
INTRODUCTION
Different genetic and epigenetic modifications have been reported in Wilms tumour (WT), a paediatric kidney malignancy [1, 2]. Interestingly, although a loss of imprinting on chromosome 11p15 is found in approximately 70% of cases, each of the genes identified to date appear to be mutated only in a relatively limited subset of cases [3]. WT1, the first WT gene to be identified, is involved in no more than 10% of sporadic cases, whereas anomalies affecting the TP53 gene are restricted to anaplastic WTs. CTNNB1 mutations, found in approximately 15% of cases, often co-occur with WT1 mutations, whereas abnormalities of the WTX gene on chromosome X were reported to be involved in 7-29% of cases [4, 5]. Even mutations involving the most recently discovered WT genes SIX1, SIX2, and microRNA processing genes, do not exceed 20% of cases, and often overlap with other genetic aberrations, mainly 11p15 anomalies [6, 7]. Thus, there is a number of sporadic cases in which no mutation in the known WT genes, are found, suggesting that further genes possibly involved in WT etiology, are still to be disclosed.
In the attempt to reduce the number of WT cases with unknown genetic cause, we aimed at using data derived from a high throughput approach, whole-genome SNP array with an average resolution of 8Kb [8, 9], to disclose further genes possibly involved in WT development. In particular, we investigated all detected focal chromosomal anomalies and, after excluding those reported as constitutionally polymorphic (http://projects.tcag.ca/variation/), seven different regions in which genes with a putative role in tumourigenesis and/or kidney organogenesis are mapped, were selected.
RESULTS
Selection of chromosome regions and genes
Following the review of the SNP-array data obtained as previously reported [8, 9], chromosome regions and genes involved in focal anomalies were identified. Among these, we selected, through literature data-mining, seven regions harbouring genes with a role in tumourigenesis and/or kidney development (Table 1).
Table 1: Selected CN aberrations observed in 96 Wilms tumours (WTs)
Chromosome region |
Length of focal anomaly |
Focal Anomaly |
Number of WTs affected by focal anomaly |
Larger anomaly |
Number of WTs affected by larger anomaly |
Total number of affected WTs (%) |
Genes of interest |
|---|---|---|---|---|---|---|---|
2p24.3 |
507kb |
CNNa and AIb |
1 primary |
CNNa and AIb |
5 primary and 1 recurrence |
12.5 |
MYCN [8] |
CNGc and AIb |
4 primary and 1 recurrence |
||||||
2q37.1 |
49kb |
HDe |
2 primary |
CNNa and AIb |
4 primary |
7.2 |
DIS3L2 and miR-562 [8] |
CNGc and AIb |
1 primary |
||||||
6q21 |
1542kb |
CNLd and LOHf |
1 primary |
CNGc and AIb |
1 primary |
15.6 |
HACE1 |
CNGc |
1 recurrence |
Supplementary Figure 1 |
|||||
CNLd and LOHf |
1 primary |
||||||
CNNa and AIb |
10 primary |
||||||
CNNa and LOHf |
1 primary |
||||||
7p14.1 |
1325kb |
CNLd and LOHf |
1 primary |
CNGc and a AIb |
1 primary |
20.8 |
GLI3 |
CNLd and AIb |
1 primary |
Supplementary Figure 2 |
|||||
CNLd and LOHf |
8 primary and 1 recurrence |
||||||
CNNa and AIb |
7 primary and 1 recurrence |
||||||
9p21.3 |
206kb |
HDe |
1 primary |
CNLd and AIb |
1 primary |
8.3 |
CDKN2A and CDKN2B |
CNNa and AIb |
4 primary and 1 recurrence |
Supplementary Figure 3 |
|||||
CNNa and LOHf |
1 primary |
||||||
16p12.1 |
780kb |
CNLd and LOH |
1 primary |
CNLd and LOHf |
1 primary |
6.2 |
PALB2 |
CNNa and AIb |
3 primary |
Supplementary Figure 4 |
|||||
CNNa and LOHf |
1 recurrence |
||||||
22q12.1 |
994kb |
CNNa and AIb |
1 primary |
CNLd and LOHf |
7 primary and 1 recurrence |
13.5 |
CHEK2 |
CNNa and LOHf |
1 primary |
Supplementary Figure 5 |
|||||
CNNa and AIb |
3 primary |
Abbreviations: aCNN: copy number neutral, bAI: allelic imbalance, cCNG: copy number gain, dCNL: copy number loss, eHD: homozygous deletion, fLOH: loss of heterozygosity
Two of these regions 2p24.3, containing the MYCN gene, and 2q37, containing the DIS3L2 and MIR562 were previously described [8, 9]. On chromosomes 2p24.3 focal overlapping anomalies ranging from 507 to 825 kb were observed in six samples. One primary WT showed allelic imbalance, four primary WTs showed both low level CN (copy number) gain and allelic imbalance. The anomaly observed in one of the latter primaries was detected also in its corresponding recurrent tumour. Further six samples showed larger allelic imbalances on chromosome 2p affecting also the MYCN region. In the DIS3L2 chromosomal region we observed two cases with homozygous deletions on 2q37 occurring within a wider region of loss of heterozygosity (LOH), leading to the complete deletion of the genes DIS3L2 and miR-562 [8]. Five other samples showed larger anomalies on chromosome 2q in overlapping regions (allelic imbalance in four cases, CN gain and allelic imbalance in one).
Five additional selected regions were mapped to chromosomes 6q, 7p, 9p, 16p and 22q. In one case, we found focal CN loss and LOH on chromosome 6q21 (chr6:103,951,059-105,493,480), involving only the HACE1 gene (Supplementary Figure 1). Anomalies spanning HACE1 were found in additional 14 cases (allelic imbalance and CN gain in one WT, CN gain in one WT, CN loss and LOH in one WT, allelic imbalance in ten WTs and LOH in one WT). In one case, we found focal CN loss and LOH on chromosome 7p14.1 (chr7:41,801,272-43,126,820), involving the GLI3 gene (Supplementary Figure 2). Other 19 WTs showed larger overlapping chromosomal anomalies on 7p (allelic imbalance and CN gain in one WT, CN loss and allelic imbalance in one WT, CN loss and LOH in nine WTs and allelic imbalance in eight WTs). In one WT sample, we found a homozygous deletion spanning approximately 206kb on chromosome 9p21.3 (chr9:21,806,162-22,012,894), involving the CDKN2A and CDKN2B genes (Supplementary Figure 3). Additional seven cases showed larger overlapping anomalies on 9p, in particular, CN loss and allelic imbalance in one WT, allelic imbalance in five WTs and LOH in one WT. In one case, we found a focal chromosomal region of CN loss and LOH spanning approximately 780kb on chromosome 16p12.1 (chr16:23,516,852-24,297,082), involving the PALB2 gene (Supplementary Figure 4). Other five WTs showed bigger anomalies on chromosome 16p affecting the same chromosomal region (CN loss and LOH was detected in one WT, allelic imbalance in three WTs and LOH in one). In one case, we found focal allelic imbalance spanning approximately 994kb on chromosome 22q12.1 (chr22:26,777,048-27,771,791) involving CHEK2 (Supplementary Figure 5). Other 12 tumours showed larger overlapping anomalies on chromosome 22q (CN loss and LOH were found in eight WTs, LOH in one WT and allelic imbalance in three WTs).
DNA sequence analysis
The previously reported MYCN c.131C>T (p.Pro44Leu) [10] hotspot was found to be mutated in seven out of 96 (ca. 7.3%) WTs.
In the remaining genes (DIS3L2, HACE1, GLI3, CDKN2A and CDKN2B, PALB2 and CHEK2), the entire coding region and corresponding intron/exon junctions were sequenced. Excluding common variants, i.e., those reported with a MAF > 0.01 in outbred populations, a total of 25 different alterations were identified, among which 16 missense, eight synonymous and a splice-site variant. Of these, 22 have been already described in genome databases with a MAF < 0.01. Of the three previously unreported variants, two were in GLI3 and one in CHEK2 (Table 2).
Table 2: Identified variations and in silico analyses
Gene |
Mutation |
Aminoacidic change |
heta/homb |
Recc |
SNP |
MAFd |
In silico prediction |
||
|---|---|---|---|---|---|---|---|---|---|
heta/homb |
SIFTe |
Polyphen2f |
Align-GVGDg |
||||||
MYCN |
c.131C>T |
p.Pro44Leu |
7/0 |
rs1057519919 |
N.A.h |
Damaging |
Probably Damaging |
||
DIS3L2 |
c.301G>T |
p.Ala101Ser |
1/0 |
rs199857926 |
1.58e-04 |
Tolerated |
Benign |
||
DIS3L2 |
c.410A>G |
p.Tyr137Cys |
1/0 |
rs201733073 |
0.001 |
Tolerated |
Benign |
||
DIS3L2 |
c.1377C>T |
p.Ser459= |
1/0 |
rs376722215 |
3.65e-05 |
||||
DIS3L2 |
c.1430T>G |
p.Leu477Arg |
1/0 |
rs201719374 |
6.50e-05 |
Damaging |
Probably Damaging |
||
DIS3L2 |
c.2424G>A |
p.Gln808= |
1/0 |
rs369113667 |
8.05e-05 |
||||
HACE1 |
c.1196A>G |
p.Asp399Gly |
3/0 |
rs34365906 |
0.005 |
Tolerated |
Benign |
||
HACE1 |
c.1406C>T |
p.Pro469Leu |
1/0 |
rs761336527 |
4.50e-05 |
Tolerated |
Benign |
||
GLI3 |
c.840C>G |
P.Ser280= |
1/0 |
rs77084911 |
0.002 |
||||
GLI3 |
c.1393G>C |
p.Gly465Arg |
4/1 |
1/0 |
rs35488756 |
0.004 |
Tolerated |
Probably Damaging |
|
GLI3 |
c.2179G>A |
p.Gly727Arg |
1/0 |
1/0 |
rs121917710 |
0.005 |
Damaging |
Probably Damaging |
|
GLI3 |
c.2240C>A |
p.Thr747Asn |
1/0 |
Tolerated |
Probably Damaging |
||||
GLI3 |
c.2961C>T |
p.Tyr987= |
1/0 |
rs528703005 |
2.80e-05 |
||||
GLI3 |
c.4374T>C |
p.Gly1458= |
1/0 |
||||||
GLI3 |
c.4554G>C |
p.Leu1518= |
1/0 |
rs769537011 |
4.06e-06 |
||||
CDKN2B |
c.360C>T |
p.Ala120= |
1/0 |
1/0 |
rs62637622 |
0.001 |
|||
PALB2 |
c.768C>T |
p.Ser256= |
1/0 |
rs45487491 |
1.38e-04 |
||||
PALB2 |
c.1408A>G |
p.Thr470Ala |
2/0 |
rs150636811 |
1.22e-05 |
Tolerated |
Benign |
C0 |
|
PALB2 |
c.2794G>A |
p.Val932Met |
1/0 |
1/0 |
rs45624036 |
0.005 |
Damaging |
Probably Damaging |
C0 |
PALB2 |
c.2816T>G |
p.Leu939Trp |
1/0 |
rs45478192 |
0.001 |
Damaging |
Probably Damaging |
C55 |
|
CHEK2 |
c.157T>A |
p.Ser53Thr |
0/1i |
rs371657037 |
4.06e-05 |
Tolerated |
Probably Damaging |
C55 |
|
CHEK2 |
c.410G>A |
p.Arg137Gln |
2/0 |
rs368570187 |
1.79e-04 |
Tolerated |
Benign |
C0 |
|
CHEK2 |
c.911T>C |
p.Met304Thr |
1/0 |
rs587782033 |
N.A.h |
Tolerated |
Probably Damaging |
C55 |
|
CHEK2 |
c.1095G>T |
splicing mut |
1/0 |
||||||
CHEK2 |
c.1312G>T |
p.Asp438Tyr |
1/0 |
rs200050883 |
3.90e-04 |
Damaging |
Probably Damaging |
C25 |
|
Abbreviations: ahet: mutation present in heterozygosity, bhom: mutation present in homozygosity, cRec: recurrence; dMAF: frequency of the minor allele in gnomAD (ALL); ehttp://sift.jcvi.org/; fhttp://genetics.bwh.harvard.edu/pph/; ghttp://agvgd.hci.utah.edu/agvgd_input.php; hN.A.: not available; imutation present in hemizygosity.
Four variants [c.1393G>C (p.Gly465Arg) and c.2179G>A (p.Gly727Arg) in GLI3, c.360C>T (p.Ala120=) in CDKN2B and c.2794G>A (p.Val932Met) in PALB2] were identified in tumours that developed recurrence and in all cases were present both in primary and recurrent samples. The c.157T>A (p.Ser53Thr) variant of the CHEK2 gene was homozygous in the tumour that showed CN neutral LOH in the corresponding chromosomal region.
All variants were found to be present in the constitutional DNA of the patients, with the exception of the c.301G>T (p.Ala101Ser) of DIS3L2. The c.1408A>G (p.Thr470Ala) of PALB2 was detected in two cases, in one of whom at somatic level only.
In silico prediction of the consequences of the amino acid changes for the 16 missense mutations are reported in Table 2. Five of them [c.1430T>G (p.Leu477Arg) of DIS3L2, c.2179G>A (p.Gly727Arg) of GLI3, c.2794G>A (p.Val932Met) and c.2816T>G (p.Leu939Trp) of PALB2 and c.1312G>T (p.Asp438Tyr) of CHEK2] were predicted to be damaging or probably damaging by SIFT and PolyPhen-2. The two latter variants were classified by the Align-GVGD algorithm [11] as scores C55, corresponding to a risk estimate of 0.65 and C25 corresponding to a risk estimate of 0.29, respectively. The CHEK2 variants c.157T>A (p.Ser53Thr) and c.911T>C (p.Met304Thr) were also scored as C55, but were predicted to be tolerated by SIFT.
Among all identified variations, only the novel c.1095G>T variant of CHEK2 was predicted to affect mRNA splicing by the ALAMUT software. This nucleotide change occurs at the last base of exon 10, and was predicted to cause the loss of the naturally occurring donor splice site. RT-PCR of the region spanning exons 8 to 12 of CHEK2, followed by sequence analysis showed the presence, in both the mutant sample and in six control WTs with no CHEK2 mutations, of the normal (full length; NM_007194) transcript and the naturally occurring mRNA isoform missing exon 10 (Δexon10; NM_145862) (Figure 1). However, in the mutant samples the level of Δexon10 transcript compared to the full-length appeared to be higher than in controls. In addition, the sequencing of the RT-PCR fragment corresponding to the full-length mRNA in the mutated WT revealed the presence of only the transcript form the wild-type allele (Figure 1). These results demonstrate that the CHEK2 c.1095G>T variant leads to loss of the normal mRNA transcript and the increase of an mRNA isoform carrying the in-frame deletion of exon 10 coding for a protein (p.Tyr337_Lys365del) missing a portion of the protein kinase domain (Supplementary Figure 6).
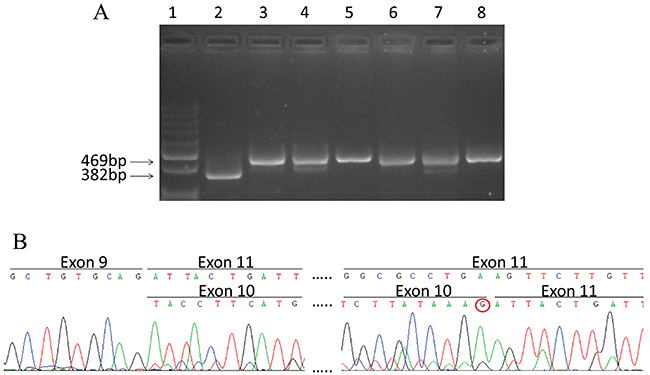
Figure 1: Splicing mutation c.1095G>T in CHEK2. (A) RT-PCR products visualized by agarose gel electrophoresis. Lane 1: Molecular size markers 100bp ladder; lane 2: cDNA from mutated WT; lane 3-8: cDNA from wild type WTs. The size of the full length transcript (469 bp) and Δexon10 transcript (382 bp) are reported. (B) cDNA sequence chromatogram of PCR products from the mutated WT, The sequence of Δexon10 and full length transcripts are reported in the upper and lower lane respectively. The absence of the mutated T nucleotide at the last position of exon 10 in the full length transcript (shown by the red circle) indicates that it originates from the wild type allele only.
MSP analysis was performed on all the 96 WTs for the promoter region of HACE1 and CDKN2A and CDKN2B. MSP of the CpG-177 island of HACE1, previously reported to be frequently methylated in WTs [12, 13], revealed the presence of PCR products from both methylated and unmethylated DNA in 81 tumours (84%), and of the product from unmethylated DNA only in the remaining 14 samples. In eight recurrent tumours, the same methylation patterns detected in the corresponding primary tumours (in seven cases both methylated and unmethylated DNA and in one case unmethylated DNA only) were observed. MSP of the CpG islands in the promoter regions of CDKN2A and CDKN2B genes revealed the absence of methylation in all samples.
DISCUSSION
As already detailed, WT is genetically heterogeneous and its pathogenesis is not completely elucidated.
In the present work, we exploited data derived from a whole-genome SNP array analysis to investigate chromosomal anomalies in 96 WTs and disclose genes possibly involved in the development of this malignancy [8].
The rationale of the study was to focus on minimal regions involved in anomalies observed in different tumours, in which to select genes with a putative role in tumourigenesis and/or organogenesis, with relevance to kidney development. Some of the selected genes had been already investigated in WTs. In the following paragraphs, our findings are discussed in detail in the context of available literature data.
Previous studies already observed focal CN gain on 2p24.3 involving MYCN in sporadic WTs [8, 14–16]. More recently, MYCN CN gains were found in 37/292 (12.7%) WTs, in 26 of which as a focal event [10]. In the present work, focal MYCN anomalies were detected in ca. 6% of cases. MYCN overexpression has been already reported in WTs, and has been proposed as a possible marker associated with prognosis, as well as with diffuse anaplasia or the presence of blastemal histology after pre-operative chemotherapy [10, 14, 17, 18]. MYCN overexpression has been also found in WTs in the absence of chromosomal gain, thus suggesting that other mechanisms (such as hypomethylation at specific regions) may operate to increase the levels of the transcript in a broader range of WTs [10, 19]. A further mechanism causing MYCN aberrant activation can be mediated by the hot spot mutation c.131C>T (p.Pro44Leu) initially identified in neuroblastoma as an acquired somatic event with predicted gain of function [20]. This mutation has been recently identified also in a small fraction (ca. 4%) of WTs [7, 10, 21]. In the present series 7% of tumours were found to carry the mutation, further supporting a role of aberrant activation of MYCN in WT.
The 2q37 chromosomal region and the DIS3L2 and miR-562 genes therein mapped had been already identified as associated with WT development in two congenital syndromic conditions, the 2q37 deletion syndrome and the Perlman syndrome [22, 23]. In fact, three cases with 2q37 deletion syndrome were reported to be affected with WT [reviewed in 22] and, furthermore, DIS3L2 mutations have been described in Perlman’s syndrome patients [23]. Previous LOH analyses in 226 sporadic WTs identified loss of this region in 4% of cases, in two of which the deletion was homozygous, and within the minimal region commonly lost only the DIS3L2 and miR-562 genes were mapped [22]. In the same study, a further 6% of cases showed allelic ratio anomalies in this region, whereas a heterozygous 19-bp deletion of miR-562 was detected in three out of 176 WTs. However, this deletion is likely to be a neutral variant (rs140596642), with a frequency in non-Finnish Europeans of 1.46%. Finally, no anomalies affecting the sequence of DIS3L2 were identified in the screening of 96 samples. In a subsequent paper, the sequencing of DIS3L2 in 40 cases of WTs led to the identification of two missense mutations, whereas MLPA analyses carried out on 20 WTs disclosed the deletion of the entire gene in two cases (in one case the deletion was homozygous) and gene partial deletions, involving only one or few exons, in four cases [23]. In our series, in addition to seven cases with chromosomal abnormalities involving the DIS3L2 gene previously identified by SNP array analysis, three cases were found to carry DIS3L2 missense variants, which in two instances were present also at germline level. Whereas the actual impact of such variants on the protein function remains to be assessed, our results are in agreement with previous data indicating a possible involvement of DIS3L2 in WT development, mainly through deletions.
HACE1 is a tumour suppressor gene involved in different types of cancers [12]. Its silencing has been shown to be mediated through hypermethylation of a CpG island (CpG-177), located upstream the transcription start site [12, 13]. In WT, genetic anomalies affecting the HACE1 gene have been previously reported in few cases. In particular, one case displayed a t(6;15)(q21;q21) translocation which led to a lower level of HACE1 expression [13], whereas another WT showed a 6q21 breakpoint that transected and truncated the HACE1 gene [24]. Furthermore, two HACE1 nonsense mutations and six different missense variations have also been reported [7, 24]. Finally, in WTs, previously published data indicated the presence of methylated CpG-177 in ca. 73% of cases and a downregulation of HACE1 expression, when compared to normal kidney, in 20/26 cases [12]. Among our samples a focal deletion involving solely the HACE1 gene was observed. In addition, the screening for HACE1 alterations led to the identification of two germline missense mutations in four samples, indicating a low frequency of gene mutations, in agreement with previous reports, and the investigation of the CpG-177 led to the identification of the presence of methylation in 81/95 (85%) samples, again in agreement with previous data. To the best of our knowledge, this paper describes the first evidence of a WT in which one allele of the HACE1 gene is deleted, and confirms the proposed role of HACE1 in WT development, mainly mediated by promoter methylation.
Mutations affecting GLI3 are responsible of different syndromic conditions, including the Greig cephalopolysyndactyly syndrome (OMIM 175700), the Pallister–Hall syndrome (OMIM 146510) and the Postaxial polydactyly type-A (OMIM 174200), but none of such conditions are associated with WT development. However, a very recent exome sequencing analysis of 58 WT cases has reported nonsense variations of GLI3 in two WTs [7]. In our samples, GLI3 sequencing led to the identification of three rare missense variants, also present at germline level, whose impact is unknown, indicating that the role of GLI3 in WT is still to be established.
Cytogenetic data reported chromosome 9 trisomy in approximately 13% of WTs and LOH studies revealed loss at the CDKN2A/2B locus, mapped to 9p21.3 region, in 4/34 (12%) cases [25]. CDKN2A mutations have been reported in two children affected with WT in a study assessing the risk of non-melanoma cancers in first-degree relatives of CDKN2A mutation carriers [26]. Moreover, a decreased expression of the CDKN2A mRNA isoform encoding the p16INK4a protein was identified in a significant fraction of WTs [27] and correlated with promoter methylation [25, 27–29]. On the contrary, another study reported the absence of methylation of the CDKN2A/p16INK4a promoter in WTs [30]. In our study, mutation screening of CDKN2A and the contiguous gene CDKN2B, both involved in a homozygous deletion observed in one case, led to the identification of a CDKN2B silent variation, already previously reported, and investigation of the promoter regions of CDKN2A and CDKN2B did not disclose the presence of methylation. While the latter findings argue against a role of CDKN2A and CDKN2B downregulation in WTs, it must be noted that our study provide the first evidence of a homozygous deletion of CDKN2A and CDKN2B in WT.
Bi-allelic germline mutations of PALB2 (also known as FANCN) are responsible for the Fanconi anemia (FA) subtype N. Patients affected with this disease show a more severe phenotype compared to other FA subtypes, including growth retardation, variable congenital malformations and predisposition to paediatric malignancies [31]. Among all described carriers of bi-allelic PALB2/FANCN mutations who developed cancer in early childhood, four had WT [32, 33]. In addition, a previous study analyzing 47 sporadic WT patients, reported nine germline missense variants in the PALB2/FANCN gene, seven of which had been previously reported [31]. Recently, a genome-wide sequencing analysis revealed seven different PALB2/FANCN mutations identified in eight of 651 WT patients [34]. In our WT cases, the screening for PALB2 mutation led to the identification of four previously described rare variants in five patients. Interestingly, two of these variants, [c.2794G>A (p.Val932Met) and c.2816T>G (p.Leu939Trp)] had been previously identified in WT patients [31]. Both variations affect structural residues belonging to a WD 40 domain that binds to the N-terminus of BRCA2 and are predicted to be damaging by both Polyphen2 and SIFT. However, these variations have been also previously described in a familial breast cancer study in which more than 4.000 alleles were screened, and it has been reported to occur with equal frequencies within cases and controls [35]. Therefore, any possible functional role of these variations has yet to be ascertained. Together with previous data, in which PALB2 anomalies have been rarely identified in WT, with the exception of WTs arising in Fanconi anemia patients, our data suggest that this gene could play a role, if any, in a restricted fraction of sporadic WTs only.
While this study was in progress, germline CHEK2 mutations in eight of 651 WT patients were reported by Gadd et al. [34]. One mutation (p.Ile157Thr) recurred in four cases and was reported to be likely pathogenic. In the present study, we identified four different CHEK2 germline missense variants in five patients. Interestingly, three of these variants [c.410G>A (p.Arg137Gln) detected in two cases, c.1312G>T (p.Asp438Tyr) and c.911T>C (p.Met304Thr)] had been previously reported in breast cancer patients [36–38]. In addition, in vitro analyses found that the p.Asp438Tyr substitution causes a 70% reduction of kinase activity of the CHEK2 protein [36, 37]. The remaining variant, c.157T>A (p.Ser53Thr) appeared to be reduced to homozygosity in the tumour DNA of the carrier consistent with a two-hit inactivation model of carcinogenesis. Finally, an additional nucleotide change, c.1095G>T, was found to affect RNA splicing. More specifically, this variant induces a relative increase in the expression of a naturally occurring form bearing the deletion of exon 10, and the loss of the full length transcript. It has been reported that the natural isoform of CHEK2 without exon 10 (Δexon10; NM_145862) loses the kinase activity of the protein [39]. Therefore, our findings suggest a loss of function of the mutant allele.
In summary, this study demonstrates that the use of SNP-array detecting minimal genomic regions commonly involved in anomalies in tumour tissues can be a useful tool to identify genes associated with cancer. In fact, while the clinical significance of the majority of genetic and epigenetic changes we were able to assess remains undetermined, this strategy allowed the identification of loss-of-function mutations in CHEK2, a gene previously reported to be involved in susceptibility to cancers of different organs, including breast, colorectum and prostate [40–42]. Therefore, whereas to the best of our knowledge no reports have assessed an increased risk of WT in carriers of germline CHEK2 mutations, our data, together with those of Gadd and colleagues [34], suggest a putative role of constitutional anomalies of this gene in WT development, which deserves further investigations.
MATERIALS AND METHODS
Patients and specimens
The study included material from 96 WTs of 85 patients previously investigated by whole-genome SNP array. In three cases bilateral disease was investigated, and in eight cases both primary and recurrent tumours were studied [8, 9]. The patients represent the first 78 cases prospectively enrolled into the AIEOP WT-2003 protocol for whom tumor samples were available, and seven relapsing cases from the previous AIEOP WT-1992 protocol. A specific informed consent to the use of biological samples for the aim of the study was obtained from the parents or legal guardian of all enrolled patients. The study has been approved by the ethics committee of IRCCS Istituto Nazionale Tumori.
DNA and RNA were purified from frozen surgical specimens from which a histological section was derived and reviewed by the pathologist for the presence of vital tumor tissue.
Mutation analysis
Sanger sequencing was performed for the MYCN hotspot mutation c.131C>T (p.Pro44Leu) [10] and for the entire coding of the DIS3L2 (Refseq NM_152383), mir562 (Refseq NR_030288), CDKN2A (p14ARF Refseq NM_058195 and p16 Refseq NM_000077), CDKN2B (Refseq NM_004936), HACE1 (Refseq NM_020771), GLI3 (Refseq NM_000168), PALB2 (Refseq NM_024675), and CHEK2 (Refseq NM_007194) genes. Primers and PCR conditions are available upon request.
Variants identified in tumour DNAs were selected if not previously identified or reported with a minor allele frequency (MAF) <0.01 in the gnomAD database linked to ensemble (http://www.ensembl.org/index.html). Selected variants were further investigated in germline DNA. The biological effect of missense mutations was predicted using the Polymorphism Phenotyping v2 (PolyPhen-2; http://genetics.bwh.harvard.edu/pph2/), the SIFT (SIFT; http://sift.jcvi.org/) tools and Align-GVGD (http://agvgd.hci.utah.edu/agvgd_input.php) for the variants identified only in PALB2 and CHEK2. Possible splicing aberrations were analyzed using Alamut (Interactive Biosoftware, Roven, France; http://www.interactive-biosoftware.com/software/alamut/overview).
Methylation analysis
Bisulfite modification was performed on 300 ng of DNA from WT samples using the EpiTect Bisulfite Kit (QIAGEN, Milan, Italy) according to the manufacturer’s instructions. The methylation status of the promoter of HACE1, CDKN2A (p14 and p16) and CDKN2B (p15) was assessed by methylation-specific PCR (MSP) using primers and conditions already described [12, 27, 43, 44]. Methylated and unmethylated DNAs (QIAGEN, Milan, Italy) were used as positive and negative controls in all MSP reactions.
Splicing analysis
To determine the effects of c.1095G>T variant on CHEK2 pre-mRNA processing, total RNA samples were isolated using the RNeasy mini Kit (QIAGEN, Milan, Italy). The cDNAs were prepared using the High Capacity cDNA Archive Kit (Applied Biosystems, Milan, Italy) following the manufacturer’s instructions. The cDNAs were PCR amplified using a forward primer in CHEK2 exon 8 (5' GATGCAGAAGATTATTATATTGTTTTGG 3') and a reverse primer in CHEK2 exon 12 (5' GAATG AAGTTGTATTTTCCACTGG 3'). The amplification products were visualized on a 3% agarose gel stained with ethidium bromide and the different PCR fragments characterized by direct sequencing.
ACKNOWLEDGMENTS
The Authors wish to thank all the AIEOP centers participating to this study.
CONFLICTS OF INTEREST
The authors declare that there is no conflicts of interest.
FUNDING
The Authors wish to thank Associazione Bianca Garavaglia ONLUS, Busto Arsizio, Varese, for financial support.
REFERENCES
1. Huff V. Wilms’ tumours: about tumour suppressor genes, an oncogene and a chameleon gene. Nat Rev Cancer. 2011; 11:111–21. https://doi.org/10.1038/nrc3002.
2. Royer-Pokora B. Genetics of pediatric renal tumors. Pediatr Nephrol. 2013; 28:13–23. https://doi.org/10.1007/s00467-012-2146-4.
3. Scott RH, Murray A, Baskcomb L, Turnbull C, Loveday C, Al-Saadi R, Williams R, Breatnach F, Gerrard M, Hale J, Kohler J, Lapunzina P, Levitt GA, et al. Stratification of Wilms tumor by genetic and epigenetic analysis. Oncotarget. 2012; 3:327–35. https://doi.org/10.18632/oncotarget.468.
4. Rivera MN, Kim WJ, Wells J, Driscoll DR, Brannigan BW, Han M, Kim JC, Feinberg AP, Gerald WL, Vargas SO, Chin L, Iafrate AJ, Bell DW, Haber DA. An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science. 2007; 315:642–45. https://doi.org/10.1126/science.1137509.
5. Perotti D, Gamba B, Sardella M, Spreafico F, Terenziani M, Collini P, Pession A, Nantron M, Fossati-Bellani F, Radice P. Functional inactivation of the WTX gene is not a frequent event in Wilms’ tumors. Oncogene. 2008; 27:4625–32. https://doi.org/10.1038/onc.2008.93.
6. Walz AL, Ooms A, Gadd S, Gerhard DS, Smith MA, Guidry Auvil JM, Meerzaman D, Chen QR, Hsu CH, Yan C, Nguyen C, Hu Y, Bowlby R, et al. Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell. 2015; 27:286–97. https://doi.org/10.1016/j.ccell.2015.01.003.
7. Wegert J, Ishaque N, Vardapour R, Geörg C, Gu Z, Bieg M, Ziegler B, Bausenwein S, Nourkami N, Ludwig N, Keller A, Grimm C, Kneitz S, et al. Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high-risk blastemal type Wilms tumors. Cancer Cell. 2015; 27:298–311. https://doi.org/10.1016/j.ccell.2015.01.002.
8. Perotti D, Spreafico F, Torri F, Gamba B, D’Adamo P, Pizzamiglio S, Terenziani M, Catania S, Collini P, Nantron M, Pession A, Bianchi M, Indolfi P, et al, and Associazione Italiana Ematologia Oncologia Pediatrica Wilms Tumor Working Group. Genomic profiling by whole-genome single nucleotide polymorphism arrays in Wilms tumor and association with relapse. Genes Chromosomes Cancer. 2012; 51:644–53. https://doi.org/10.1002/gcc.21951.
9. Spreafico F, Ciceri S, Gamba B, Torri F, Terenziani M, Collini P, Macciardi F, Radice P, Perotti D. Chromosomal anomalies at 1q, 3, 16q, and mutations of SIX1 and DROSHA genes underlie Wilms tumor recurrences. Oncotarget. 2016; 7:8908–15. https://doi.org/10.18632/oncotarget.6950.
10. Williams RD, Chagtai T, Alcaide-German M, Apps J, Wegert J, Popov S, Vujanic G, van Tinteren H, van den Heuvel-Eibrink MM, Kool M, de Kraker J, Gisselsson D, Graf N, et al. Multiple mechanisms of MYCN dysregulation in Wilms tumour. Oncotarget. 2015; 6:7232–43. https://doi.org/10.18632/oncotarget.3377.
11. Tavtigian SV, Byrnes GB, Goldgar DE, Thomas A. Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications. Hum Mutat. 2008; 29:1342–54. https://doi.org/10.1002/humu.20896.
12. Zhang L, Anglesio MS, O’Sullivan M, Zhang F, Yang G, Sarao R, Mai PN, Cronin S, Hara H, Melnyk N, Li L, Wada T, Liu PP, et al. The E3 ligase HACE1 is a critical chromosome 6q21 tumor suppressor involved in multiple cancers. Nat Med. 2007; 13:1060–69. https://doi.org/10.1038/nm1621.
13. Anglesio MS, Evdokimova V, Melnyk N, Zhang L, Fernandez CV, Grundy PE, Leach S, Marra MA, Brooks-Wilson AR, Penninger J, Sorensen PH. Differential expression of a novel ankyrin containing E3 ubiquitin-protein ligase, Hace1, in sporadic Wilms’ tumor versus normal kidney. Hum Mol Genet. 2004; 13:2061–74. https://doi.org/10.1093/hmg/ddh215.
14. Williams RD, Al-Saadi R, Chagtai T, Popov S, Messahel B, Sebire N, Gessler M, Wegert J, Graf N, Leuschner I, Hubank M, Jones C, Vujanic G, Pritchard-Jones K, and Children’s Cancer and Leukaemia Group, and SIOP Wilms’ Tumour Biology Group. Subtype-specific FBXW7 mutation and MYCN copy number gain in Wilms’ tumor. Clin Cancer Res. 2010; 16:2036–45. https://doi.org/10.1158/1078-0432.CCR-09-2890.
15. Schaub R, Burger A, Bausch D, Niggli FK, Schäfer BW, Betts DR. Array comparative genomic hybridization reveals unbalanced gain of the MYCN region in Wilms tumors. Cancer Genet Cytogenet. 2007; 172:61–65. https://doi.org/10.1016/j.cancergencyto.2006.08.010.
16. Williams RD, Al-Saadi R, Natrajan R, Mackay A, Chagtai T, Little S, Hing SN, Fenwick K, Ashworth A, Grundy P, Anderson JR, Dome JS, Perlman EJ, et al. Molecular profiling reveals frequent gain of MYCN and anaplasia-specific loss of 4q and 14q in Wilms tumor. Genes Chromosomes Cancer. 2011; 50:982–95. https://doi.org/10.1002/gcc.20907.
17. Zirn B, Hartmann O, Samans B, Krause M, Wittmann S, Mertens F, Graf N, Eilers M, Gessler M. Expression profiling of Wilms tumors reveals new candidate genes for different clinical parameters. Int J Cancer. 2006; 118:1954–62. https://doi.org/10.1002/ijc.21564.
18. Wittmann S, Wunder C, Zirn B, Furtwängler R, Wegert J, Graf N, Gessler M. New prognostic markers revealed by evaluation of genes correlated with clinical parameters in Wilms tumors. Genes Chromosomes Cancer. 2008; 47:386–95. https://doi.org/10.1002/gcc.20544.
19. Nisen PD, Zimmerman KA, Cotter SV, Gilbert F, Alt FW. Enhanced expression of the N-myc gene in Wilms’ tumors. Cancer Res. 1986; 46:6217–22.
20. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, Kim J, Lawrence MS, Lichenstein L, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013; 45:279–84. https://doi.org/10.1038/ng.2529.
21. Rakheja D, Chen KS, Liu Y, Shukla AA, Schmid V, Chang TC, Khokhar S, Wickiser JE, Karandikar NJ, Malter JS, Mendell JT, Amatruda JF. Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat Commun. 2014; 2:4802. https://doi.org/10.1038/ncomms5802.
22. Drake KM, Ruteshouser EC, Natrajan R, Harbor P, Wegert J, Gessler M, Pritchard-Jones K, Grundy P, Dome J, Huff V, Jones C, Aldred MA. Loss of heterozygosity at 2q37 in sporadic Wilms’ tumor: putative role for miR-562. Clin Cancer Res. 2009; 15:5985–92. https://doi.org/10.1158/1078-0432.CCR-09-1065.
23. Astuti D, Morris MR, Cooper WN, Staals RH, Wake NC, Fews GA, Gill H, Gentle D, Shuib S, Ricketts CJ, Cole T, van Essen AJ, van Lingen RA, et al. Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat Genet. 2012; 44:277–84. https://doi.org/10.1038/ng.1071.
24. Slade I, Stephens P, Douglas J, Barker K, Stebbings L, Abbaszadeh F, Pritchard-Jones K, Cole R, Pizer B, Stiller C, Vujanic G, Scott RH, Stratton MR, Rahman N, and FACT collaboration. Constitutional translocation breakpoint mapping by genome-wide paired-end sequencing identifies HACE1 as a putative Wilms tumour susceptibility gene. J Med Genet. 2010; 47:342–47. https://doi.org/10.1136/jmg.2009.072983.
25. Natrajan R, Warren W, Messahel B, Reis-Filho JS, Brundler MA, Dome JS, Grundy PE, Vujanic G, Pritchard-Jones K, Jones C. Complex patterns of chromosome 9 alterations including the p16INK4a locus in Wilms tumours. J Clin Pathol. 2008; 61:95–102. https://doi.org/10.1136/jcp.2007.047159.
26. Mukherjee B, Delancey JO, Raskin L, Everett J, Jeter J, Begg CB, Orlow I, Berwick M, Armstrong BK, Kricker A, Marrett LD, Millikan RC, Culver HA, et al, and GEM Study Investigators. Risk of non-melanoma cancers in first-degree relatives of CDKN2A mutation carriers. J Natl Cancer Inst. 2012; 104:953–56. https://doi.org/10.1093/jnci/djs221.
27. Arcellana-Panlilio MY, Egeler RM, Ujack E, Pinto A, Demetrick DJ, Robbins SM, Coppes MJ. Decreased expression of the INK4 family of cyclin-dependent kinase inhibitors in Wilms tumor. Genes Chromosomes Cancer. 2000; 29:63–69. https://doi.org/10.1002/1098-2264(2000)9999:9999%3C::AID-GCC1006%3E3.0.CO;2-L.
28. Morris MR, Hesson LB, Wagner KJ, Morgan NV, Astuti D, Lees RD, Cooper WN, Lee J, Gentle D, Macdonald F, Kishida T, Grundy R, Yao M, et al. Multigene methylation analysis of Wilms’ tumour and adult renal cell carcinoma. Oncogene. 2003; 22:6794–801. https://doi.org/10.1038/sj.onc.1206914.
29. Wagner KJ, Cooper WN, Grundy RG, Caldwell G, Jones C, Wadey RB, Morton D, Schofield PN, Reik W, Latif F, Maher ER. Frequent RASSF1A tumour suppressor gene promoter methylation in Wilms’ tumour and colorectal cancer. Oncogene. 2002; 21:7277–82. https://doi.org/10.1038/sj.onc.1205922.
30. Faussillon M, Monnier L, Junien C, Jeanpierre C. Frequent overexpression of cyclin D2/cyclin-dependent kinase 4 in Wilms’ tumor. Cancer Lett. 2005; 221:67–75. https://doi.org/10.1016/j.canlet.2004.08.010.
31. Adank MA, Segers H, van Mil SE, van Helsdingen YM, Ameziane N, van den Ouweland AM, Wagner A, Meijers-Heijboer H, Kool M, de Kraker J, Waisfisz Q, van den Heuvel-Eibrink MM. Fanconi anemia gene mutations are not involved in sporadic Wilms tumor. Pediatr Blood Cancer. 2010; 55:742–44. https://doi.org/10.1002/pbc.22588.
32. Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, Neveling K, Kelly P, Seal S, Freund M, Wurm M, Batish SD, Lach FP, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007; 39:162–64. https://doi.org/10.1038/ng1947.
33. Serra A, Eirich K, Winkler AK, Mrasek K, Göhring G, Barbi G, Cario H, Schlegelberger B, Pokora B, Liehr T, Leriche C, Henne-Bruns D, Barth TF, Schindler D. Shared copy number variation in simultaneous nephroblastoma and neuroblastoma due to fanconi anemia. Mol Syndromol. 2012; 3:120–30. https://doi.org/10.1159/000341935.
34. Gadd S, Huff V, Walz AL, Ooms AH, Armstrong AE, Gerhard DS, Smith MA, Auvil JM, Meerzaman D, Chen QR, Hsu CH, Yan C, Nguyen C, et al. A Children’s Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat Genet. 2017; 49:1487–94. https://doi.org/10.1038/ng.3940.
35. Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T, Jayatilake H, McGuffog L, Hanks S, et al, and Breast Cancer Susceptibility Collaboration (UK). PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007; 39:165–67. https://doi.org/10.1038/ng1959.
36. Sodha N, Mantoni TS, Tavtigian SV, Eeles R, Garrett MD. Rare germ line CHEK2 variants identified in breast cancer families encode proteins that show impaired activation. Cancer Res. 2006; 66:8966–70. https://doi.org/10.1158/0008-5472.CAN-06-1990.
37. Bell DW, Kim SH, Godwin AK, Schiripo TA, Harris PL, Haserlat SM, Wahrer DC, Haiman CA, Daly MB, Niendorf KB, Smith MR, Sgroi DC, Garber JE, et al. Genetic and functional analysis of CHEK2 (CHK2) variants in multiethnic cohorts. Int J Cancer. 2007; 121:2661–67. https://doi.org/10.1002/ijc.23026.
38. Le Calvez-Kelm F, Lesueur F, Damiola F, Vallée M, Voegele C, Babikyan D, Durand G, Forey N, McKay-Chopin S, Robinot N, Nguyen-Dumont T, Thomas A, Byrnes GB, et al, and Breast Cancer Family Registry. Rare, evolutionarily unlikely missense substitutions in CHEK2 contribute to breast cancer susceptibility: results from a breast cancer family registry case-control mutation-screening study. Breast Cancer Res. 2011; 13:R6. https://doi.org/10.1186/bcr2810.
39. Staalesen V, Falck J, Geisler S, Bartkova J, Børresen-Dale AL, Lukas J, Lillehaug JR, Bartek J, Lønning PE. Alternative splicing and mutation status of CHEK2 in stage III breast cancer. Oncogene. 2004; 23:8535–44. https://doi.org/10.1038/sj.onc.1207928.
40. Apostolou P, Papasotiriou I. Current perspectives on CHEK2 mutations in breast cancer. Breast Cancer (Dove Med Press). 2017; 9:331–35. https://doi.org/10.2147/BCTT.S111394.
41. Liu C, Wang QS, Wang YJ. The CHEK2 I157T variant and colorectal cancer susceptibility: a systematic review and meta-analysis. Asian Pac J Cancer Prev. 2012; 13:2051–55. https://doi.org/10.7314/APJCP.2012.13.5.2051.
42. Hale V, Weischer M, Park JY. CHEK2 (*) 1100delC mutation and risk of prostate cancer. Prostate Cancer. 2014; 2014:294575. https://doi.org/10.1155/2014/294575.
43. Esteller M, Tortola S, Toyota M, Capella G, Peinado MA, Baylin SB, Herman JG. Hypermethylation-associated inactivation of p14(ARF) is independent of p16(INK4a) methylation and p53 mutational status. Cancer Res. 2000; 60:129–33.
44. Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996; 93:9821–26. https://doi.org/10.1073/pnas.93.18.9821.