
INTRODUCTION
Breast implant-associated anaplastic large cell lymphoma (BIA-ALCL) is a rare form of T-cell lymphoma that occurs after implantation of breast prostheses typically after a relatively long latency [1, 2]. BIA-ALCL is notable for its aggressive histological appearance but a paradoxical predominance of clinical presentation with early stage disease and a relatively favourable prognosis when compared with its systemic counterpart [3, 4]. Whilst the underlying cause of BIA-ALCL is unknown, a possible contribution from chronic antigen stimulation by a unique implant-biofilm associated microbiome has been hypothesized [5].
Genomic and functional characterization of systemic ALK-negative anaplastic large cell lymphoma (sALCL) has revealed the importance of STAT3 activation, MYC expression, PRDM1/TP53 abnormalities and recurrent structural variants involving the DUSP22 and TP63 loci [6–9]. By contrast, the genomic landscape of BIA-ALCL and its relevant pathogenic drivers are significantly less well characterized. Whole exome sequencing of two cases and targeted sequence variant detection of case series to date have demonstrated activating mutations in the JAK/STAT pathway in a proportion of cases [10–12]. We aimed to extend the understanding of this rare lymphoma by performing comprehensive genomic characterization by targeted sequence variant detection, whole genome copy number assessment, T-cell receptor locus structural variant detection and T-cell receptor repertoire sequencing on a cohort of cases of BIA-ALCL.
RESULTS
Patient cohort
Thirteen patients were referred for diagnostic NGS in the study period as part of investigation of newly diagnosed BIA-ALCL. Eleven out of thirteen patients had sufficient quality and quantity of DNA for NGS analysis. All eleven patients were female and the median age of the cohort was 42 years (range 29–59). Eight patients had stage IA (T1N0M0), two patients had stage IB (T2N0M0) and one patient had stage IIA (T4N0M0) disease by MD Anderson TNM staging [13].
Sequence variant detection
Pathogenic sequence variants detected in the eleven cases are listed in Table 1. Pathogenic activating STAT3 mutations were detected in 7 out of 11 cases (64%). In one of the four STAT3 wildtype patients, we detected a truncating mutation of SOCS1 which is predicted to result in activation of the JAK/STAT pathway through loss of negative regulation [14]. Another STAT3 wildtype patient had an activating JAK1 mutation detected as previously described [10]. Finally, one STAT3 wildtype patient (BALCL11) had a novel sequence variant detected in PTPN1 (Thr263Ile). PTPN1 encodes PTP1B which dephosphorylates tyrosine residues and through this mechanism is involved in regulation of intracellular signalling pathways including the JAK/STAT pathway [15]. This variant causes a change in an amino acid which is critical for protein function and is predicted to be deleterious by multiple in silico predictors [16]. Loss of PTPN1 function through mutation has been shown to result in JAK/STAT activation in primary mediastinal B-cell lymphoma and Hodgkin lymphoma [17]. Therefore, in ten out of eleven cases (91%) we observed direct evidence of JAK/STAT activation attributable to sequence variants alone.
Table 1: Sequence variant, copy number changes and TRB characteristics from eleven cases of breast implant associated anaplastic large cell lymphoma (BIA-ALCL)
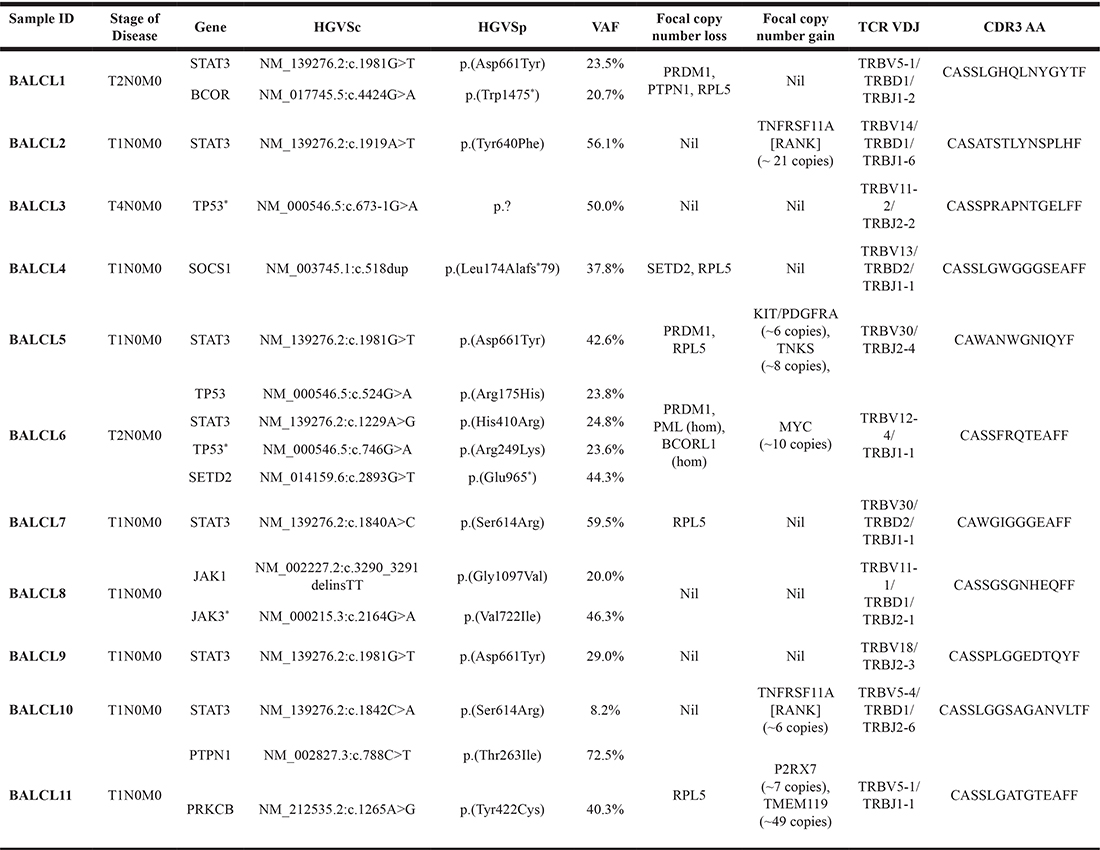
*confirmed germline, Abbreviations: hom – homozygous, VAF (variant allele frequency).
Patient BALCL11 also had an acquired sequence variant detected in PRKCB (Tyr422Cys). This is a previously undescribed variant that results in an amino acid change in a residue which stabilizes the interaction between PRKCB and diacyglycerol (DAG). Mutations in similarly critical residues have been shown to result in protein kinase β (PKCβ) pathway activation in adult T-cell leukemia/lymphoma (ATLL) [18].
Three pathogenic TP53 variants were detected in two patients. Both patients with TP53 mutations had breast implants inserted after undergoing mastectomy for breast carcinoma. Moreover, both patients had a positive family history of breast cancer. Subsequent testing of germline samples from these patients confirmed the germline origin of the two TP53 variants (c.673-1G>A and Arg249Lys) identified in both cases, BALCL3 and BALCL6 respectively. The TP53 c.673-1G>A mutation affects the canonical splice on the intron 6/exon 7 boundary with functional validation of a splicing defect and has been observed in multiple Li-Fraumeni kindreds [19–21]. The TP53 Arg249Lys mutation has not been described in Li-Fraumeni kindreds, however it has been observed multiple times as an acquired variant in diverse malignancies as well as being categorised as non-functional in the TP53 IARC database [22].
Copy number assessment
We focussed our genome wide copy number analysis on recurrent abnormalities (i.e. those occurring in more than one patient) as well as focal (generally <5 Mb or involving single coding genes) deletions and amplifications as these are most likely to provide insights into lymphoma biology. Recurrent and focal copy number changes are shown in Table 1.
A recurrent focally deleted region on chromosome 1p was observed in five cases. Deletions in these five cases involved 1p21-22 with a minimal deleted region (MDR) between nucleotides 92340000 and 94450000 on chromosome 1 (human reference sequence GRCh37 [hg19]; Figure 1). This MDR contains the recently characterised haploinsufficient tumor suppressor gene RPL5 [23].
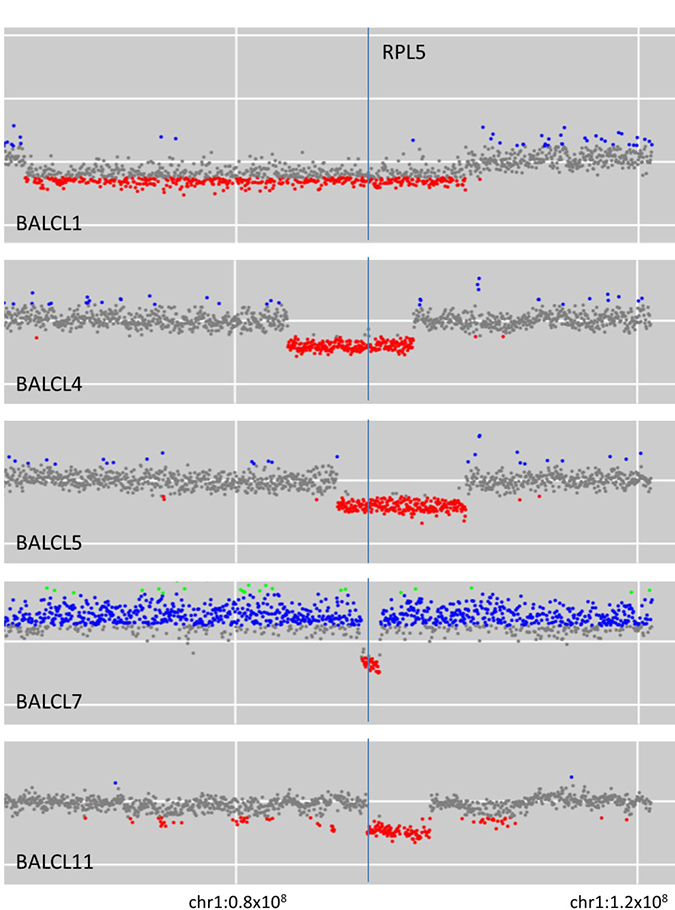
Figure 1: Five cases of breast implant-associated anaplastic large cell lymphoma with deletion on chromosome 1p involving RPL5.
TP53 and PRDM1 (encoding BLIMP-1) loci were specifically assessed due to the presence of recurrent losses in sALCL [24]. PRDM1 copy number loss was observed in three out of eleven cases (27%) but no TP53 copy number losses were detected.
Focal high-level amplifications were detected in five cases. In two of these cases the focal amplification involved 18q21 (Figure 2). In both cases the amplified genomic segment involved TNFRSF11A (which encodes Receptor Activator of Nuclear Factor κB [RANK]). Immunohistochemistry (IHC) staining for RANK was performed on one of the cases where formalin-fixed, paraffin-embedded (FFPE) tumor material was available (BALCL10). Strong membrane staining of tumor cells was observed (Figure 3A).
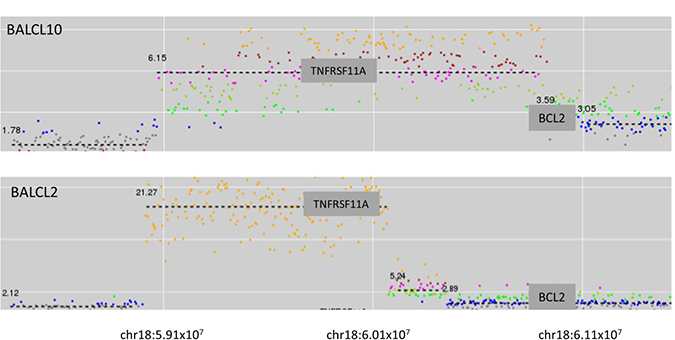
Figure 2: Two cases of breast implant-associated anaplastic large cell lymphoma with high level amplification of TNFRSF11A (RANK).
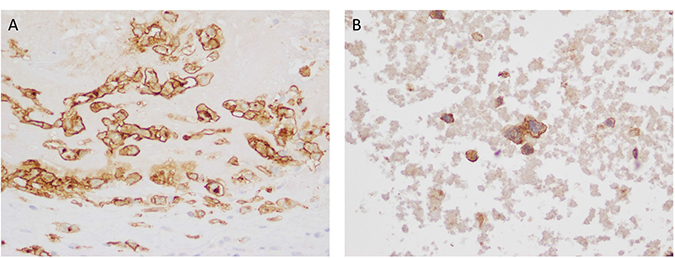
Figure 3: (A) RANK immunohistochemistry in a case of breast implant-associated anaplastic large cell lymphoma (BIA-ALCL) with TNFRSF11A (RANK) amplification (×400) (B) PDGFRA immunohistochemistry in a case of BIA-ALCL with PDGFRA amplification.
In one case a high level focal amplification on 4q12 involving both PDGFRA and KIT was detected. IHC for PDGFRA showed strong membrane staining of tumor cells (Figure 3B). This patient also had a focal amplification of 8p23.1 involving TNKS, the gene encoding Tankyrase – a poly ADP-ribose polymerase (PARP) enzyme involved in Wnt/β-catenin pathway.
In BALCL11 a copy number amplification (approximately 50 copies) was detected involving TMEM119, a recently characterised oncogene implicated in transforming growth factor beta (TGF-β) signalling in osteosarcoma [25]. This patient also had a copy number amplification (approximately 7 copies) involving P2RX7 which encodes the P2X7 purine-receptor and which is associated with complex immune effects including involvement in NLRP3 inflammasome assembly, T-cell survival and differentiation [26].
A focal amplification of MYC (approximately 10 copies) was observed in BALCL6. No structural variants involving the TRA, TRB, TRG or TRD loci were detected. A summary of recurrent genomic abnormalities are shown in Figure 4.
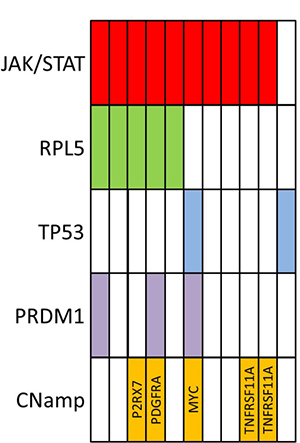
Figure 4: Summary of recurrent genomic findings from 11 cases of breast implant-associated large lymphoma (CNamp–copy number amplification).
T-cell repertoire sequencing
TRB amplicon deep sequencing was performed on all patients. A dominant clonal rearrangement was detected in TRB in all cases. The VDJ family usage and CDR3 amino acid sequences for TRB sequencing are shown in Table 1. The CDR3 sequences of the dominant clones were queried against a curated database of TCR sequences associated with known antigen specificity using VDJdb [27]. The dominant clonal TRB sequences were not predicted to have affinity with known antigens in the VDJdb database. Seroma T-cell repertoire was analysed after removal of the dominant clonal sequence (i.e. leaving residual sequences representing the non-malignant T-cell infiltrate [confirmed through different V/D/J-family usage]). The non-malignant seroma T-cell repertoire was found to have decreased diversity when compared to normal polyclonal T-cells obtained from peripheral blood of healthy volunteers (normalized Shannon Index 0.843 vs 0.959, p = 0.0044).
DISCUSSION
We have described the genomic findings of a cohort of BIA-ALCL which has provided insights into the pathobiology of this rare entity. Aberrant STAT3 signalling is a central pathogenic abnormality in sALCL and has also been observed to be strongly activated in BIA-ALCL cell lines [6, 28, 29]. There are multiple genomic routes through which STAT3 activation can be achieved in sALCL including activating sequence variants in JAK1/STAT3 (in approximately 20–30% of cases) and translocations involving ROS1 and TYK2 [6]. In contrast, we observed direct evidence of JAK/STAT activation attributable to sequence variants alone in ten out of eleven cases in our BIA-ALCL cohort - predominantly through activating STAT3 mutations (in seven patients). Therefore whilst aberrant STAT3 activation appears to be ubiquitous in both in BIA-ALCL and sALCL, the genomic mechanism by which this signalling aberration is achieved appears to be different. Moreover, the spectrum of STAT3 mutations observed in our cohort also differed from those previously described in sALCL [6] with three of the seven STAT3 mutations occurring outside the typical Tyr640 (and surrounding codons). We observed two STAT3 Ser614Arg mutations as well as one mutation in the DNA-binding domain (STAT3 His410Arg). The explanation for the predilection of JAK/STAT3 activation in BIA-ALCL through STAT3 activating mutations is not clear. Whilst JAK1/STAT3 sequence variants have previously been described in BIA-ALCL, the frequency in our cohort is significantly higher than previously observed [11, 30].
BIA-ALCL has been described in the context of pathogenic germline TP53 abnormalities in two separate case reports to date [31, 32]. In our cohort, we observed two further cases with confirmed pathogenic germline TP53 mutations. Both these patients had breast implant insertion after surgery for breast cancer and had a family history of breast cancer. In one of these cases a second TP53 mutation (Arg175His) was acquired in the patient’s tumor consistent with Knudson’s two-hit paradigm of tumorigenesis. Despite the description of four cases in total to date with germline TP53 abnormalities, the overall number of patients with germline TP53 mutations undergoing breast implant insertion is unclear and therefore the precise risk in this population cannot be determined. However given the observed association to date and the increase in risk of these patients to a variety of malignancies it is a phenomenon that warrants further monitoring and study.
We have described a novel recurrent copy number loss on chromosome 1p in our cohort of BIA-ALCL. The minimally deleted region occurring at 1p22 was approximately 2 Mb in size and is the same area that has been recently observed to be recurrently deleted in multiple myeloma [33]. Experimental evidence and investigation of deletions in this area have indicated that the most likely coding gene target of the deletion is RPL5, a gene encoding a ribosomal protein which forms part of the 60S ribosomal subunit and which shows decreased expression in deleted cases [23, 33]. Evidence to date suggests that RPL5 (along with numerous other ribosomal protein genes [RPGs]) is a haploinsufficient tumor suppressor gene which is frequently deleted in a range of cancers including glioblastoma multiforme, melanoma and breast cancer [23]. In a large cancer dataset analysis, RPG deletions were associated with TP53 mutation and/or TP53 copy number loss [34]. It has been hypothesized that deletion of RPGs (such as RPL5) results in ribosomal biogenesis stress which triggers TP53 activation and cell death, therefore a selection against these deletions in the face of an intact TP53 pathway has been proposed [34]. Interestingly in our cohort, we detected RPL5 deletions exclusively in patients that were TP53 wildtype by mutational analysis and copy number (Figure 4). Although this may seem contradictory to the negative selection hypothesis, in the majority of cases of sALCL it has been observed that the TP53 pathway is abnormal as assessed by positive TP53 immunohistochemical staining, however, this is typically not due to recurrent mutation or copy number loss [35]. Therefore, ALCL may represent a permissive environment for RPL5 deletion due to the high prevalence of multifactorial TP53 dysfunction. Of note, abnormal TP53 activation in response to DNA damage by radiation and cytotoxic agents has been observed in BIA-ALCL cell lines [36]. Whilst the rate of TP53 pathway abnormalities in BIA-ALCL is not known, the observation of two patients with germline TP53 mutations and recurrent deletion in RPL5 is supportive evidence of the biological importance of an aberrant TP53 pathway in BIA-ALCL.
In addition, RPL5 has been shown to be involved in suppression of MYC expression [37] and RPL5 knockdown is associated with increased MYC expression in breast cancer cell lines [23]. MYC dysregulation (driven by IRF4 signalling) has been recognised as a central pathogenic abnormality in sALCL [7]. The observation of recurrent RPL5 deletions in our cohort as well as the observation of a high-level MYC copy number gain observed in BALCL6 provides evidence of the potential importance of MYC aberration in this disease.
We also detected novel focal high level amplifications involving multiple oncogenes of potential relevance to lymphoma pathogenesis including TNFRSF11A, PDGFRA, TMEM119 and P2RX7. TNFRSF11A encodes Receptor Activator of Nuclear Factor κ B (RANK) and ligation of this receptor by RANKL results in NF-κB activation. RANKL plays a role in normal breast physiology, where it is produced by mammary cells in response to progesterone and has been shown to be a key paracrine factor that activates mammary stem/progenitor cells [38, 39]. The RANKL/RANK signaling pathway has also been implicated in progesterone-mediated breast tumorigenesis and BRCA1-mutated breast cancer [40, 41]. In metastatic breast cancer, tumor infiltrating regulatory T-cells (Treg) [42] may also contribute to local RANKL production. Thus the breast is a recognized tissue responsible for local RANKL secretion, where RANKL/RANK signalling (which activates the NF-κB pathway) induces pro-survival and growth signals. Invariant NKT cells also contribute to RANKL production in the microenviroment in multiple myeloma [43]. The finding that RANK can be over-expressed in BIA-ALCL has potential therapeutic implications, given the availability of monoclonal antibody RANKL inhibitors that are currently in clinical use for the treatment of osteoporosis and prevention of skeletal related events in metastatic breast cancer [44, 45].
The observation of PDGFRA amplification and confirmed overexpression is of interest given that, in ALK+ sALCL, ALK has been shown to induce the expression of PDGFR via JUNB and JUN which subsequently contributes to the malignant behaviour of ALK+ ALCL [46]. Moreover, inhibition of PDGFRA and PDGFRB by imatinib has been used in a patient with refractory ALK+ ALCL with reported efficacy [46]. The detection of PDGFRA amplification and overexpression in our patient supports PDGFRA as another potentially targetable lesion.
It is well-established that the majority of cases of ALK-negative sALCL do not express a T-cell receptor despite having rearranged TRG and TRB loci [47]. We sequenced TRB in our cohort and show that, similar to ALK-negative sALCL, all cases of BIA-ALCL harboured a rearranged TRB locus. One hypothesis of BIA-ALCL development is chronic antigen stimulation by microorganisms contained in a biofilm over the implant. Whilst our observation of restricted diversity in the non-malignant T-cell compartment is supportive of this hypothesis, this could be further investigated more directly with stimulation of BIA-ALCL cells with candidate antigens.
In summary, we have comprehensively genomically characterized eleven cases of BIA-ALCL. Our genomic data demonstrate that the aberrant signalling pathways in BIA-ALCL are analogous to those in sALCL including abnormalities of the TP53, MYC and JAK/STAT3 pathways. However, the observed set of genomic abnormalities that achieve dysregulation of these pathways appears to be unique in BIA-ALCL with highly recurrent activating STAT3 mutations and recurrent deletions of 1p22 involving RPL5 which have not been identified in sALCL to date. In addition, our genomic data also implicates abnormalities of TGF-β, PKC, Wnt/β-catenin pathway and inflammosome signalling as areas of future study. Finally, we have made the important observation of high-level amplification of TNFRSF11A and PDGFRA in a proportion of cases which are potentially targetable by available therapeutics. These findings provide a basis for future functional studies to clarify the roles of the pathways implicated by these genomic aberrations in the pathogenesis of BIA-ALCL.
MATERIALS AND METHODS
Patient cohort
Consecutive cases of BIA-ALCL were identified from July 2015 to July 2018 that had been referred for diagnostic molecular testing at the Peter MacCallum Cancer Centre Molecular Haematology Laboratory (Melbourne, Australia). All patients consented for genomic testing as part of their tumor diagnostic workup as per institutional guidelines, as well as with approval by the ethics committee of the Peter MacCallum Cancer Centre (03/09) and research was conducted in accordance with the Helsinki Declaration of 1975, as revised in 2008. The diagnosis of BIA-ALCL was confirmed by expert pathology review in each case. All testing was performed on fresh seroma fluid with adequate purity (>40%) of tumor cells confirmed by either cytological assessment or by multiparameter flow cytometry.
Sequence variant, copy number change and structural variant detection by next generation sequencing (NGS)
Tumor samples were sequenced with the Peter MacCallum Cancer Centre (PMCC) PanHaem Panel as described previously [48]. Briefly, the PMCC PanHaem panel is a hybridisation based NGS panel targeting genes associated with haematological malignancy (Supplementary Table 1), the entire IGH/TRA/TRB/TRG and TRD loci and genome-wide copy number assessment. Two cases (BALCL7 and BALCL8) had undergone whole exome sequencing with the variant and copy number findings described previously [10].
T-cell repertoire sequencing
T-cell receptor beta (TRB) loci deep amplicon sequencing was performed using LymphoTrack TRB (Invivoscribe, San Diego, CA, USA) as per manufacturer’s instructions. Sequence assembly from FASTQs, annotation and error correction was performed by MiXCR [49] with secondary analysis (diversity assessment, VDJ family usage analysis) performed by VDJtools (ver 1.1.9) [50]. T-cell repertoire diversity was assessed by the normalized Shannon-Wiener entropy calculated as -Σi(piLnpi)/LnN (where pi is the frequency of each species and N is the total number of the species).
Immunohistochemistry
For RANK immunohistochemical analysis, formalin-fixed paraffin embedded sections from BIA-ALCL patients were dewaxed in xylene prior to rehydration using graded ethanol concentrations. Antigen retrieval and immunostaining were performed as described [51] using monoclonal mouse anti-human RANK antibody (N-1H8; Amgen Inc.). For PDGFRA immunohistochemical analysis, monoclonal mouse anti-PDGFR-α (C-9) (Santa Cruz, sc-398206, dilution 1:100) was incubated overnight at 4° C following antigen retrieval with Dako Low pH 3 in 1 buffer at 97° C for 20 minutes in a Dako PT Link (Agilent Technologies, K8005). Visualization was performed on a Dako Autostainer48 using Envison Flex detection kit (Agilent Technologies, K5007).
ACKNOWLEDGMENTS
We gratefully acknowledge the funding support of the Snowdome Foundation, The Wilson Centre for Lymphoma Genomics and Vision Super.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
REFERENCES
1. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J. WHO classification of Tumours of Haematopoietic and Lymphoid Tissues. (Lyon, France: International Agency for Research on Cancer). 2017.
2. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD, Jaffe ES. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016; 127:2375–90. https://doi.org/10.1182/blood-2016-01-643569.
3. Clemens MW, Medeiros LJ, Butler CE, Hunt KK, Fanale MA, Horwitz S, Weisenburger DD, Liu J, Morgan EA, Kanagal-Shamanna R, Parkash V, Ning J, Sohani AR, et al. Complete Surgical Excision Is Essential for the Management of Patients With Breast Implant-Associated Anaplastic Large-Cell Lymphoma. J Clin Oncol. 2016; 34:160–8. https://doi.org/10.1200/JCO.2015.63.3412.
4. Miranda RN, Aladily TN, Prince HM, Kanagal-Shamanna R, de Jong D, Fayad LE, Amin MB, Haideri N, Bhagat G, Brooks GS, Shifrin DA, O'Malley DP, Cheah CY, et al. Breast implant-associated anaplastic large-cell lymphoma: long-term follow-up of 60 patients. J Clin Oncol. 2014; 32:114–20. https://doi.org/10.1200/JCO.2013.52.7911.
5. Hu H, Johani K, Almatroudi A, Vickery K, Van Natta B, Kadin ME, Brody G, Clemens M, Cheah CY, Lade S, Joshi PA, Prince HM, Deva AK. Bacterial Biofilm Infection Detected in Breast Implant-Associated Anaplastic Large-Cell Lymphoma. Plast Reconstr Surg. 2016; 137:1659–69. https://doi.org/10.1097/PRS.0000000000002010.
6. Crescenzo R, Abate F, Lasorsa E, Tabbo F, Gaudiano M, Chiesa N, Di Giacomo F, Spaccarotella E, Barbarossa L, Ercole E, Todaro M, Boi M, Acquaviva A, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell. 2015; 27:516–32. https://doi.org/10.1016/j.ccell.2015.03.006.
7. Weilemann A, Grau M, Erdmann T, Merkel O, Sobhiafshar U, Anagnostopoulos I, Hummel M, Siegert A, Hayford C, Madle H, Wollert-Wulf B, Fichtner I, Dorken B, et al. Essential role of IRF4 and MYC signaling for survival of anaplastic large cell lymphoma. Blood. 2015; 125:124–32. https://doi.org/10.1182/blood-2014-08-594507.
8. Feldman AL, Dogan A, Smith DI, Law ME, Ansell SM, Johnson SH, Porcher JC, Ozsan N, Wieben ED, Eckloff BW, Vasmatzis G. Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood. 2011; 117:915–9. https://doi.org/10.1182/blood-2010-08-303305.
9. Parrilla Castellar ER, Jaffe ES, Said JW, Swerdlow SH, Ketterling RP, Knudson RA, Sidhu JS, Hsi ED, Karikehalli S, Jiang L, Vasmatzis G, Gibson SE, Ondrejka S, et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood. 2014; 124:1473–80. https://doi.org/10.1182/blood-2014-04-571091.
10. Blombery P, Thompson ER, Jones K, Arnau GM, Lade S, Markham JF, Li J, Deva A, Johnstone RW, Khot A, Prince HM, Westerman D. Whole exome sequencing reveals activating JAK1 and STAT3 mutations in breast implant-associated anaplastic large cell lymphoma anaplastic large cell lymphoma. Haematologica. 2016; 101:e387–90. https://doi.org/10.3324/haematol.2016.146118.
11. Di Napoli A, Jain P, Duranti E, Margolskee E, Arancio W, Facchetti F, Alobeid B, Santanelli di Pompeo F, Mansukhani M, Bhagat G. Targeted next generation sequencing of breast implant-associated anaplastic large cell lymphoma reveals mutations in JAK/STAT signalling pathway genes, TP53 and DNMT3A. Br J Haematol. 2018; 180:741–4. https://doi.org/10.1111/bjh.14431.
12. Oishi N, Brody GS, Ketterling RP, Viswanatha DS, He R, Dasari S, Mai M, Benson HK, Sattler CA, Boddicker RL, McPhail ED, Bennani NN, Harless CA, et al. Genetic subtyping of breast implant-associated anaplastic large cell lymphoma. Blood. 2018; 132:544–547. https://doi.org/10.1182/blood-2017-12-821868.
13. Clemens MW, Horwitz SM. NCCN Consensus Guidelines for the Diagnosis and Management of Breast Implant-Associated Anaplastic Large Cell Lymphoma. Aesthet Surg J. 2017; 37:285–9. https://doi.org/10.1093/asj/sjw259.
14. Yoshimura A, Nishinakamura H, Matsumura Y, Hanada T. Negative regulation of cytokine signaling and immune responses by SOCS proteins. Arthritis Res Ther. 2005; 7:100–10. https://doi.org/10.1186/ar1741.
15. Mei W, Wang K, Huang J, Zheng X. Cell Transformation by PTP1B Truncated Mutants Found in Human Colon and Thyroid Tumors. PLoS One. 2016; 11:e0166538. https://doi.org/10.1371/journal.pone.0166538.
16. Xiao P, Wang X, Wang HM, Fu XL, Cui FA, Yu X, Wen SS, Bi WX, Sun JP. The second-sphere residue T263 is important for the function and catalytic activity of PTP1B via interaction with the WPD-loop. Int J Biochem Cell Biol. 2014; 57:84–95. https://doi.org/10.1016/j.biocel.2014.10.004.
17. Gunawardana J, Chan FC, Telenius A, Woolcock B, Kridel R, Tan KL, Ben-Neriah S, Mottok A, Lim RS, Boyle M, Rogic S, Rimsza LM, Guiter C, et al. Recurrent somatic mutations of PTPN1 in primary mediastinal B cell lymphoma and Hodgkin lymphoma. Nat Genet. 2014; 46:329–35. https://doi.org/10.1038/ng.2900.
18. Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J, Totoki Y, Chiba K, Sato-Otsubo A, Nagae G, Ishii R, Muto S, Kotani S, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet. 2015; 47:1304–15. https://doi.org/10.1038/ng.3415.
19. Bougeard G, Limacher JM, Martin C, Charbonnier F, Killian A, Delattre O, Longy M, Jonveaux P, Fricker JP, Stoppa-Lyonnet D, Flaman JM, Frebourg T. Detection of 11 germline inactivating TP53 mutations and absence of TP63 and HCHK2 mutations in 17 French families with Li-Fraumeni or Li-Fraumeni-like syndrome. J Med Genet. 2001; 38:253–7.
20. Zebisch A, Lal R, Muller M, Lind K, Kashofer K, Girschikofsky M, Fuchs D, Wolfler A, Geigl JB, Sill H. Acute myeloid leukemia with TP53 germ line mutations. Blood. 2016; 128:2270–2. https://doi.org/10.1182/blood-2016-08-732610.
21. Heitzer E, Lax S, Lafer I, Muller SM, Pristauz G, Ulz P, Jahn S, Hogenauer C, Petru E, Speicher MR, Geigl JB. Multiplex genetic cancer testing identifies pathogenic mutations in TP53 and CDH1 in a patient with bilateral breast and endometrial adenocarcinoma. BMC Med Genet. 2013; 14:129. https://doi.org/10.1186/1471-2350-14-129.
22. Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007; 28:622–9. https://doi.org/10.1002/humu.20495.
23. Fancello L, Kampen KR, Hofman IJ, Verbeeck J, De Keersmaecker K. The ribosomal protein gene RPL5 is a haploinsufficient tumor suppressor in multiple cancer types. Oncotarget. 2017; 8:14462–78. https://doi.org/10.18632/oncotarget.14895.
24. Boi M, Rinaldi A, Kwee I, Bonetti P, Todaro M, Tabbo F, Piva R, Rancoita PM, Matolcsy A, Timar B, Tousseyn T, Rodriguez-Pinilla SM, Piris MA, et al. PRDM1/BLIMP1 is commonly inactivated in anaplastic large T-cell lymphoma. Blood. 2013; 122:2683–93. https://doi.org/10.1182/blood-2013-04-497933.
25. Jiang ZH, Peng J, Yang HL, Fu XL, Wang JZ, Liu L, Jiang JN, Tan YF, Ge ZJ. Upregulation and biological function of transmembrane protein 119 in osteosarcoma. Exp Mol Med. 2017; 49:e329. https://doi.org/10.1038/emm.2017.41.
26. Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 Receptor in Infection and Inflammation. Immunity. 2017; 47:15–31. https://doi.org/10.1016/j.immuni.2017.06.020.
27. Shugay M, Bagaev DV, Zvyagin IV, Vroomans RM, Crawford JC, Dolton G, Komech EA, Sycheva AL, Koneva AE, Egorov ES, Eliseev AV, Van Dyk E, Dash P, et al. VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res. 2018; 46:D419–D27. https://doi.org/10.1093/nar/gkx760.
28. Kadin ME, Deva A, Xu H, Morgan J, Khare P, MacLeod RA, Van Natta BW, Adams WP Jr, Brody GS, Epstein AL. Biomarkers Provide Clues to Early Events in the Pathogenesis of Breast Implant-Associated Anaplastic Large Cell Lymphoma. Aesthet Surg J. 2016; 36:773–81. https://doi.org/10.1093/asj/sjw023.
29. Lechner MG, Megiel C, Church CH, Angell TE, Russell SM, Sevell RB, Jang JK, Brody GS, Epstein AL. Survival signals and targets for therapy in breast implant-associated ALK—anaplastic large cell lymphoma. Clin Cancer Res. 2012; 18:4549–59. https://doi.org/10.1158/1078-0432.CCR-12-0101.
30. Letourneau A, Maerevoet M, Milowich D, Dewind R, Bisig B, Missiaglia E, de Leval L. Dual JAK1 and STAT3 mutations in a breast implant-associated anaplastic large cell lymphoma. Virchows Arch. 2018; 473:505–511. https://doi.org/10.1007/s00428-018-2352-y.
31. Lee YS, Filie A, Arthur D, Fojo AT, Jaffe ES. Breast implant-associated anaplastic large cell lymphoma in a patient with Li-Fraumeni syndrome. Histopathology. 2015; 67:925–7. https://doi.org/10.1111/his.12737.
32. Pastorello RG, D'Almeida Costa F, Osório CABT, Makdissi FBA, Bezerra SM, de Brot M, Campos AHJFM, Soares FA, Vassallo J. Breast implant-associated anaplastic large cell lymphoma in a Li-FRAUMENI patient: a case report. Diagn Pathol. 2018; 13:10. https://doi.org/10.1186/s13000-018-0688-x.
33. Hofman IJF, van Duin M, De Bruyne E, Fancello L, Mulligan G, Geerdens E, Garelli E, Mancini C, Lemmens H, Delforge M, Vandenberghe P, Wlodarska I, Aspesi A, et al. RPL5 on 1p22.1 is recurrently deleted in multiple myeloma and its expression is linked to bortezomib response. Leukemia. 2017; 31:1706–14. https://doi.org/10.1038/leu.2016.370.
34. Ajore R, Raiser D, McConkey M, Joud M, Boidol B, Mar B, Saksena G, Weinstock DM, Armstrong S, Ellis SR, Ebert BL, Nilsson B. Deletion of ribosomal protein genes is a common vulnerability in human cancer, especially in concert with TP53 mutations. EMBO Mol Med. 2017; 9:498–507. https://doi.org/10.15252/emmm.201606660.
35. Rassidakis GZ, Thomaides A, Wang S, Jiang Y, Fourtouna A, Lai R, Medeiros LJ. p53 gene mutations are uncommon but p53 is commonly expressed in anaplastic large-cell lymphoma. Leukemia. 2005; 19:1663–9. https://doi.org/10.1038/sj.leu.2403840.
36. Wang H, Xie C, Li S, George EV, Reeves W, Epstein AL, Yang L. Dysregulation, But Not Mutation Of p53 Signaling Pathway In Breast Implant-Associated Anaplastic Large Cell Lymphoma Cell Lines. Blood. 2013; 122:4879.
37. Liao JM, Zhou X, Gatignol A, Lu H. Ribosomal proteins L5 and L11 co-operatively inactivate c-Myc via RNA-induced silencing complex. Oncogene. 2014; 33:4916–23. https://doi.org/10.1038/onc.2013.430.
38. Fata JE, Kong YY, Li J, Sasaki T, Irie-Sasaki J, Moorehead RA, Elliott R, Scully S, Voura EB, Lacey DL, Boyle WJ, Khokha R, Penninger JM. The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell. 2000; 103:41–50.
39. Tanos T, Sflomos G, Echeverria PC, Ayyanan A, Gutierrez M, Delaloye JF, Raffoul W, Fiche M, Dougall W, Schneider P, Yalcin-Ozuysal O, Brisken C. Progesterone/RANKL is a major regulatory axis in the human breast. Sci Transl Med. 2013; 5:182ra55. https://doi.org/10.1126/scitranslmed.3005654.
40. Dougall WC. Molecular pathways: osteoclast-dependent and osteoclast-independent roles of the RANKL/RANK/OPG pathway in tumorigenesis and metastasis. Clin Cancer Res. 2012; 18:326–35. https://doi.org/10.1158/1078-0432.CCR-10-2507.
41. Nolan E, Vaillant F, Branstetter D, Pal B, Giner G, Whitehead L, Lok SW, Mann GB, Rohrbach K, Huang LY, Soriano R, Smyth GK, Dougall WC, et al; Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab). RANK ligand as a potential target for breast cancer prevention in BRCA1-mutation carriers. Nat Med. 2016; 22:933–9. https://doi.org/10.1038/nm.4118.
42. Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, Karin M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature. 2011; 470:548–53. https://doi.org/10.1038/nature09707.
43. Spanoudakis E, Papoutselis M, Terpos E, Dimopoulos MA, Tsatalas C, Margaritis D, Rahemtulla A, Kotsianidis I, Karadimitris A. Overexpression of RANKL by invariant NKT cells enriched in the bone marrow of patients with multiple myeloma. Blood Cancer J. 2016; 6:e500. https://doi.org/10.1038/bcj.2016.108.
44. Cummings SR, San Martin J, McClung MR, Siris ES, Eastell R, Reid IR, Delmas P, Zoog HB, Austin M, Wang A, Kutilek S, Adami S, Zanchetta J, et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009; 361:756–65. https://doi.org/10.1056/NEJMoa0809493.
45. Lipton A, Fizazi K, Stopeck AT, Henry DH, Brown JE, Yardley DA, Richardson GE, Siena S, Maroto P, Clemens M, Bilynskyy B, Charu V, Beuzeboc P, et al. Superiority of denosumab to zoledronic acid for prevention of skeletal-related events: a combined analysis of 3 pivotal, randomised, phase 3 trials. Eur J Cancer. 2012; 48:3082–92. https://doi.org/10.1016/j.ejca.2012.08.002.
46. Laimer D, Dolznig H, Kollmann K, Vesely PW, Schlederer M, Merkel O, Schiefer AI, Hassler MR, Heider S, Amenitsch L, Thallinger C, Staber PB, Simonitsch-Klupp I, et al. PDGFR blockade is a rational and effective therapy for NPM-ALK-driven lymphomas. Nat Med. 2012; 18:1699–704. https://doi.org/10.1038/nm.2966.
47. Bonzheim I, Geissinger E, Roth S, Zettl A, Marx A, Rosenwald A, Muller-Hermelink HK, Rudiger T. Anaplastic large cell lymphomas lack the expression of T-cell receptor molecules or molecules of proximal T-cell receptor signaling. Blood. 2004; 104:3358–60. https://doi.org/10.1182/blood-2004-03-1037.
48. Ryland GL, Jones K, Chin M, Markham J, Aydogan E, Kankanige Y, Caruso M, Guinto J, Dickinson M, Prince HM, Yong K, Blombery P. Novel genomic findings in multiple myeloma identified through routine diagnostic sequencing. J Clin Pathol. 2018; 71:895–899. https://doi.org/10.1136/jclinpath-2018-205195.
49. Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, Chudakov DM. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods. 2015; 12:380–1. https://doi.org/10.1038/nmeth.3364.
50. Shugay M, Bagaev DV, Turchaninova MA, Bolotin DA, Britanova OV, Putintseva EV, Pogorelyy MV, Nazarov VI, Zvyagin IV, Kirgizova VI, Kirgizov KI, Skorobogatova EV, Chudakov DM. VDJtools: Unifying Post-analysis of T Cell Receptor Repertoires. PLoS Comput Biol. 2015; 11:e1004503. https://doi.org/10.1371/journal.pcbi.1004503.
51. Branstetter DG, Nelson SD, Manivel JC, Blay JY, Chawla S, Thomas DM, Jun S, Jacobs I. Denosumab induces tumor reduction and bone formation in patients with giant-cell tumor of bone. Clin Cancer Res. 2012; 18:4415–24. https://doi.org/10.1158/1078-0432.CCR-12-0578.