
INTRODUCTION
DNA mismatch repair (MMR) deficiency is a well characterised form of genetic instability in cancer [1–5], characterized by a failure to repair DNA replication-associated errors. A defective MMR system leads to the persistence of mismatched mutations across the genome, particularly in regions of repetitive DNA (microsatellites), leading to microsatellite instability (MSI). MSI causes the production of truncated protein products, resulting in the development of life-threatening malignancies [1–9].
Constitutional Mismatch Repair Deficiency (CMMRD) (also known as Biallelic Mismatch Repair Deficiency: BMMRD) is a hereditary cancer predisposition that presents in infancy or young adulthood at an incidence of approximately 1 per million patients [10]. CMMRD occurs as a result of mutations in well characterised MMR genes including mutS homolog 2 (MSH2); mutL homolog 1 (MLH1); mutS homolog 6 (MSH6); post-meiotic segregation increased 2 (PMS2); and post-meiotic segregation increased 1 (PMS1) [11, 12]. The major function of these genes are to eliminate the mismatch of base-base insertions and deletions that occur as a consequence of DNA polymerase errors during DNA synthesis (Figure 1) [13]. Single-nucleotide variations (SNV) result from errors during base pair incorporation whilst slippages of the polymerase result in insertions and deletions [14]. MMR genes act to promote genome stabilization through correcting these errors, ensuring the fidelity of genetic recombination and the initiation of apoptosis in response to DNA damage (Figure 1). MMR genes have been extensively studied and their contribution to disease has been reviewed in several reports [11, 15].
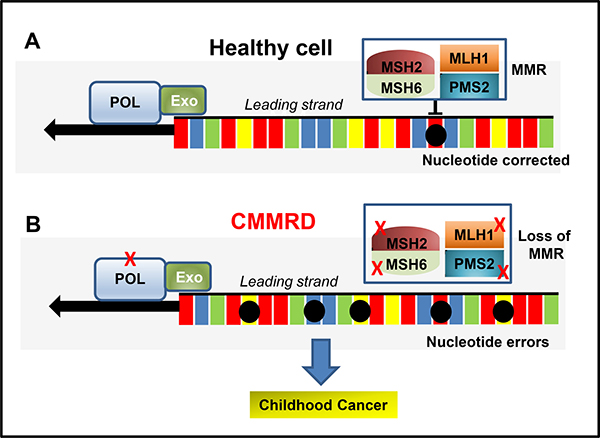
Figure 1: (A) Schematic of MMR in a healthy cell (adapted from [12]). The proofreading capability of the DNA polymerases (POL) and the MMR system recognises and prevent errors (black circles) during DNA replication. (B) CMMRD. Inherited MMR defects (X) that lead to a loss of MMR function/expression lead to accumulated mutations and a predisposition to cancer during adulthood. When a combination of mutations affect POL and MMR function, the accumulation of mutations become more rapid and the onset of cancer occurs in young children (CMMRD).
Heterozygous (monoallelic) mutations in MMR genes can impair MMR functionality resulting in a cancer condition termed Lynch Syndrome (LS), previously known as hereditary non-polyposis colorectal cancer (HNPCC) syndrome [9, 11, 16–19]. LS is characterized by gastrointestinal and genitourinary cancers during adulthood and represents 1–7% of all cases of colorectal cancer (CRC) [9, 11, 18]. For LS to be defined, germline mutations in at least one of the repair genes must be identified [17]. In contrast, biallelic germline mutations in the MMR genes that cause LS leads to CMMRD. The estimated carrier frequencies for mutations in the MMR-genes are 1 in 1946 for MLH1, 1 in 2841 for MSH2, 1 in 758 for MSH6 and 1 in 714 for PMS2 [20, 21]. If both parents have LS, the CMMRD risk to the siblings is 25% chance of having CMMRD, 25% chance of no LS mutations and a 50% chance of LS (Figure 2). Individuals with CMMRD develop a large variety of malignant neoplasms during early life with the majority of sufferer’s failing to reaching adulthood [8, 20, 22–27].
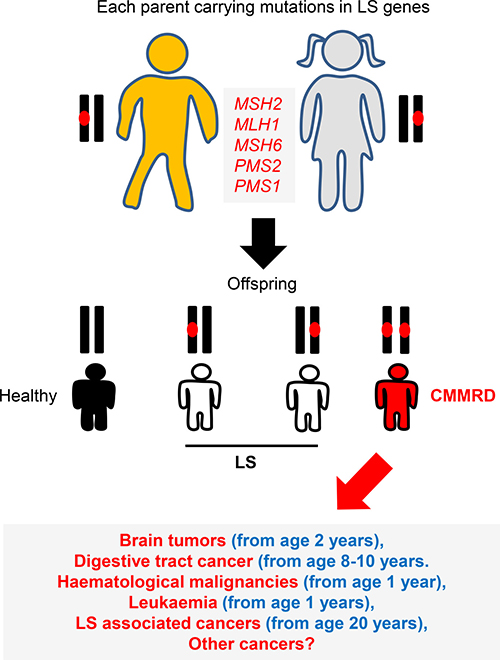
Figure 2: CMMRD genetics. LS is an autosomal dominant disorder caused by defects in one of DNA MMR genes. Siblings of two parents with LS can develop CMMRD (biallelic MMR mutations). The spectrum of cancers observed for CMMRD are more severe than those found in LS. Up to 50% of children develop brain tumours, around 50% digestive tract cancers and approximately 33% develop haematological malignancies.
HISTORY
Nearly twenty years ago, case reports presented a phenotype of offspring from consanguineous marriages within LS families (both carried MLH1 mutations) [28]. The offspring developed malignancies during early childhood. Of note, the individuals displayed clinical features reminiscent of neurofibromatosis type 1 (NF1) (tumor formation on nerve tissues), now commonly associated with CMMRD [28]. Since those studies, close to 200 paediatric and young adult CMMRD cancer cases have been reported in at least one of the MMR genes involved in LS [10, 29]. Contrary to traditional LS, CMMRD patients lack expression of the MMR protein(s) in both cancer and normal tissue (27). This recessively inherited condition is now fully recognised as a distinct childhood cancer predisposition syndrome [1, 30–33].
Although unproven at the molecular level, Jacques Turcot is attributed to have described the first cases of CMMRD in siblings with colorectal adenomatous polyps, colorectal carcinoma and malignant brain tumours [34]. ‘Turcot syndrome’ classically refers to a combination of colorectal polyposis and primary tumors of the Central Nervous System (CNS) [34–47]. CMMRD was referred to under Turcot syndrome for many years, until it was noted that this definition was too restrictive, as the manifestation of CMMRD also includes early-onset hematologic malignancies and cafe-au-lait spots suggestive of NF1 [31, 48]. Other non-neoplastic features have now emerged that are indicative of CMMRD in paediatric cancer patients, but until recently the CMMRD diagnostic criteria was lacking [49, 50]. The ability to accurately diagnose CMMRD is of critical importance to patient treatment and care, particularly in Arab and developing countries due to their high prevalence of consanguinity and increased susceptibility to this syndrome.
CMMRD DIAGNOSIS
The rapid identification of CMMRD is crucial for patient management and for afflicted family members. However, given the complex nature of CMMRD, diagnosis is often delayed or in some instances, not-stated. A major reason for the lack of CMMRD awareness amongst pediatric oncologists can be explained by the diagnostic difficulties that result from the lack of clear disease-specific clinical features that combine the full spectrum of CMMRD tumors. This has, on occasion, resulted in the refusal of standard CMMRD treatment due to families being unconvinced by the initial diagnostic criteria [5]. To address this issue, a newly established European consortium named “Care for CMMRD” developed a scoring system of clinical criteria used to confirm CMMRD diagnosis (summarized in Table 1). However, these guidelines are however difficult to follow due to the rarity of CMMRD cases and the diversity of cancer presentation [11, 12]. Various clinical characteristics that suggest CMRRD include any child or young adult with an LS associated tumor, hypermutated tumors, adenomatous polyposis, pediatric cancer in the setting of consanguinity, loss of MMR protein expression in normal and tumor tissues, and café au lait spots without NF1 diagnosis. It is also recommended to consider CMMRD syndrome in individuals with brain cancer, leukemia or lymphoma that lack a history of radiation exposure. As with the majority of autosomal recessive diseases, the index case of CMMRD typically lacks a family history of cancer that would raise a suspicion of CMMRD. Many of the cases identified to date are thus ascertained only after another sibling becomes affected by cancer.
Table 1: Diagnostic scoring criteria for CMMRD from the European consortium “Care for CMMRD” [10]
Indications for CMMRD-Testing |
More than 3 points |
|---|---|
Maligancies or pre-malignancies: one is mandatory. If more than one is present add points |
|
LS carcinoma* at age less than 25 years |
3 points |
Multiple bowel adenomas at age less than 25 years and absence of APC/MUTYH or a single grade dysplasic adenoma (also at age less than 25 years). |
3 points |
WHO grade III or IV glioma at age less than 25 years |
2 points |
NHL of T-cell lineage or sPNET at age less than 18 years |
2 points |
Any malignancy in a patient under 18 years |
1 point |
Additional features: if more than one of the following are present add points |
|
Clinical NF1 diagnosis or more than 2 hyper/hypo-skin pigmentations (greater than 1 cm) |
2 points |
Diagnosis of LS in a 1st and/or 2nd degree relative |
2 points |
LS carcinoma*, high grade glioma, sPNET, or NHL |
1 point |
Sibling with a childhood cancer |
1 point |
Multiple pilomatricomas present |
2 points |
One pilomatricoma present |
1 point |
Agenesis of the corpus callosum or non-therapy induced cavernoma |
1 point |
Consanguineous parents# |
1 point |
Deficiency/reduced levels of IgG2/4 and or IgA |
1 point |
LS cancers: CRC, endometrial, small bowel, renal pelvis, ureter, biliary tract, bladder, stomach.
#more common in Arab/Developing countries.
The evolution of sequencing technologies has benefited diagnosis as the rapid detection of characteristic CMMRD mutational patterns can be obtained from patient blood and tumor samples [51, 52]. This can be used simultaneously to facilitate downstream germline testing [25, 51, 52] and be used to offer genetic counseling to families with “at-risk” siblings. [8]. This information can also provide personalized targeted treatment programs, once the genetic basis of the CMMRD tumor is understood [52, 53].
CMMRD TUMOR SPECTRUM
The prognosis of CMMRD is poor; current statistics suggest over 50% of patients develop malignant brain tumours, 40% develop digestive tract tumours and 30% develop haematological malignancies, all during childhood [27]. The most frequent CMMRD cancers are brain gliomas (diagnosed at an average age of 9.5 years), non-Hodgkin’s lymphomas (diagnosed at 5 years) and colorectal cancers (CRCs) (diagnosed at 16 years) [10]. The cancer spectrum is related to the nature of the MMR gene mutated; patients with MSH6 and/or PMS2 mutations develop brain tumours within 10 years of life and over 40% of patients homozygous for PMS2 mutations develop second primary malignancies [29]. By comparison, patients homozygous for MLH1/MSH2 mutations are less likely to develop second primary malignancies (22%). This difference is because patients with homozygous PMS2 mutations often survive their first malignancy, unlike MLH1/MSH2 patients who develop more aggressive haematological malignancies [10]. Consanguinity of the parents and/or homozygosity for a founder mutation is observed in over 50% of CMMRD-cancers [10, 17] the most common of which are reviewed in [21].
CMMRD SURVEILLANCE
CMMRD tumors in the CNS are often observed during infancy, and as such MRI scanning is generally performed in the first two years of life. Brain MRI is recommended at diagnosis and every 6 months thereafter [54, 55]. Repeated CT scanning of the brain is not recommended due to the possible induction of tumours due to radiation [54]. For digestive tract cancers (including CRC), colonoscopy is used for surveillance. Colonic polyps have been reported in CMMRD patients as early as 6 years of age, so surveillance is generally initiated at this age [56]. Once polyps are identified, colonoscopy is performed every 6 months under general anaesthesia. Flat and non-polypoid lesions can be missed so colonoscopy is recommended for their detection [27, 54]. Colectomy is considered in patients with high-grade dysplasia as these present a significant risk of carcinoma [54]. Small bowel polyps develop later in life so upper endoscopy is recommended at 8 years of age in these patients. For the detection of duodenal cancers, upper GI endoscopy is performed at the same time as colonoscopy. Lymphoid and other hematologic malignancies represent the third most common malignancies in CMMRD, but effective tools for diagnosis are still lacking [57]. Information on the natural history of CMMRD lymphomas is sparse and the natural course of the disease may differ from sporadic cases [54]. These typically present with tumours and clinical manifestations a month prior to diagnosis. Standard surveillance is performed every six months including repeated blood counts and abdominal ultrasounds [27, 54]. This strategy is useful to assess the natural history of the lymphoma.
A modification to current protocols now includes the implementation of whole body MRI to be implemented once a year at 6 years of age (when anaesthesia is not needed). This is not recommended as a replacement for ultrasound and brain MRI, but may present benefits for CMMRD diagnosis as the spectrum of CMMRD cancers continues to increase [54].
CURRENT CMMRD TREATMENT
Children with CMMRD have a high risk of developing multiple cancers and early diagnosis does not guarantee detection at a curable stage [49, 50]. Preventive treatment strategies would represent a major advance for CMMRD therapy and the benefits of novel long-term conventional therapies such as acetylsalicylic acid (aspirin) are emerging [58]. As CMMRD is rare, information on its optimal therapeutic strategies are limited and current knowledge of treatment regimens and their outcomes are limited to individual case reports, often with variable disease phenotypes [29, 36, 39, 42, 46, 59–70]. Chemotherapy remains a frontline treatment, but toxicity is a major issue in children. Selection of appropriate chemotherapy drugs [71–74] is generally based on their toxicity profile [75, 76] and the knowledge of tumor resistance [71].
CHEMOTHERAPY
Many frequently used chemotherapeutic agents require a functional MMR system to initiate tumor damage. Accordingly, MMR deficient cells are frequently resistant to chemotherapeutics [77–95]. This includes resistance to mercaptopurine and temozolomide, drugs commonly used to treat hematopoietic and glioma cancers, respectively [55, 96–115]. MMR resistance is exemplified by temozolomide, the drug of choice for glioblastomas multiforme (GBM), a highly malignant CMMRD-brain tumor [24, 54, 116, 117]. The response to temozolomide is limited in CMMRD-related GBM and its use is now avoided due to its known ability to increase the accumulation of somatic mutations in patients, increasing the risk of secondary tumors [15, 71, 118]. No obvious lack of efficacy of other therapeutic agents such as alkylating agents has been reported, but effective chemotherapeutic therapies for CMMRD cancer are still lacking, and new effective therapies are urgently required [54].
CMMRD IMMUNOTHERAPY
The ultrahypermutation phenotype (≥ 100 mut/Mb compared to <10/MB in other childhood cancers) of CMMRD tumors does offer some opportunities for new approaches to treatment [117, 119]. When microsatellites in gene-encoding regions are mutated in CMMRD, the numerous frameshifts lead to the production of truncated and functionally inactive proteins that are frequently processed into mutanome-derived epitopes (termed neoantigens) that are presented to cytotoxic T lymphocytes (CTLs) [71]. Mutations in the exonuclease domain of the catalytic subunit of DNA polymerase epsilon (POLE) also exhibit such ultra mutated genomes (Figure.1). Neoantigen loads in CMMRD are substantially higher than in other cancer patients without the condition making these tumors more likely to be recognized by the immune system [120, 121]. Tumors that are recognized by the immune system have an improved prognosis. For example, greater densities of tumor-infiltrating lymphocytes are observed in cases of CRC with MSI [56, 122]. This improves their prognosis when compared to microsatellite stable CRCs and alters the response to chemotherapeutics. The use of NGS technologies can therefore guide treatment regimens and identify hypermutated tumors (≥100 mut/Mb) most likely to respond to immunotherapy. Meier and coworkers also recently performed signature extractions from 215 human CRC cases and 289 gastric adenocarcinomas revealing three novel MMR-associated signatures that strongly discriminate MS stable and unstable tumors, the knowledge of which can dictate treatment planning following identification [123].
CHECKPOINT INHIBITORS
In the presence of malignant tumors, immunoreactivity becomes compromised due to tumor induced immunosuppression. PD-L1 is overexpressed in many cancers and acts as a binding site for PD1. Binding of PD1 to PD-L1 within the tumor activates PD1 signalling, which in turn inhibits T cell activation allowing the tumor to evade immune attack [124, 125]. Inhibiting the interaction of PD1 and PD-L1 can thus enhance the anti-cancer T cell response and promote anti-tumor activity. This knowledge has been used to develop immunotherapies termed checkpoint inhibitors, that counteract the actions of proteins that impede the immune response to cancer. Blocking PD-1 in CMMRD tumors produces a significant clinical response and CMMRD tumors are more responsive to PD-1 blockers than MMR proficient tumors [117, 124, 125]. When PD1- blockers were used to treat children with CMMRD with recurrent GBM, shrinking of tumors through MRI was observed, indicating a successful clinical response [117]. Other recent data have demonstrated the effectiveness of checkpoint inhibitors in the treatment of some non-Hodgkin lymphomas [57]. This is of interest to CMMRD patients that often have non-Hodgkin’s lymphoma in addition to other cancers [26].
NEOANTIGEN VACCINATION
Vaccination with neoantigens is another promising approach for CMMRD cancer and neoantigen-loaded cell vaccinations are in clinical trials for CRC patients with MSI. Preliminary data from these trials suggests they are safe and well tolerated [126, 127]. Strong immune responses against neoantigen vaccines have been observed in LS patients that already show neoantigen-specific immune responses [126]. This holds promise for the use of adjuvant or preventive neoantigen-based vaccinations for CMMRD. However, the limited number of CMMRD patients means the identification of commonly mutated microsatellites is challenging. In addition, CMMRD vaccinations must be approached with caution since all cells of a CMMRD patient are MMR deficient [17–19] leading to a risk of autoimmune disease. It is likely that the combination of check-point inhibitors and neoantigen vaccination will hold the most promise for CMMRD. Indeed, our own recent studies highlight the benefits of incorporating genomic and/or molecular testing for CMMRD into routine paediatric oncology, whereby clinical care can identify a subset of patients likely to benefit from targeted treatment regimens, dependent on their MMR mutation status [53].
SUMMARY
CMMRD has emerged a rare childhood cancer syndrome and as such, consistency regarding its diagnosis and optimal treatment strategies have been challenging. The spectrum of CMMRD-associated tumours is broad and as sequencing technologies and thus diagnosis evolves, these are likely to expand further. The severity of CCMRD is well understood; CMMRD-patients possess a high risk of multiple cancers during childhood and typically do not survive to later life. Urgent CMMRD therapies are therefore required and hope in this area has been provided through immunotherapy interventions, including check-point-inhibitors and/or neoantigen vaccinations. The promise of anti-cancer immunotherapy must now be combined with our knowledge of the underlying genetic basis of CMMRD-tumors to produce personalised and targeted CMMRD treatment regimens. These will benefit current CMMRD patients, and future CMMRD-afflicted children.
Abbreviations
MMR: Mismatch repair; CMMRD: Constitutional Mismatch Repair Deficiency; LS: Lynch Syndrome; HNPCC: Hereditary Non-Polyposis Colorectal Cancer; CRC: Colorectal Cancer; BMMRD: Biallelic Mismatch Repair Deficiency; MSH2: mutS homolog 2; MLH1: mutL homolog 1; MSH6: mutS homolog 6; PMS2: post-meiotic segregation increased 2; PMS1: post-meiotic segregation increased 1; SNV: single-nucleotide variations; MSI: Microsatellite Instability; NF1: Neurofibromatosis type 1; GMB: glioblastoma multiforme; CTLs: Cytotoxic T lymphocytes; PD1: Programmed Cell Death Protein 1.
ACKNOWLEDGMENTS AND FUNDING
King Abdulaziz City for Science and Technology and the Saudi Human Genome Project for the technical support. This study was supported by King Fahad Medical City and Sanad Paediatric Cancer foundation.
CONFLICTS OF INTEREST
The author declares that she has no competing interests.
REFERENCES
1. Leicher LW, Lammertink MHA, Offerman SR, Morreau H, de Jong MM, de Groot JWB, van Westreenen HL, Vasen HFA, de Vos Tot Nederveen Cappel WH. Consequences of testing for mismatch repair deficiency of colorectal cancer in clinical practice. Scand J Gastroenterol. 2018; 53:632–636.
2. Haraldsdottir S, Roth R, Pearlman R, Hampel H, Arnold CA, Frankel WL. Mismatch repair deficiency concordance between primary colorectal cancer and corresponding metastasis. Fam Cancer. 2016; 15:253–260.
3. Chika N, Fukuchi M, Suzuki O, Ito T, Yamamoto A, Ishiguro T, Kumagai Y, Ishibashi K, Mochiki E, Ishida H. [Incidence and Characteristics of Mismatch Repair Protein Deficiency in Elderly Gastric Cancer Patients]. [Article in Japanese]. Gan To Kagaku Ryoho. 2016; 43:1298–1300.
4. Andrici J, Farzin M, Sioson L, Clarkson A, Watson N, Toon CW, Gill AJ. Mismatch repair deficiency as a prognostic factor in mucinous colorectal cancer. Mod Pathol. 2016; 29:266–274.
5. Urganci N, Genc DB, Kose G, Onal Z, Vidin OO. Colorectal Cancer due to Constitutional Mismatch Repair Deficiency Mimicking Neurofibromatosis I. Pediatrics. 2015; 136:e1047–1050.
6. Levi Z, Kariv R, Barnes-Kedar I, Goldberg Y, Half E, Morgentern S, Eli B, Baris HN, Vilkin A, Belfer RG, Niv Y, Elhasid R, Dvir R, et al. The gastrointestinal manifestation of constitutional mismatch repair deficiency syndrome: from a single adenoma to polyposis-like phenotype and early onset cancer. Clin Genet. 2015; 88:474–478.
7. Xiao X, Melton DW, Gourley C. Mismatch repair deficiency in ovarian cancer—molecular characteristics and clinical implications. Gynecol Oncol. 2014; 132:506–512.
8. Bruwer Z, Algar U, Vorster A, Fieggen K, Davidson A, Goldberg P, Wainwright H, Ramesar R. Predictive genetic testing in children: constitutional mismatch repair deficiency cancer predisposing syndrome. J Genet Couns. 2014; 23:147–155.
9. Poulogiannis G, Frayling IM, Arends MJ. DNA mismatch repair deficiency in sporadic colorectal cancer and Lynch syndrome. Histopathology. 2010; 56:167–179.
10. Wimmer K, Kratz CP, Vasen HF, Caron O, Colas C, Entz-Werle N, Gerdes AM, Goldberg Y, Ilencikova D, Muleris M, Duval A, Lavoine N, Ruiz-Ponte C, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium ‘care for CMMRD’ (C4CMMRD). J Med Genet. 2014; 51:355–365.
11. Silva FC, Valentin MD, Ferreira Fde O, Carraro DM, Rossi BM. Mismatch repair genes in Lynch syndrome: a review. Sao Paulo Med J. 2009; 127:46–51.
12. Hsieh P, Yamane K. DNA mismatch repair: molecular mechanism, cancer, and ageing. Mech Ageing Dev. 2008; 129:391–407.
13. Sijmons RH, Hofstra RM. Review: Clinical aspects of hereditary DNA Mismatch repair gene mutations. DNA Repair (Amst). 2016; 38:155–162.
14. Rattray AJ, Strathern JN. Error-prone DNA polymerases: when making a mistake is the only way to get ahead. Annu Rev Genet. 2003; 37:31–66.
15. Fedier A, Fink D. Mutations in DNA mismatch repair genes: implications for DNA damage signaling and drug sensitivity (review). Int J Oncol. 2004; 24:1039–1047.
16. Medina-Arana V, Delgado L, Bravo A, Martin J, Fernandez-Peralta AM, Gonzalez-Aguilera JJ. Tumor spectrum in lynch syndrome, DNA mismatch repair system and endogenous carcinogens. J Surg Oncol. 2012; 106:10–16.
17. Peltomaki P. Update on Lynch syndrome genomics. Fam Cancer. 2016; 15:385–393.
18. Blount J, Prakash A. The changing landscape of Lynch syndrome due to PMS2 mutations. Clin Genet. 2018; 94:61–69.
19. Cox VL, Saeed Bamashmos AA, Foo WC, Gupta S, Yedururi S, Garg N, Kang HC. Lynch Syndrome: Genomics Update and Imaging Review. Radiographics. 2018; 38:483–499.
20. Baris HN, Barnes-Kedar I, Toledano H, Halpern M, Hershkovitz D, Lossos A, Lerer I, Peretz T, Kariv R, Cohen S, Half EE, Magal N, Drasinover V, et al. Constitutional Mismatch Repair Deficiency in Israel: High Proportion of Founder Mutations in MMR Genes and Consanguinity. Pediatr Blood Cancer. 2016; 63:418–427.
21. Ponti G, Castellsague E, Ruini C, Percesepe A, Tomasi A. Mismatch repair genes founder mutations and cancer susceptibility in Lynch syndrome. Clin Genet. 2015; 87:507–516.
22. Wimmer K, Kratz CP. Constitutional mismatch repair-deficiency syndrome. Haematologica. 2010; 95:699–701.
23. Li L, Hamel N, Baker K, McGuffin MJ, Couillard M, Gologan A, Marcus VA, Chodirker B, Chudley A, Stefanovici C, Durandy A, Hegele RA, Feng BJ, et al. A homozygous PMS2 founder mutation with an attenuated constitutional mismatch repair deficiency phenotype. J Med Genet. 2015; 52:348–352.
24. Bakry D, Aronson M, Durno C, Rimawi H, Farah R, Alharbi QK, Alharbi M, Shamvil A, Ben-Shachar S, Mistry M, Constantini S, Dvir R, Qaddoumi I, et al. Genetic and clinical determinants of constitutional mismatch repair deficiency syndrome: report from the constitutional mismatch repair deficiency consortium. Eur J Cancer. 2014; 50:987–996.
25. Hoell JI, Gombert M, Ginzel S, Loth S, Landgraf P, Kafer V, Streiter M, Prokop A, Weiss M, Thiele R, Borkhardt A. Constitutional mismatch repair-deficiency and whole-exome sequencing as the means of the rapid detection of the causative MSH6 defect. Klin Padiatr. 2014; 226:357–361.
26. Ramachandra C, Challa VR, Shetty R. Constitutional mismatch repair deficiency syndrome: Do we know it. Indian J Hum Genet. 2014; 20:192–194.
27. Vasen HF, Ghorbanoghli Z, Bourdeaut F, Cabaret O, Caron O, Duval A, Entz-Werle N, Goldberg Y, Ilencikova D, Kratz CP, Lavoine N, Loeffen J, Menko FH, et al. Guidelines for surveillance of individuals with constitutional mismatch repair-deficiency proposed by the European Consortium “Care for CMMR-D” (C4CMMR-D). J Med Genet. 2014; 51:283–293.
28. Wang Q, Lasset C, Desseigne F, Frappaz D, Bergeron C, Navarro C, Ruano E, Puisieux A. Neurofibromatosis and early onset of cancers in hMLH1-deficient children. Cancer Res. 1999; 59:294–297.
29. Ramchander NC, Ryan NA, Crosbie EJ, Evans DG. Homozygous germ-line mutation of the PMS2 mismatch repair gene: a unique case report of constitutional mismatch repair deficiency (CMMRD). BMC Med Genet. 2017; 18:40.
30. Menko FH, Kaspers GL, Meijer GA, Claes K, van Hagen JM, Gille JJ. A homozygous MSH6 mutation in a child with cafe-au-lait spots, oligodendroglioma and rectal cancer. Fam Cancer. 2004; 3:123–127.
31. Trimbath JD, Petersen GM, Erdman SH, Ferre M, Luce MC, Giardiello FM. Cafe-au-lait spots and early onset colorectal neoplasia: a variant of HNPCC? Fam Cancer. 2001; 1:101–105.
32. Auclair J, Leroux D, Desseigne F, Lasset C, Saurin JC, Joly MO, Pinson S, Xu XL, Montmain G, Ruano E, Navarro C, Puisieux A, Wang Q. Novel biallelic mutations in MSH6 and PMS2 genes: gene conversion as a likely cause of PMS2 gene inactivation. Hum Mutat. 2007; 28:1084–1090.
33. Hegde MR, Chong B, Blazo ME, Chin LH, Ward PA, Chintagumpala MM, Kim JY, Plon SE, Richards CS. A homozygous mutation in MSH6 causes Turcot syndrome. Clin Cancer Res. 2005; 11:4689–4693.
34. Turcot J, Despres JP, St Pierre F. Malignant tumors of the central nervous system associated with familial polyposis of the colon: report of two cases. Dis Colon Rectum. 1959; 2:465–468.
35. Dipro S, Al-Otaibi F, Alzahrani A, Ulhaq A, Al Shail E. Turcot syndrome: a synchronous clinical presentation of glioblastoma multiforme and adenocarcinoma of the colon. Case Rep Oncol Med. 2012; 2012:720273.
36. Chung HJ, Oh ST, Kim JG, Kang WK. Turcot syndrome: a case report in an unsuspected setting. J Gastrointest Surg. 2012; 16:411–414.
37. Lusis EA, Travers S, Jost SC, Perry A. Glioblastomas with giant cell and sarcomatous features in patients with Turcot syndrome type 1: a clinicopathological study of 3 cases. Neurosurgery. 2010; 67:811–817; discussion 817.
38. Sjursen W, Bjornevoll I, Engebretsen LF, Fjelland K, Halvorsen T, Myrvold HE. A homozygote splice site PMS2 mutation as cause of Turcot syndrome gives rise to two different abnormal transcripts. Fam Cancer. 2009; 8:179–186.
39. Sanou R, Anxionnat R, Taillandier L, Bigard MA, Regent D, Bracard S. [Turcot syndrome: a case report]. [Article in French]. J Radiol. 2009; 90:842–844.
40. Giunti L, Cetica V, Ricci U, Giglio S, Sardi I, Paglierani M, Andreucci E, Sanzo M, Forni M, Buccoliero AM, Genitori L, Genuardi M. Type A microsatellite instability in pediatric gliomas as an indicator of Turcot syndrome. Eur J Hum Genet. 2009; 17:919–927.
41. Baehring J, Hui P, Piepmeier J, Bannykh SI. Anaplastic oligoastrocytoma in Turcot syndrome. J Neurooncol. 2009; 95:293–298.
42. Sarin S, Bernath A. Turcot syndrome (glioma polyposis): a case report. South Med J. 2008; 101:1273–1274.
43. Niizuma K, Fujimura M, Kumabe T, Tominaga T. Malignant transformation of high-grade astrocytoma associated with neurocysticercosis in a patient with Turcot syndrome. J Clin Neurosci. 2007; 14:53–55.
44. Lebrun C, Olschwang S, Jeannin S, Vandenbos F, Sobol H, Frenay M. Turcot syndrome confirmed with molecular analysis. Eur J Neurol. 2007; 14:470–472.
45. Kleinerman R, Marino J, Loucas E. Muir-Torre Syndrome / Turcot Syndrome overlap? A patient with sebaceous carcinoma, colon cancer, and a malignant astrocytoma. Dermatol Online J. 2012; 18:3.
46. Dora V, Diego G, Romulo B, Natalia O. First case report of turcot syndrome type 1 in Colombia. Case Rep Oncol Med. 2012; 2012:356384.
47. Miyaki M, Nishio J, Konishi M, Kikuchi-Yanoshita R, Tanaka K, Muraoka M, Nagato M, Chong JM, Koike M, Terada T, Kawahara Y, Fukutome A, Tomiyama J, et al. Drastic genetic instability of tumors and normal tissues in Turcot syndrome. Oncogene. 1997; 15:2877–2881.
48. Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, Krush AJ, Berk T, Cohen Z, Tetu B, Burger PC, Wood PA, Taqi F, et al. The molecular basis of Turcot's syndrome. N Engl J Med. 1995; 332:839–847.
49. Bodo S, Colas C, Buhard O, Collura A, Tinat J, Lavoine N, Guilloux A, Chalastanis A, Lafitte P, Coulet F, Buisine MP, Ilencikova D, Ruiz-Ponte C, et al. Diagnosis of Constitutional Mismatch Repair-Deficiency Syndrome Based on Microsatellite Instability and Lymphocyte Tolerance to Methylating Agents. Gastroenterology. 2015; 149:1017–29.e3.
50. Gorovoy IR, de Alba Campomanes A. A potential life-saving diagnosis—recognizing Turcot syndrome. J AAPOS. 2014; 18:186–188.
51. Adam R, Spier I, Zhao B, Kloth M, Marquez J, Hinrichsen I, Kirfel J, Tafazzoli A, Horpaopan S, Uhlhaas S, Stienen D, Friedrichs N, Altmuller J, et al. Exome Sequencing Identifies Biallelic MSH3 Germline Mutations as a Recessive Subtype of Colorectal Adenomatous Polyposis. Am J Hum Genet. 2016; 99:337–351.
52. Spier I, Kerick M, Drichel D, Horpaopan S, Altmuller J, Laner A, Holzapfel S, Peters S, Adam R, Zhao B, Becker T, Lifton RP, Holinski-Feder E, et al. Exome sequencing identifies potential novel candidate genes in patients with unexplained colorectal adenomatous polyposis. Fam Cancer. 2016; 15:281–288.
53. AlHarbi M, Ali Mobark N, AlMubarak L, Aljelaify R, AlSaeed M, Almutairi A, Alqubaishi F, Hussain ME, Balbaid AAO, Said Marie A, AlSubaie L, AlShieban S, AlTassan N, et al. Durable Response to Nivolumab in a Pediatric Patient with Refractory Glioblastoma and Constitutional Biallelic Mismatch Repair Deficiency. Oncologist. 2018 Aug 13. https://doi.org/10.1634/theoncologist.2018-0163. [Epub ahead of print].
54. Tabori U, Hansford JR, Achatz MI, Kratz CP, Plon SE, Frebourg T, Brugieres L. Clinical Management and Tumor Surveillance Recommendations of Inherited Mismatch Repair Deficiency in Childhood. Clin Cancer Res. 2017; 23:e32–e37.
55. Proske J, Walter L, Bumes E, Hutterer M, Vollmann-Zwerenz A, Eyupoglu IY, Savaskan NE, Seliger C, Hau P, Uhl M. Adaptive Immune Response to and Survival Effect of Temozolomide- and Valproic Acid-induced Autophagy in Glioblastoma. Anticancer Res. 2016; 36:899–905.
56. Guastadisegni C, Colafranceschi M, Ottini L, Dogliotti E. Microsatellite instability as a marker of prognosis and response to therapy: a meta-analysis of colorectal cancer survival data. Eur J Cancer. 2010; 46:2788–2798.
57. Merryman RW, Armand P, Wright KT, Rodig SJ. Checkpoint blockade in Hodgkin and non-Hodgkin lymphoma. Blood Adv. 2017; 1:2643–2654.
58. Leenders EKSM, Westdorp H, Brüggemann RJ, Loeffen J, Kratz C, Burn J, Hoogerbrugge N, Jongmans MCJ. Cancer prevention by aspirin in children with Constitutional Mismatch Repair Deficiency (CMMRD). Eur J Hum Genet. 2018; 26:1417–23.
59. Peserico L, Merli GA, Cortesi N, Canossi GC, Crisi G. The Turcot syndrome. Case report. Ital J Neurol Sci. 1981; 2:391–393.
60. Chowdhary UM, Boehme DH, Al-Jishi M. Turcot syndrome (glioma polyposis). Case report. J Neurosurg. 1985; 63:804–807.
61. Takayama H, Nakagawa K, Onozuka S, Ibata Y, Shinomiya Y, Haruyama K, Toya S. Nonfamilial turcot syndrome presenting with astrocytoma—case report. Neurol Med Chir (Tokyo). 1989; 29:606–609.
62. Schneider T, Victor S. The Turcot syndrome (glioma polyposis) and its neurosurgical significance. Case report. Neurosurg Rev. 1993; 16:327–330.
63. Fracasso P, Caviglia R, Grassi A, Lapenta R, Stigliano V, Franco D, Casale V. Turcot syndrome: case report and nosological aspects. J Exp Clin Cancer Res. 1997; 16:433–435.
64. McLaughlin MR, Gollin SM, Lese CM, Albright AL. Medulloblastoma and glioblastoma multiforme in a patient with Turcot syndrome: a case report. Surg Neurol. 1998; 49:295–301.
65. Tamiya T, Hamazaki S, Ono Y, Tokunaga K, Matsumoto K, Furuta T, Ohmoto T. Ganglioglioma in a patient with Turcot syndrome. Case report. J Neurosurg. 2000; 92:170–175.
66. Edouard A, Rat C, Edoh-Koffi P, Rivierez M, Landau-Ossondo M, Lombard F, Smadja D. [Turcot syndrome: the first case reported from Martinique]. [Article in French]. Gastroenterol Clin Biol. 2002; 26:797–798.
67. Okamoto H, Mineta T, Nakahara Y, Ichinose M, Shiraishi T, Tabuchi K. Molecular analysis of astrocytoma associated with Turcot syndrome type 1—case report. Neurol Med Chir (Tokyo). 2004; 44:124–128.
68. Ilencikova D. [Constitutional mismatch repair-deficiency syndrome (CMMR-D) - a case report of a family with biallelic MSH6 mutation]. [Article in Slovak]. Klin Onkol. 2012; 25:S34–38.
69. Daou B, Zanello M, Varlet P, Brugieres L, Jabbour P, Caron O, Lavoine N, Dhermain F, Willekens C, Beuvon F, Malka D, Lechapt-Zalcmann E, Abi Lahoud G. An Unusual Case of Constitutional Mismatch Repair Deficiency Syndrome With Anaplastic Ganglioglioma, Colonic Adenocarcinoma, Osteosarcoma, Acute Myeloid Leukemia, and Signs of Neurofibromatosis Type 1: Case Report. Neurosurgery. 2015; 77:E145–152; discussion E152.
70. Corbera-Hincapie M, Beasley GL. A Challenging Treatment Decision for a Rare Association: Case Report of Familial Turcot Syndrome With Fistulizing Crohn’s Disease. Front Pediatr. 2018; 6:83.
71. Hunter C, Smith R, Cahill DP, Stephens P, Stevens C, Teague J, Greenman C, Edkins S, Bignell G, Davies H, O’Meara S, Parker A, Avis T, et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006; 66:3987–3991.
72. Ramirez LY, Huestis SE, Yap TY, Zyzanski S, Drotar D, Kodish E. Potential chemotherapy side effects: what do oncologists tell parents. Pediatr Blood Cancer. 2009; 52:497–502.
73. Cordelli DM, Masetti R, Zama D, Toni F, Castelli I, Ricci E, Franzoni E, Pession A. Central Nervous System Complications in Children Receiving Chemotherapy or Hematopoietic Stem Cell Transplantation. Front Pediatr. 2017; 5:105.
74. Schmiegelow K, Muller K, Mogensen SS, Mogensen PR, Wolthers BO, Stoltze UK, Tuckuviene R, Frandsen T. Non-infectious chemotherapy-associated acute toxicities during childhood acute lymphoblastic leukemia therapy. F1000Res. 2017; 6:444.
75. Liu Y, Gu F, Liang J, Dai X, Wan C, Hong X, Zhang K, Liu L. The efficacy and toxicity profile of metronomic chemotherapy for metastatic breast cancer: A meta-analysis. PLoS One. 2017; 12:e0173693.
76. Saletta F, Seng MS, Lau LM. Advances in paediatric cancer treatment. Transl Pediatr. 2014; 3:156–182.
77. Fink D, Aebi S, Howell SB. The role of DNA mismatch repair in drug resistance. Clin Cancer Res. 1998; 4:1–6.
78. Legrand M, Chan CL, Jauert PA, Kirkpatrick DT. Role of DNA mismatch repair and double-strand break repair in genome stability and antifungal drug resistance in Candida albicans. Eukaryot Cell. 2007; 6:2194–2205.
79. Xu J, Zhu W, Xu W, Cui X, Chen L, Ji S, Qin Y, Yao W, Liu L, Liu C, Long J, Li M, Yu X. Silencing of MBD1 reverses pancreatic cancer therapy resistance through inhibition of DNA damage repair. Int J Oncol. 2013; 42:2046–2052.
80. Bharthuar A, Saif Ur Rehman S, Black JD, Levea C, Malhotra U, Mashtare TL, Iyer R. Breast cancer resistance protein (BCRP) and excision repair cross complement-1 (ERCC1) expression in esophageal cancers and response to cisplatin and irinotecan based chemotherapy. J Gastrointest Oncol. 2014; 5:253–258.
81. Joshi PM, Sutor SL, Huntoon CJ, Karnitz LM. Ovarian cancer-associated mutations disable catalytic activity of CDK12, a kinase that promotes homologous recombination repair and resistance to cisplatin and poly(ADP-ribose) polymerase inhibitors. J Biol Chem. 2014; 289:9247–9253.
82. Duan S, Tsai Y, Keng P, Chen Y, Lee SO, Chen Y. IL-6 signaling contributes to cisplatin resistance in non-small cell lung cancer via the up-regulation of anti-apoptotic and DNA repair associated molecules. Oncotarget. 2015; 6:27651–27660. https://doi.org/10.18632/oncotarget.4753.
83. Jain R, Jamal S, Goyal S, Wahi D, Singh A, Grover A. Resisting the Resistance in Cancer: Cheminformatics Studies on Short- Path Base Excision Repair Pathway Antagonists Using Supervised Learning Approaches. Comb Chem High Throughput Screen. 2015; 18:881–891.
84. Chen KH, Yuan CT, Tseng LH, Shun CT, Yeh KH. Case report: mismatch repair proficiency and microsatellite stability in gastric cancer may not predict programmed death-1 blockade resistance. J Hematol Oncol. 2016; 9:29.
85. Modi S, Kir D, Giri B, Majumder K, Arora N, Dudeja V, Banerjee S, Saluja AK. Minnelide Overcomes Oxaliplatin Resistance by Downregulating the DNA Repair Pathway in Pancreatic Cancer. J Gastrointest Surg. 2016; 20:13–23; discussion 23–14.
86. Sadik H, Korangath P, Nguyen NK, Gyorffy B, Kumar R, Hedayati M, Teo WW, Park S, Panday H, Munoz TG, Menyhart O, Shah N, Pandita RK, et al. HOXC10 Expression Supports the Development of Chemotherapy Resistance by Fine Tuning DNA Repair in Breast Cancer Cells. Cancer Res. 2016; 76:4443–4456.
87. Wang S, Liu F, Zhu J, Chen P, Liu H, Liu Q, Han J. DNA Repair Genes ERCC1 and BRCA1 Expression in Non-Small Cell Lung Cancer Chemotherapy Drug Resistance. Med Sci Monit. 2016; 22:1999–2005.
88. Haricharan S, Ellis MJ. Defects in mismatch repair: the Achilles heel of estrogen receptor positive breast cancer with intrinsic endocrine therapy resistance. Oncoscience. 2017; 4:77–78. https://doi.org/10.18632/oncoscience.363.
89. Jiang J, Lu Y, Li Z, Li L, Niu D, Xu W, Liu J, Fu L, Zhou Z, Gu Y, Xia F. Ganetespib overcomes resistance to PARP inhibitors in breast cancer by targeting core proteins in the DNA repair machinery. Invest New Drugs. 2017; 35:251–259.
90. Ye L, Jiang T, Shao H, Zhong L, Wang Z, Liu Y, Tang H, Qin B, Zhang X, Fan J. miR-1290 Is a Biomarker in DNA-Mismatch-Repair-Deficient Colon Cancer and Promotes Resistance to 5-Fluorouracil by Directly Targeting hMSH2. Mol Ther Nucleic Acids. 2017; 7:453–464.
91. Anurag M, Punturi N, Hoog J, Bainbridge MN, Ellis MJ, Haricharan S. Comprehensive profiling of DNA repair defects in breast cancer identifies a novel class of endocrine therapy resistance drivers. Clin Cancer Res. 2018; 24:4887–4899.
92. Cruz C, Castroviejo-Bermejo M, Gutierrez-Enriquez S, Llop-Guevara A, Ibrahim YH, Gris-Oliver A, Bonache S, Morancho B, Bruna A, Rueda OM, Lai Z, Polanska UM, Jones GN, et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann Oncol. 2018; 29:1203–1210.
93. Farhane Z, Bonnier F, Howe O, Casey A, Byrne HJ. Doxorubicin kinetics and effects on lung cancer cell lines using in vitro Raman micro-spectroscopy: binding signatures, drug resistance and DNA repair. J Biophotonics. 2018; 11.
94. Mills A, Zadeh S, Sloan E, Chinn Z, Modesitt SC, Ring KL. Indoleamine 2,3-dioxygenase in endometrial cancer: a targetable mechanism of immune resistance in mismatch repair-deficient and intact endometrial carcinomas. Mod Pathol. 2018; 31:1282–1290.
95. Pajic M, Froio D, Daly S, Doculara L, Millar E, Graham PH, Drury A, Steinmann A, de Bock CE, Boulghourjian A, Zaratzian A, Carroll S, Toohey J, et al. miR-139-5p Modulates Radiotherapy Resistance in Breast Cancer by Repressing Multiple Gene Networks of DNA Repair and ROS Defense. Cancer Res. 2018; 78:501–515.
96. Boggs DH, Robins HI, Langer CJ, Traynor AM, Berkowitz MJ, Mehta MP. Strategies to prevent brain metastasis in high-risk non-small-cell lung cancer: lessons learned from a randomized study of maintenance temozolomide versus observation. Clin Lung Cancer. 2014; 15:433–440.
97. Jiang G, Jiang AJ, Xin Y, Li LT, Cheng Q, Zheng JN. Progression of O-methylguanine-DNA methyltransferase and temozolomide resistance in cancer research. Mol Biol Rep. 2014; 41:6659–6665.
98. Cao KI, Lebas N, Gerber S, Levy C, Le Scodan R, Bourgier C, Pierga JY, Gobillion A, Savignoni A, Kirova YM. Phase II randomized study of whole-brain radiation therapy with or without concurrent temozolomide for brain metastases from breast cancer. Ann Oncol. 2015; 26:89–94.
99. Farace C, Oliver JA, Melguizo C, Alvarez P, Bandiera P, Rama AR, Malaguarnera G, Ortiz R, Madeddu R, Prados J. Microenvironmental Modulation of Decorin and Lumican in Temozolomide-Resistant Glioblastoma and Neuroblastoma Cancer Stem-Like Cells. PLoS One. 2015; 10:e0134111.
100. Jaiswal AS, Panda H, Law BK, Sharma J, Jani J, Hromas R, Narayan S. NSC666715 and Its Analogs Inhibit Strand-Displacement Activity of DNA Polymerase beta and Potentiate Temozolomide-Induced DNA Damage, Senescence and Apoptosis in Colorectal Cancer Cells. PLoS One. 2015; 10:e0123808.
101. Kim SS, Rait A, Kim E, DeMarco J, Pirollo KF, Chang EH. Encapsulation of temozolomide in a tumor-targeting nanocomplex enhances anti-cancer efficacy and reduces toxicity in a mouse model of glioblastoma. Cancer Lett. 2015; 369:250–258.
102. Lacin S, Esin E, Karakas Y, Yalcin S. Metastatic medullary thyroid cancer: a dramatic response to a systemic chemotherapy (temozolomide and capecitabine) regimen. Onco Targets Ther. 2015; 8:1039–1042.
103. Lai IC, Shih PH, Yao CJ, Yeh CT, Wang-Peng J, Lui TN, Chuang SE, Hu TS, Lai TY, Lai GM. Elimination of cancer stem-like cells and potentiation of temozolomide sensitivity by Honokiol in glioblastoma multiforme cells. PLoS One. 2015; 10:e0114830.
104. Rosiere R, Gelbcke M, Mathieu V, Van Antwerpen P, Amighi K, Wauthoz N. New dry powders for inhalation containing temozolomide-based nanomicelles for improved lung cancer therapy. Int J Oncol. 2015; 47:1131–1142.
105. Smith MA, Reynolds CP, Kang MH, Kolb EA, Gorlick R, Carol H, Lock RB, Keir ST, Maris JM, Billups CA, Lyalin D, Kurmasheva RT, Houghton PJ. Synergistic activity of PARP inhibition by talazoparib (BMN 673) with temozolomide in pediatric cancer models in the pediatric preclinical testing program. Clin Cancer Res. 2015; 21:819–832.
106. Yang L, Wang Y, Guo H, Guo M. Synergistic Anti-Cancer Effects of Icariin and Temozolomide in Glioblastoma. Cell Biochem Biophys. 2015; 71:1379–1385.
107. Zhang J, Hummersone M, Matthews CS, Stevens MF, Bradshaw TD. N3-substituted temozolomide analogs overcome methylguanine-DNA methyltransferase and mismatch repair precipitating apoptotic and autophagic cancer cell death. Oncology. 2015; 88:28–48.
108. Pietrantonio F, de Braud F, Milione M, Maggi C, Iacovelli R, Dotti KF, Perrone F, Tamborini E, Caporale M, Berenato R, Leone G, Pellegrinelli A, Bossi I, et al. Dose-Dense Temozolomide in Patients with MGMT-Silenced Chemorefractory Colorectal Cancer. Target Oncol. 2016; 11:337–343.
109. Halperin EC, Herndon J, Schold SC, Brown M, Vick N, Cairncross JG, Macdonald DR, Gaspar L, Fischer B, Dropcho E, Rosenfeld S, Morowitz R, Piepmeier J, et al. A phase III randomized prospective trial of external beam radiotherapy, mitomycin C, carmustine, and 6-mercaptopurine for the treatment of adults with anaplastic glioma of the brain. CNS Cancer Consortium. Int J Radiat Oncol Biol Phys. 1996; 34:793–802.
110. Balis FM, Holcenberg JS, Poplack DG, Ge J, Sather HN, Murphy RF, Ames MM, Waskerwitz MJ, Tubergen DG, Zimm S, Gilchrist GS, Bleyer WA. Pharmacokinetics and pharmacodynamics of oral methotrexate and mercaptopurine in children with lower risk acute lymphoblastic leukemia: a joint children’s cancer group and pediatric oncology branch study. Blood. 1998; 92:3569–3577.
111. Lokich J, Moore C, Anderson N, Bern M, Coco F, Dow E, Zipoli T. Phase I-II trial of 14 day infusional 6-mercaptopurine in advanced colorectal cancer. Eur J Cancer. 1998; 34:584–585.
112. Alvarez Delgado A, Perez Garcia ML, Fradejas Salazar PM, de la Coba Ortiz C, Rodriguez Perez A. [Invasive cancer of the cervix in a patient undergoing chronic treatment with 6-mercaptopurine for Crohns disease]. [Article in Spanish]. Gastroenterol Hepatol. 2003; 26:52–53.
113. Bostrom BC, Sensel MR, Sather HN, Gaynon PS, La MK, Johnston K, Erdmann GR, Gold S, Heerema NA, Hutchinson RJ, Provisor AJ, Trigg ME, Children’s Cancer G. Dexamethasone versus prednisone and daily oral versus weekly intravenous mercaptopurine for patients with standard-risk acute lymphoblastic leukemia: a report from the Children’s Cancer Group. Blood. 2003; 101:3809–3817.
114. Tolar J, Bostrom BC, La MK, Sather HN. Intravenous 6-mercaptopurine decreases salvage after relapse in childhood acute lymphoblastic leukemia: a report from the Children’s Cancer Group study CCG 1922. Pediatr Blood Cancer. 2005; 45:5–9.
115. Jacobs SS, Stork LC, Bostrom BC, Hutchinson R, Holcenberg J, Reaman GH, Erdmann G, Franklin J, Neglia JP, Steinberg SM, Balis FM, Adamson PC, Children’s Oncology G, National Cancer I. Substitution of oral and intravenous thioguanine for mercaptopurine in a treatment regimen for children with standard risk acute lymphoblastic leukemia: a collaborative Children’s Oncology Group/National Cancer Institute pilot trial (CCG-1942). Pediatr Blood Cancer. 2007; 49:250–255.
116. Durno CA, Aronson M, Tabori U, Malkin D, Gallinger S, Chan HS. Oncologic surveillance for subjects with biallelic mismatch repair gene mutations: 10 year follow-up of a kindred. Pediatr Blood Cancer. 2012; 59:652–656.
117. Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, Durno C, Krueger J, Cabric V, Ramaswamy V, Zhukova N, Mason G, Farah R, et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J Clin Oncol. 2016; 34:2206–2211.
118. Scott RH, Mansour S, Pritchard-Jones K, Kumar D, MacSweeney F, Rahman N. Medulloblastoma, acute myelocytic leukemia and colonic carcinomas in a child with biallelic MSH6 mutations. Nat Clin Pract Oncol. 2007; 4:130–134.
119. Nebot-Bral L, Brandao D, Verlingue L, Rouleau E, Caron O, Despras E, El-Dakdouki Y, Champiat S, Aoufouchi S, Leary A, Marabelle A, Malka D, Chaput N, Kannouche PL. Hypermutated tumours in the era of immunotherapy: The paradigm of personalised medicine. Eur J Cancer. 2017; 84:290–303.
120. Wang RF, Wang HY. Immune targets and neoantigens for cancer immunotherapy and precision medicine. Cell Res. 2017; 27:11–37.
121. Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017; 17:209–222. Erratum in: Targeting neoantigens to augment antitumour immunity. [Nat Rev Cancer. 2017].
122. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010; 138:2073–2087.e3.
123. Meier B, Volkova NV, Hong Y, Schofield P, Campbell PJ, Gerstung M, Gartner A. Mutational signatures of DNA mismatch repair deficiency in C. elegans and human cancers. Genome Res. 2018; 28:666–675.
124. Ma W, Gilligan BM, Yuan J, Li T. Current status and perspectives in translational biomarker research for PD-1/PD-L1 immune checkpoint blockade therapy. J Hematol Oncol. 2016; 9:47.
125. Tan S, Zhang CW, Gao GF. Seeing is believing: anti-PD-1/PD-L1 monoclonal antibodies in action for checkpoint blockade tumor immunotherapy. Signal Transduct Target Ther. 2016; 1:16029.
126. Schwitalle Y, Kloor M, Eiermann S, Linnebacher M, Kienle P, Knaebel HP, Tariverdian M, Benner A, von Knebel Doeberitz M. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology. 2008; 134:988–997.
127. Kloor M, Michel S, von Knebel Doeberitz M. Immune evasion of microsatellite unstable colorectal cancers. Int J Cancer. 2010; 127:1001–1010.