
INTRODUCTION
Lung cancer is the most frequently diagnosed cancer and the leading cause of cancer death worldwide, the most common cancer diagnosed in men with more than 1.5 million persons annually [1, 2]. These numbers are a stark reminder of the inadequacy of present preventative and therapeutic interventions for this disease epidemic. Prevention of lung cancer is of utmost importance because treatment of lung cancer remains woefully inadequate and represents a massive social and financial burden on healthcare systems costing over $12 billion in the United States in 2010 alone [3]. Because smoking cessation strategies have yet proven insufficient, development of effective and inexpensive pharmacological means to prevent and treat lung cancer worldwide is needed. Preventative agents with therapeutic activity would be of particular importance since present systemic therapies for lung cancer including chemotherapy, targeted signaling inhibitors and novel immunotherapies fail to cure the vast majority of patients, and are plagued with obstacles including toxicity, narrow spectrum of activity, de-novo or acquired resistance and inordinate costs [4–7].
Increased risk of carcinogenesis, genetic instability, resistance to therapy, and poor survival outcomes are strongly linked to loss of normal functions of p53, a stress-responsive tumor suppressor protein (encoded by the human TP53 gene) that normally halts cell cycling and activates DNA repair mechanism in response to DNA damage caused by cigarette smoke genotoxins. TP53 is mutated in 94%, of small cell lung cancer (SCLC) [8], 94% of squamous (SqCA) [9], and 54% of adenocarcinoma (AdCA) [10] subtypes of non-small cell lung cancer (NSCLC). The former two types of lung cancer, i.e. SCLC and SqCA are mainly treated with chemotherapy while subsets of adenocarcinoma that have targetable oncogenic driver mutations such as EGFR, ROS1, or ALK rearrangements are treated with Tyrosine Kinase Inhibitors(TKIs) [11, 12]. Because TP53 also normally activates apoptosis of cells with irreparable genotoxic injury, its loss allows the survival of genetically damaged cells. As these damaged cells accumulate, they develop increasing genetic instability that accelerates carcinogenesis [13, 14]. In response to genotoxic stress, p53 also activates stress-responsive xenobiotic detoxification and drug efflux mechanisms that protect cells from acute injury through metabolic detoxification and removal of genotoxins. Long-term exposure to genotoxic stress in persons addicted to cigarettes eventually overwhelms these defenses because rapidly proliferating cells with genomic instability can eventually acquire critical DNA lesions that permit pre-cancerous cells to cross the threshold of malignant transformation. Though genoprotective xenobiotic defenses exert beneficial effects by reducing exposure to genotoxins on normal or pre-malignant cells, their anti-apoptotic activity also permits survival and accumulation of pre-malignant cells that harbor increasing numbers of cancer promoting DNA lesions. After malignant transformation, the cancer-protective effects of xenobiotic defenses actually become cancer promoting since their anti-apoptotic activities promote survival of cancer stem cells and mediate drug-resistance through anti-apoptotic, drug-metabolism, and drug-efflux processes.
Glutathione (GSH, γ-glutamyl-cysteinyl-glycine) is a sulfhydryl-containing tripeptide utilized by the mercapturic acid pathway (MPy) of GSH-mediated metabolism of genotoxic electrophilic xenobiotics (E) or reactive mutagens generated upon mono-oxygenation of xenobiotics by cytochromes P450. Glutathione S-transferases (GST) catalyze the next reaction, conjugation of GSH with electrophilic genotoxins (E) to form glutathione-electrophile thioether conjugates (GS-E) that are removed from cells through active transport by the down-stream GS-E transporters (the next step of MPy), and subsequently metabolized and excreted by the kidney as mercapturic acids [15–17]. Many phenolic antioxidants have been shown to exert protective effects against carcinogenic chemical in numerous experimental models of cancer. These antioxidants exert their cancer preventative activities though free radical scavenging and through activation of endogenous biological antioxidant enzyme and suppression of pro-inflammatory/pro-oxidant pathways such as NFκB. The cancer-preventative effect of antioxidant on augmenting endogenous genoprotective defenses characteristically include increased GST expression and decreased p450 expression. Indeed, this characteristic feature has proven to an excellent screen for identifying cancer-preventative antioxidants [18]. Despite success in short-lived animal models, phenolic antioxidants have not yet proven to be effective in preventives of lung cancer in humans [19]. The dual role of GSTs in cancers, ameliorating carcinogenic mutations but protecting mutated cells from undergoing apoptosis may be a reason for failure of many antioxidants in lung cancer prevention trials. The contrasting effect of stress-defense mechanisms on normal vs. transformed cells underlies a fundamental problem with phytochemical candidates for lung cancer prevention because their genoprotective effects are mediated by inducing biological antioxidant mechanisms, particularly glutathione-linked enzymes of the mercapturic acid pathway (MPy). In support of this model, the expression of GSTs is increased in lung cancer portends poor survival and resistance to chemotherapy as well as radiotherapy. Furthermore, GST inhibitors are in development as anticancer agents to modify treatment resistance and shortened survival associated with high GST expression in cancer patients [20]. All antioxidants are also oxidant at higher concentrations [21, 22], thus at high concentrations they can suppress GSTs; this would serve a therapeutic function. At low concentrations, however, they could reduce oxidative damage though GST induction and be cancer preventative. Because early lung cancers have previously not been readily detectable at an early curable stage, the effectiveness of low-dose antioxidants in lung cancer prevention could be underestimated by inclusion of patients with preclinical malignancy.
The U.S Preventive Services Task Force recently approved Lung Cancer Screening with low-dose CT for patients aged 55 to 88 who had a smoking habit of 30 pack-years or had quit within the past 15 years. The advent of effective CT-based lung cancer screening has resulted in increasingly frequent identification of very small (2–4 mm) lesions for which biopsies are not feasible, thus it is unclear whether an antioxidant would be best used as a primary preventative (low-dose) or secondary preventative (high dose) [22–27]. Screening will also lead to detection of more preinvasive lesions like Squamous Dysplasia and atypical adenomatous hyperplasia. Antioxidants like 2HF may be useful in these selected types of patients to prevent progression to aggressive lung cancer.
We have previously shown that the phenolic antioxidant 2-hydroxyflavanone (2HF) displays anticancer activity in RCC in a VHL-dependent manner and is associated with marked suppression of GST expression [22, 25, 27]. These studies also showed that 2HF could also inhibit the catalytic activity of GST. Some of the most potent physiological inhibitors of GSTs are GS-E, which can inhibit other enzymes GSH-linked enzymes, inhibiting efflux of GS-E can globally impair GSH-linked antioxidant and anti-apoptotic defenses utilized by cancer cells for survival [15–17]. We previously showed that GS-E transport the Ral-binding protein-1 (a 76 kDa splice variant protein referred to as RLIP76, or simply Rlip, encoded by the human RALBP1 gene) is a high-capacity ATP-dependent transporter responsible for the bulk of GS-E removal. An existential necessity of Rlip for survival of cancer cells is suggested by studies showing that Rlip deficiency strongly prevents benzo[a] pyrene induced carcinogenesis [17]. The magnitude of this cancer preventative effect was evident in recently reported studies showing that Rlip haploinsufficiency is sufficient to prevent spontaneous carcinogenesis in TP53 null mice [28]. Depleting Rlip by antisense or siRNA, or inhibition by anti-Rlip antibodies causes regression, not simply a growth delay, of syngeneic melanoma, and allogeneic xenografts of human neuroblastoma, and lung as well as kidney, prostate, pancreas, and colon cancer [29]. Thus, Rlip targeting should also have a therapeutic role in established malignancy.
We studied the effects of 2HF in lung cancer cell lines and found that 2HF displays strong anticancer activity in both small cell and non-small cell histologies of lung cancer associated and present evidence of direct interaction of 2HF with Rlip.
RESULTS
Rlip is expressed human lung cancer
The expression of the Rlip analyzed showed remarkable differences amongst the human adenocarcinoma of lung tissue sections and adjacent normal tissue. Using immunohistochemistry (IHC), tissues affected with adenocarcinoma showed significantly strong Rlip expression (Figure 1A) in cancerous tissues. On the contrary, adjacent normal tissue showed basal Rlip expression. Quantification of Rlip expression (Figure 1B) in tissues using image pro, reveals a significant difference (p = 0.0386) in staining intensity of Rlip between cancerous tissue and the adjacent normal tissue. Thus, higher Rlip expression may be responsible for drug resistance in lung cancer patients.
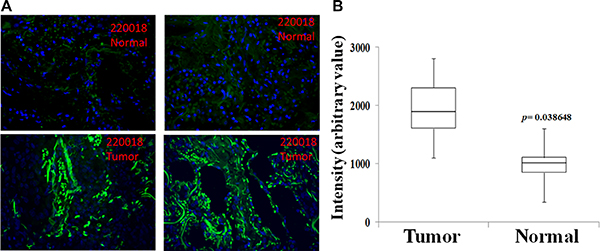
Figure 1: Immunohistochemistry analysis of Rlip protein expression in human adenocarcinoma of lung tissue sections. Unidentified lung cancer and adjacent normal lung tissue were analyzed by IHC using anti-Rlip antibodies and representative photomicrographs are shown (A). IHC quantification of Rlip expression in tissues was performed using image pro (B) (n = 8).
2HF inhibited growth and survival of lung cancer cell lines
Concentration dependent growth inhibitory effects of 2HF was also observed in the TP53 wild-type squamous cell histology H520 and H358 NSCLC cell line, and well as in the TP53 mutant H1417 and H1618 SCLC cell lines. The two SCLC cell lines were somewhat more sensitive to 2HF (average GI50 21 μM) than the NSCLC cell lines (average GI50 52 μM, p < 0.01) (Figure 2A). Clonogenic assays confirmed that 2HF inhibited lung cancer cell survival as shown for the H358 and H520 NSCLC cell lines (54 and 46% inhibition, respectively, p < 0.001). The immortalized non-malignant human lung bronchioepithelial cell line HLBEC grew significantly slower than the malignant cell lines as expected and its survival was not significantly affected by 2HF in clonogenic assays (Figure 2B). The GI50 of 2HF towards these lung cancer cell lines ranged from 20 to 80 μM, in a concentration range observed for many ‘generally regarded as safe’ natural compounds that are sufficiently non-toxic to achieve these concentrations in-vivo upon oral dosing, even in humans [23]. Importantly, 2HF inhibited the growth of ALK (anaplastic lymphoma kinase) gene-rearranged H3122 and H2228 adenocarcinoma NSCLC cell lines. This is significant because ALK rearranged adenocarcinomas are resistant to the commonly used EGFR kinase inhibitors. Furthermore, the H2228 cell line displays acquired resistance to ALK-inhibition, GI50 of 2HF being 84 ± 5 vs 62 ± 5 μM for H2228 and H3122, respectively (p < 0.01) (Figure 2C).
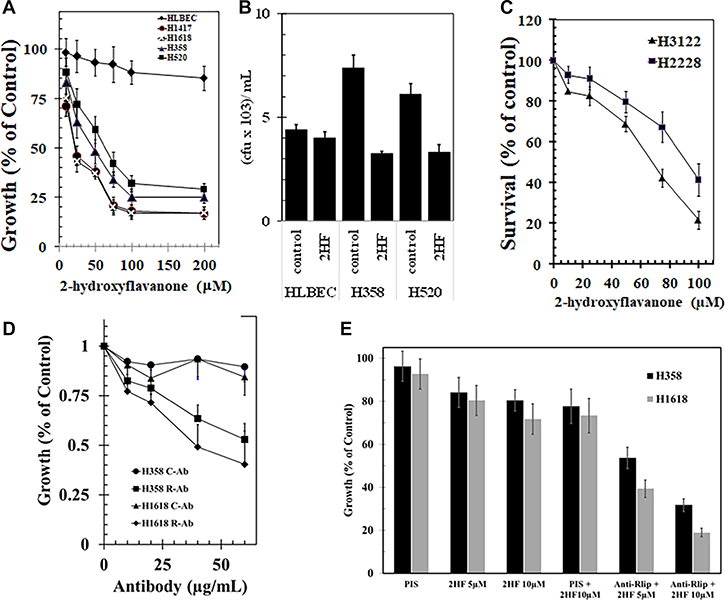
Figure 2: Anticancer activity of 2-hydroxyflavonone and effect of Rlip expression in lung cancer cell lines. We examined the differential toxicity of 2HF in non-malignant human lung bronchioepithelial cells (HLBEC) compared with the H1417 and H1618 SCLC and the H358 and H520 NSCLC cell lines (A). The cytotoxicity of 2HF on the H358 and H520 NSCLC cell lines was further confirmed by a colony-forming assay is shown (B). The growth inhibitory activity of 2HF toward ALK-rearrangement harboring H3122 and H2228 cell line (C). The growth inhibitory effect of polyclonal rabbit-anti-human anti-Rlip IgG was determined by MTT assays on H358 or H1618 lung cancer cells treated with varying antibody concentration (0–60 μg/mL) at 72 h after addition of antibody to cells in log-phase of growth (D). We performed the effects of anti-RLIP76 IgG and 2HF (5 and 10 μM conc.) and their combination on the survival of H1618 (SCLC) and H358 (NSCLC) cells using MTT assay (E). Control antibodies were the purified IgG fraction from pre-immune serum. MTT assays were performed according to our previously published standardized methods. Anti-Rlip and pre-immune IgG fractions were purified from rabbit sera provided by Alpha Diagnostics (San Antonio, TX).
2HF enhanced growth effect of Rlip inhibition
We have previously shown that anti-Rlip antibodies bind to a cell surface epitope of Rlip and exert potent and specific growth-inhibitory and pro-apoptotic effects against a wide variety of human SCLC and NSCLC cell lines in-vitro and in-vivo [30]. We reasoned that 2HF would potentiate the effects of these antibodies. Using MTT assays, we confirmed that the polyclonal anti-Rlip antibodies used in present studies exhibited similar growth inhibitory effects towards SCLC and NSCLC as previously observed (Figure 2D). Sub-GI50 concentrations of 2HF (5 and 10 μM) inhibited growth of H1618 SCLC by 21 ± 8% and 28 ± 7%, respectively and of H358 by 14 ± 5% and 19 ± 5%, respectively. At 10 μg/ml anti-RLIP76 IgG itself inhibited growth of H1618 by 20 ± 7% and H358 by 16 ± 5%, respectively. Together, 10 μg/mL anti-RLIP76 IgG and 5 or 10 μM 2HF exerted supra-additive growth inhibition (61 ± 4% and 81 ± 6% in H1618; 46 ± 3% & 68 ± 4% in H358) (Figure 2E).
2HF reduced protein level and inhibited the transport activity of Rlip
We found that cellular content of Rlip protein was significantly reduced by 25 μM 2HF. At 24 h after treatment with 25 μM of 2HF, the expression of RLIP76 was reduced to approximately half. This effect was manifested similarly in the H358 NSCLC and H1618 SCLC cell line, earlier than the appearance of significant apoptosis (Figure 3A). The anthracycline chemotherapy drug doxorubicin (DOX) is a model transported substrate (allocrite) for transport by purified recombinant Rlip reconstituted into lecithin: cholesterol artificial liposomes [31]. In this assay, purified Rlip protein from NSCLC has been shown to display higher specific activity for DOX transport than that purified from SCLC cell lines [30]. To determine whether 2HF inhibited Rlip transport activity, we measured DOX transport activity of Rlip protein purified from H358 and H1618 cell lines and reconstituted into artificial liposomes. Control liposomes prepared in the absence of Rlip protein displayed no uptake of 14C-DOX, whereas ATP-dependent uptake was clearly evident in liposomes reconstituted with Rlip protein from SCLC or NSCLC cell lines. Specific activity of transport (nmol/min/mg protein) was greater for Rlip from NSCLC than SCLC as previously observed [30]. At 10 μg/mL, anti-Rlip IgG inhibited transport whereas pre-immune IgG did not. At 20 μM, 2HF reduced DOX transport by 22 and 25% for NSCLC and SCLC, respectively. This concentration of 2HF synergistically enhanced the transport inhibition by anti-Rlip antibodies, to 93 and 95% by Rlip from NSCLC and SCLC respectively (Figure 3B and 3C).
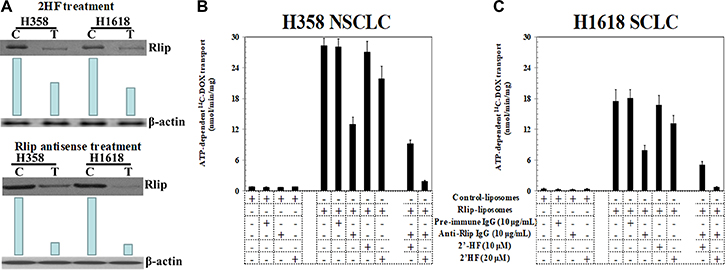
Figure 3: Effects of 2HF on expression and activity of Rlip. Western-blot analyses for RLIP76 were performed on control (C) as well as 25 μM of 2HF treated (T) H358 and H1618 lung cancer cells. Aliquots of 50 μg crude membrane fraction were loaded in SDS-PAGE, transblotted, and probed with anti-Rlip IgG, and developed bands were quantified by scanning densitometry. β-actin expression was used as loading control. Depletion of RLIP76 in lung cancer cells by 2HF was comparable to the results with RLIP76-antisense (10 μg/mL conc.) mediated knockdown of RLIP76 (A). Effects of anti-RLIP76 IgG (10 μg/mL conc.) and 2HF (10 and 20 μM conc.) and their combination on the transport of 14C-DOX in purified reconstituted vesicles prepared from H1618 and H358 cells were also performed by rapid-filtration technique using 250 ng protein per 30 μL reaction mixture (B and C).
Docking of 2HF at the DNP-SG binding site of Rlip
Rlip protein bound to the dinitrophenyl-S-glutathione (DNP-SG) affinity resin could specifically eluted by with 50 and 100 μM 2HF (Figure 4A). Specificity of elution was evident because the same major Rlip protein bands were observed in the 2HF eluate as in the DNP-SG eluate. Additionally, a marked increase in elution of Rlip protein by 2HF was observed in the presence of ATP, a finding identical to that with DNP-SG (Figure 4A). Elution pattern of eluted Rlip peptides resembles with our previous purifications, that they have been confirmed by mass to be fragments of Rlip that 38 kDA is DNP-SG ATPase (the former designation of Rlip) [32–34]. Because the concentrations of 2HF that effectively eluted Rlip by displacing DNP-SG are similar to those necessary for cytotoxicity in NSCLC cell lines, it was reasonable to conjecture that 2HF and DNP-SG shared a common binding site on Rlip. We explored this possibility by defining the DNP-SG binding site of Rlip through site-directed mutagenesis. Point mutants of Rlip were created at residues Y231, R232, S234, A264, S265, K268, I344, V345 and the allocrite-stimulated ATPase activity was determined (Table 1).
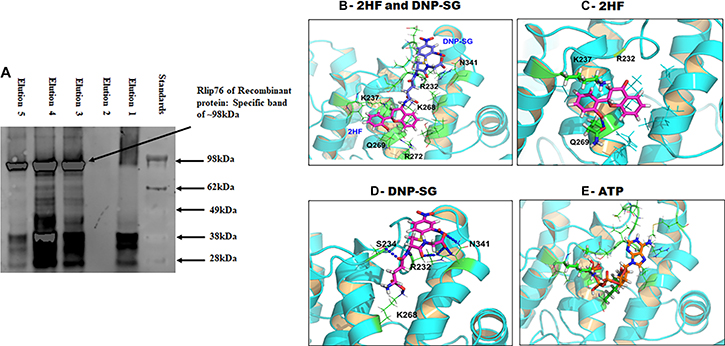
Figure 4: Docking of 2HF, DNPSG and ATP to RLIP76. (A) Binding interaction of RLIP with 2HF was demonstrated by elution of recombinant RLIP with elution buffer containing 2HF. (B) shows binding modes of 2HF and DNP-SG at the R232 site. (C) 2HF forms a hydrogen bond with Q269 and cation-π interaction with K237. (D) DNP-SG forms hydrogen bond/salt bridge network with R232/S234/K268/N341. (E) ATP also prefers binding at the ATP-binding pocket of Rlip forming 5 H-Bonds to G235, K237, S265, K268 and R272. Details are given in the Materials and Methods section.
Table 1: ATPase activity of Rlip and its mutants*
Protein |
specific activity (nmol/min/mg) |
|
|---|---|---|
DOX |
DNP-SG |
|
RLIP76 |
275 ± 7 |
220 ± 15 |
M1 |
145 ± 1 |
95 ± 11 |
M2 |
105 ± 20 |
75 ± 4 |
M3 |
150 ± 1 |
175 ± 4 |
M4 |
205 ± 16 |
60 ± 7 |
*Mutants designated as M1 through M4 were mutated at the following residues: M1 (Y231A;R232A;V233K,S234A); M2 (A264R;S265A;L267A;K268A); M3 (Q340A;N341A); M4 (I344A;V345A)
We used three-dimensional NMR structure of the human RLIP76 RhoGAP-Ral binding domain with 265 amino-acid residues (RCSB protein databank PDB ID 2 mbg [35]) as model to dock the small molecules by using our in-house developed All-Around Docking (AAD) method [36] which can predict the best docking site of a ligand, together with the induce fit docking (IFD) method that allows side-chain flexible for model refinement (Schrödinger, New York, NY). The three-dimensional structural docking model revealed that 2HF and DNP-SG can both bind to the R232 site in similar proximity (Figure 4B). 2HF may form one hydrogen bond (HB) with Q269 and cation-π interaction with K237 (Figure 4C), and DNP-SG forms HBs/salt-bridge with R232, S234, K268 and N341 (Figure 4D). The ATP molecule is also predicted to bind this site (Figure 4E), forming HB network with G235, K237, S265, K268 and R272.
CDNB co-treatment can reduced DOX efflux and GI50 of 2HF
We have shown that 2HF inhibits Rlip expression, Rlip transport activity and DOX efflux in cancer cells. CDNB (1-chloro-2,4-dinitrobenzene) is an alkylating agent previously used for treatment of skin metastases of melanoma through intralesional injection [37]. It is used as a model substrate for GST, which conjugates it with GSH to form S-(2,4-dinitrophenyl)-glutathione (DNP-SG) ex-vivo [38, 39] and is used to measure GST activity. Furthermore, conjugation of GSH in cells with CDNB causes over 98% depletion of GSH in cancer cell lines as well as in-vivo lymphocytes (GSH- measurement method; sulfasalazine). GS-E are known to be highly effective competitive inhibitors of transport of other allocrites by Rlip [40]. Therefore, we conjectured that DNP-SG formed in cancer cells would increase Rlip-inhibitory activity of 2HF and synergistically potentiate its anticancer activity. Consistent with this assertion, CDNB lowered the GI50 of 2HF significantly (p < 0.05) in H358 cells (Table 2). Furthermore, the time-dependent efflux of DOX was impaired significantly by CDNB as well as 2HF. The half life of DOX removal from cells was determined 12 min, 16 min and 18 min for 1 μM DOX, 1 μM DOX+50 μM 2HF and 1 μM DOX+50 μM 2HF+20 μM CDNB, respectively (Figure 5).
Table 2: GI50 of CDNB, 2HF and 2HF in presence of CDNB on H358 lung cancer cells
H358 NSCLC |
|
|---|---|
Substrate |
GI50 (μM) |
CDNB |
2.83 ± 0.148 |
2HF |
25.44 ± 0.671 |
2HF in presence of 1 μM CDNB |
8.25 ± 0.786 |
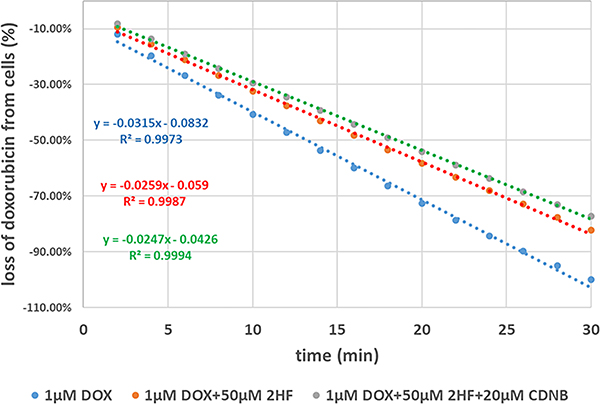
Figure 5: Effects of 2HF and/or CDNB on DOX efflux. DOX efflux studies were performed in presence and absence of 2HF and CDNB as described in Materials and Methods section. Loss of DOX was calculated and rate constant was determine to demonstrate inhibition of DOX efflux in H358 cells.
2HF reduced expression of genes responsive to Rlip depletion
We have previously observed [41] that Rlip deficiency causes differential expression changes in other GSH-linked stress defense enzymes. 2HF inhibited the transport activity of Rlip and reduced its expression at the protein and mRNA level. Therefore, we determine the effects of 2HF (50 and 100 μM) on adenocarcinoma (A549-p53 WT) and squamous cells (H226-p53MUT), on expression of several key MPy mRNA (Figure 6). Based on the effects of 2HF on mercapturic acid pathway gene mRNA expression in cell lines, we identified a set of seven genes that predicted opposite survival effects in adeno and squamous carcinoma: worsened in adeno and improved in squamous. This gene set predicted survival better than p53 status.
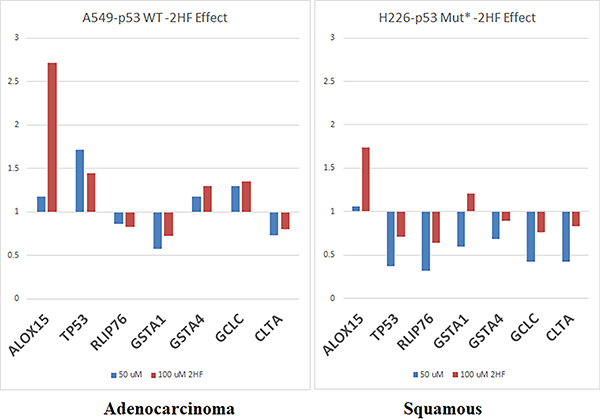
Figure 6: Effect of 2HF on selected MPy gene expression. Expression level of MPy genes was determined by qPCR. A549 and H226 cells grown in 100 mM tissue culture Petri dishes were treated with vehicle, 50 μM or 100 μM 2HF for 24 h. Total RNA and then cDNA was prepared from harvested cells using standard methods described in Materials and Methods section. qPCR was performed using gene-specific primers.
2HF inhibited NSCLC and SCLC xenografts in nude mice
H1618 and H358 lung cancer cells bearing animals with established s.c. implanted tumors (~20 mm2) were treated with 2HF (7 mg/kg b.w.) in corn oil by oral gavage on alternate days. At this dose, 2HF was well tolerated without any overt toxicity or weight loss (Figure 7A). The product of bi-dimensional tumor measurements revealed significant growth inhibition of H358 and H1618 cell lines (Figure 7B). Excised tumors from 2HF treated mice were visibly smaller and tumor weight was significantly lower (Figure 7C and 7D). At day 60, the tumor cross-sectional area and tumor-weight of mice bearing H1618 and H358 cells was significantly lower in 2HF treated group as compared to the vehicle only (corn-oil) treated group (H1618: area 47.4 ± 6.2 mm2 vs. 129 ± 11 mm2 and weight 0.71 ± 0.1 g vs. 2.01 ± 0.23 g, p < 0.001; H358: area 53.5 ± 6.8 mm2 vs. 137 ± 11 mm2 and weight 0.92 ± 0.12 g vs. 2.32 ± 0.21 g, p < 0.001). Euthanasia was performed at 24 h after the last dose of 2HF and steady-state 2HF levels in the serum by LC-MS were found to be 5.2 and 3.8 μM in the H1618 and H358 mice, respectively. 2HF metabolites were not measured. Taken together, these results showed that orally administered 2HF is systemically absorbed and exhibits antitumor activity against an SCLC and an NSCLC human cell line in a mouse xenograft model without overt toxicity.
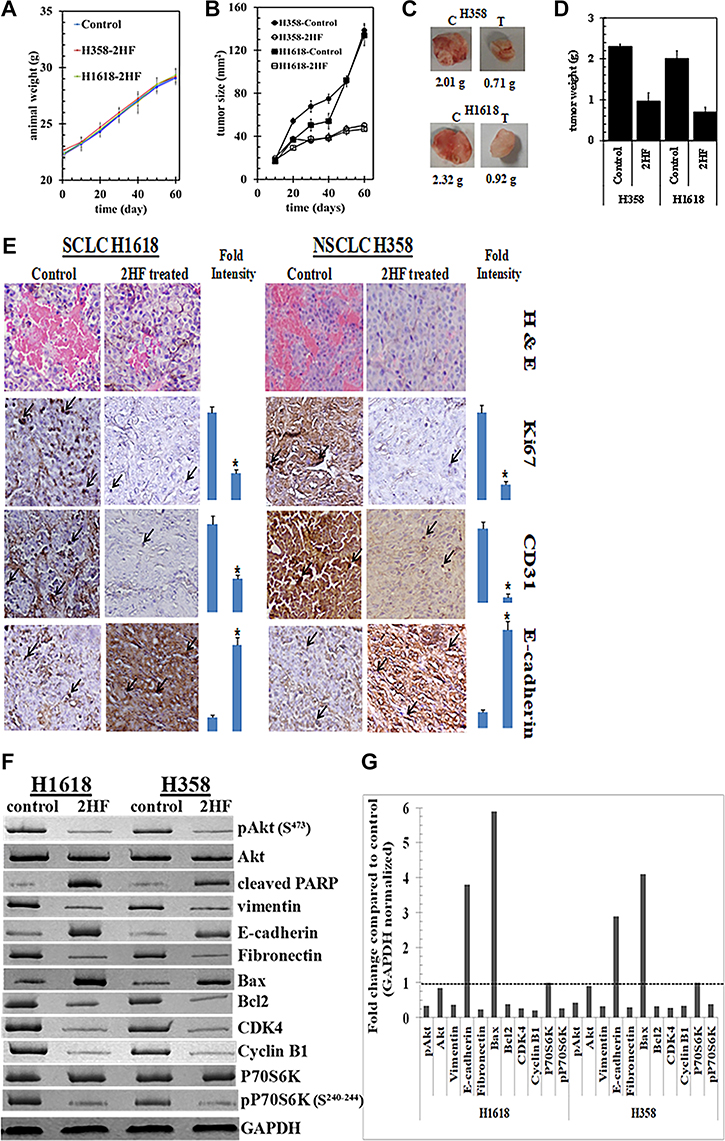
Figure 7: Anticancer activity of 2HF towards SCLC and NSCLC. Hsd: Athymic nude nu/nu mice were obtained from Harlan, Indianapolis, IN. Ten 10-weeks-old mice were divided into two groups of 5 animals (treated with corn oil (vehicle), and 2HF compound 7 mg/kg b.w.). All animals were injected with 1 × 106 H1618 cells suspensions in 100 μL of PBS, subcutaneously into one flank of each mouse. Treatment was started 10 days after the implantation to see palpable tumor growth. Treatment consisted of 0.2 mg of 2HF/mice in 200 μl corn oil by oral gavage alternate day. Control groups were treated with 200 μL corn oil by oral gavage alternate day. Experimental details are given in the Materials and Methods section. Animals were examined daily for signs of tumor growth and body weights were recorded. Animal weights (A) and product of bi-dimensional tumor measurements (B), photographs of dissected tumors (C) and tumor weights (D) are shown. Control and 2HF treated lung cancer bearing nude mice tumor sections were used for histopathologic analyses. Presented are H&E stained sections, IHC analyses for Ki67 expression (marker of cellular proliferation), CD31 (angiogenesis marker), and E-cadherin (tumor suppressor) from tumors in mice of control and 2HF-treated groups. Statistical significance of difference was determined by two-tailed Student’s t test. p < 0.001, 2HF-treated compared with control. Immuno-reactivity is evident as a dark brown stain, whereas non-reactive areas display only the background color. Sections were counterstained with Hematoxylin (blue). Photomicrographs at 40x magnification were acquired using Olympus DP 72 microscope. Percent staining was determined by measuring positive immuno-reactivity per unit area. Arrows represent the area for positive staining for an antigen. The intensity of antigen staining was quantified by digital image analysis using Pro Plus software. Bars represent mean + S.E. (n = 5); *p < 0.001 compared with control (E). Western-blot analyses of signaling proteins in H1618 and H358 human lung cancer tumor tissue lysates in control and 2HF treated experimental groups. GAPDH was used as a loading control (F). The bar diagrams represent the fold change in the levels of proteins as compared to controls as determined by scanning densitometry. Dotted line represents no significant change as observed with control (G).
2HF treated tumors exhibited broad anticancer signaling effects in tumors
Immunohistochemical examination of paraffin-embedded tumor xenograft sections revealed that 2HF treatment was associated with decreased expression of the proliferation marker Ki67 and the angiogenesis marker CD31. E-Cadherin, a protein that inversely correlates with invasion and growth of many epithelial cancers, was increased (Figure 7E). Western-blot analyses of the tumor tissue lysates from 2HF treated mice revealed reduction in cell growth promoting signals such as AKT and p70S6K, proliferation signals such as CDK4 and cyclin B1. Increased apoptosis was evident from changes in BH3 domain apoptosis regulatory proteins, with the anti-apoptotic Bcl-2 being reduced and pro-apoptotic Bax being increased. Epithelial-mesenchymal transition signaling was also reduced as indicated by reduction in vimentin and fibronectin and increase in E-cadherin. Thus, we conclude that 2HF induces apoptosis, inhibits cell cycling and growth signaling by modulating the expressions of BCL2 (B-cell lymphoma 2), BAX (BCL2 Associated X, Apoptosis Regulator), CDK4 (Ser/Thr-kinase component of cyclin D), cyclin B1, PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha), AKT (Protein kinase B) and p70S6K (phosphoprotein 70 ribosomal protein S6 kinase) in lung tumor.
DISCUSSION
In previous studies, we found that the anticancer activity of 2′-hydroxyflavanone (2HF), a citrus flavonoid, against renal carcinogenesis is associated with the inhibition of glutathione S-transferases (GSTs), the first step enzyme of the mercapturic acid pathway (MPy). Since the MPy enzymes are upregulated in lung cancer, we evaluated the anti-carcinogenic activity of 2HF in SCLC and NSCLC cell lines. We showed by immunohistochemistry that Rlip protein is present in human lung cancer and its level are increased in lung cancer cells compared with surrounding normal tissue. Inhibition of epithelial-mesenchymal transition was indicated by increased E-cadherin and reduced fibronectin and vimentin. 2HF reduced mercapturic acid pathway activity by reducing the expression as well as transport activity of the Rlip. Inhibition of doxorubicin efflux by 2HF increased its accumulation in the cells, but 2HF did not add to the transport inhibitory effect of anti-Rlip antibodies alone. Binding of Rlip to 2HF was evident from successful purification of Rlip by 2HF affinity chromatography. Consistent with increased drug accumulation, combined treatment with 1-chloro-2,4-dinitrobenzene, which is converted to a glutathione conjugate in cells, reduced the GI50 of 2HF by an order of magnitude. We have previously shown that Rlip is a major determinant of the inherent chemoresistance of NSCLC compared with SCLC and results of present studies showing that 2HF potentiated the apoptotic effect of anti-RLIP76 antibodies in SCLC as well as NSCLC in cell culture support our previous results. Orally administered 2HF inhibited growth of xenografts of SCLC and NSCLC cell lines in nu/nu mice. The anticancer activity of 2HF in-vitro was preferentially directed towards malignant cell lines and was confirmed in these in-vivo xenograft studies where we found excellent tolerability and lack of significant toxicity of orally administered 2HF.
Our previously published studies have consistently shown greater activity of Rlip-targeting in SCLC as compared with NSCLC [42]. Mimicking human disease, SCLC cell lines are generally more sensitive to many clinically available chemotherapy drugs (doxorubicin, vincristine, cyclophosphamide and others), than NSCLC [42–44]. Chemosensitivity of SCLC results in a rapid response, but relapse with refractory cancer occurs in the vast majority due to residual resistant populations of treatment-resistant cancer stem-cells [45]. SCLC cell lines are known to have frequent loss of p53 in SCLC [46] and p53 deficiency is known to cause promoter demethylation of cancer stem cell genes [47]. Taken together with our recent studies showing that Rlip depletion in Tp53–/– mice nearly completely prevents the hypomethylation of the promoters of many stem cell genes [28], suggest that 2HF could overcome cancer stem-cell mediated therapy resistance of p53-mutated SCLC. Treatment resistant stem cell have also been demonstrated in lung SqCA [48], thus effects of 2HF on cancer stem-cell gene could translate into clinical activity in this subtype of NSCLC. Consistent with our previous studies in multiple histologies of cancer [22, 23, 30, 49–52], we found that Rlip targeting exerts anticancer effects regardless of the presence of normal p53. This is relevant to lung AdCA, nearly half of which lack genetic lesions in p53.
We have previously shown that 2HF can inhibit expression as well as the catalytic activity of GST [26, 53], the first committed step of the MPy. Present studies show that 2HF also inhibits both expression and transport activity of Rlip, the next and rate-limiting step of the Mpy. Synergy between 2HF and anti-Rlip antibodies in their anti-cancer activities supports the importance of Rlip and the MPy in the mechanisms of action of 2HF. Regulation of redox-regulating genes by Rlip, and the regulation of Rlip by transcription factors involved in oxidative stress response suggests a possible mechanism for the anticancer activity of 2HF because Rlip inhibition would have a broad impact on the generation of pro-apoptotic oxidative metabolites contributing to anticancer activity of 2HF [54–56].
Similar to several other plant phenols [57], the anticancer activity of 2HF seem to be associated with pleiotropic effects on multiple cancer signaling pathways that regulate cell growth and proliferation, epithelial-mesenchymal transition (EMT), apoptosis, and angiogenesis. The cellular concentration of E-cadherin was increased by 2HF exposure, which was paralleled by a decrease in vimentin and fibronectin, paradigmatic for inhibition of EMT [58]. E-cadherin is considered a suppressor of invasion and growth of many epithelial cancers because of its role in inhibition of EMT and promoting normal epithelial phenotype [59]. EMT is intimately linked to a higher availability of growth factors, such as insulin like growth factor (IGF) [60], membrane metalloproteases (MMPs) [61], and an increase in vascularity [62]. This is paralleled by a repression of certain transcription factors [63] and cytoskeletal proteins [64] and finally by the development of matrix independent growth and corresponding tumor cell progression and metastasis formation [65]. In correlative studies the occurrence of EMT has been associated with a more aggressive phenotype, the resistance to chemotherapy and biologic agents and a poor prognosis [66]. In addition, we demonstrated a striking reduction of fibronectin. Fibronectin, which is assembled in the extracellular matrix, is involved in several biologic functions, including in cell adhesion, growth, migration, and differentiation. It is increased in NSCLC subtypes and has been linked to cross pathway activation and radiation-resistance [67]. Since EMT signaling plays a key role in maintenance of cancer stem cells, our findings predict that 2HF can be useful in overcoming the treatment resistance conferred by the persistence of highly resistant cancer stem-cells.
Bax and Bcl-2 are both key members of the Bcl-2 family of proteins that are functionally linked with mitochondria and intimately involved in the late signaling phase of intrinsic apoptotic cell death [68]. Increased Bcl-2 and reduced Bax have been linked to the malignant phenotype, and correlated with survival advantage [69]; in normal bronchioepithelial cells, the opposite is true. Early studies of the Bcl-2 family have suggested that p53 loss up-regulates Bcl-2 and is associated with the loss of Bax transactivation and Bax has been shown to regulate apoptosis in lung cancer patients [70]. Chemotherapy resistance of p53-mutated tumors has been attributed to the loss of transactivation of anti-apoptotic Bax and more recently increased levels of Bcl-2 have been linked to a poor outcome in SCLC [71]. Both cell lines we investigated carry genetically altered p53, H358 having a homozygous deletion of the p53 gene [72] and H1618 having a p53 exon 248 mutation [73]. When compared to untreated animals, 2HF demonstrate the down-regulation of anti-apoptotic Bcl-2 which is paralleled by an increased in pro-apoptotic Bax.
The PI3K/Akt/mTOR pathway governs the growth of cancer cells and has been the subject of intense interest in cancer therapy. Phosphorylated Akt has been described to be a negative prognosticator in lung cancer [74] and an increased level can be observed early in lung cancer carcinogenesis [75] and in therapeutic resistance [76–78]. In addition, activation of the pP70S6 Kinase is as an important distal event in the intracellular signal transduction of several growth factor pathways, including the insulin like growth factor pathway. It should be noted that Rlip targeting alone blocks the same signaling pathway in other cancers [79], and perhaps more remarkably, that this pathway is actually activated by Rlip depletion in non-malignant cells [80]. The fundamentally opposite effect of Rlip on this pathway may underlie low toxicity towards non-malignant cells. Additionally, greater expression of Rlip in cancer compared with normal tissue suggest a greater dependence of cancer cells on Rlip and could also contribute to improved selectivity towards lung cancer cells.
In addition to inhibiting the PIK3CA/AKT/p70S6K pathway that promotes the growth of cells, we found the cell cycling and proliferation were also affected, as evidenced by reduction in CCNB1 and CDK4 protein. Both proteins are intimately related to cell cycle progression and an increase in their levels has been linked to an adverse prognosis in lung cancer [81, 82]. Cyclin B1 in conjunction with cdc2 (Cdk1) forms a complex called the ‘mitotic promoting factor’ (MPF), which crucial for G2/M transition [83]. We and others have previously shown that Rlip also interacts directly with cdc2 (CDK1) [84]. Reductions of cyclin B-CDK1 complex levels are related to cell cycle arrest at G2 and finally DNA fragmentation [85]. It is felt that this is one of the major molecular processes accounting for the anti-neoplastic effect of p53 associated cisplatin activity. Intriguingly, this mechanism is different from the DOX-induced apoptosis in SqCA, which is associated with heightened levels of cyclin B1 in the process of mitotic catastrophe. In addition to its value as a negative prognostic marker, CDK4 has been more recently targeted in other malignancies with clinical success [82, 85].
MATERIALS AND METHODS
Reagents
MTT and 2HF were obtained from sigma (St Louis, MO). CD31, Ki67, cyclin B1, CDK4, cleaved PARP, vimentin, Akt, pAKT (S473), fibronectin, Bax, Bcl2, P70S6K, pP70S6K (S240–244), PI3K, and E-cadherin antibodies were purchased from Santa Cruz Biotechnology (Columbus, OH) and Cell Signaling Technologies (Danvers, MA). 14C-DOX (doxorubicin: specific activity 58 mCi/mmol) was purchased from NEN Life Sciences (Boston, MA). Matrigel was procured from BD Biosciences (San Jose, CA). TUNEL fluorescence and avidin/biotin complex (ABC) detection kits were purchased from Promega (Madison, WI) and Vector (Burlingame, CA), respectively. Ninety-six well nitrocellulose membrane plates (pore size 0.45 μm) used in transport studies were purchased from the Millipore Co. (Bedford, MA). The source of anti-RLIP76 antibodies and DNP-SG were the same as previously described [32]. All other reagents were of analytical grade.
Cell lines and cultures
Human SCLC (H1417 and H1618) and NSCLC (H358 and H520) were purchased from ATCC, Manassas, VA. Human lung bronchoepithelial (HLBE) cells were a generous gift from Dr. John D. Minna, University of Texas Southwestern Medical Center, Dallas, TX. All cells were cultured at 37°C in a humidified atmosphere of 5% CO2 in RPMI-1640 medium supplemented with 10% FBS and 1% P/S solution except HLBEC, which grow in K-SFM. The cells were immediately expanded and frozen after being obtained from ATCC and restarted every 3 to 4 months from a frozen vial of the same batch of cells and no additional authentication was done on these cells. All cells lines were free of Mycoplasma infection tested by PCR.
RLIP76 expression in lung cancer
Immunohistochemistry of human adenocarcinoma of lung tissue sections was performed on 5-μm thick serial sections prepared from formalin-fixed paraffin-embedded tissue, using monoclonal mouse anti-RalBP1 IgG from Santa Cruz (sc-48337). Tissue sections were de-paraffinized in xylene and rehydrated in graded alcohol. This was followed by antigen retrieval (5 mL antigen un-masking reagent + 1 mL EDTA (0.5 M) in 500 mL water) by steaming. The slides were placed in incubation chamber and blocked with 1% human serum albumin at room temperature for 1 h. The anti-Ralbp1 IgG (from Santa Cruz) was added (1:25 dilution) and incubated at 4°C overnight and the slides were washed in 1x PBS+ 0.2% Triton X-100 by incubating for 5 min at shaker with gentle shaking. Slides were stained using the ABC universal kit (Vector) following manufactures instructions, counterstained with hematoxylin, and mounted. All images were acquired at a fixed distance, using the same channel settings (exposure times, laser intensities) using Olympus BH2 microscope at Anatomical Pathology Lab at the City of Hope. All human subject studies were approved by the COH (IRB#11175) and carried out in accordance with their guidelines.
Drug sensitivity (MTT) assay
Cell density measurements were performed using a hemacytometer to count intact cells resistant to staining with trypan blue. Approximately 20,000 cells were plated into each well of 96-well flat-bottomed micro-titer plates. After 12 h incubation at 37°C, medium containing either pre-immune IgG or anti-RLIP76 IgG (ranging 0–60 μg/mL final conc.) were added to cells followed by incubation for 24 h, then 2HF (ranging 0–200 μM) were added to cells. After 48 h incubation, 20 μL of 5 mg/mL MTT were introduced to each well and incubated for 2 h of exposure. The plates were centrifuged and the medium was decanted. Cells were dissolved in 100 μL DMSO with gentle shaking for 2 h at room temperature, followed by measurement of OD570 [41]. Eight replicate wells in each of three separate measurements. For GI50 determination of CDNB and 2HF ±CDNB in H358 cells, approximately 5,000 cells were plated as above in 96 well plate. After 12 h cells were treated with 0–15 μM CDNB or 0–150 μM 2HF and 1 μM CDNB+0–150 μM 2HF. After 48 h MTT assay was performed as described above.
Colony formation assay
Cell survival was evaluated using a standard colony-forming assay. 1 × 105 cells / mL were incubated with 2HF (50 μM) for 24 h and aliquots of 50 and 100 μL were added to 60 mm size Petri dishes containing 4 mL culture medium. After 10 days, adherent colonies were stained with 0.5% methylene blue for 30 min and counted using Innotech Alpha Imager HP [86].
DOX efflux study
1.5 × 105 H358 cells were plated in chamber slides. After overnight incubation at 37°C in a 5% CO2 atmosphere, cells were exposed with 1 μM DOX for 16 h followed by treatment with vehicle, 20 μM 2HF or 20 μM 2HF+20 μM CDNB for 4 h. Cells were gently washed, added 2 ml of PBS and placed the chamber slides on a magnetic stirrer with magnetic bar in each chamber. Every 2 min for 30 min, 50 μL aliquot of the effluent from each well was taken and placed in a black 96 well flat bottom plate. Fluorescence was measured by using of infinite M1000Pro (Tecan) with 500/550 nm for excitation and emission wavelength setting.
Docking studies
The predicting binding modes of 2HF, DNP-SG and ATP molecule in complex with RLIP76 were calculated by using our in-house developed All-Around Docking method [36] and the Glide XP docking tools of Schrödinger suite of software [87]. The human RLIP76 RhoGAP-Ral domain three-dimensional NMR structure were obtained from protein data bank with 265 amino-acid residues (RCSB protein databank PDB ID 2 mbg [35]). First, we performed All-Around Docking for small molecules 2HF, DNP-SG and ATP to search the human RLIP76 protein three-dimensional surface for the best docking site(s). Thereafter with the induced fit docking, which allows side-chain of the pocket residues to be flexible for more accurate computation of the binding modes, the complex models were refined with docking score as –6.7 for DNP-SG, –5.1 for 2HF and –9.8 for ATP.
Effect of 2HF on RLIP76 expression
H358 and H1618 cells were treated with 25 μM of 2HF for 24 h at 37°C followed by washing with PBS, re-suspended in the medium, and allowed to recover for 2 h. Cells were solubilized in lysis buffer and centrifuged at 105,000 × g for 1 h at 4°C, and the supernatant was collected. 50 μg of proteins were subjected to SDS-PAGE followed by Western-blot analyses using anti-RLIP76 IgG.
Quantitative (real-time) PCR
A panel of transcripts encoding enzymes involved in 4-HNE metabolism and in general antioxidant defense was quantitated by PCR. Total RNA was isolated from A549 and H226 cells treated with 50 or 100 μM 2HF for 24 h using TRIZOL reagent. DNase-treated total RNA was used for first DNA strand synthesis (superscript II, Invitrogen). Real-time quantitative PCR (QPCR) amplification reactions were performed with the SYBR Green qPCR kit (Roche) using 5 μL of cDNA and 0.3 μM gene-specific primers.
Purification of RLIP76
DNPSG-affinity purification of RLIP76 from SCLC (H1417 & H1618) and NSCLC (H358 & H520) cells was carried out as described previously [50]. Purifications were monitored by measuring transport activity. To demonstrate 2HF competitively bind to RLIP, we eluted recombinant RLIP with elution buffers containing 2HF with or without ATP. Eluted protein was separated on NuPAGE 4–12% Bis-Tris Protein Gels (Invitrogen) followed by transfer on nitro cellulose membrane. Purified protein was identified using anti Rlip antibody. Compositions of elution buffer used in purification are as follows: Elution buffer1: 10 mM ATP+ 10 mM MgCl2+ 0.025% C12E9+ 0.2 mM DNP-SG in Lysis buffer (Original) [32–34]; Elution buffer2: Original lysis buffer with no ATP, No DNP-SG; Elution buffer3: 1 mM ATP+ 50 μM of 2HF (no DNP-SG); Elution buffer4: 1 mM ATP+ 100 μM of 2HF (no DNP-SG); Elution buffer5: no ATP + 100 μM 2HF (no DNP-SG)).
Reconstitution of purified Rlip into artificial liposomes
Purified Rlip from lung cancer cells was dialyzed against reconstitution buffer (10 mM Tris-HCl, pH 7.4, 4 mM MgCl2, 1 mM EGTA, 100 mM KCl, 40 mM sucrose, 2.8 mM β-mercaptoethanol, 0.05 mM BHT, and 0.025% polidocanol), and reconstituted into artificial asolectin-cholesterol liposomes. An aqueous emulsion of soybean asolectin (40 mg/mL) and cholesterol (10 mg/mL) was prepared in the reconstitution buffer by sonication, from which a 100 μL aliquot was added to dialyzed purified RLIP76 protein to achieve a final concentration of 40 μg/mL. After sonication of the resulting mixture for 30 s at 50 W, 200 mg of SM-2 Bio-beads pre-equilibrated with reconstitution buffer (without polidocanol) were added to initiate vesiculation, and after 4 h incubation at 4°C, SM-2 beads were removed by centrifugation at 3000 × g and the vesicles (proteoliposomes) were collected. Control-liposomes were prepared using an equal amount of crude protein from E. coli not expressing Rlip [32]. The size of reconstituted vesicles was examined by electron microscopy and intra-vesicular volume was estimated by 14C-inulin trapping [32].
Transport studies in Rlip-proteoliposomes
Transport studies of 14C-DOX in reconstituted vesicles were performed by rapid-filtration technique as described by us using 250 ng protein per 30 μL reaction mixture. ATP-dependent uptake of 14C-DOX (specific activity 8.6 × 104 cpm/nmol, use 3.6 μM final concentration) was determined by subtracting the radioactivity (cpm) of the control without ATP from that of the experimental containing ATP, and the transport of DOX was calculated in terms of nmol/min/mg protein. Liposomes prepared without addition of Rlip were used for controls. Each determination was performed in triplicate [32].
Transport inhibition by 2HF and anti-Rlip IgG
Purified reconstituted liposomes (250 ng protein/30 μL reaction mixture) were incubated separately with either 2HF (0–50 μM final concentration) or anti-Rlip IgG (0–60 μg/ml final concentration) or both, for 30 min at room temperature. In one of the controls, IgG was excluded while the other control was treated with an equal amount of pre-immune IgG. After incubation, the ATP-dependent transport of 14C-DOX was measured by using a 96 well-plate filtration manifold to separate the extra-vesicular drug from that taken up by the vesicles. Uptake was measured in Rlip-proteoliposomes and control liposomes, in absence or presence of 2HF, anti-Rlip IgG and 4 mM ATP at a fixed time point of 5 min, at 37°C.
In vivo xenograft studies
Hsd: Athymic nude nu/nu mice were obtained from Harlan, Indianapolis, IN, and were acclimated for a week before beginning the experiment. All animal experiments were carried out in accordance with a protocol approved by the Institutional Animal Care and Use Committee. Ten 10-weeks-old mice were divided into two groups of 5 animals (treated with vehicle only i.e. corn oil, and 2HF 7 mg/kg b.w.). All 10 animals were injected with 1 × 106 H1618-SCLC suspensions in 100 μL of PBS, subcutaneously into one flank of each mouse. At the same time, animals were randomized into treatment groups. Treatment was started 10 days after the H1618 cells implantation when tumors are ~20 mm2 in diameter (palpable tumor growth). Treatment consisted of 7 mg of 2HF/kg b.w., in 200 μL corn oil by oral gavage alternate day. Control groups were treated with corn oil only. Similar protocol was followed for H358 NSCLC xenografts studies. Animals were examined daily for signs of tumor growth. Tumors were measured in two dimensions using calipers and body weights were recorded. Each mouse in every group was monitored on alternate days for signs of distress and areas of swelling or redness. Photographs of animals were taken at day 1, day 10, day 20, day 30, and day 60 after subcutaneous injection, are shown for all groups. Photographs of tumors were also taken at day 60.
Assessment of angiogenesis, proliferation, and apoptosis
Lung tumors (control as well as 2HF treated) were harvested from mice bearing tumors and used for histopathologic analyses. Tumor samples fixed in buffered formalin for 12 h were processed conventionally for paraffin-embedded tumor sections (5 μm thick). Hematoxylin and Eosin (H&E) staining was performed on paraffin-embedded tumor sections. Histopathologic analyses with anti-E cadherin, anti-CD31, and anti-Ki67 IgG, were also performed, using Universal ABC detection kit (Vector). Immuno-reactivity is evident as a dark brown stain, whereas non-reactive areas display only the background color. Sections were counterstained with Hematoxylin (blue). Photomicrographs at 40x magnification were acquired using Olympus DP 72 microscope.
Analysis of cancer-signaling by western blot
The effect of 2HF treatment on signaling proteins in tumors was determined using 28,000 × g supernatant fraction of a 10% homogenate. For Western blotting, ~ 50 μg protein loaded per lane was subjected to SDS-PAGE and transferred onto the nitrocellulose membrane. Horseradish peroxidase (HRP)-conjugated anti-mouse and anti-rabbit secondary antibodies and primary antibodies towards Akt, pAKT (S473), PARP, vimentin, E-cadherin, Fibronectin, Bcl2, Bax, CDK4, Cyclin B1, P70S6K, pP70S6K (S240–244) and GAPDH were purchased from Santa Cruz Biotechnology, Inc. Detection was using chemiluminescence ECL kit (Amersham Life Sciences). GAPDH was used as loading control and representative results from one of several experiments are shown.
Statistical analyses
All data were evaluated with a two-tailed unpaired student’s t test and are expressed as the mean ± SD. The statistical significance of differences between control and treatment groups was determined by ANOVA followed by multiple comparison tests. Changes in tumor size and body weight during the course of the experiments were visualized by scatter plot. Differences were considered statistically significant when the p value was less than 0.05.
CONCLUSIONS
Flavonoids are compounds ubiquitously present in many food sources [88]. These naturally appearing compounds have been linked to multiple biologic functions, such as anti-proliferative, pro-apoptotic, anti-inflammatory, anti-invasive and anti-angiogenic effects [26]. Present studies demonstrate that this naturally abundant, orally bioavailable and nontoxic compound exhibits tumor growth inhibitory activity SCLC and NSCLC activity by inhibiting cancer signaling and MPy, making it an attractive candidate in therapy and prevention of lung cancer.
Author contributions
Conception, design and study supervision: S. Awasthi; Development of methodology: S. Awasthi, S.P. Singh, S.S Singhal; Acquisition of data: S.S. Singhal, S.P. Singh, S. Awasthi, Y-C. Yuan, J. Singhal; Analysis and interpretation of data: S. Awasthi, S.S. Singhal, L. Nagaprashantha, D. Berz, S.P. Singh; Writing, review, and/or edit of the manuscript: S. Awasthi S.S. Singhal, H. Igid, V. Tonk, W. Green, D. Berz; S. P. Singh, Y. Awasthi.
CONFLICTS OF INTEREST
Authors do not have any competing financial interests that influence on behavior or content or from perception of such potential influences, could undermine the objectivity, integrity or perceived value of this publication.
GRANT SUPPORT
This work was supported in part by DoD (W81XWH-18-1-0534), USPHS grant CA 77495, and the funds from Perricone Family Foundation, Los Angeles, CA to SA.
REFERENCES
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61:69–90. https://doi.org/10.3322/caac.20107.
2. Santos-Alves E, Marques-Aleixo I, Rizo-Roca D, Torrella JR, Oliveira PJ, Magalhães J, Ascensão A. Exercise modulates liver cellular and mitochondrial proteins related to quality control signaling. Life Sci. 2015; 135:124–30. https://doi.org/10.1016/j.lfs.2015.06.007.
3. Mariotto AB, Yabroff KR, Shao Y, Feuer EJ, Brown ML. Projections of the cost of cancer care in the United States: 2010–2020. J Natl Cancer Inst. 2011; 103:117–28. https://doi.org/10.1093/jnci/djq495.
4. Karp DD, Lee SJ, Shaw Wright GL, Johnson DH, Johnston MR, Goodman GE, Clamon GH, Okawara GS, Marks R, Ruckdeschel JC, and MDACC Thoracic. A phase III, intergroup, randomized, double-blind, chemoprevention trial of selenium (Se) supplementation in resected stage I non-small cell lung cancer (NSCLC). J Clin Oncol. 2010 (Suppl 18); 28:CRA7004. https://doi.org/10.1200/jco.2010.28.18_suppl.cra7004.
5. Goldstraw P, Crowley J, Chansky K, Giroux DJ, Groome PA, Rami-Porta R, Postmus PE, Rusch V, Sobin L, and International Association for the Study of Lung Cancer International Staging Committee, and Participating Institutions. The IASLC Lung Cancer Staging Project: proposals for the revision of the TNM stage groupings in the forthcoming (seventh) edition of the TNM Classification of malignant tumours. J Thorac Oncol. 2007; 2:706–14. https://doi.org/10.1097/JTO.0b013e31812f3c1a.
6. Heon S, Johnson BE. Adjuvant chemotherapy for surgically resected non-small cell lung cancer. J Thorac Cardiovasc Surg. 2012; 144:S39–42. https://doi.org/10.1016/j.jtcvs.2012.03.039.
7. Gadgeel SM, Ramalingam SS, Kalemkerian GP. Treatment of lung cancer. Radiol Clin North Am. 2012; 50:961–74. https://doi.org/10.1016/j.rcl.2012.06.003.
8. George J, Lim JS, Jang SJ, Cun Y, Ozretić L, Kong G, Leenders F, Lu X, Fernández-Cuesta L, Bosco G, Müller C, Dahmen I, Jahchan NS, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015; 524:47–53. https://doi.org/10.1038/nature14664.
9. Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012; 489:519–25. https://doi.org/10.1038/nature11404. Erratum in: Nature. 2012 Nov 8;491(7423):288. Rogers, Kristen [corrected to Rodgers, Kristen].
10. Jordan EJ, Kim HR, Arcila ME, Barron D, Chakravarty D, Gao J, Chang MT, Ni A, Kundra R, Jonsson P, Jayakumaran G, Gao SP, Johnsen HC, et al. Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov. 2017; 7:596–609. https://doi.org/10.1158/2159-8290.CD-16-1337.
11. Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, Geater SL, Orlov S, Tsai CM, Boyer M, Su WC, Bennouna J, Kato T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013; 31:3327–34. https://doi.org/10.1200/JCO.2012.44.2806.
12. Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, Wu YL, Thomas M, O’Byrne KJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013; 368:2385–94. https://doi.org/10.1056/NEJMoa1214886.
13. Lane DP. Cancer. p53, guardian of the genome. Nature. 1992; 358:15–16. https://doi.org/10.1038/358015a0.
14. Zilfou JT, Lowe SW. Tumor suppressive functions of p53. Cold Spring Harb Perspect Biol. 2009; 1:a001883. https://doi.org/10.1101/cshperspect.a001883.
15. Awasthi S, Singhal SS, Srivastava SK, Torman RT, Zimniak P, Bandorowicz-Pikula J, Singh SV, Piper JT, Awasthi YC, Pikula S. ATP-Dependent human erythrocyte glutathione-conjugate transporter. I. Purification, photoaffinity labeling, and kinetic characteristics of ATPase activity. Biochemistry. 1998; 37:5231–38. https://doi.org/10.1021/bi972130z.
16. Awasthi S, Srivastava SK, Ahmad F, Ahmad H, Ansari GA. Interactions of glutathione S-transferase-pi with ethacrynic acid and its glutathione conjugate. Biochim Biophys Acta. 1993; 1164:173–78. https://doi.org/10.1016/0167-4838(93)90245-M.
17. Singhal SS, Wickramarachchi D, Yadav S, Singhal J, Leake K, Vatsyayan R, Chaudhary P, Lelsani P, Suzuki S, Yang S, Awasthi YC, Awasthi S. Glutathione-conjugate transport by RLIP76 is required for clathrin-dependent endocytosis and chemical carcinogenesis. Mol Cancer Ther. 2011; 10:16–28. https://doi.org/10.1158/1535-7163.MCT-10-0699.
18. Rahmani AH, Al Zohairy MA, Aly SM, Khan MA. Curcumin: a potential candidate in prevention of cancer via modulation of molecular pathways. Biomed Res Int. 2014; 2014:761608. https://doi.org/10.1155/2014/761608.
19. Zhou Y, Zheng J, Li Y, Xu DP, Li S, Chen YM, Li HB. Natural Polyphenols for Prevention and Treatment of Cancer. Nutrients. 2016; 8:515. https://doi.org/10.3390/nu8080515.
20. van Gisbergen MW, Cebula M, Zhang J, Ottosson-Wadlund A, Dubois L, Lambin P, Tew KD, Townsend DM, Haenen GR, Drittij-Reijnders MJ, Saneyoshi H, Araki M, Shishido Y, et al. Chemical Reactivity Window Determines Prodrug Efficiency toward Glutathione Transferase Overexpressing Cancer Cells. Mol Pharm. 2016; 13:2010–25. https://doi.org/10.1021/acs.molpharmaceut.6b00140.
21. Dalasanur Nagaprashantha L, Adhikari R, Singhal J, Chikara S, Awasthi S, Horne D, Singhal SS. Translational opportunities for broad-spectrum natural phytochemicals and targeted agent combinations in breast cancer. Int J Cancer. 2018; 142:658–70. https://doi.org/10.1002/ijc.31085.
22. Chikara S, Nagaprashantha LD, Singhal J, Horne D, Awasthi S, Singhal SS. Oxidative stress and dietary phytochemicals: role in cancer chemoprevention and treatment. Cancer Lett. 2018; 413:122–34. https://doi.org/10.1016/j.canlet.2017.11.002.
23. Singhal J, Nagaprashantha L, Chikara S, Awasthi S, Horne D, Singhal SS. 2′-Hydroxyflavanone: A novel strategy for targeting breast cancer. Oncotarget. 2017; 8:75025–37. https://doi.org/10.18632/oncotarget.20499.
24. Singhal J, Nagaprashantha LD, Vatsyayan R, Singhal A, Awasthi S, Singhal SS. Didymin induces apoptosis by inhibiting N-Myc and upregulating RKIP in neuroblastoma. Cancer Prev Res (Phila). 2012; 5:473–83. https://doi.org/10.1158/1940-6207.CAPR-11-0318.
25. Singhal SS, Singhal J, Figarola JL, Riggs A, Horne D, Awasthi S. 2′-Hydroxyflavanone: A promising molecule for kidney cancer prevention. Biochem Pharmacol. 2015; 96:151–58. https://doi.org/10.1016/j.bcp.2015.04.022.
26. Nagaprashantha LD, Vatsyayan R, Singhal J, Lelsani P, Prokai L, Awasthi S, Singhal SS. 2′-hydroxyflavanone inhibits proliferation, tumor vascularization and promotes normal differentiation in VHL-mutant renal cell carcinoma. Carcinogenesis. 2011; 32:568–75. https://doi.org/10.1093/carcin/bgr021.
27. Nagaprashantha LD, Singhal J, Li H, Warden C, Liu X, Horne D, Awasthi S, Salgia R, Singhal SS. 2′-Hydroxyflavanone effectively targets RLIP76-mediated drug transport and regulates critical signaling networks in breast cancer. Oncotarget. 2018; 9:18053–68. https://doi.org/10.18632/oncotarget.24720.
28. Awasthi S, Tompkins J, Singhal J, Riggs AD, Yadav S, Wu X, Singh S, Warden C, Liu Z, Wang J, Slavin TP, Weitzel JN, Yuan YC, et al. Rlip depletion prevents spontaneous neoplasia in TP53 null mice. Proc Natl Acad Sci U S A. 2018; 115:3918–23. https://doi.org/10.1073/pnas.1719586115.
29. Leake K, Singhal J, Nagaprashantha LD, Awasthi S, Singhal SS. RLIP76 regulates PI3K/Akt signaling and chemo-radiotherapy resistance in pancreatic cancer. PLoS One. 2012; 7:e34582. https://doi.org/10.1371/journal.pone.0034582.
30. Singhal SS, Wickramarachchi D, Singhal J, Yadav S, Awasthi YC, Awasthi S. Determinants of differential doxorubicin sensitivity between SCLC and NSCLC. FEBS Lett. 2006; 580:2258–64. https://doi.org/10.1016/j.febslet.2006.03.038.
31. Singhal SS, Singhal J, Cheng J, Pikuła S, Sharma R, Zimniak P, Awasthi YC, Awasthi S. Purification and functional reconstitution of intact ral-binding Gtpase activating protein, RLIP76, in artificial liposomes. Acta Biochim Pol. 2001; 48:551–62.
32. Awasthi S, Cheng J, Singhal SS, Saini MK, Pandya U, Pikula S, Bandorowicz-Pikula J, Singh SV, Zimniak P, Awasthi YC. Novel function of human RLIP76: ATP-dependent transport of glutathione conjugates and doxorubicin. Biochemistry. 2000; 39:9327–34. https://doi.org/10.1021/bi992964c.
33. Awasthi S, Cheng JZ, Singhal SS, Pandya U, Sharma R, Singh SV, Zimniak P, Awasthi YC. Functional reassembly of ATP-dependent xenobiotic transport by the N- and C-terminal domains of RLIP76 and identification of ATP binding sequences. Biochemistry. 2001; 40:4159–68. https://doi.org/10.1021/bi002182f.
34. Singhal SS, Sehrawat A, Mehta A, Sahu M, Awasthi S. Functional reconstitution of RLIP76 catalyzing ATP-dependent transport of glutathione-conjugates. Int J Oncol. 2009; 34:191–99.
35. Rajasekar KV, Campbell LJ, Nietlispach D, Owen D, Mott HR. The structure of the RLIP76 RhoGAP-Ral binding domain dyad: fixed position of the domains leads to dual engagement of small G proteins at the membrane. Structure. 2013; 21:2131–42. https://doi.org/10.1016/j.str.2013.09.007.
36. Yu DD, Andrali SS, Li H, Lin M, Huang W, Forman BM. Novel FXR (farnesoid X receptor) modulators: potential therapies for cholesterol gallstone disease. Bioorg Med Chem. 2016; 24:3986–93. https://doi.org/10.1016/j.bmc.2016.06.039.
37. Dunagin WG, Millikan LE. Dinitrochlorobenzene immunotherapy for verrucae resistant to standard treatment modalities. J Am Acad Dermatol. 1982; 6:40–45. https://doi.org/10.1016/S0190-9622(82)80199-0.
38. Pabst MJ, Habig WH, Jakoby WB. Glutathione S-transferase A. A novel kinetic mechanism in which the major reaction pathway depends on substrate concentration. J Biol Chem. 1974; 249:7140–47.
39. Habig WH, Pabst MJ, Jakoby WB. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J Biol Chem. 1974; 249:7130–39.
40. Awasthi S, Sharma R, Singhal SS, Zimniak P, Awasthi YC. RLIP76, a novel transporter catalyzing ATP-dependent efflux of xenobiotics. Drug Metab Dispos. 2002; 30:1300–10. https://doi.org/10.1124/dmd.30.12.1300.
41. Singhal SS, Awasthi YC, Awasthi S. Regression of melanoma in a murine model by RLIP76 depletion. Cancer Res. 2006; 66:2354–60. https://doi.org/10.1158/0008-5472.CAN-05-3534.
42. Singhal SS, Yadav S, Singhal J, Zajac E, Awasthi YC, Awasthi S. Depletion of RLIP76 sensitizes lung cancer cells to doxorubicin. Biochem Pharmacol. 2005; 70:481–88. https://doi.org/10.1016/j.bcp.2005.05.005.
43. Stuckler D, Singhal J, Singhal SS, Yadav S, Awasthi YC, Awasthi S. RLIP76 transports vinorelbine and mediates drug resistance in non-small cell lung cancer. Cancer Res. 2005; 65:991–98.
44. Singhal SS, Singhal J, Nair MP, Lacko AG, Awasthi YC, Awasthi S. Doxorubicin transport by RALBP1 and ABCG2 in lung and breast cancer. Int J Oncol. 2007; 30:717–25. https://doi.org/10.3892/ijo.30.3.717.
45. Phi LT, Sari IN, Yang YG, Lee SH, Jun N, Kim KS, Lee YK, Kwon HY. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018; 2018:5416923. https://doi.org/10.1155/2018/5416923.
46. Gibbons DL, Byers LA, Kurie JM. Smoking, p53 mutation, and lung cancer. Mol Cancer Res. 2014; 12:3–13. https://doi.org/10.1158/1541-7786.MCR-13-0539.
47. Tovy A, Spiro A, McCarthy R, Shipony Z, Aylon Y, Allton K, Ainbinder E, Furth N, Tanay A, Barton M, Oren M. p53 is essential for DNA methylation homeostasis in naïve embryonic stem cells, and its loss promotes clonal heterogeneity. Genes Dev. 2017; 31:959–72. https://doi.org/10.1101/gad.299198.117.
48. Weiss DJ, Kolls JK, Ortiz LA, Panoskaltsis-Mortari A, Prockop DJ. Stem cells and cell therapies in lung biology and lung diseases. Proc Am Thorac Soc. 2008; 5:637–67. https://doi.org/10.1513/pats.200804-037DW.
49. Vatsyayan R, Chaudhary P, Lelsani PC, Singhal P, Awasthi YC, Awasthi S, Singhal SS. Role of RLIP76 in doxorubicin resistance in lung cancer. Int J Oncol. 2009; 34:1505–11.
50. Singhal SS, Singhal J, Sharma R, Singh SV, Zimniak P, Awasthi YC, Awasthi S. Role of RLIP76 in lung cancer doxorubicin resistance: I. The ATPase activity of RLIP76 correlates with doxorubicin and 4-hydroxynonenal resistance in lung cancer cells. Int J Oncol. 2003; 22:365–75. https://doi.org/10.3892/ijo.22.2.365.
51. Sharma R, Singhal SS, Srivastava SK, Bajpai KK, Frenkel EP, Awasthi S. Glutathione and glutathione linked enzymes in human small cell lung cancer cell lines. Cancer Lett. 1993; 75:111–19. https://doi.org/10.1016/0304-3835(93)90195-F.
52. Awasthi S, Singhal SS, Singhal J, Cheng J, Zimniak P, Awasthi YC. Role of RLIP76 in lung cancer doxorubicin resistance: II. Doxorubicin transport in lung cancer by RLIP76. Int J Oncol. 2003; 22:713–20. https://doi.org/10.3892/ijo.22.4.713.
53. Bode AM, Dong Z. Signal transduction and molecular targets of selected flavonoids. Antioxid Redox Signal. 2013; 19:163–80. https://doi.org/10.1089/ars.2013.5251.
54. Sehrawat A, Yadav S, Awasthi YC, Basu A, Warden C, Awasthi S. P300 regulates the human RLIP76 promoter activity and gene expression. Biochem Pharmacol. 2013; 85:1203–11. https://doi.org/10.1016/j.bcp.2013.02.012.
55. Sun Z, Chin YE, Zhang DD. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol Cell Biol. 2009; 29:2658–72. https://doi.org/10.1128/MCB.01639-08.
56. Jinesh GG, Kamat AM. RalBP1 and p19-VHL play an oncogenic role, and p30-VHL plays a tumor suppressor role during the blebbishield emergency program. Cell Death Discov. 2017; 3:17023. https://doi.org/10.1038/cddiscovery.2017.23.
57. Al-Rimawi F, Rishmawi S, Ariqat SH, Khalid MF, Warad I, Salah Z. Anticancer Activity, Antioxidant Activity, and Phenolic and Flavonoids Content of Wild Tragopogon porrifolius Plant Extracts. Evid Based Complement Alternat Med. 2016; 2016:9612490. https://doi.org/10.1155/2016/9612490.
58. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009; 119:1420–28. https://doi.org/10.1172/JCI39104.
59. Cao M, Seike M, Soeno C, Mizutani H, Kitamura K, Minegishi Y, Noro R, Yoshimura A, Cai L, Gemma A. MiR-23a regulates TGF-β-induced epithelial-mesenchymal transition by targeting E-cadherin in lung cancer cells. Int J Oncol. 2012; 41:869–75. https://doi.org/10.3892/ijo.2012.1535.
60. Morali OG, Delmas V, Moore R, Jeanney C, Thiery JP, Larue L. IGF-II induces rapid beta-catenin relocation to the nucleus during epithelium to mesenchyme transition. Oncogene. 2001; 20:4942–50. https://doi.org/10.1038/sj.onc.1204660.
61. Tester AM, Ruangpanit N, Anderson RL, Thompson EW. MMP-9 secretion and MMP-2 activation distinguish invasive and metastatic sublines of a mouse mammary carcinoma system showing epithelial-mesenchymal transition traits. Clin Exp Metastasis. 2000; 18:553–60. https://doi.org/10.1023/A:1011953118186.
62. Aokage K, Ishii G, Ohtaki Y, Yamaguchi Y, Hishida T, Yoshida J, Nishimura M, Nagai K, Ochiai A. Dynamic molecular changes associated with epithelial-mesenchymal transition and subsequent mesenchymal-epithelial transition in the early phase of metastatic tumor formation. Int J Cancer. 2011; 128:1585–95. https://doi.org/10.1002/ijc.25500.
63. Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, Ali S, Abbruzzese JL, Gallick GE, Sarkar FH. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009; 69:2400–07. https://doi.org/10.1158/0008-5472.CAN-08-4312.
64. Wei L, Yang Y, Zhang X, Yu Q. Anchorage-independent phosphorylation of p130(Cas) protects lung adenocarcinoma cells from anoikis. J Cell Biochem. 2002; 87:439–49. https://doi.org/10.1002/jcb.10322.
65. Nagaprashantha LD, Vatsyayan R, Lelsani PC, Awasthi S, Singhal SS. The sensors and regulators of cell-matrix surveillance in anoikis resistance of tumors. Int J Cancer. 2011; 128:743–52. https://doi.org/10.1002/ijc.25725.
66. Liu S, Miao Y, Fan C, Liu Y, Yu J, Zhang Y, Dai S, Wang E. Clinicopathologic correlations of liver kinase B1, E-cadherin, and N-cadherin expression in non-small cell lung cancer. Appl Immunohistochem Mol Morphol. 2013; 21:334–40. https://doi.org/10.1097/PAI.0b013e31826b128b.
67. Han S, Khuri FR, Roman J. Fibronectin stimulates non-small cell lung carcinoma cell growth through activation of Akt/mammalian target of rapamycin/S6 kinase and inactivation of LKB1/AMP-activated protein kinase signal pathways. Cancer Res. 2006; 66:315–23. https://doi.org/10.1158/0008-5472.CAN-05-2367.
68. Cory S, Huang DC, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003; 22:8590–607. https://doi.org/10.1038/sj.onc.1207102.
69. Porebska I, Wyrodek E, Kosacka M, Adamiak J, Jankowska R, Harłozińska-Szmyrka A. Apoptotic markers p53, Bcl-2 and Bax in primary lung cancer.
70. Shabnam MS, Srinivasan R, Wali A, Majumdar S, Joshi K, Behera D. Expression of p53 protein and the apoptotic regulatory molecules Bcl-2, Bcl-XL, and Bax in locally advanced squamous cell carcinoma of the lung. Lung Cancer. 2004; 45:181–88. https://doi.org/10.1016/j.lungcan.2004.01.021.
71. Halasova E, Adamkov M, Matakova T, Vybohova D, Antosova M, Janickova M, Singliar A, Dobrota D, Jakusova V. Expression of Ki-67, Bcl-2, Survivin and p53 Proteins in Patients with Pulmonary Carcinoma. Adv Exp Med Biol. 2013; 756:15–21. https://doi.org/10.1007/978-94-007-4549-0_3.
72. Takahashi T, Nau MM, Chiba I, Birrer MJ, Rosenberg RK, Vinocour M, Levitt M, Pass H, Gazdar AF, Minna JD. p53: a frequent target for genetic abnormalities in lung cancer. Science. 1989; 246:491–94. https://doi.org/10.1126/science.2554494.
73. Winter SF, Minna JD, Johnson BE, Takahashi T, Gazdar AF, Carbone DP. Development of antibodies against p53 in lung cancer patients appears to be dependent on the type of p53 mutation. Cancer Res. 1992; 52:4168–74.
74. David O, Jett J, LeBeau H, Dy G, Hughes J, Friedman M, Brody AR. Phospho-Akt overexpression in non-small cell lung cancer confers significant stage-independent survival disadvantage. Clin Cancer Res. 2004; 10:6865–71. https://doi.org/10.1158/1078-0432.CCR-04-0174.
75. Tsao AS, McDonnell T, Lam S, Putnam JB, Bekele N, Hong WK, Kurie JM. Increased phospho-AKT (Ser(473)) expression in bronchial dysplasia: implications for lung cancer prevention studies. Cancer Epidemiol Biomarkers Prev. 2003; 12:660–64.
76. Burris HA 3rd. Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemother Pharmacol. 2013; 71:829–42. https://doi.org/10.1007/s00280-012-2043-3.
77. Gadgeel SM, Wozniak A. Preclinical rationale for PI3K/Akt/mTOR pathway inhibitors as therapy for epidermal growth factor receptor inhibitor-resistant non-small-cell lung cancer. Clin Lung Cancer. 2013; 14:322–32. https://doi.org/10.1016/j.cllc.2012.12.001.
78. Papadimitrakopoulou V. Development of PI3K/AKT/mTOR pathway inhibitors and their application in personalized therapy for non-small-cell lung cancer. J Thorac Oncol. 2012; 7:1315–26. https://doi.org/10.1097/JTO.0b013e31825493eb.
79. Fox JL, MacFarlane M. Targeting cell death signalling in cancer: minimising ‘Collateral damage’. Br J Cancer. 2016; 115:5–11. https://doi.org/10.1038/bjc.2016.111.
80. Awasthi S, Singhal SS, Yadav S, Singhal J, Drake K, Nadkar A, Zajac E, Wickramarachchi D, Rowe N, Yacoub A, Boor P, Dwivedi S, Dent P, et al. RLIP76 is a major determinant of radiation sensitivity. Cancer Res. 2005; 65:6022–28. https://doi.org/10.1158/0008-5472.CAN-05-0968.
81. Soria JC, Jang SJ, Khuri FR, Hassan K, Liu D, Hong WK, Mao L. Overexpression of cyclin B1 in early-stage non-small cell lung cancer and its clinical implication. Cancer Res. 2000; 60:4000–04.
82. Wu A, Wu B, Guo J, Luo W, Wu D, Yang H, Zhen Y, Yu X, Wang H, Zhou Y, Liu Z, Fang W, Yang Z. Elevated expression of CDK4 in lung cancer. J Transl Med. 2011; 9:38. https://doi.org/10.1186/1479-5876-9-38.
83. Pines J, Hunter T. Human cyclin A is adenovirus E1A-associated protein p60 and behaves differently from cyclin B. Nature. 1990; 346:760–63. https://doi.org/10.1038/346760a0.
84. Singhal SS, Yadav S, Vatsyayan R, Chaudhary P, Borvak J, Singhal J, Awasthi S. Increased expression of cdc2 inhibits transport function of RLIP76 and promotes apoptosis. Cancer Lett. 2009; 283:152–58. https://doi.org/10.1016/j.canlet.2009.03.033.
85. Dickson MA, Tap WD, Keohan ML, D’Angelo SP, Gounder MM, Antonescu CR, Landa J, Qin LX, Rathbone DD, Condy MM, Ustoyev Y, Crago AM, Singer S, Schwartz GK. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J Clin Oncol. 2013; 31:2024–28. https://doi.org/10.1200/JCO.2012.46.5476.
86. Singhal J, Singhal SS, Yadav S, Suzuki S, Warnke MM, Yacoub A, Dent P, Bae S, Sharma R, Awasthi YC, Armstrong DW, Awasthi S. RLIP76 in defense of radiation poisoning. Int J Radiat Oncol Biol Phys. 2008; 72:553–61. https://doi.org/10.1016/j.ijrobp.2008.06.1497.
87. Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006; 49:6177–96. https://doi.org/10.1021/jm051256o.
88. Romagnolo DF, Selmin OI. Flavonoids and cancer prevention: a review of the evidence. J Nutr Gerontol Geriatr. 2012; 31:206–38. https://doi.org/10.1080/21551197.2012.702534.