
INTRODUCTION
Cancer genomes are riddled with single base substitutions (SBSs), the predominant genetic alteration found in cancer cells [1–5]. Mutations in cancer genomes are not always randomly scattered, but can occur in discreet patterns. Large-scale sequencing efforts have uncovered patterns of spatially close and strand-coordinated mutations that likely occur at the same time [6–8]. Discovery of the molecular mechanisms facilitating accumulation of these SBSs, and selective expansion of cells carrying them, is clarifying the processes of accumulation of mutations that favor cell survival, proliferation, and invasiveness.
Knowledge of endogenous enzymatic sources of mutations has been elusive, but cancer genome and exome sequencing endeavors have exposed striking cancer-specific mutation signatures that suggested previously unconsidered candidates. The most pervasive of these signatures, observed in at least 16 of the most common forms of cancer [6], is defined as an abundance of C > T and/or C > G mutations at TC dinucleotides (mutated base underlined) [6–10]. Because of the specificity for TC motifs, this signature has been attributed to the APOBEC family of cytidine deaminases, enzymes that are unable to edit double-stranded DNA (dsDNA) and exhibit exquisite specificity for single-stranded DNA (ssDNA) substrates [11]. The frequency of SBSs reportedly due to APOBEC enzymes is positively correlated with expression level of APOBEC3A (A3A) and/or APOBEC3B (A3B) mRNA in cancerous cells and tissues [13–18]. Thus, A3A and A3B are the likely family members responsible for these signature mutations. Induction of dsDNA breaks has been shown to stimulate the production of APOBEC-mediated mutations [7, 12], and APOBEC TC context mutations often colocalize with rearrangement breakpoints in cancer cells. These mutations are also frequently found within known cancer driver genes [4, 10, 13, 14].
In spite of the evidence for APOBEC enzymatic activity in generating the TC motif hypermutation seen in cancer, previous analyses reveal that numerous tumors with high A3B mRNA expression exhibit very few mutations [13, 14], suggesting that APOBEC activation alone is not sufficient cause for the hypermutation profile. Another requirement is availability of APOBEC target ssDNA. ssDNA increases in cells as a result of transcription, replication, and recombination. When these processes encounter stress, abnormally long ssDNA regions accumulate and dsDNA breaks form, leading to production of ssDNAs during repair [19, 20]. For example, replication stress can cause uncoupling of polymerase and helicase activities due to an obstruction in the replicating DNA, which can lead to abnormally long stretches of ssDNA. This ssDNA accumulation recruits repair proteins to remove the obstruction. During this process the stalled forks are cleaved by endonucleases to produce dsDNA breaks and repaired by homologous recombination, which involves resection of dsDNA break ends by exonucleases, a process that creates ssDNA [19, 20]. Frequently, this type of damage activates one of the cell cycle checkpoints and arrests the cell cycle, providing time for faithful DNA repair or induction of cellular senescence [21]. If the damage is too extensive, cells instead undergo apoptosis [22].
We have recently described a source of endogenous DNA replication stress and dsDNA breaks that neither activates cell cycle checkpoints nor induces apoptosis; in other words, this damage is checkpoint “blind.” Through ongoing research on functions of common chromosome fragile site (CFS)-associated genes, we found that loss of Fhit protein expression causes a nucleotide imbalance, specifically a decrease in dTTP pools due to lack of sufficient thymidine kinase 1 (TK1) protein activity. The nucleotide imbalance leads to replication fork stalling, collapse, and dsDNA breaks [23, 24]. This checkpoint-blind replication stress leads to downstream global chromosomal instability [25]. Among the various types of genome instability observed in Fhit−/− mouse tissues and derived cell lines, we observed large increases in SBSs, including robust increases in C > T mutations (Paisie et al., in review). Alterations at the FRA3B CFS, centered within the FHIT gene locus, leads to partial or full loss of FHIT gene and protein expression in >50% of sporadic cancers [26–29], many of which exhibit APOBEC signature mutations [6, 14].
Because of the dependence of APOBEC enzymes for ssDNA substrates and the observation that many cancers with the APOBEC signature are Fhit negative, we hypothesized that Fhit protein loss creates an environment of ongoing DNA damage ripe for APOBEC hypermutation. To test this hypothesis we analyzed The Cancer Genome Atlas (TCGA) data for lung adenocarcinoma, tumors which exhibit frequent loss of Fhit expression and display the APOBEC signature, and determined that FHIT-low/APOBEC-high lung cancers exhibit the highest numbers of C > T and C > G mutations with the TC dinucleotide signature. In vitro Fhit-deficient/A3B-high cells displayed an increased frequency of C > T mutations in an amplified TP53 DNA fragment; inhibition of Fhit loss-induced replication stress and dsDNA breaks via thymidine supplementation decreased the mutation frequency below that of Fhit-normal/A3B-high cells. Fhit protein loss occurs in the earliest prenoplastic lesions of multiple types of cancer [26–29]. Thus, we have proposed that Fhit loss-induced replication stress, and resulting dsDNA break accumulation in preneoplasias, initiates the genome instability that can lead, through progressive waves of APOBEC-mediated mutation and clonal expansion, to sporadic cancers.
RESULTS
APOBEC mRNA overexpression shows correlation with increased mutations
Using publically available TCGA RNA expression and exome sequencing datasets, we first determined A3A and A3B mRNA expression levels in each tumor sample in order to then examine the correlation of A3B and A3A mRNA overexpression (A3B-, A3A-high) with SBS mutations in lung adenocarcinomas. Only A3B was overexpressed in this patient cohort and thus we continued our analysis with A3B only (Figure 1A, B). With the expression datasets, we stratified the tumor cohort, by mRNA expression levels of A3B, into three roughly equal groups and compared mutation loads among tumors with low, intermediate, and high APOBEC expression. Increasing A3B mRNA levels correlated with increased total mutation load per exome and an increase in C > A, C > G, and C > T mutations (Figure 1C, D). C > T mutations were the dominant SBS found in this tumor cohort (Figure 1D). C > A mutations, a signature of environmental exposure to tobacco carcinogens, are common in lung cancer DNAs [6]. Examination of the local sequence context for C > T transitions and C > G transversions in the samples showed that these SBSs are largely focused at nucleotides that match the preferred APOBEC target TC dinucleotide context (Figure 1E), suggesting that A3B is a major enzymatic source of mutation in this tumor cohort. However, a fraction of the lung cancers with high A3B mRNA had very few mutations per exome; 33% of A3B-high tumors exhibited fewer than 242 mutations per exome, the average mutation load per exome for A3B-low tumors. This suggests that APOBEC overexpression is not sufficient to induce hypermutation.
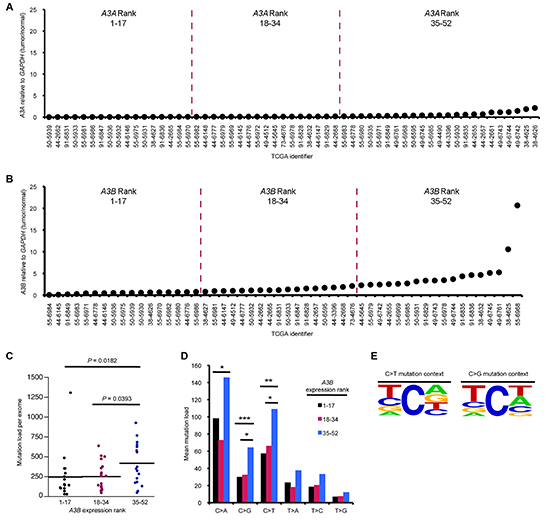
Figure 1: APOBEC overexpression correlates with hypermutation in lung adenocarcinoma. RNA-seq and DNA-seq mutation data sets for lung cancers were downloaded from TCGA. A3A (A) and A3B (B) mRNA levels (tumor/normal) in the indicated TCGA lung adenocarcinoma samples. Tumor DNAs were grouped into low, middle and upper thirds based on A3B mRNA expression, such that tumors 1–17 express the lowest levels of A3B mRNA while samples 35–52 express the highest levels. (C) Mutation load per exome in tumor DNAs with different A3B mRNA expression levels. (D) Mean mutation load for specific transition and transversion mutations in A3B groups. (E) 5’ & 3’-flanking nucleotides of C > T transitions and C > G transversions in lung adenocarcinoma DNAs. Frequency plots were generated using www.weblogo.berkeley.edu [35]. Font size is proportional to nucleotide frequency and nucleotides at the top of the column occur most frequently. P values in (C) and (D) are from Mann-Whitney U test. (*P < 0.05, **P < 0.02, ***P < 0.009).
FHIT-low/APOBEC-high tumors display the highest frequency of SBSs
Because a subset of APOBEC-high tumors was resistant to APOBEC-induced mutations, we hypothesized that these tumors may produce a low level of dsDNA breaks and ssDNA vs those enriched with mutations. Fhit loss has been shown to increase replication stress and DNA breaks in the types of cancers displaying APOBEC signature mutations, so we next determined if stratifying the same lung cancers by FHIT mRNA levels, in addition to APOBEC levels, might suggest an explanation for the lack of mutations in some APOBEC high tumors and enrichment of mutations in others. Tumors with FHIT expression level <0.5 (tumor/normal) were considered FHIT-low (Figure 2A), as it is known that loss of one FHIT allele causes haploinsufficiency for previously studied Fhit functions [30]. FHIT-low lung adenocarcinoma DNAs displayed a moderate, but not significant, increase in total mutation load per exome and mutations at individual nucleotides (Figure 2B, C). The individual nucleotide mutation pattern in FHIT-low tumors mimicked the increase in C > A, C > G, and C > T seen with A3B overexpression but to a more modest level (Figure 2C). We then divided the A3B-high tumors into two groups: A3B-high/FHIT-low and A3B-high/FHIT-normal. A3B-high tumors with low FHIT showed significant increases in total mutations per exome and in C > A and C > T mutations, as well as increased C > G mutations (Figure 3A, B). This suggests cooperation of low FHIT expression with A3B overexpression to allow pronounced hypermutation. Alone, both A3B overexpression and FHIT loss lead to increased mutation frequency. However, hypermutation by A3B is more significant in A3B-high/FHIT-low tumors than in tumors with A3B overexpression alone. When examining total mutation load per exome in FHIT-low cancers, increased A3B expression was associated with a significant increase in both total mutation load and C > A, C > G, and C > T mutations (Figure 3C, D). However, when examining lung cancers with normal FHIT expression level (FHIT-normal), this association was lost (Figure 3E, F); ie, A3B overepxression had no effect on total mutation load or individual point mutations in tumors with normal FHIT expression levels. This suggests that DNA in cells that have lost Fhit caretaker function is more sensitive to APOBEC-induced mutagenesis, while genomic DNA from FHIT positive cells are protected from APOBEC activity. Because a positive association between A3B-high/FHIT-low tumor status and mutation load was seen in the lung adenocarcinoma data (R = 0.5076, P = 0.00218), we also analyzed tumor DNAs from a heterogenous breast cancer TCGA cohort to determine the mutation load in A3B-high/FHIT-low tumor DNAs (not shown). Due to the mixed subtype nature of the breast tumor cohort, the clear trend toward increases in mutations due to the A3B-high/FHIT-low status did not reach significance. We believe that a study including more breast cancers stratified by specific tumor subtype will provide important information about association of A3B hypermutation not only with Fhit loss but also with specific clinical features. For example, HER2-overexpressing breast cancers have demonstrated enrichment for APOBEC mutagenesis [14] and both HER2 positive and Triple Negative breast cancer subtypes exhibit reduced Fhit protein expression in most tumors [31].
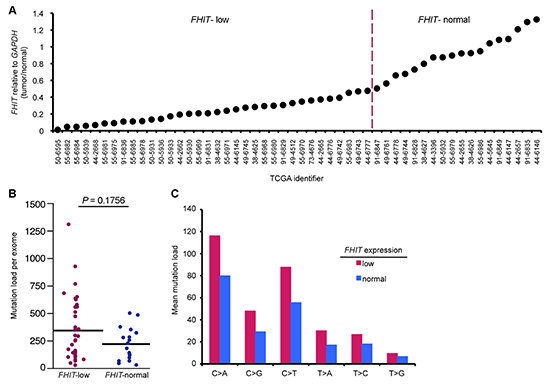
Figure 2: Low FHIT expression alone correlates with modest hypermutation in lung adenocarcinoma DNAs. (A) FHIT mRNA levels (tumor/normal) in the indicated TCGA lung adenocarcinoma samples. Tumor DNAs were grouped into FHIT-normal and FHIT-low lung tumor DNAs (>0.5 T/N-normal, <0.5 T/N-low). (B) Mutation load per exome in FHIT-normal and FHIT-low tumor DNAs. (C) Mean mutation load for specific point mutations in FHIT-normal and FHIT-low tumor DNAs. P values in (B) are from Mann-Whitney U test. (*P < 0.05, **P < 0.02, ***P < 0.009).
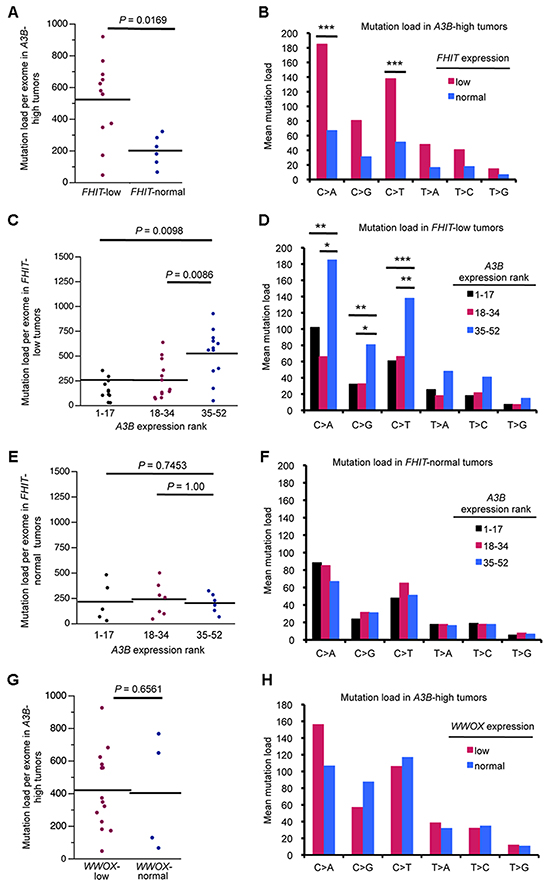
Figure 3: A3B-high/FHIT-low lung adenocarcinoma DNAs exhibit pronounced hypermutation. (A) Total mutation load per exome in FHIT-normal and FHIT-low lung tumor DNAs (>0.5 T/N-normal, <0.5 T/N-low) in A3B-high tumors. (B) Mean mutation load for specific point mutations in A3B-high lung tumor DNAs with normal and low FHIT mRNA expression. (C) Mutation load per exome in FHIT-low tumor DNAs stratified by A3B mRNA, grouped into lower, middle and upper thirds. (D) Mean mutation load for specific point mutations in FHIT-low tumor DNAs stratified by A3B mRNA. Mutation load per exome (E) and mean mutation load for specific point mutations (F) in FHIT-normal tumor DNAs stratified by A3B mRNA. (G) Total mutation load per exome in WWOX-normal and WWOX-low lung tumor DNAs (>0.5 T/N-normal, <0.5 T/N-low) in A3B-high tumors. (H) Mean mutation load for specific point mutations in A3B-high lung tumor DNAs with normal and low WWOX mRNA expression. P values in all panels are from Mann-Whitney U test. (*P < 0.05, **P < 0.02, ***P < 0.009).
Expression level of a second fragile gene, WWOX, does not correlate with APOBEC hypermutation in lung tumors
To differentiate the consequences of reduced gene expression of specific CFS-encoded genes, we repeated our lung cancer analysis, focusing on another tumor suppressor gene product, WWOX, expression of which is highly reduced in a large fraction of lung cancers. Although Wwox protein is a tumor suppressor, it has not been shown to have genome caretaker function, as is the case for Fhit. In the TCGA lung tumor cohort, tumors with low WWOX (<0.5 tumor/normal) expression displayed no increase in total mutations per exome or increased C > A, C > G, or C > T mutations indicative of APOBEC hypermutation, neither by stratification by WWOX alone (Supplementary Figure 1), nor by stratification by WWOX level in A3B-high tumors (Figure 3G, H). This indicated that WWOX expression level does not affect the ability of A3B to mutate genomic DNA. This suggests that loss of CFS gene products in general does not affect APOBEC activity but rather that loss of Fhit caretaker function is key to enhancing APOBEC mutagenesis.
Fhit loss-induced DNA damage and A3B overexpression cooperate to cause mutations in vitro
To validate the in silico correlations found in our TCGA analysis, we sought to determine if Fhit-low/A3B-high protein expressing cells accumulate C > T mutations in TP53 DNA more frequently than Fhit-expressing cells. Both Fhit loss and A3B overexpression have previously been associated with mutated p53 [13, 25]. For our in vitro model system we used a TREx-293 clone [13] that stably expresses a doxycycline-inducible A3B-GFP gene (Figure 4D), graciously given to us by the Reuben Harris lab. 293 cells normally express moderate levels of Fhit, so we silenced Fhit with siRNA to produce A3B-high/Fhit-normal cells and A3B-high/Fhit-low cells (Figure 4A, D). In this model, FHIT silencing led to a decrease in TK1 protein expression (Figure 4A); Fhit loss-induced down-modulation of TK1 expression causes nucleotide imbalance and subsequent replication stress [23]. FHIT silencing also led to an increase in γH2AX foci (Figure 4B, C), indicating increased dsDNA breaks in this model due to Fhit loss and subsequent TK1 reduction. Supplementation of Fhit-silenced cells with 10 μM thymidine, the substrate for TK1, which has previously been shown to rescue cells from Fhit loss-induced replication stress [23], decreased the mean fluorescence intensity of γH2AX (Figure 4B, C), indicating rescue from Fhit loss-induced DNA damage. We then performed nested PCR amplification for a 235 bpTP53 DNA fragment using DNA from cells with the following conditions of protein expression: A3B-low (uninduced)/Fhit-normal, A3B-high (induced)/Fhit-normal, and A3B-high (induced)/Fhit-low (silenced). The PCR products were then cloned into pcDNA3 plasmid and DNAs of individual clones were sequenced. Induction of A3B alone (A3B-high/Fhit-normal cells), caused an increase in the percent of clones displaying C > T mutations from 6.7% to 11.1% over A3B-low/Fhit-normal cells, with an average of 0.47 C > T mutations per kb (Figure 4E). A3B-high/Fhit-low clones showed the highest frequency of C > T mutations, with 17.8% of clones exhibiting these mutations (Figure 4D). This corresponded to an increase to 0.85 mutations per kb (Figure 4E). In order to confirm that, mechanistically, Fhit loss-induced dsDNA breaks were responsible for the increased incidence of C > T mutations, we supplemented A3B-high/Fhit-low cells with 10 μM thymidine in order to rescue the cells from Fhit loss-induced replication stress and DNA damage. Following thymidine supplementation, the percentage of A3B-high/Fhit-low clones with C > T mutations decreased to 8.9% (0.38 mutations per kb), less than the percentage for A3B-high/Fhit-normal clones (Figure 4E). These in vitro experiments confirm the correlations in the TCGA data and support the hypothesis that Fhit loss and A3B overexpression cooperate to cause hypermutation that could facilitate cancer initiation and/or progression.
DISCUSSION
Most cancers, whether familial or sporadic, exhibit genome instability, which underlies development of suppressor and oncogene mutations that drive progression to lethal cancer stages [1, 2]. The results of this study suggest a logical pathway to the nucleotide context-specific hypermutation genotype observed in many cancers. The hypothesis stems from several parallel findings: 1) that loss of the fragile FHIT gene product, Fhit, which occurs early in development of most human cancers, results in replication fork stress due to dTTP imbalance, production of dsDNA breaks and thus ssDNA during DNA repair, and extensive genome instability in all in vitro and in vivo models tested [23–25]; 2) that the activation/overexpression in cancers of A3B and/or A3A cytidine deaminase enzymes, known previously to be involved in C > T mutations in ssDNA substrates of viral genomes, could be the cause of the hypermutation observed in many of the most common cancers [6, 14]. Thus, we sought to demonstrate Fhit loss-induced DNA damage as a cooperating factor necessary to enhance A3A/B-induced hypermutation.
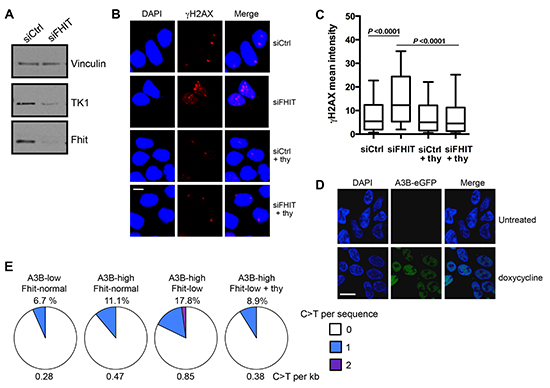
Figure 4: TP53 DNA amplified from A3B-high/Fhit-low TREx 293 cells exhibit an increased frequency of C > T mutations. (A) Western blot analysis of TK1 expression in siFHIT transfected TREx-293 cells. Representative blot of >3 independent experiments is shown. (B) Representative fields of TREx-293 cells imaged for γH2AX 2 days after siFHIT siRNA transfection (40x), bar 5 μm. (C) Box plots for mean fluorescence intensities of γH2AX for data pooled from three independent experiments. (D) Representative fields of cells imaged for A3B–eGFP, 3 days after induction with 1 μg/ml doxycycline (100x), bar 10μm. (E) Pie charts illustrating the frequency of C > T mutations per clone in designated conditions of expression. C > T mutations in TP53 detected by sequencing cloned, nested PCR products, 3 days after A3B induction and FHIT knock-down (n = 45 sequences per condition). C > T mutations occurring at GpC dinucleotides were excluded to avoid inclusion of mutations due to PCR error [37].
Analysis of TCGA datasets for lung adenocarcinoma revealed that the APOBEC signature was prevalent in this type of cancer, for both C > T and C > G mutations. A3B, but not A3A, was overexpressed in this cohort. Tumors with both low FHIT expression and A3B overexpression showed a significantly higher number of C > T and C > G mutations than those with A3B overexpression alone. Furthermore, in FHIT-normal tumor DNAs, A3B overexpression had no effect on the frequency of total mutations or C > T/C > G mutations, indicating that functional Fhit protein impedes APOBEC-induced hypermutation. We further showed in vitro, that silencing Fhit expression in A3B-overexpressing cells increased the occurrence of C > T mutations in the TP53 gene, validating the association between Fhit loss and A3B mutations in the TCGA datasets. The increase from 0.47 mutations/kb in A3B-high/Fhit-normal clones to 0.85/kb in A3B-high/Fhit-low clones equals 378 additional A3B-mediated mutations/Mb. This mutation frequency was decreased following thymidine supplementation, supporting the claim that it is Fhit loss-induced replication stress and subsequent dsDNA breaks that increase A3B-mediated mutagenesis. Interestingly, C > A mutations were also enriched in A3B-high/FHIT-low lung adenocarcinomas. FHIT gene deletions occur very frequently in response to tobacco carcinogens; thus, this mutation signature is likely a result of carcinogen exposure due to smoking.
These findings imply that loss of Fhit function is a limiting step for the formation of the APOBEC-mutational signature. Loss of Fhit caretaker function causes spontaneous replication stress [23–25], generating transient dsDNA breaks with each round of DNA replication and increased ssDNA levels during DNA repair of these breaks. Upon A3B overexpression, cytidine nucleotides within these areas of ongoing dsDNA repair would be deaminated by A3B, leading to increased genomic uracil. Removal of genomic uracil has been shown to produce clusters of C > T and/or C > G mutations after subsequent rounds of DNA replication and/or aberrant genomic uracil excision repair activities [32, 33]. Aberrant uracil excision repair occurs when base excision repair activities are ‘hijacked’ by the mismatch repair pathway [34], which requires more DNA resection than BER and consequently more exposed ssDNA to act as a template for A3B. This model for hypermutation due to synergism of Fhit loss and A3B overexpression is outlined in Figure 5, and is likely to apply to multiple types of cancers.
The discovery of cytidine deaminase enzymes such as A3A and/or A3B as a major source of mutation is ground-breaking not only in revealing a mechanism of somatic hypermutation in cancer genomes, but in revealing possible therapeutic targets for common forms of cancer, targets that spare normal cells because: 1) APOBEC enzymes are oftentimes over-expressed in cancer cells and not normal cells [13] and 2) they are non essential enzymes, so transitory or temporary inhibition of their activity may have little effect on normal cells [9]. In light of our findings, it will be useful to stratify cancer cohorts by both FHIT and A3B expression to more accurately predict which cancers are most susceptible to APOBEC-induced hypermutation. Further analysis of larger patient cohorts and more types of cancers, especially those with associated clinical features available for correlative studies, could tell us if APOBEC activity is associated with specific clinical features.
The current study shows that DNA damage/repair-generated APOBEC substrates are a prominent feature of Fhit loss and are major facilitators of A3B activity. Might there be other sources of damage-induced substrate DNAs during cancer development, such as mutated familial caretaker genes, oncogene mutation and possibly even cancer therapeutics? In response to cytotoxic and radiation treatment many cancer cells are eliminated, but some cells, including cancer stem cells, escape DNA damage-induced death and become resistant to further treatment. These resistant cells likely produce enhanced ssDNA levels due to DNA damage caused by the therapy. If this therapy-induced abundance of ssDNA occurs in cancers with activated A3B, A3B hypermutation may contribute to development of resistance to cancer therapy, a major hurdle to conquering this disease, highlighting the importance of clues to DNA mutation mechanisms and underscoring the importance of correlating APOBEC activity with synergizing factors and clinical features in order to better approach cancer prevention and treatment strategies.
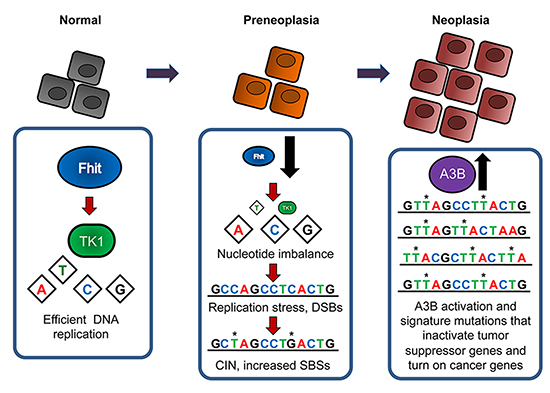
Figure 5: Fhit loss-induced DNA damage model for APOBEC-mediated hypermutation. In normal cells, Fhit protein expression positively regulates thymidine kinase 1 (TK1) expression, thus ensuring sufficient intracellular dTTP pool levels for efficient DNA replication. Deletions or other alterations in FHIT alleles occur due to FRA3B fragility causing loss of Fhit protein expression in early preneoplastic lesions. Subsequent decreases in TK1 and dTTP pools causes checkpoint-blind replication stress, resulting in DSBs. DSBs promote the acquisition of chromosomal instability (CIN), including single base substitutions (SBSs). When A3B becomes activated, the moderate level of SBS mutations in Fhit negative cells becomes exacerbated due to mutagenic effects of A3B on damaged DNA substrates, thus increasing the likelihood of mutations in tumor suppressor genes and/or oncogenes that are observed in neoplastic cells.
MATERIALS AND METHODS
Cell lines, mouse tissues and reagents
T-REx-293 clones expressing doxycycline-inducible A3B-eGFP were obtained from the Harris lab [13] and were cultured in DMEM with 10% FBS, 1% pen/strep, 50 μg/ml hygromycin, and 20 μg/ml blasticidin. A3B-eGFP was induced with 1 μg/ml doxycycline for 3 days.
Analysis of TCGA sequence data
Somatic mutations and RNA-seq expression data for lung and breast tumor and corresponding normal samples were downloaded from The Cancer Genome Atlas (TCGA) Data Matrix. Gene expression values were mined from RNA-seq data sets. APOBEC3A (A3A), APOBEC3B (A3B), FHIT, and WWOX expression values were normalized to the expression of GAPDH for each tumor and normal sample. Relative A3A, A3B, FHIT and WWOX expression levels were determined via comparison of tumor samples to corresponding normal samples (tumor/normal). Samples were divided into 2 groups for FHIT expression (and WWOX): low (<0.5) and normal (>0.5). Samples were also stratified based on A3B expression levels and divided into three roughly equal groups so that the group with the lowest assigned numbers express the lowest level of gene expression. Duplicate mutations were removed and the total number of mutations per exome was determined as well as the number of mutations at individual nucleotides. Mutation local trinucleotide context sequences for C > T transitions and C > G transversions were used to determine mutation context via http://www.weblogo.berkeley.edu and displayed as weblogo motifs [35].
PCR and clone sequencing
DNA was collected from T-REx-293 clones [13] expressing doxycycline-inducible A3B-eGFP, with and without FHIT silencing via siRNAs (ON-TARGET plus siRNAs, Thermo). Outer amplification for nested PCR for a 235 bp portion of TP53 was performed as follows: 95°C for 5 min followed by 30 cycles (95°C for 1 min, 60°C for 30 sec, 68°C for 30 sec). Inner PCR, with 1/25 of the previous reaction as DNA input, was performed using the following program: 87°C for 5 min followed by 30 cycles (87°C for 1 min, 60°C for 30 sec, 68°C for 30 sec). The PCR products were analyzed by gel electrophoresis with ethidium bromide, purified (Machery-Nagel), sticky-end cloned into pcDNA3 vector, sequenced with T7 primer (IDT), and aligned and analyzed with Finch TV software (Geospiza Inc.).
Western blot analysis
Cells were lysed with RIPA buffer (Thermo Scientific) supplemented with Halt Protease Cocktail Inhibitors (Thermo Scientific). Proteins were separated by SDS gel electrophoresis, transferred to nylon membranes and immunoblotted with antisera against Fhit [36], TK1 (Santa Cruz sc-134475), γH2AX (Millipore 05-636), and vinculin (Abcam ab18058).
Immunofluorescence
Cells were grown on glass coverslips in 6 well plates (Fisher), fixed with 4% paraformaldehyde, permeabilized with 0.1% triton X, and blocked in 1% BSA. Slides were washed and coverslips mounted using Fluoro-Gel II with Dapi (Electron Microscope Sciences). Images were acquired at room temperature with an Olympus FV1000 spectral confocal microscope, a UPLFLN 100X objective lens, NA 1.30, and with Olympus FLOWVIEW acquisition software.
Statistics
All p-values were determined from Mann Whitney U test.
ACKNOWLEDGEMENTS
The results shown here are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. We would like to thank Rueben S. Harris for his gracious donation of the A3B-eGFP inducible cells that were used for the in vitro experiments in this manuscript. This research was supported by: a Predoctoral fellowship from the OSUMC (to CEW), an F31 fellowship from NCI (to JCS), a T32 fellowship from NCI (to MSS) and grants from the NCI (CA120516 and CA154200 to KH).
REFERENCES
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674.
2. Loeb LA. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991; 51:3075–3079.
3. Nik-Zainal S, Alexandrov LB, Wedge DC, Loo PV, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, Menzies A, Martin S, Leung K, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012; 149:979–993.
4. Stephens P, Edkins S, Davies H, Greenman C, Cox C, Hunter C, Bignell G, Teague J, Smith R, Stevens C, O’Meara S, Parker A, Tarpey P, et al. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nat Genet. 2005; 37:590–592.
5. Drier Y, Lawrence MS, Carter SL, Stewart C, Gabriel SB, Lander ES, Meyerson M, Beroukhim R, Getz G. Somatic rearrangements across cancer reveal classes of samples with distinct patterns of DNA breakage and rearrangement-induced hypermutability. Genome Res. 2013; 23:228–235.
6. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale A, Boyault S, Burkhardt B, Butler AP, et al. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–421.
7. Taylor BJ, Nik-Zainal S, Wu YL, Stebbings LA, Raine K, Campbell PJ, Rada C, Stratton MR, Neuberger MS. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. Elife. 2013; 16:e00534.
8. Lada AG, Dhar A, Boissy R, Hirano M, Rubel A, Rogozin TB, Pavlov YI. AID/APOBEC cytosine deaminase induces genome-wide kataegis. Biol Direct. 2012; 7:47.
9. Smith HC, Bennett RP, Kizilyer A, McDougall WM, Prohaska KM. Functions and regulation of the APOBEC family of proteins. Semin Cell Dev Biol. 2012; 23:258–268.
10. Beale RC, Peterson-Mahrt SK, Watt IN, Harris RS, Rada C, Neuberger MS. Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: correlation with mutation spectra in vivo. J Mol Biol. 2012; 337:585–596.
11. Chelico L, Pham P, Goodman MF. Mechanisms of APOBEC3G-catalyzed processive deamination of deoxycytidine on single-stranded DNA. Nat Struct Mol Biol. 2009; 16:454–455, author reply 455–456.
12. Poltrovastsky V, Heacock M, Kissling GE, Prasad R, Wilson SH. Mutagenesis depdendent upon the combination of activation-induced deaminase expression and double-strand break. Mol Immunol. 2010; 48:164–170.
13. Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, Yee D, Temiz NA, Donohue DE, et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013; 494:366–370.
14. Roberts SA, Lawrence MS, Klimczak LJ, Grim SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, Harris S, Shah RR, Resnick MA, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013; 45:970–976.
15. Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet. 2013; 45:977–983.
16. Sasaki H, Suzuki A, Tatematsu T, Shitara M, Hikosaka Y, Okuda K, Moriyama S, Yano M, Fujii Y. APOBEC3B gene overexpression in non-small-cell lung cancer. Biomed Rep. 2014; 2:392–395.
17. Leonard B, Hart SN, Burns MB, Carpenter MA, Temiz NA, Rathore A, Vogel RI, Nikas JB, Law EK, Brown WL, Li Y, Zhang Y, Maurer MJ, et al. APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 2013; 73:222–231.
18. Nik-Zainal S, Wedge DC, Alexandrov LB, Petljak M, Butler AP, Bolli N, Davies HR, Knappskog S, Martin S, Papaemmanuli E, Ramakrishna M, Shlien A, Simonic I, et al. Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat Genet. 2014; 46:487–491.
19. Cimprich KA, Cortez D. ATR: essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008; 9:616–27.
20. Jazayeri A, Falck J, Lukas C, Bartek J, Smith G, Lucas J, Jackson SP. ATM-and cell cycle-dependent regulation of ATR in response to DNA double strand breaks. Nat Cell Biol. 2006; 8:37–45.
21. Di Fagagna FA. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008; 8:512–522.
22. Wang JYJ. DNA damage and apoptosis. Nature. 2011; 8:1047–1048.
23. Saldivar JC, Miuma S, Bene J, Hosseini SA, Shibata H, Sun J, Wheeler LJ, Matthews CK, Huebner K. Initiation of genome instability and preneoplastic process through loss of Fhit expression. PLoS Genet. 2012; 8:e1003077.
24. Saldivar JC, Shibata H, Huebner K. Pathology and biology associated with the fragile FHIT gene and gene product. J Cell Biochem. 2013; 109:858–865.
25. Miuma S, Saldivar JC, Karras JR, Waters CE, Paisie CA, Wang Y, Jin V, Sun J, Druck T, Zhang J, Huebner K. Fhit deficiency-induced global genome instability promotes selective mutation and clonal expansion. PLoS ONE. 2013; 8:e80730.
26. Sozzi G, Pastorino U, Moiraghi L, Tagliabue E, Pezzella F, Chirelli C, Tornielli S, Sard L, Huebner K, Pierotti MA, Croce CM, Pilotti S. Loss of FHIT function in lung cancer and preinvasive bronchial lesions. Cancer Res. 1998; 58:5032–5037.
27. Cirombella R, Montrone G, Stoppacciaro A, Giglio S, Volinia S, Graziano P, Huebner K, Vecchione A. Fhit loss in lung preneoplasia: relation to DNA damage response checkpoint activation. Cancer Lett. 2010; 291:230–236.
28. Guler G, Uner A, Guler N, Han SY, Illiopoulos D, McCue P, Huebner K. Concordant loss of fragile gene expression early in breast cancer development. Pathol Int. 2005; 55:471–478.
29. Terry G, Ho L, Londesborough P, Duggan C, Hanby A, Cuzick J. The expression of FHIT, PCNA, and EGFR in benign and malignant breast lesions. Br J Cancer. 2007; 96:110–117.
30. Fong LY, Fidanza V, Zanesi N, Lock LF, Siracusa LD, Mancini R, Siprashvili Z, Ottey M, Martin SE, Druck T, McCue PA, Croce CM, Huebner K. Muir-Torre-like syndrome in Fhit-deficient mice. Proc Natl Acad Sci USA. 2000; 97:4742–4747.
31. Guler G, Himmetoglu C, Jimenez RE, Geyer SM, Wang WP, Costinean S, Pilarski RT, Morrison C, Suren D, Liu J, Chen J, Kamal J, Shapiro CL, et al. Aberrant expression of DNA damage response proteins is associated with breast cancer subtype and clinical features. Breast Cancer Res Treat. 2011; 129:421–432.
32. Shinohara M, Io K, Shindo K, Matsui M, Sakamoto T, Tada K, Kobayashi M, Kadowaki N, Takaori-Kondo A. APOBEC3B can impair genomic stability by inducing base substitutions in genomic DNA in human cells. Sci Rep. 2012; 2:806.
33. Simonelli V, Narciso L, Dogliotti E, Fortini P. Base excision repair intermediates are mutagenic in mammalian cells. Nucleic Acids Res. 2005; 33:4404–4411.
34. Chen J, Miller BF, Furano AV. Repair of naturall occurring mismatches can induce mutations in flanking DNA. eLife. 2014; 3:eo2001. Doi: 10.7554/eLife.02001.
35. Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: A sequence logo generator. Genome Res. 2004; 14:1188–1190.
36. Druck T, Hadaczek P, Fu TB, Ohta M, Siprashvili Z, Baffa R, Negrini M, Kastury K, Veronese ML, Rosen D, Rothstein J, McCue P, Cotticelli MG, et al. Structure and expression of the human FHIT gene in normal and tumor cells. Cancer Res. 1997; 57:504–512.
37. Suspene R, Caval V, Henry M, Bouzidi MS, Wain-Hobson S, Vartanian JP. Erroneous indentification of APOBEC3-edited chromosomal DNA in cancer genomics. Br J Cancer. 2014; 110:2615–2622.