to be elucidated how the IDH1/2 mutant-generated oncometabolite, 2-HG, regulates the expression of TAGLN2 and how TAGLN2 in turn drives glioma cell proliferation and invasion.
Here, we have shown that TAGLN2 functions as an oncogene in IDH1/2 WT gliomas and is expressed at significantly higher levels in IDH1/2 WT gliomas due to TAGLN2 promoter hypermethylation and subsequent gene silencing in IDH1/2 mutant gliomas. We also found higher mRNA expression of TAGLN2 in GBM compared to IDH1/2 WT gliomas of lower grades. An in-depth understanding of the role and regulation of TAGLN2 in gliomas may be useful in identifying novel therapeutic vulnerabilities in TAGLN2-associated signaling pathways driving proliferation, invasion and therapeutic resistance mechanisms in gliomas.
RESULTS
IDH1/2 WT LGGs have increased TAGLN2 expression compared to IDH1/2 mutant gliomas and TAGLN2 mRNA expression correlates with glioma grade
In order to better understand the biological mechanisms responsible for differences in clinical outcomes between patients harboring IDH1/2 WT and IDH1/2 mutant gliomas, we performed mRNA profiling to compare gene expression between IDH1/2 WT (n=7) and IDH1/2 mutant (n=51) grade II and III gliomas from our institutional cohort. Among the 175 significantly differentially expressed genes, we identified TAGLN2 as an upregulated gene in IDH1/2 WT grade II and III gliomas compared to IDH1/2 mutant tumors (p-value=7.73x10-5; FDR=0.053) in our institutional cohort (Figure 1A). We confirmed the up-regulation of TAGLN2 mRNA levels in IDH1/2 WT tumors (n=95) compared to IDH1/2 mutant tumors (n=418) using the publicly available TCGA LGG database (p-value < 0.0001; FDR=< 0.0001) (Figure 1A).
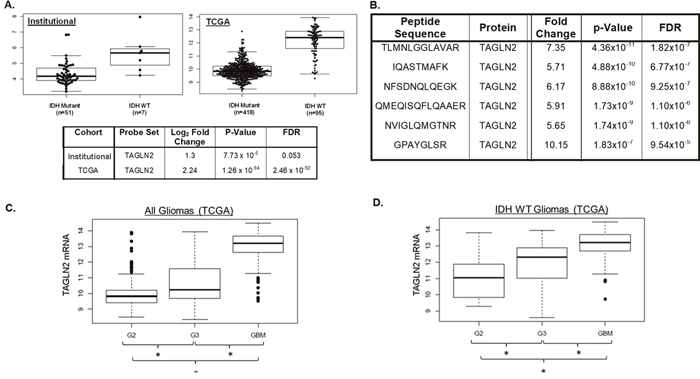
Figure 1: IDH1/2 WT LGGs have increased TAGLN2 expression compared to IDH1/2 mutant gliomas and TAGLN2 mRNA expression correlates with glioma grade. (A) TAGLN2 mRNA was expressed at significantly higher levels in IDH1/2 WT tumors compared to IDH1/2 mutant tumors in both our institutional (p-value=7.73x10-5; FDR=0.053) and the validation TCGA (p-value <0.0001; FDR < 0.0001) cohorts. (B) Mass spectrometry identified higher TAGLN2 protein expression in IDH1/2 WT compared to IDH1/2 mutant LGG from our institutional cohort. Six different peptides corresponding to TAGLN2 protein were expressed at significantly up-regulated in IDH1/2 WT compared to IDH1/2 mutant tumors. (C) Publicly available TAGLN2 mRNA expression of all Grade II (G2, n=249), III (G3, n=265) and IV (GBM, n=153) gliomas from the TCGA database are significantly different (p<0.0001). (D) TAGLN2 expression in IDH1/2 WT Grade II (n=21), III (n=52) and IV (n=133) tumors from TCGA data was significantly different (p=5.136x10-20).
In addition, we performed proteomic profiling of an institutional discovery cohort of 40 LGGs (7 IDH1/2 WT and 33 IDH1/2 mutant) that identified 120 differentially expressed peptides in IDH1/2 WT vs. IDH1/2 mutant tumors that mapped to 65 proteins (FDR p value <0.10). Interestingly, 6 of the 120 significantly differentially regulated peptides corresponded to TAGLN2 protein (Figure 1B). These six TAGLN2 peptides were significantly upregulated in IDH1/2 WT compared to IDH1/2 mutant LGGs with fold-changes ranging from 5.65 to 10.15 (Figure 1B). We validated these findings in an additional institutional cohort of IDH1/2 WT (n=7) versus IDH1/2 mutant (n=23) grade II and III gliomas. We identified 36 significantly differentially regulated proteins in common with our discovery cohort, including 3 peptides corresponding to TAGLN2 (Supplemental Figure 1). Unfortunately, TAGLN2 was not included in the reverse-phase protein lysate array (RPPA) used in the TCGA analysis and therefore TAGLN2 protein levels could not be validated in tumors from the TCGA cohort [8].
Because TAGLN2 mRNA and protein were both overexpressed in IDH1/2 WT LGGs and ~90% of GBMs carry IDH1/2 WT, we assessed TAGLN2 mRNA expression levels in malignant GBMs. We compared TAGLN2 gene expression levels among all grade II (n=249) and grade III (n=265) gliomas as well as GBMs (n=153) included in the TCGA database. Our analyses showed that not only was TAGLN2 expressed at significantly higher levels in GBMs compared to LGGs, but TAGLN2 was found to be significantly associated with glioma grade (p< 0.0001) (Figure 1C). In order to confirm that this correlation was not due to the association between grade and IDH1/2 status, we also evaluated TAGLN2 mRNA levels in IDH1/2 WT gliomas of all grades. TAGLN2 mRNA levels were significantly different between IDH1/2 WT grade II (n=21), grade III (n=52) and grade IV (n=133) gliomas from the TCGA database (p< 0.0001) (Figure 1D). It is important to note, however, that TAGLN2 gene expression data in TCGA GBM and LGG cohorts were acquired at different times, which may also account for differences in TAGLN2 gene expression.
Down-regulation of TAGLN2 expression in IDH1/2 mutant gliomas correlates with promoter hypermethylation
While the association between the hypermethylator phenotype and IDH1/2 mutation in gliomas has been well-established, the majority of individual genes involved remain to be identified. Since TAGLN2 was down-regulated in IDH1/2 mutant gliomas, we examined our global methylation data to determine whether increased promoter methylation may account for decreased TAGLN2 expression in these tumors. Indeed, 60% (n=9/15) of the 15 CpG sites predicted to correspond to the TAGLN2 promoter showed significantly higher levels of methylation (FDR<0.05) in IDH1/2 mutant tumors. The heatmap in Figure 2A depicts methylation intensity of each of the 15 CpG sites corresponding to TAGLN2 on the array. These results were validated using methylation data from the publicly available TCGA cohort (Figure 2B).
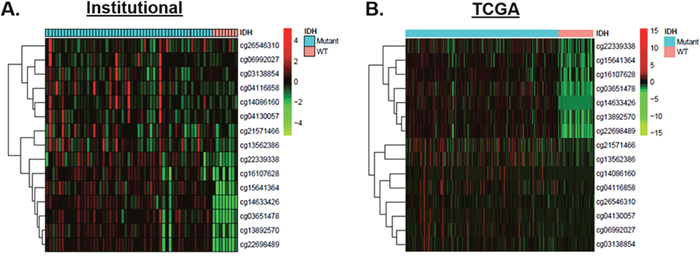
Figure 2: Increased TAGLN2 promoter hypermethylation accounts for decreased TAGLN2 expression in IDH1/2 mutant gliomas. (A) Promoter methylation was detected in IDH1/2 mutant tumors (n=54, cyan) and IDH1/2 WT tumors (n=8, salmon) from our institutional cohort using 15 CpG TAGLN2 promoter methylation sites included on the Illumina HM-450K array. IDH1/2 mutant showed significantly higher levels of methylation (FDR<0.05) in the majority of CpG islands corresponding to TAGLN2 (n=11), as demonstrated by the heat map. Low methylation levels are denoted in green and high methylation levels are denoted in red. (B) Methylation results were validated using methylation data from the publicly available TCGA cohort.
Poor prognosis associated with TAGLN2 expression is dependent on IDH1/2 mutation status in gliomas:
Since TAGLN2 has been shown to be a poor prognostic biomarker in other cancers and may play a role in glioma progression, we determined whether TAGLN2 expression was associated with overall survival (OS) of patients in both institutional and TCGA glioma cohorts. As shown in the Kaplan-Meier curve in Figure 3A, patients with high TAGLN2 mRNA levels (median split) trended toward worse OS in our institutional cohort of 58 grade II/III patients (HR 3.74, 0.77-18.09, p=0.079). In the LGG-TCGA validation cohort (n=513), patients with TAGLN2 mRNA above the median had significantly worse OS (HR 2.6, 1.6-4.0, p= 3.1x10-5) than patients with mRNA levels below the median (Figure 3B). While the trend was similar between TAGLN2 expression and OS in both LGG cohorts, our institutional cohort did not reach statistical significance, which may be due to the limited number of survival events in this cohort.
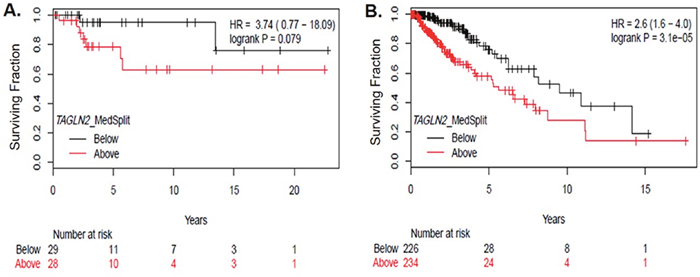
Figure 3: TAGLN2 mRNA levels are associated with poor survival among gliomas. (A) TAGLN2 mRNA levels above the median (detected by Affymetrix Clariom D array) trended toward poor OS in lower grade gliomas from our institutional cohort (HR 3.74, p=0.079), as shown by the Kaplan Meier curve. (B) In the TCGA validation dataset, high TAGLN2 mRNA levels above the median were significantly associated with poor survival (HR= 2.6, p=0.00003).
In order to further assess the prognostic significance of TAGLN2, we categorized TAGLN2 mRNA expression as above or below median and correlated expression with clinical/pathologic characteristics from our institutional and TCGA LGG cohorts (Table 1). The TCGA analysis confirmed our previous results that increased TAGLN2 expression was significantly associated with IDH1/2 WT and higher grade gliomas (Table 1). Other clinical-pathologic factors known to be associated with IDH1/2 WT gliomas, such as 1p/19q non-codeletion status, astrocytoma histology and older age, were also identified to significantly correlate with TAGLN2 expression in the TCGA analysis (Table 1). While there was a trend toward increased TAGLN2 mRNA expression in gliomas that are IDH1/2 WT, 1p/19q non-codeleted and astrocytomas in our institutional cohort (Table 1), an inadequate sample number could again explain why the association did not reach statistical significance.
Table 1: Clinical-pathological characteristics of patients analyzed for TAGLN2 mRNA expression in institutional and TCGA LGG cohorts
Clinical Factor |
Institutional Cohort |
TCGA Cohort |
||||
|---|---|---|---|---|---|---|
TAGLN2 mRNA |
Above median |
Below median |
P |
Above median |
Below median |
P |
Age |
42 |
42 |
0.539 |
45 |
38 |
<0.001 |
Gender |
0.038 |
0.182 |
||||
Female |
4 (13.8%) |
12 (41.4) |
101 (39/1%) |
100 (38.8%) |
||
Male |
25 (86.2%) |
17 (58.6%) |
134 (51.9%) |
122 (47.3%) |
||
Unknown |
N/A |
N/A |
23 (8.9%) |
36 (14.0%) |
||
Histology |
0.278 |
<0.001 |
||||
Astrocytoma |
7 (24.1%) |
4 (13.8%) |
106 (41.1%) |
63 (24.4%) |
||
Mixed |
13 (44.8%) |
13 (44.8%) |
56 (21.7%) |
58 (22.5%) |
||
Oligodendroglioma |
7 (24.1%) |
12 (41.4%) |
73 (28.3%) |
101 (39.1%) |
||
Unknown |
2 (6.9%) |
N/A |
||||
Grade |
0.263 |
<0.001 |
||||
II |
12 (41.4%) |
7 (24.1%) |
92 (35.7%) |
124 (48.1%) |
||
III |
17 (58.6%) |
22 (75.9%) |
143 (55.4%) |
98 (38.0%) |
||
Unknown |
N/A |
N/A |
23 (8.9%) |
36 (14.0%) |
||
IDH1/2 Mutation Status |
0.102 |
<0.001 |
||||
IDH1/2 Mutant |
23 (79.3%) |
28 (96.6%) |
168 (65.1%) |
251 (97.3%) |
||
IDH1/2 WT |
6 (20.7%) |
1 (3.4%) |
88 (34.1%) |
6 (2.3%) |
||
Unknown |
N/A |
N/A |
2 (0.8%) |
1 (0.4%) |
||
1p/19q Status |
0.114 |
0.005 |
||||
Co-deleted |
11 (37.9%) |
18 (62.1%) |
69 (26.7%) |
100 (38.8%) |
||
Non-co-deleted |
18 (62.1%) |
11 (37.9%) |
189 (73.3%) |
158 (61.2%) |
||
A multivariate analysis of the TCGA data was also performed to determine whether TAGLN2 is a prognostic biomarker independent of other clinical variables. However, after adjusting for age, gender, histology, grade, IDH1/2 mutation status and 1p/19q co-deletion status in our multivariate analysis, TAGLN2 expression was no longer significantly associated with OS (Table 2), which may be due to its strong association with IDH1/2 mutation status and older age (Table 1). Since IDH1/2 mutation status has also shown to be highly associated with younger age in adults with LGGs [7], IDH1/2 mutation status also lost significance in our multivariate analysis. Unfortunately, there were insufficient survival events available from the institutional cohort to perform a multivariate analysis comparing TAGLN2 expression with OS.
Table 2: Multi-variable analysis of clinical-pathological factors with OS from low(er) grade gliomas in the TCGA cohort
Variable |
HR |
95% CI |
P |
|---|---|---|---|
Gender |
1.035 |
0.58-1.85 |
0.907 |
Age at diagnosis (continuous) |
1.049 |
1.022-1.077 |
<0.001* |
Histology |
|||
Astrocytoma |
0.98 |
0.48-2.00 |
0.957 |
Oligodendroglioma |
0.55 |
0.23-1.28 |
0.165 |
IDH1/2 WT (non-mutated) |
2.37 |
0.75-7.50 |
0.142 |
1p/19q Non-codeleted |
2.25 |
0.84-6.05 |
0.108 |
TAGLN2 mRNA |
1.25 |
0.83-1.86 |
0.285 |
TAGLN2 regulates proliferation of IDH1/2 WT GBM cells in vitro
Since TAGLN2 was shown to be associated with poor prognosis and tumor progression, we evaluated the role of TAGLN2 in cell proliferation in vitro. Our in vitro studies were conducted using high grade glioma GBM30 neurospheres (primary) and U87 MG established cell lines since we found that GBMs (which are nearly all IDH1/2 WT) have significantly higher TAGLN2 levels than LGGs (Figure 1). In addition, to our knowledge, no established LGG cell lines currently exist, with the exception of BT142 cells, which do not harbor an IDH1 wild-type allele [31].
GBM30 (primary GBM neurospheres) cells stably expressing TAGLN2 shRNA or scrambled shRNA control were generated for our knock-down in vitro studies. The level of stable knock-down of TAGLN2 protein was detected by Western blot analysis (Figure 4A). GBM30 neurospheres were trypsinized and counted 24, 72 and 120 hours after plating in the presence of trypan blue for the exclusion of non-viable cells. As shown in Figure 4B, there was a moderate but significant decrease in cell proliferation at 72 and 120 hours in GBM30 cells in which TAGLN2 expression was markedly knocked down using 2 different TAGLN2-shRNA compared to the control shRNA (p<0.05). Similar results were obtained when TAGLN2 expression was transiently knocked down by RNAi in established GBM cell lines U87 MG and LN18 (Supplemental Figure 2).
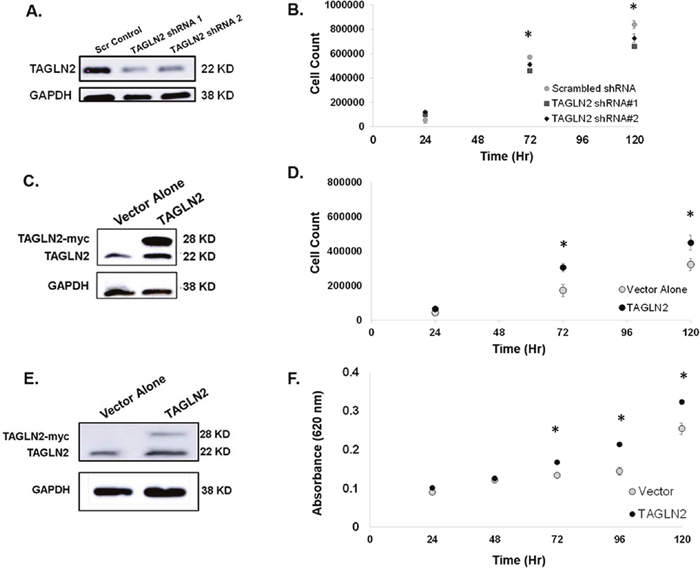
Figure 4: TAGLN2 regulates proliferation of IDH1/2 WT GBM cells in vitro. (A) GBM30 neurospheres stably expressing TAGLN2 shRNA and corresponding scrambled shRNA control were generated and the level of stable TAGLN2 knock-down detected by Western blot is shown. (B) GBM30 neurospheres with stable knock-down of TAGLN2 or scrambled shRNA control were counted at 24, 72, and 1120 hours after plating. Knock-down of TAGLN2 resulted in significantly decreased cell counts (p<0.05). (C) GBM30 neurospheres and (D) U87 MG glioma cells stably overexpressing TAGLN2 and corresponding vector control were generated and the level of stable TAGLN2 overexpression was detected by Western blot. Of note, endogenous TAGLN2 (22 Kda) and exogenous TAGLN2-myc (28 kDa) are shown. (E) GBM30 neurospheres stably overexpressing TAGLN2 resulted in significantly increased cell proliferation compared to vector control at 72 and 120 hours (p<0.05). (F) U87 MG cells stably overexpressing TAGLN2 resulted in increased cell proliferation compared to the vector alone. Experiments were performed twice with six replicates each. *, statistically significant difference in proliferation.
Since TAGLN2-knockdown compromised the proliferation of GBM cells, we also evaluated whether TAGLN2 overexpression was sufficient to increase cell proliferation. We generated stable GBM30 and U87 MG cell lines overexpressing TAGLN2 (tagged with Myc-epitope). Figure 4C and 4E show increases in the level of endogenous (22 kDa) and Myc-tagged exogenous (28 kDa) TAGLN2 expression in both GBM30 and U87 MG cells, respectively. We counted cells with trypan blue for non-adherent GBM30 neurospheres and performed methylene blue assay for U87 MG cells. In both cell models, TAGLN2 overexpression resulted in a significant increase in cell proliferation after 72 hours of incubation (p<0.05) (Figure 4D and 4F).
TAGLN2 regulates invasion of IDH1/2 WT GBM cells in vitro
Since invasion into the surrounding brain parenchyma is a hallmark of gliomas and GBMs that contributes to local recurrence and worse prognosis, we assessed whether TAGLN2 is involved in invasion by glioma cells. The ability of GBM30 neurospheres to invade through an extracellular matrix after shRNA-mediated stable knock-down of TAGLN2 was compared to each respective cell type stably transfected with scrambled shRNA control. Figure 5A shows a decreased average number of cells invading through the matrix after knock-down of TAGLN2 in GBM30 cells, respectively. As previously shown (Figure 4), there were minimal differences in cell proliferation before 24 hours, further suggesting that the number of invading cells were due to invasion capability and not cell proliferation. These experiments were also performed with siRNA-mediated transient knock-down of TAGLN2 in U87 MG and LN18 cells and similar results were obtained (Supplemental Figure 2).
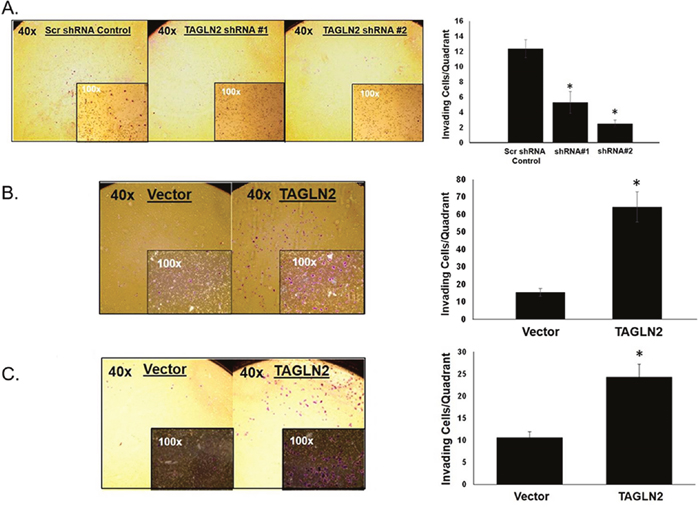
Figure 5: TAGLN2 regulates invasion of IDH1/2 WT GBM cells in vitro. Since TAGLN2 has been shown to play a role in invasion and metastases of other cancer types, the invasive ability of (A) GBM30 neurospheres with stable shRNA-mediated knock-down of TAGLN2 were compared to their respective scrambled shRNA control. GBM30 cells showed a decrease in average number of cells invading through the matrix after knock-down of TAGLN2 compared to control. In contrast, (B) GBM30 neurospheres and (C) U87 MG glioma cells with stable overexpression of TAGLN2 showed an increase in average number of cells invading through the matrix compared to vector control. Experiments were performed three times with triplicate invasion assays. *, statistically significant difference in invading cells (p<0.05). Photographs are representative images at 40x and 100x magnification.
Since knockdown of TAGLN2 expression compromised the invasion of GBM cells, we also evaluated whether TAGLN2 overexpression increased invasion. GBM30 neurospheres and U87 MG cells stably overexpressing TAGLN2 or vector control were plated on invasion chambers. As shown in Figure 5B and 5C, TAGLN2 overexpression significantly increased the number of invading cells by approximately 2.5 to three-fold in glioma cells (p<0.05).
TAGLN2 protein expression is higher in U87 MG (IDH1/2 WT) parental cells compared to U87 MG cells overexpressing IDH1 R132H mutant protein due to hypomethylation of TAGLN2 promoter
Since TAGLN2 was found to be expressed at significantly lower mRNA and protein levels in IDH1/2 mutant gliomas from both institutional and TCGA patient cohorts, we confirmed these findings in vitro by evaluating TAGLN2 protein levels in a commercially available U87 MG isogenic cell line overexpressing IDH1R132H mutant protein (ATCC, Manassas, VA), which will be referred to as IDH1 mutant U87 MG cells. We compared levels of TAGLN2 protein in U87 MG cells overexpressing mutant IDH1 to U87 IDH1/2 WT MG parental cells by Western blot. Similar to our patient-derived glioma tissues, cell lines expressing mutant IDH1 had decreased TAGLN2 protein levels compared to U87 MG IDH1/2 WT glioma cells (Figure 6A). Since our methylation data in glioma patients suggested that TAGLN2 expression might be silenced in IDH1/2 mutant LGGs by promoter hypermethylation, we confirmed these results in vitro by treating IDH1 mutant U87 MG cells with 5-aza-2′-deoxycytidine (5-AZA), an inhibitor of DNA methyltransferase that results in DNA demethylation [32]. Cells were treated with increasing concentrations of 5-AZA (1, 5 and 10 μM) for 96 hours and TAGLN2 protein levels were evaluated by Western blot analysis. As shown in Figure 6B, treatment with 5-AZA resulted in increased TAGLN2 protein expression in an almost dose-dependent manner in IDH1 mutant U87 MG cells. Treatment of IDH1/2 WT U87 MG with 5-AZA did not change TAGLN2 protein levels (data not shown). These results suggest that TAGLN2 expression may be decreased due to its promoter hypermethylation-mediated transcriptional downregulation in IDH1/2 mutant gliomas.
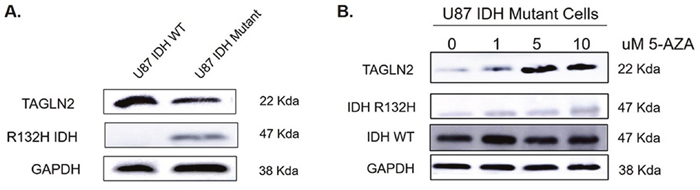
Figure 6: TAGLN2 protein expression is higher in U87 MG (IDH1/2 WT) parental cells compared to U87 MG cells overexpressing IDH1 R132H mutant protein due to hypomethylation of TAGLN2 promoter. (A) TAGLN2 protein levels were compared in U87 MG IDH1/2 WT parental cells and a commercially available U87 MG isogenic cell line overexpressing IDH1 with a heterozygous R132H mutation by Western blot analysis. TAGLN2 protein was decreased in IDH1 mutant cells compared to IDH1/2 WT cells. (B) U87 MG IDH1 mutant cells were treated with increasing concentrations of 5-azacytidine (5-AZA) demethylating agent and TAGLN2 protein was evaluated by Western blot. 5-AZA resulted in increasing TAGLN2 protein levels expression in a dose-dependent manner.
DISCUSSION
It has been well-established that IDH1/2 WT gliomas demonstrate worse prognoses compared to gliomas with IDH1/2 mutations [4]. While multiple explanations have been proposed, the underlying biological mechanisms responsible for these survival differences remain poorly understood. In this study, we performed mRNA and proteomic profiling of IDH1/2 WT versus IDH1/2 mutant Grade II and III gliomas in order to better understand gene expression differences contributing to worse overall survival in IDH1/2 WT gliomas. Our analyses identified TAGLN2 as one of the significantly up-regulated genes in IDH1/2 WT compared to IDH1/2 mutant low(er) grade gliomas. Subsequently, we tested the hypothesis that increased TAGLN2 expression may contribute to worse prognoses in IDH1/2 WT gliomas by investigating the role and regulation of TAGLN2 regulation in both LGGs and GBMs.
While in agreement with a recent study showing that TAGLN2 is a poor prognostic marker for gliomas, our study is the first to report that TAGLN2 is a negative prognostic factor associated with IDH1/2 WT gliomas and its regulation in gliomas is highly dependent on IDH1/2 mutation status. Previous studies have demonstrated that reorganization of the methylome and the resulting hypermethylator phenotype in gliomas is established by the presence of mutant IDH1/2 [33, 34]. However, the individual genes involved in G-CIMP and their associated effects on oncogenesis remain uncertain. We are the first to identify TAGLN2 as a gene likely to be involved in the hypermethylator phenotype and further show that TAGLN2 undergoes epigenetic regulation in IDH1/2 mutant gliomas. Our global methylation analysis of low(er) grade gliomas from our institutional cohort showed that CpG sites corresponding to the TAGLN2 promoter are more highly methylated in IDH1/2 mutant compared to IDH1/2 WT gliomas. Therefore, our data suggest that epigenetic silencing by promoter hypermethylation likely accounts for decreased TAGLN2 mRNA and protein levels observed in IDH1/2 mutant gliomas. Our in vitro studies confirmed that overexpression the protein of IDH1R132H in U87 MG GBM cells resulted in decreased TAGLN2 expression compared to U87 MG IDH1/2 WT parental cells and further showed that treatment of IDH1 mutant cells with 5-AZA, a DNA methyltransferase inhibitor, rescued TAGLN2 expression after demethylation. Such an association between the IDH1/2 mutation and TAGLN2 expression is also demonstrated by our multivariate analysis that showed TAGLN2 as a negative prognostic factor, though highly dependent on IDH1/2 mutation status.
While our study is limited by the availability of only one established heterozygous IDH1 mutant glioma cell line, the epigenetic regulation of transgelin, a homologue of TAGLN2, has been reported in multiple other cancer types. While these homologues share 64% amino acid sequence homology and similar functions, transgelin is mainly expressed in smooth muscle cells and fibroblasts whereas TAGLN2 is expressed primarily in epithelial cells. Transgelin has been reported to be regulated by promoter hypermethylation in breast [35], colorectal [36], malignant peripheral nerve sheath tumors [37] and hepatocellular carcinomas [38]. Moreover, promoter methylation has also been shown to regulate transgelin expression during normal physiologic processes, including epigenetic silencing of transgelin in endometrial stroma during reorganization of the actin cytoskeleton in preparation for pregnancy [39]. Similar to our data, Logan et al. [39] showed that treatment with 5-AZA demethylating agent rescued transgelin expression in endometrial stromal cells.
While TAGLN2 expression appears to be regulated at the epigenetic level in IDH mutant gliomas, we also found significant differences in TAGLN2 mRNA expression among different grades of IDH WT gliomas, suggesting that TAGLN2 expression may also be regulated during glioma progression in a 2HG-independent fashion. Our results are in agreement with a recent study that found higher levels of multiple transgelin peptides in stem cells isolated from high grade gliomas compared to low grade gliomas [40]. Moreover, Han et al. [22] reported that TGF-β signaling increases TAGLN2 expression, which in turn regulates the epithelial-mesenchymal transition (EMT) leading to acquisition of a mesenchymal phenotype and increased cell invasion [41]. Taken together, these results suggest that TAGLN2 may be involved in glioma progression.
A more invasive phenotype is believed to contribute to worse prognoses in IDH1/2 WT LGGs compared to IDH1/2 mutant gliomas [42–46], among other proposed mechanisms. Infiltration into the surrounding brain parenchyma is a hallmark of gliomas and GBMs that allows cancer cells to escape surgical resection and radiation, often resulting in local recurrence after aggressive therapy. Beiko et al. [42] concluded that IDH mutant astrocytomas were more amenable to gross surgical resection due to minimal invasion compared to IDH WT astrocytomas [42], therefore reducing the risk of local recurrence. Moreover, Price et al. [44] found that IDH mutant GBMs were less invasive than IDH WT GBMs based on MRI diffusor-tensor imaging, a technique believed to delineate tumor margins more accurately than conventional MRI. Another study further showed IDH mutant cells to have a less invasive phenotype than IDH WT cells in vivo and in vitro [45]. Our results show that TAGLN2 significantly contributes to invasion and proliferation in IDH WT gliomas, however the mechanism remains poorly understood.
Glioma cell invasion is a multi-step process that requires several changes to occur within the cell, including gaining cell motility through reorganization of the cytoskeleton, detachment from the primary tumor, adhesion to the extracellular matrix (ECM) and degradation of the ECM [47, 48]. However, the role of TAGLN2 in this complex process remains to be elucidated. A recently published study provided evidence that TAGLN2 is capable of regulating the actin cytoskeleton in response to extracellular signals when localized to the cell membrane in airway smooth muscle cells [49]. Indeed, TAGLN2 is believed to contribute to invasion by remodeling the actin cytoskeleton and forming cell protrusions, referred to as invadopodia, that promote cell motility [30]. Other studies have demonstrated that TAGLN2 and its homologues have the ability to localize to the nucleus and regulate expression of invasion-related genes that facilitate detachment from tumor cells as well as adhesion to and degradation of the ECM [22]. Transgelin and its homologues have been shown to regulate expression of critical invasion-related genes, including adherens junctions (E-cadherin and B-catenin), tight junctions (occludin), intermediate filament proteins (vimentin), adhesion glycoproteins (fibronectin), proteases (MMP-9, MMP-2) and chemokine receptors (CXCR4) [17, 21, 30, 50–52]. Furthermore, Han et al. [30] suggested that TAGLN2 may promote EMT and invasion in glioma cells by up-regulating expression of mesenchymal markers, such as N-cadherin, Snail, Slug and Twist. TAGLN2 therefore appears to regulate glioma invasion at multiple levels.
In summary, we have shown that TAGLN2 expression is silenced by promoter hypermethylation and therefore likely involved in the glioma hypermethylator phenotype of IDH1/2 mutant gliomas. Moreover, we have shown that TAGLN2 is highly expressed in IDH1/2 WT gliomas and GBMs and appears to function as an oncogene. This differential regulation of TAGLN2 provides further insight into the genetic, epigenetic and oncogenic differences between IDH1/2 WT and mutant gliomas. Taken together, our data demonstrate that TAGLN2 protein functions as an oncogene and may serve as a novel therapeutic target for effectively treating IDH1/2 WT LGG and GBM.
MATERIALS AND METHODS
Glioma databases
Sixty-seven Grade II and III formalin-fixed paraffin embedded (FFPE) glioma specimens in our institutional cohort were provided by the University of Freiburg. Corresponding clinical data including age at diagnosis, gender, KPS, histology and overall survival (OS) were also provided. OS was defined as the time from pathologic glioma diagnosis (date of surgery) to date of death or last documented date of follow-up. Clinical data for Grade II, III and IV gliomas included in The Cancer Genome Atlas (TCGA) research database as well as their corresponding mRNA expression, promoter methylation, IDH1/2 mutation and 1p/19q co-deletion analysis were obtained from the publicly available data portal at https://cancergenome.nih.gov.
Briefly, IDH1/2 mutation status for our institutional cohort was determined by next-generation sequencing with a customized IonTorrent panel (ThermoFisher Scientific, Waltham, MA) that included amplicons covering the coding regions (>98% of bases) of IDH1 and IDH2 [53, 54]. Patients were deemed IDH1/2 mutant if mutations were present at IDH1R132 or IDH2R172 positions. 1p/19q co-deletion status was determined by the Infinium Human Methylation 450K array (Illumina) and/or the Affymetrix Oncoscan v2.0 platform (Thermofisher Scientific, Waltham, MA). A reference from the CopyNumber450kData package was used. For 1p/19q status, the ranges chr1 15865-100000000 and chr19 40000000-59095508 bp, respectively were analyzed. A median log R ratio of -0.2 was used to call deletions of each of these regions. Samples were omitted if deemed of poor quality (due to large variation in the by probe log R ratios) based on the plots. Co-deletion status was determined by the Nexus calls for the Oncoscan data, as well as visual inspection of the segmentation plots for each sample.
Microarray analysis
Total RNA was extracted from FFPE glioma tissues with the miRNAeasy kit (Qiagen, Hilden, Germany). Only RNA with a 260/280 ratio of at least 1.8 measured by Nanodrop was used for the microarray analysis. cRNA was hybridized to the Affymetrix HTA ClariomD array (Thermofisher Scientific, Waltham, MA). Normalizaton (SST-RMA) and summarization were carried out using the Affymetrix transcriptome analysis console software. Differential gene expression between IDH1/2 WT and IDH1/2 mutant gliomas was determined by LIMMA analysis [55]. The cox regression model was employed to identify associations between gene expression and overall survival.
Proteomics
Proteins were comparatively evaluated between IDH1/2 mutant and WT tumors by mass spectrometry as previously described in Bassett et al. [56]. Briefly, proteins were digested with trypsin and LysC and resulting peptides were analyzed by mass spectrometry (completed by CWRU Center for Proteomics). LC-MS/MS data collection was performed using a UPLC system (NanoAcquity, Waters) interfaced to an Orbitrap ProVelos Elite MS system (ThermoFisher Scientific). All peptide precursor ions across all chromatographic analyses were clustered using Rosetta Elucidator software. Peptide and protein identifications were integrated from the protein database search engine output (MASCOT, Matrix Science Inc.) [56].
Methylation
DNA was extracted from FFPE tissues using a combination of Recoverall Total Nucleic Acids Isolation (ThermoFisher Scientific) and Epicentre Masterpure DNA Purification (Illumina) kits. DNA quantity was assessed using the Qubit dsDNA High Sensitivity Assay Kit (ThermoFisher Scientific). Approximately 250 ng of DNA was submitted to the University of Southern California Epigenome Center (Los Angeles, CA) and run on the Illumina Human Methylation-450K Array. Data were processed using the R package “minfi” with hg19 annotation. Data were Noob normalized and M-value [57] transformed. Probes failing in > 10% (by Illumina’s detection p-value) of samples were removed (n=1301). Samples failing across > 10% of probes were removed (n=0). SNP containing probes were removed (n=17, 534).
Cell culture
U87 MG high grade glioma cell lines (American Type Culture Collection (ATCC), Manassas, VA, USA) were maintained in DMEM media (Gibco, Life Technologies, Gaithersburg, MD) with 10% Fetal Bovine Serum (FBS) and 1% penicillin/streptomycin. An isogenic line derived from overexpression of IDH1R132H in U87 MG parental cells was purchased from ATCC (Manassas, VA, USA). This cell line has a heterozygous C395G>A knock-in mutation encoding IDH1R132H protein expression generated by using the CRISPR/Cas9 gene editing technology. GBM30 glioma neurospheres were a generous gift from Balveen Kaur’s lab and have been previously characterized [58, 59]. They were cultured in neurobasal medium (Gibco, Life Technologies, Gaithersburg, MD) supplemented with 20% B27 (Invitrogen, Carlsbad, CA), 100 ug/ml penicillin/ streptomycin, 20 ng/ml epidermal growth factor (EGF), and 20 ng/ml basic fibroblast growth factor. Cells were maintained at 37 °C in 5% CO2.
Development of stable cell lines
Stable TAGLN2 Knock-down: Short hairpin RNA (shRNA) targeting TAGLN2 were transfected into cells using Lipofectamine 2000 (ThermoFisher Scientific, Waltham, MA) per the manufacturer’s protocol and TAGLN2 knock-down efficiency was measured 48 hours after transfection. For GBM30 cells, shRNA targeting TAGLN2 was purchased from Origene with the following sequences: shRNA#1: CTGTGTGCAGCGGACGCTGATGAATCTGG and shRNA#2: GGCGTCTCAGGCAGGCATGACTGGCT ACG. Control cells were transfected with scrambled shRNA control in the pGFP-V-RS shRNA vector (Origene, Rockville, MD). shRNA #3 targeting human TAGLN2 was purchased from Sigma and transfected into U87 MG cells with the following sequence: CCGGGAACGTGATCGGGTTACAGATCTCGAGATCTGTAACCCGATCACGTTCTTTTTTG (Sigma, St. Louis, MO). Transfection with MISSION pLKO.1-puro Non-Mammalian shRNA Control Plasmid DNA was used for the corresponding scrambled shRNA control (Sigma, St. Louis, MO). Different TAGLN2-targeted shRNAs with greater knock-down efficiency were required for GBM30 cells due to their lower transfection rates. Stable cell lines were maintained in selection media supplemented with puromycin (2 μg/mL) (Sigma, St. Louis, MO).
Stable TAGLN2 overexpression
Human TAGLN2 (Myc-DDK-tagged) in pCMV6-entry vector (Origene, Rockville, MD) was transfected into GBM30, U87 MG IDH1/2 WT and U87 MG glioma cells overexpressing mutant IDH1R132H using Lipofectamine 2000 (Thermo Scientific, Waltham, MA) according to the manufacturer’s protocol. Control cells were transfected with pCMV6-entry vector alone. Stable cell lines were maintained in selection media supplemented with G418 at 400μ/mL (Sigma, St. Louis, MO).
Western blot
Cells (5 × 105) were seeded in 60 mm plates, washed with PBS, trypsinized and transferred to a microcentrifuge tube. After centrifuging and washing in PBS, cells were lysed in RIPA buffer (Sigma, St. Louis, MO) containing phosphatase and protease inhibitor cocktail and 1 mmol/L phenylmethanesulfonyl fluoride (Thermo Scientific, Waltham, MA). Protein concentrations were measured using the BSA protein assay (Pierce, Thermo Scientific, Waltham, MA) per protocol. Equal amounts of protein were separated with 10% SDS-PAGE gel electrophoresis and transferred to Hybond ECL nitrocellulose membranes (GE Healthcare, Chicago, IL). After blocking for 1 hour in Tris-buffered saline containing 0.1% Tween 20 and 5% milk, the membranes were probed with various primary antibodies: anti-TAGLN2 polyclonal antibody (Abcam, Cambridge, United Kingdom), anti-IDH1 R132H mutant antibody (Cell Signaling Technologies, Danvers, MA) and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) monoclonal antibody (American Research Products, Waltham, MA). Membrane was then probed with secondary anti-rabbit IgG conjugated to horseradish peroxidase (HRP) and secondary anti-mouse IgG-HRP (Cell Signaling Technologies, Danvers, MA). Immunoreactivity was detected by enhanced chemiluminescent (ECL) kit (Amersham Biosciences Co., Little Chalfont, United Kingdom).
Proliferation assays
U87 MG stable cells (2×103 cells) were seeded in a 96 well-plate. Six replicates were seeded for each of five time points, including 24, 48, 72, 96 and 120 hours. At each time point, cells were washed with PBS twice and fixed with 4% paraformaldehyde for 20 minutes. Cells were stained with 1% methylene blue for 25 minutes. Methylene blue was dissolved in an acetic acid solution and absorbance at 620 nm was measured using a spectrophotometer. Since the neurospheres are non-adherent, we performed cell proliferation assays by counting cells at 24, 72 and 120 hours using Trypan blue to exclude non-viable cells. Briefly, 1×105 dissociated GBM30 cells were seeded in a 24 well plate with serum-free media for three different time points, including 24, 72 and 120 hours. At each time point, cells were dissociated, counted and compared between the vector control and the stable cell lines. Cell count (absorbance) at each time point was compared between stable cell lines with TAGLN2 knock-down/overexpression relative to their respective control cells using t-test (p<0.05). Experiments were performed three times with 4 replicates.
Invasion assay
Matrigel invasion chambers (Corning, Corning, NY) were incubated in serum-free media two hours before seeding cells. 2 × 105 U87 MG and GBM30 cells were seeded onto matrigel invasion chambers. Cells were incubated in chamber over 24 hours at 37 °C in 5% CO2. After incubation, cells were fixed and stained in 0.5% crystal violet with methanol for 20 minutes and washed in PBS. Average number of invading cells in each quadrant was averaged and compared between stable cell lines with TAGLN2 knock-down/overexpression relative to their respective control cells using t-test (p<0.05). Experiments were performed three times with duplicates. Photos of stained cells in invasion matrix were obtained using the EVOS XL Core microscope (ThermoFisher Scientific, Waltham, MA).
Statistics
For two group univariable survival comparisons, Kaplan Meier curves were plotted and the log-rank test applied. For multivariable and survival associations with continuous variables, Cox regression was used. For two group comparisons, two sample t-tests were employed, and for multiple group analyses ANOVA was used followed by pairwise comparisons using Tukey’s post-hoc test. For high dimensional analyses, FDR < 0.1 was considered significant, while for conformational analyses a raw P<0.05 was considered to be statistically significant. Heat maps were produced using Ward clustering on correlations.
Abbreviations
Low(er) grade gliomas (LGG), glioblastoma (GBM), transgelin-2 (TAGLN2), isocitrate dehydrogenase (IDH), wild-type (WT), World Health Organization (WHO), α-ketoglutarate (α-KG), 2-hydroxyglutarate (2-HG), transgelin-1 (TAGLN1), transgelin-3 (TAGLN3), 5-azacytidine (5-AZA).
ACKNOWLEDGMENTS AND FUNDING
This work was supported by National Cancer Institute R01CA169368, R01CA108633, R01CA188228, R01CA188500, 1RC2CA148190 and U10CA180850; A Brain Tumor Funders Collaborative Grant; and The Ohio State University Comprehensive Cancer Center Award (all to A.C.). We would also like to thank the CWRU Center for Proteomics for their contributions to our proteomics work.
CONFLICTS OF INTEREST
The authors have nothing to disclose.
REFERENCES
1. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352: 987-96. https://doi.org/10.1056/NEJMoa043330.
2. Koshy M, Villano JL, Dolecek TA, Howard A, Mahmood U, Chmura SJ, Weichselbaum RR, McCarthy BJ. Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J Neurooncol. 2012; 107: 207-12. https://doi.org/10.1007/s11060-011-0738-7.
3. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008; 321: 1807-12. https://doi.org/10.1126/science.1164382.
4. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009; 360: 765-73. https://doi.org/10.1056/NEJMoa0808710.
5. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016; 131: 803-20. https://doi.org/10.1007/s00401-016-1545-1.
6. Siegal T. Clinical Relevance of Prognostic and Predictive Molecular Markers in Gliomas. Adv Tech Stand Neurosurg. 2016; 43: 91-108. https://doi.org/10.1007/978-3-319-21359-0_4.
7. Cohen AL, Holmen SL, Colman H. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep. 2013; 13: 345. https://doi.org/10.1007/s11910-013-0345-4.
8. Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, Morozova O, Robertson AG, Noushmehr H, Laird PW, et al; Cancer Genome Atlas Research Network. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med. 2015; 372: 2481-98. https://doi.org/10.1056/NEJMoa1402121.
9. Aibaidula A, Chan AK, Shi Z, Li Y, Zhang R, Yang R, Li KK, Chung NY, Yao Y, Zhou L, Wu J, Chen H, Ng HK. Adult IDH wild-type lower-grade gliomas should be further stratified. Neuro Oncol. 2017; 19: 1327-37. https://doi.org/10.1093/neuonc/nox078.
10. Waitkus MS, Diplas BH, Yan H. Isocitrate dehydrogenase mutations in gliomas. Neuro Oncol. 2016; 18: 16-26. https://doi.org/10.1093/neuonc/nov136.
11. Ducray F, Marie Y, Sanson M. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009; 360: 2248-9; author reply 2249.
12. Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, Felsberg J, Wolter M, Mawrin C, Wick W, Weller M, Herold-Mende C, Unterberg A, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 2009; 118: 469-74. https://doi.org/10.1007/s00401-009-0561-9.
13. Losman JA, Kaelin WG. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes & Development. 2013; 27: 836-52. https://doi.org/10.1101/gad.217406.113.
14. Dvorakova M, Nenutil R, Bouchal P. Transgelins, cytoskeletal proteins implicated in different aspects of cancer development. Expert Rev Proteomics. 2014; 11: 149-65. https://doi.org/10.1586/14789450.2014.860358.
15. Meng T, Liu L, Hao R, Chen S, Dong Y. transgelin-2: A potential oncogenic factor. Tumour Biol. 2017; 39: 1010428317702650. https://doi.org/10.1177/1010428317702650.
16. Jo S, Kim HR, Mun Y, Jun CD. transgelin-2 in immunity: Its implication in cell therapy. J Leukoc Biol. 2018; 104:903-910. https://doi.org/10.1002/JLB.MR1117-470R.
17. Lin Y, Buckhaults PJ, Lee JR, Xiong H, Farrell C, Podolsky RH, Schade RR, Dynan WS. Association of the actin-binding protein transgelin with lymph node metastasis in human colorectal cancer. Neoplasia. 2009; 11: 864-73.
18. Zhou H, Zhang Y, Chen Q, Lin Y. AKT and JNK Signaling Pathways Increase the Metastatic Potential of Colorectal Cancer Cells by Altering Transgelin Expression. Dig Dis Sci. 2016; 61: 1091-7. https://doi.org/10.1007/s10620-015-3985-1.
19. Rao D, Kimler BF, Nothnick WB, Davis MK, Fan F, Tawfik O. Transgelin: a potentially useful diagnostic marker differentially expressed in triple-negative and non-triple-negative breast cancers. Hum Pathol. 2015; 46: 876-83. https://doi.org/10.1016/j.humpath.2015.02.015.
20. Cai J, Chen S, Zhang W, Zheng X, Hu S, Pang C, Lu J, Xing J, Dong Y. Salvianolic acid A reverses paclitaxel resistance in human breast cancer MCF-7 cells via targeting the expression of transgelin 2 and attenuating PI3 K/Akt pathway. Phytomedicine. 2014; 21: 1725-32. https://doi.org/10.1016/j.phymed.2014.08.007.
21. Yakabe K, Murakami A, Kajimura T, Nishimoto Y, Sueoka K, Sato S, Nawata S, Sugino N. Functional significance of transgelin-2 in uterine cervical squamous cell carcinoma. J Obstet Gynaecol Res. 2016; 42: 566-72. https://doi.org/10.1111/jog.12935.
22. Wu X, Dong L, Zhang R, Ying K, Shen H. Transgelin overexpression in lung adenocarcinoma is associated with tumor progression. Int J Mol Med. 2014; 34: 585-91. https://doi.org/10.3892/ijmm.2014.1805.
23. Jin H, Cheng X, Pei Y, Fu J, Lyu Z, Peng H, Yao Q, Jiang Y, Luo L, Zhuo H. Identification and verification of transgelin-2 as a potential biomarker of tumor-derived lung-cancer endothelial cells by comparative proteomics. J Proteomics. 2016; 136: 77-88. https://doi.org/10.1016/j.jprot.2015.12.012.
24. Leung WK, Ching AK, Chan AW, Poon TC, Mian H, Wong AS, To KF, Wong N. A novel interplay between oncogenic PFTK1 protein kinase and tumor suppressor TAGLN2 in the control of liver cancer cell motility. Oncogene. 2011; 30: 4464-75. https://doi.org/10.1038/onc.2011.161.
25. Shi YY, Wang HC, Yin YH, Sun WS, Li Y, Zhang CQ, Wang Y, Wang S, Chen WF. Identification and analysis of tumour-associated antigens in hepatocellular carcinoma. Br J Cancer. 2005; 92: 929-34. https://doi.org/10.1038/sj.bjc.6602460.
26. Sun Y, He W, Luo M, Zhou Y, Chang G, Ren W, Wu K, Li X, Shen J, Zhao X, Hu Y. Role of transgelin-2 in diabetes-associated pancreatic ductal adenocarcinoma. Oncotarget. 2017; 8: 49592-604. https://doi.org/10.18632/oncotarget.17519.
27. Zhou L, Zhang R, Zhang L, Sun Y, Yao W, Zhao A, Li J, Yuan Y. Upregulation of transgelin is an independent factor predictive of poor prognosis in patients with advanced pancreatic cancer. Cancer Sci. 2013; 104: 423-30. https://doi.org/10.1111/cas.12107.
28. Nohata N, Sone Y, Hanazawa T, Fuse M, Kikkawa N, Yoshino H, Chiyomaru T, Kawakami K, Enokida H, Nakagawa M, Shozu M, Okamoto Y, Seki N. miR-1 as a tumor suppressive microRNA targeting TAGLN2 in head and neck squamous cell carcinoma. Oncotarget. 2011; 2: 29-42. https://doi.org/10.18632/oncotarget.213.
29. Pei J, Li P, Zhang ZY, Zhang HL, Gao YH, Wang DY, Zheng Y, Xu X, Cui JZ. Effect of TAGLN2 in the regulation of meningioma tumorigenesis and development. Eur Rev Med Pharmacol Sci. 2018; 22: 307-13. https://doi.org/10.26355/eurrev_201801_14173.
30. Han MZ, Xu R, Xu YY, Zhang X, Ni SL, Huang B, Chen AJ, Wei YZ, Wang S, Li WJ, Zhang Q, Li G, Li XG, et al. TAGLN2 is a candidate prognostic biomarker promoting tumorigenesis in human gliomas. J Exp Clin Cancer Res. 2017; 36: 155. https://doi.org/10.1186/s13046-017-0619-9.
31. Luchman HA, Stechishin OD, Dang NH, Blough MD, Chesnelong C, Kelly JJ, Nguyen SA, Chan JA, Weljie AM, Cairncross JG, Weiss S. An in vivo patient-derived model of endogenous IDH1-mutant glioma. Neuro Oncol. 2012; 14: 184-91. https://doi.org/10.1093/neuonc/nor207.
32. Creusot F, Acs G, Christman JK. Inhibition of DNA methyltransferase and induction of Friend erythroleukemia cell differentiation by 5-azacytidine and 5-aza-2'-deoxycytidine. J Biol Chem. 1982; 257: 2041-8.
33. Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, Thompson CB, Kaufman A, Guryanova O, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012; 483: 479-83. https://doi.org/10.1038/nature10866.
34. Ichimura K. Molecular pathogenesis of IDH mutations in gliomas. Brain Tumor Pathol. 2012; 29: 131-9. https://doi.org/10.1007/s10014-012-0090-4.
35. Sayar N, Karahan G, Konu O, Bozkurt B, Bozdogan O, Yulug IG. Transgelin gene is frequently downregulated by promoter DNA hypermethylation in breast cancer. Clin Epigenetics. 2015; 7: 104. https://doi.org/10.1186/s13148-015-0138-5.
36. Zhao L, Wang H, Deng YJ, Wang S, Liu C, Jin H, Ding YQ. Transgelin as a suppressor is associated with poor prognosis in colorectal carcinoma patients. Modern Pathology. 2009; 22: 786-96. https://doi.org/10.1038/modpathol.2009.29.
37. Park GH, Lee SJ, Yim H, Han JH, Kim HJ, Sohn YB, Ko JM, Jeong SY. TAGLN expression is upregulated in NF1-associated malignant peripheral nerve sheath tumors by hypomethylation in its promoter and subpromoter regions. Oncology Reports. 2014; 32: 1347-54. https://doi.org/10.3892/or.2014.3379.
38. Hirasawa Y, Arai M, Imazeki F, Tada M, Mikata R, Fukai K, Miyazaki M, Ochiai T, Saisho H, Yokosuka O. Methylation status of genes upregulated by demethylating agent 5-aza-2 '-deoxycytidine in hepatocellular carcinoma. Oncology. 2006; 71: 77-85. https://doi.org/10.1159/000100475.
39. Logan PC, Ponnampalam AP, Rahnama F, Lobie PE, Mitchell MD. The effect of DNA methylation inhibitor 5-Aza-2'-deoxycytidine on human endometrial stromal cells. Hum Reprod. 2010; 25: 2859-69. https://doi.org/10.1093/humrep/deq238.
40. Taghipour M, Omidvar A, Razmkhah M, Ghaderi A, Mojtahedi Z. Comparative Proteomic Analysis of Tumor Mesenchymal-Like Stem Cells Derived from High Grade versus Low Grade Gliomas. Cell J. 2017; 19: 250-8. https://doi.org/10.22074/cellj.2016.4179.
41. Iwadate Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol Lett. 2016; 11: 1615-20. https://doi.org/10.3892/ol.2016.4113.
42. Beiko J, Suki D, Hess KR, Fox BD, Cheung V, Cabral M, Shonka N, Gilbert MR, Sawaya R, Prabhu SS, Weinberg J, Lang FF, Aldape KD, et al. IDH1 mutant malignant astrocytomas are more amenable to surgical resection and have a survival benefit associated with maximal surgical resection. Neuro Oncol. 2014; 16: 81-91. https://doi.org/10.1093/neuonc/not159.
43. Cui DM, Ren J, Shi JL, Feng LJ, Wang K, Zeng T, Jin Y, Gao L. R132H mutation in IDH1 gene reduces proliferation, cell survival and invasion of human glioma by downregulating Wnt/beta-catenin signaling. Int J Biochem Cell Biol. 2016; 73: 72-81. https://doi.org/10.1016/j.biocel.2016.02.007.
44. Price SJ, Allinson K, Liu H, Boonzaier NR, Yan JL, Lupson VC, Larkin TJ. Less Invasive Phenotype Found in Isocitrate Dehydrogenase-mutated Glioblastomas than in Isocitrate Dehydrogenase Wild-Type Glioblastomas: A Diffusion-Tensor Imaging Study. Radiology. 2017; 283: 215-21. https://doi.org/10.1148/radiol.2016152679.
45. Bralten LB, Kloosterhof NK, Balvers R, Sacchetti A, Lapre L, Lamfers M, Leenstra S, de Jonge H, Kros JM, Jansen EE, Struys EA, Jakobs C, Salomons GS, et al. IDH1 R132H decreases proliferation of glioma cell lines in vitro and in vivo. Ann Neurol. 2011; 69: 455-63. https://doi.org/10.1002/ana.22390.
46. Hu H, Wang Z, Liu Y, Zhang C, Li M, Zhang W, Wang K, Cai J, Cheng W, Huang H, Jiang T. Genome-wide transcriptional analyses of Chinese patients reveal cell migration is attenuated in IDH1-mutant glioblastomas. Cancer Lett. 2015; 357: 566-74. https://doi.org/10.1016/j.canlet.2014.12.018.
47. Paw I, Carpenter RC, Watabe K, Debinski W, Lo HW. Mechanisms regulating glioma invasion. Cancer Lett. 2015; 362: 1-7. https://doi.org/10.1016/j.canlet.2015.03.015.
48. Onishi M, Ichikawa T, Kurozumi K, Date I. Angiogenesis and invasion in glioma. Brain Tumor Pathol. 2011; 28: 13-24. https://doi.org/10.1007/s10014-010-0007-z.
49. Yin LM, Xu YD, Peng LL, Duan TT, Liu JY, Xu Z, Wang WQ, Guan N, Han XJ, Li HY, Pang Y, Wang Y, Chen Z, et al. transgelin-2 as a therapeutic target for asthmatic pulmonary resistance. Sci Transl Med. 2018; 10. https://doi.org/10.1126/scitranslmed.aam8604.
50. Nair RR, Solway J, Boyd DD. Expression cloning identifies transgelin (SM22) as a novel repressor of 92-kDa type IV collagenase (MMP-9) expression. J Biol Chem. 2006; 281: 26424-36. https://doi.org/10.1074/jbc.M602703200.
51. Lee EK, Han GY, Park HW, Song YJ, Kim CW. Transgelin Promotes Migration and Invasion of Cancer Stem Cells. Journal of Proteome Research. 2010; 9: 5108-17. https://doi.org/10.1021/pr100378z.
52. Zheng XW, Chen SY, Yang QT, Cai JX, Zhang WP, You HS, Xing JF, Dong YL. Salvianolic acid A reverses the paclitaxel resistance and inhibits the migration and invasion abilities of human breast cancer cells by inactivating transgelin 2. Cancer Biology & Therapy. 2015; 16: 1407-14. https://doi.org/10.1080/15384047.2015.1070990.
53. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nature Biotechnology. 2011; 29: 24-6. https://doi.org/10.1038/nbt.1754.
54. Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in Bioinformatics. 2013; 14: 178-92. https://doi.org/10.1093/bib/bbs017.
55. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43: e47. https://doi.org/10.1093/nar/gkv007.
56. Bassett EA, Palanichamy K, Pearson M, McElroy JP, Haque SJ, Bell EH, Chakravarti A. Calpastatin phosphorylation regulates radiation-induced calpain activity in glioblastoma. Oncotarget. 2018; 9: 14597-607. https://doi.org/10.18632/oncotarget.24523.
57. Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010; 11: 587. https://doi.org/10.1186/1471-2105-11-587.
58. Van Brocklyn JR, Wojton J, Meisen WH, Kellough DA, Ecsedy JA, Kaur B, Lehman NL. Aurora-A inhibition offers a novel therapy effective against intracranial glioblastoma. Cancer Res. 2014; 74: 5364-70. https://doi.org/10.1158/0008-5472.CAN-14-0386.
59. Jaime-Ramirez AC, Dmitrieva N, Yoo JY, Banasavadi-Siddegowda Y, Zhang J, Relation T, Bolyard C, Wojton J, Kaur B. Humanized chondroitinase ABC sensitizes glioblastoma cells to temozolomide. J Gene Med. 2017; 19. https://doi.org/10.1002/jgm.2942.