
INTRODUCTION
Triple negative breast cancer (TNBC) is a subtype of breast cancer (BC) and is defined by the lack of expression of three receptors: the estrogen receptor (ER), progesterone receptor (PR), and the human epidermal growth factor receptor 2 (HER2) [1]. TNBC accounts for roughly 15% of all breast cancer cases but represents over 50% of mortality seen in breast cancer [2-4]. Therefore, current endocrine and HER2-targeted therapies are not viable for TNBCs, and the only treatment option available is chemotherapy [2, 4-6]. Although, TNBC patients tend to have higher clinical response rates to chemotherapy, they also have higher rates of distant recurrence and a worse overall prognosis than women with other breast cancer subtypes [7, 8]. TNBC is associated with health disparity because it is more common in premenopausal women of color (African-American women (AA) and Hispanic women (HS)) than Caucasian American women (CA) [3, 9]. Population based studies show that AA women with TNBC have a higher incidence, disease stage, and metastasis than CA [10, 11]. Therefore finding a new molecular target and/or treatment is the upmost importance within this patient population and in women with breast cancer in general.
Estrogens promote progression of ER-alpha (ERα) positive cancers, effected by the binding of estradiol to ERα [12]. ERα is predominantly localized in the nucleus and positive staining by immunohistochemical (IHC) detection, helps plan patient management [13]. Recent reports have shown a second ER, ERβ, is expressed in TNBC [14, 15]. ERα and ERβ are encoded by two different genes but share 96% homology in the DNA-binding domain and 60% homology in the ligand binding domain of ERα. ERβ’s role in BC progression remains to be elucidated, however, some studies have shown ERβ positivity is a biomarker related to a more aggressive clinical outcome [15] and correlates with Ki-67, a proliferation marker [14-16]. Early studies suggest ERβ levels are higher in BCs in AA as compared to CA, and may play a role in TNBC progression [17-20]. Several ERβ isoforms occur in BC, including ERβ1, ERβ2, ERβ4, and ERβ5. Only ERβ1 retains an intact ligand binding domain which makes it a preferred clinical target [21-23]. ERβ occurs in the nucleus and in the cytoplasm and can be activated by both genomic or indirectly by nongenomic pathways [21, 24-26]. Recent reports indicate that ERβ target genes are enriched in genes that regulate cell death/survival, cell movement, cell development, growth and proliferation, as well as genes involved in the Wnt/β-catenin and G1/S cell cycle phase checkpoint pathways [27-30]. Furthermore, the exact role of ERβ, especially when expressed alone is not well studied. The first priority is to identity new prognostic and therapeutic targets that can identity and treat TNBC. We predict that ERβ may be such a target in TNBC.
Emerging evidence indicates that metabolic factors, such as insulin-like growth factor (IGF-1 and IGF-2) pathways enhance the progression of BC [31-37]. IGF-2 occurs in two forms, precursor (pIGF-2) and mature (mIGF-2) and plays a role in BC proliferation and inhibition of apoptosis [38-40]. Under normal conditions IGF-2 is bound and sequestered [41-44], but overexpression of IGF-2 is associated with increased BC formation [43, 45]. Most human cancers over express IGF-1 receptor (IGF-1R) and IGF-2 is a known ligand for the respective receptor and can bind to IGF-2 receptor (IGF-2R), which sequesters IGF-2 [31-33, 35, 40]. Disparities with IGF-2 expression is also apparent, with AA having higher expression when compared to CA and could contribute to clinical outcome [46]. IGF-2 is expressed both in the BC stromal and epithelial compartments, and corresponds with both stromal and epithelial BC cell proliferation [33] [47-50]. ER signaling pathways are reported to intersect with IGF pathways, with estrogen promoting increased BC production of IGF2 [32]. In this study, we predict ERβ and/or IGF-2 may be such targets in TNBC.
Here, we report our findings on the importance of ERβ expression in TNBC progression. We investigated ERβ expression in different patient subtypes of breast cancer along with a panel of TNBC cell lines. We performed immunoblotting, quantitative-real-time PCR (qPCR), cell proliferation, migration/invasion, enzyme-linked immunosorbent assay (ELISA), and apoptosis studies on cell lines when ERβ is activated and/or inhibited. ERβ activation or inhibition resulted in significant differences in protein expression, migration/invasion, IGF2 expression, and apoptosis. This study also identified that the activation of ERβ activates the IR/IGF-1R signaling pathway via IGF2. We also show that activation of ERβ activated the MAPK signaling pathway.
RESULTS
ERβ is associated with decrease in relapse-free survival in TNBC
The roles of ERβ in breast cancer, let alone TNBC development and progression are not clearly elucidated. A database was established using mRNA expression data downloaded from gene expression omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/), the cancer genome Atlas (TCGA) (http://cancergenome.nih.gov/), European genome-phenome archive (EGA) (https://ega.crg.eu/), and PubMed (http://www.pubmed.com) repositories to identify datasets with published mRNA expression and clinical data. Kaplan–Meier survival analysis was performed to validate the prognostic value. We determined the relapse-free survival (RFS) status between ERβ and TNBC, using the public database KMPlot [51, 52], which consisted of 3,951 breast cancer patients. KMPlot were used because it is a database integrating mRNA expression data and clinical information derived from four independent datasets of breast cancer. It is an easy-to-use bioinformatic tool capable of performing survival analyses (Months) for the identification and validation of prognostic mRNAs in breast cancer, and it allows for a real-time analysis to evaluate the prognostic value of those mRNAs in breast cancer. When accounted for TNBC patients only (restricted analysis to subtype hormone receptor negative: ERα-, PR-, HER2-), high ERβ expression is significantly correlated with lower overall RFS (P = 0.007) (Figure 1A). When patients were given chemotherapy as a whole (Figure 1B) or adjuvant chemotherapy (Figure 1C), high ERβ expression is significantly correlated with lower relapse free survival (RFS) (P = 0.0315 and 0.0415 respectively) and a trend with high ERβ expression is seen to coincide with lower distance metastasis free survival. These results suggest that high ERβ expression in TNBC patients might play a role in the progression of TNBC.
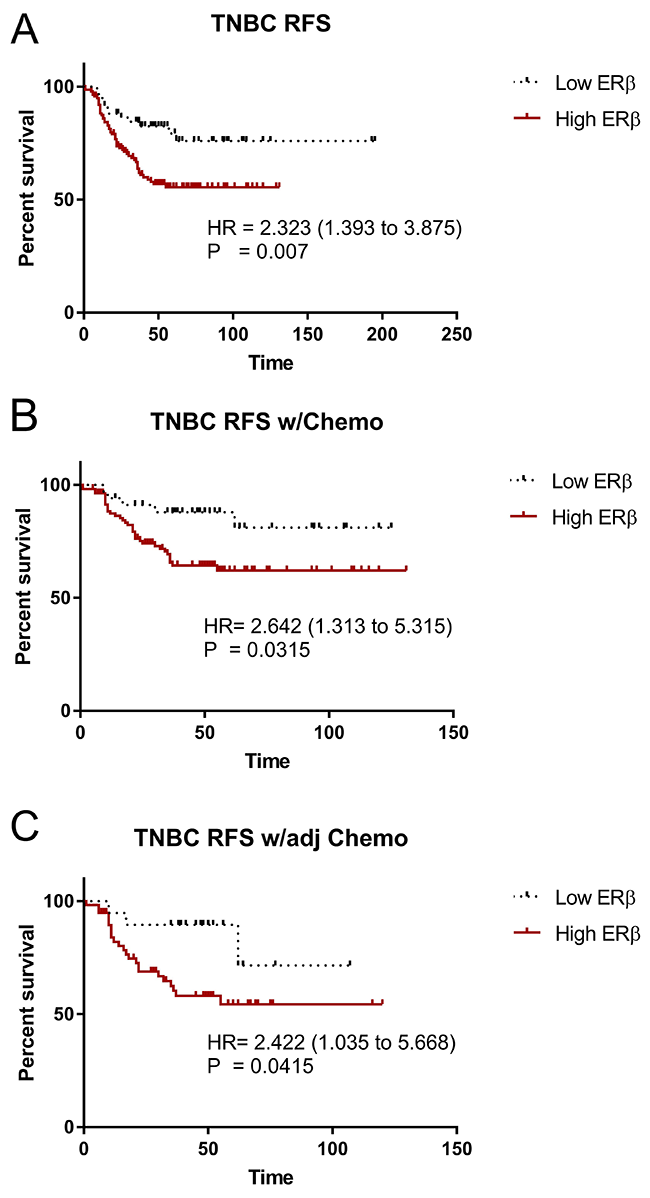
Figure 1: ERβ is associated with decrease relapse free survival in TNBC. mRNA level of ERβ in TNBC was obtained from a public database, KMPlot. Kaplan–Meier survival analysis was used to assess the association between ERβ level and relapse-free survival (RFS). (A) RFS in all TNBC between Low ERβ and High ERβ groups, (B) RFS in TNBC underwent chemotherapy (Chemo) between Low ERβ and High ERβ groups, and (C) RFS in TNBC with adjuvant chemotherapy (adj Chemo) between Low ERβ and High ERβ groups. Log-rank test was used to assess the statistical significance between the groups and P<0.05 was considered as significance.
ERβ expression in breast cancer cell lines
We assessed ERβ expression in a panel of several breast cancer cell lines from our lab. The cell lines we have represent the major subtypes of breast cancer: Luminal A (MCF7, T47D), Luminal B (BT474), HER2+ (SKBR3), and TNBC (BT549, HCC38, HCC70, HCC1806, HCC1937, HS578T, MBA157, MBA231, MBA453, MBA468). Luminal A cell lines will were taken as controls. Under normal growth conditions (10% FBS with phenol red), when compared to luminal A cell lines, all TNBC cell lines had increased expression of ERβ (Figure 2A right). Quantification of the bands are shown in Figure 2A left. Quantification of cellular lines were used to select 5 TNBC cell lines that represents African-American ethnicity (HCC70, HCC1806, MBA468) and Caucasian ethnicity (MBA231, BT549) for further studies. Selected cell lines were then analyzed for ERβ mRNA expression under 2% charcoal dextran stripped FBS in phenol red free media. Our data showed that all of the TNBC cells have at least a two fold increase in ERβ mRNA expression (Figure 2B) when compared to MCF7 cells. The respective cell lysates were subjected to gel electrophoresis and immunoblotting with ERβ and ERα antibodies. Results are shown in Figure 2C. TNBC cells show higher ERβ gene and protein expression.
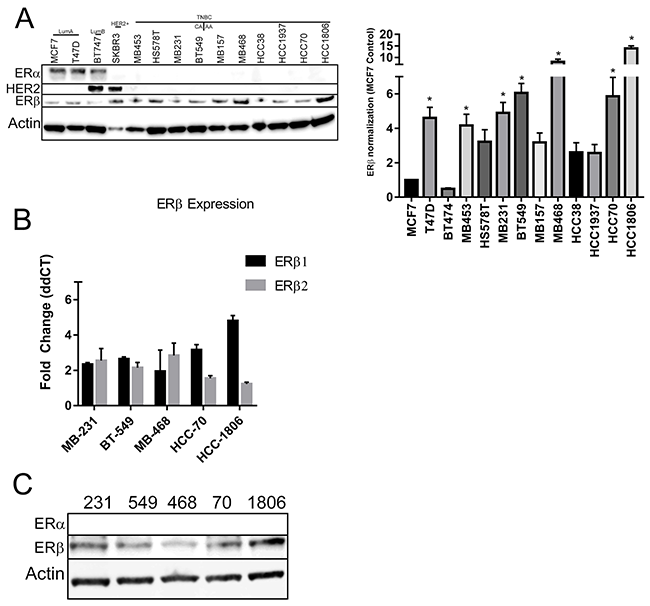
Figure 2: ERβ is expressed in TNBC cell lines. (A) The indicated panel of breast cancer cell were maintained in growth media with 10% FBS and protein was extracted until the cells reach to 80% confluence. Western blot analyses were performed with ERα, ERβ1 and HER2 antibodies and β-actin antibody was used for loading control (A-right). The western blot of ERβ1 was repeated 3 times and the bands intensities were adjusted to β-actin. The bar graph (A-left) showed the mean intensities from three independent bots and SD for the indicated cell lines. “*” indicated p<0.05 compared to the intensity from MCF-7 cells and determined by one-way ANOVA. (B) The indicated TNBC cells were grown in phenol red free media with 5% charcoal treated FBS until 80% confluence and RNA was extracted. RT-qPCR was performed with ERβ1 and ERβ2 primers and adjusted to GAPDH. The bar graph presented as fold changed of ERβ1 and ERβ2 levels in the indicated TNBC cells compared to MCF-7 cells. Each bar indicated mean±SD from three experiments. (C) The indicated TNBC cells were grown in phenol red media with 5% charcoal treated FBS until 80% confluence and protein was extracted. Western blot analyses with ERα and ERβ1 antibodies were performed and β-actin was used for loading control.
ERβ activation increases migration and invasion in TNBC cells
To determine the effect of ERβ activation on cell migration, a wound healing assay on the TNBC cell lines were performed. TNBC cell lines were treated with known estrogen receptor agonist and/or antagonist, 20nM Diarylpropionitrile (DPN, ERβ agonist) and 100nM 4-[2-phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]-pyrimidin-3-yl]phenol (PHTPP, ERβ antagonist) alone [53, 54]. TNBC cells were grown in 2-well inserts that gave a uniform scratch of 500μm in either control media (2% charcoal stripped FBS is phenol red free media), 20μM ERβ agonist (DPN), or 100μM ERβ antagonist (PHTPP). Images captured at 0, 4, 8, 12, and 24h after incubation using phase-contrast microscope. The rate of migration was measured by quantifying the total distance that the cells moved from the edge of the scratch toward the center of the scratch at 24h when compared to 0h. Activation of ERβ by DPN resulted in a significant increase in would closure in our TNBC cell lines (Figure 3A-3D). PHTPP treatment resulted in a significant decrease in wound closure in our MB468 (Figure 3D) and HCC1806 (Figure 3F) when compared to control. These results indicated that ERβ activation in TNBC cells results in increased cell migration.
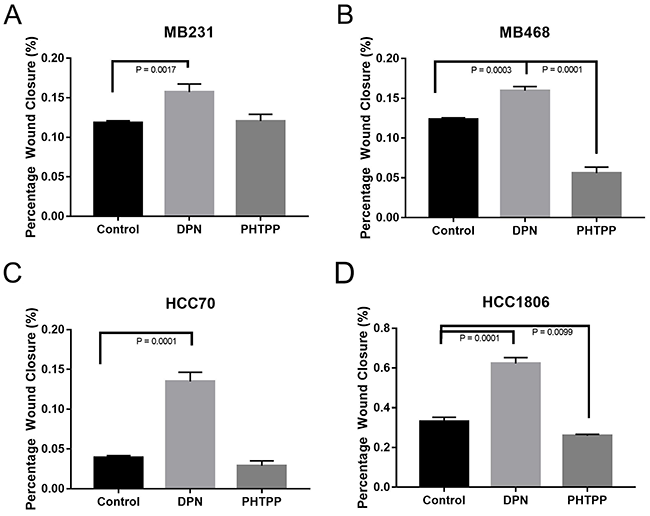
Figure 3: Activation of ERβ increases migration of TNBC cells. The cells were seeded in 8.5cm3 dishes with a 2 well silicone insert and treated with or without DPN or PHTPP for 24 hrs as described in methods. The wound closure was monitored and images were taken and analyzed from three different rears for each conditions. The bar graphs for MB231 (A), MB468 (B), HCC70 (C) and HCC1806 (D) presented as percentage wound closures (mean ±SD) for each conditions. P-values between the indicated conditions were determined by one-way ANOVA.
We then wanted to investigate what role activation of ERβ on cellular invasion of TNBC cells. To determine the effect that ERβ may have on invasion, TNBC cells were grown in the Corning BioCoat Matrigel Invasion Chamber. Cells were treated with either DPN or PHTPP in the upper-chamber for 24 hours, and the cell invasion was measured. Cells were fixed, stained with crystal violet, and quantification of invaded cells was done. Treatment of DPN significantly increased invasion in all cell lines (P < 0.042) (Figure 4A-4E), except MB231 where we saw a significant decrease in invasion when treated with DPN (Figure 4B), and, similarly, the treatment with PHTPP significantly decreased cellular invasion of BT549 and HCC1806, respectively (P < 0.04, Figure 4B, 4E).
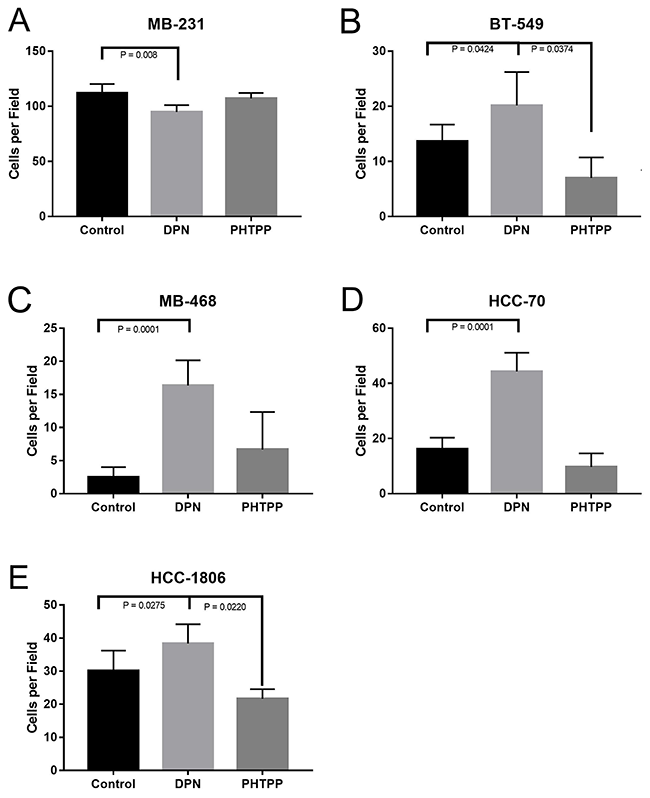
Figure 4: Activation of ERβ increases invasion of TNBC cells. The cellswere plated in top chamber of matrigel invasion chamber in phenol red free media with or without DPN or PHTPP and bottom chamber was filled with medium containing CSFBS as described in methods. After 24 hrs number of cell invasion was assessed. The total numbers of invaded cells were counted from 4 independent areas for each conditions and all experiments were independently repeated three times. The bar graphs for MB231 (A) BT549 (B) MB468 (C) and HCC70 (D), HCC-1806 (E) showed invaded cell numbers (mean ±SD) under each conditions. P-values between the indicated conditions were determined by one-way ANOVA.
ERβ activation increases proliferation in TNBC cells
The data presented here shows that activation of ERβ is involved in the migration and invasion of TNBC cells. We then wanted to investigate the effect of ERβ activation on cellular proliferation. To determine the effect of the activation of ERβ on TNBC cells we treated TNBC cells with DPN, PHTPP, or both over a 3 day period. Cell proliferation was measured by CellTiter-Glo. Quantification of cell proliferation was then measured by relative luciferase units (RLU), and then normalized to control to get fold change. Proliferation rate of TNBC cells were compared to control (Luminal A – MCF7 cells). Under normal conditions over a three day period we saw that all TNBC cell lines had a significant increase, at least a 1.5fold, in cellular proliferation when compared to control MCF7 cells (Figure 5A). We saw the highest proliferation rates in mesenchymal-like TNBC cells (MB231/BT549) vs basal-like subtype except for MB-468 cells. Next, we determined the proliferation rate over a three day period, when treated with either DPN and/or PHTPP. We saw that treatment with PHTPP significantly decreased cellular proliferation over a three day period and DPN treatment increased cellular proliferation (Figure 5B-5F). When TNBC cell lines were treated with both agonist and antagonist returned proliferation back to control levels. This increase in cellular proliferation could be due to ERβ down regulation of cell cycle inhibitors p21 and p27kip (Figure 5I). This data highlights that ERβ promotes cell proliferation when expressed in TNBC.
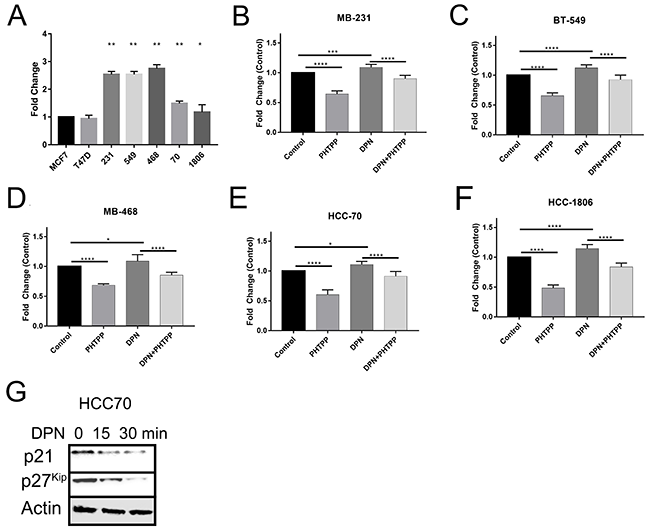
Figure 5: Activation of ERβ increases cell proliferation. The cells were treated with or without DPN, PHTPP, or DPN+PHTPP for 3 days and cell proliferation was measured by The CellTiter-Glo® Luminescent Cell Viability Assay as described in methods. Proliferation rate of different TNBC cells without treatment (A), MB231 (B), BT549 (C) MB468 (D) HCC70 (E) and HCC1806 (F) cells with and without treated with DPN, PHTPP and DPN+PHTPP were determined by compared to MCF7 cells. The bar graphs indicated the mean fold-changes with SD from 3 independent measurements and each measurements contained 6 repeated values for each condition. The p-values were determined by One-Way ANOVA (*P < 0.01, **P < 0.001, ***P < 0.0009, ****P < 0.0001). (G) HCC70 cells were treated with DNP for the indicated time and protein was extracted. Western blot analysis was performed with p21 and p27kip antibodies and β-actin was used as loading control.
ERβ activation upregulates IGF2 and the IR
Since previous data has shown that activation of ERβ resulted in increased proliferation of TNBC cell lines, we then wanted to determine by what mechanism is ERβ regulating cell growth. When we treated our cell lines with DPN and/or PHTPP we saw an upregulation in IGF2 mRNA (Figure 6A) and IGF2 and insulin receptor (IR) protein expression by DPN (Figure 6B). TNBC cells treated with PHTPP resulted in decreased IGF2 protein expression (Figure 6C). When TNBC cell lines were treated with DPN or PHTPP, a significant increase in IGF2 secretion by DPN and a decrease in IGF2 secretion by PHTPP in TNBC cell lines (Figure 6D). We then looked at what pathways were activated by ERβ. The data showed that the MAPK pathway and the IR pathway were activated in HCC1806 when compared to MCF7 cells and growth inhibition by DPN seen in MCF7 was due to the inhibition of the cell cycle through p27kip (Figure 6E). This data highlights that activation of ERβ increases IGF2 secretion that can then act on the IR/IGF1R signaling pathway to control cell growth.
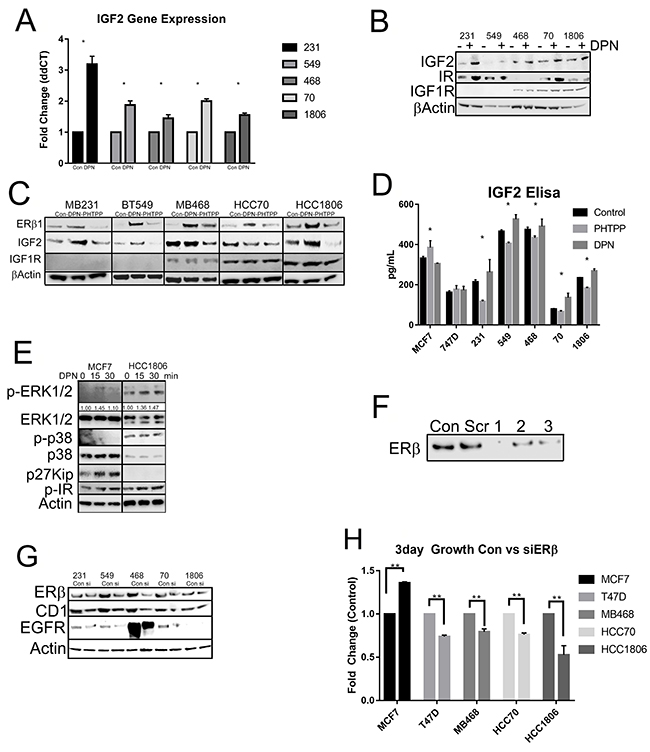
Figure 6: Activation of ERβ upregulates IGF2 and activates the IR/IGF1R/MAPK pathways. (A) The indicated cells were treated with or without DPN. mRNA level of IGF2 was determined by RT-qPCR and adjusted for GAPDH. The bar graph presented fold changes of IGF2 level in cells treated with DPN compared to non-treated cells (control). Each bar indicated mean ±SD from 3 measurements and “*” indicated p<0.05 determined by one-way ANOVA. (B) Total protein was extracted from the indicated cells treated with or without DPN. Western blot analysis was performed with antibodies for IGF2, insulin receptor (IR) and IGFIR. β-actin was used for loading control. (C) The cells were treated with or without DPN, PHTPP or DPN+PHTPP as descripted in methods and total protein was extracted. Protein levels of ERβ1, IGF2 and IGFIR were determined by Western Blot analysis and β-actin was used as loading control. (D) The cells were maintained in serum free media and treated with or without DPN or PHTPP. Secreted protein level of IGF2 in the media was measured by IGF2 ELISA kit according to manufacture instruction as described in methods. The bars indicated mean level of IGF2 with SD from duplicated measurements and “*” indicated P<0.05 for the differences in level of IGF2 between the indicated cells and MCF7 cells determined by one-way ANOVA. (E) The cells were treated with DPN from 0 to 30 min and protein was extracted. Western blot analysis was performed with antibodies for P-ERK1/2, ERK1/2, P-p38, p38 and p27kip. β-actin was used as loading control. The level of P-ERK1/2 was further quantified with β-actin and showed under membrane of P-ERK1/2. (F) T47D cells were treated with 3 different siRNA sequences of ERβ and ERβ1 expression was determined Western Blot analysis with antibody for ERβ1 was used to assess efficiency of the siRNAs. (G) The cells were treated with pooled siRNA sequences for ERβ and the level of ERβ was determined by Western blot. The protein levels of cyclin D1 (CD1) and EGFR were also determined by Western blot analysis. β-actin was used as loading control. (H) The cells were treated with or without siRNA for ERβ and cell proliferation was determined by The CellTiter-Glo® Luminescent Cell Viability Assay as described in methods. The bars indicated fold changes (mena ±SD) of cells treated with siRNA compared to cells without treating with siRNA (control). “**” indicated P<0.001 between the indicated groups determined by one-way ANOVA.
Knockdown of ERβ results in decreased proliferation
We then wanted to determine what role knockdown of ERβ had on cellular proliferation. We used scrambled and three pools of siRNA towards ERβ. Figure 6F shows the knockdown efficiency with the scramble and three different pools of siRNA. We saw that each siRNA was able to knockdown ERβ in T47D cells (Figure 6C). The pooled siRNA were transfected in our TNBC cell lines and through western blot determined the effect of ERβ knockdown on protein expression. Knockdown of ERβ was able to downregulate Cyclin D1 (CD1), EGFR, and ERβ (Figure 6G). We then looked at cellular proliferation. Knockdown of ERβ had a significant decrease in cellular proliferation (Figure 6H). We then wanted to determine what role ERβ plays in apoptosis. Apoptosis was determined by measuring the uptake of annexin in control cells (untreated), compared to cells with ERβ knockdown. The stained cells were analyzed by flow cytometry. Images represent 4 independent flow cytometer runs. As shown in Figure 7A-7E, a significant increase in apoptosis was seen in ERβ knocked down cells (Figure 7).
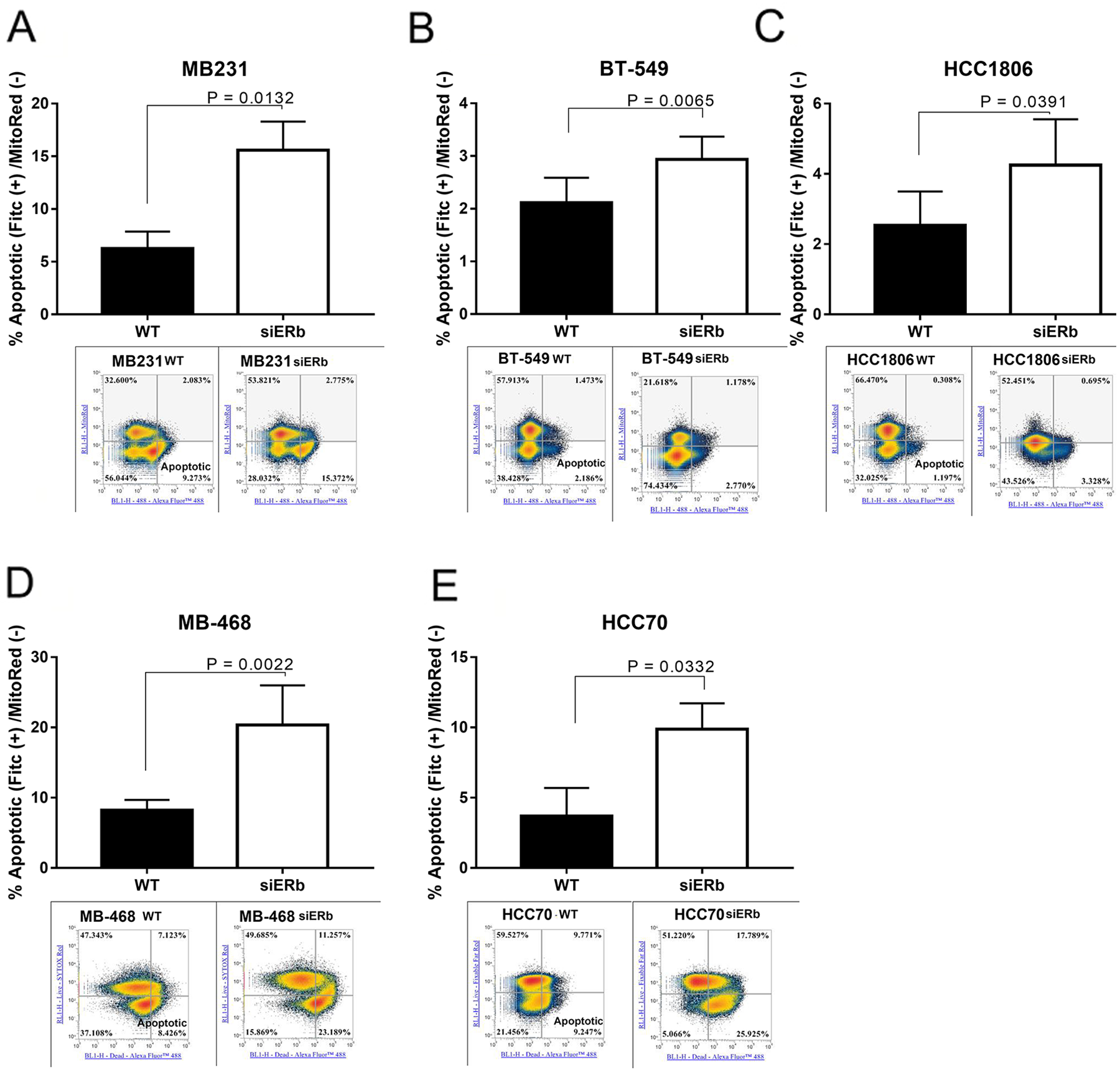
Figure 7: Knockdown of ERβ results in increased apoptosis. TNBC cells were knockdown for ERβ and then subjected to flow cytometer as described in methods Representative images of flow cytometry data were presented. Percentage of apoptotic cells in (A) MB231, (B) BT-549, (C) MB-468, (D) HCC-70, and (E) HCC-1806 were showed in bar graphs as mean ±SD. The statistical differences between the indicated groups were determined by paired t-test.
ERβ expression in clinical samples using tissue microarray
We have seen that through the public database that high ERβ expression is correlated with lower RFS, and in TNBC cell lines that activation of ERβ resulted in increased cellular proliferation, migration, and invasion. We then wanted to see ERβ expression in TNBC across ethnicity. The make-up of our patient population is located in Table 1. As illustrated in Figure 8A, we had archival TNBC tissue specimens from Asian (AS, commercially purchased from US Biomax), African-American (AA), Hispanic (His), and Caucasian (CA) samples from the patient cohort in South Los Angeles. When compared to AS, AA and His patients had higher ERβ and IGF2 expression (Figure 8A and Table 2). When separated by tumor type, both ERβ (Figure 8B) and IGF2 (Figure 8C) are expressed in similar levels within TNBC and Lum A cancers. When separated by race, looking at TNBC only, we see that ERβ and IGF2 are expressed significantly higher in AA and His when compared to CA (P < 0.02) (Figure 8D). We then looked at the association between ERβ and IGF2 and saw that, within TNBC, IGF2 and ERβ expression correlated with each other (Figure 8E) and that IGF2 was associated with increased ERβ expression (Table 3). This data highlights that African American and Hispanic patients with TNBC had higher ERβ and IGF2 expression. This co-expression may result in decreased overall survival of TNBC patients.
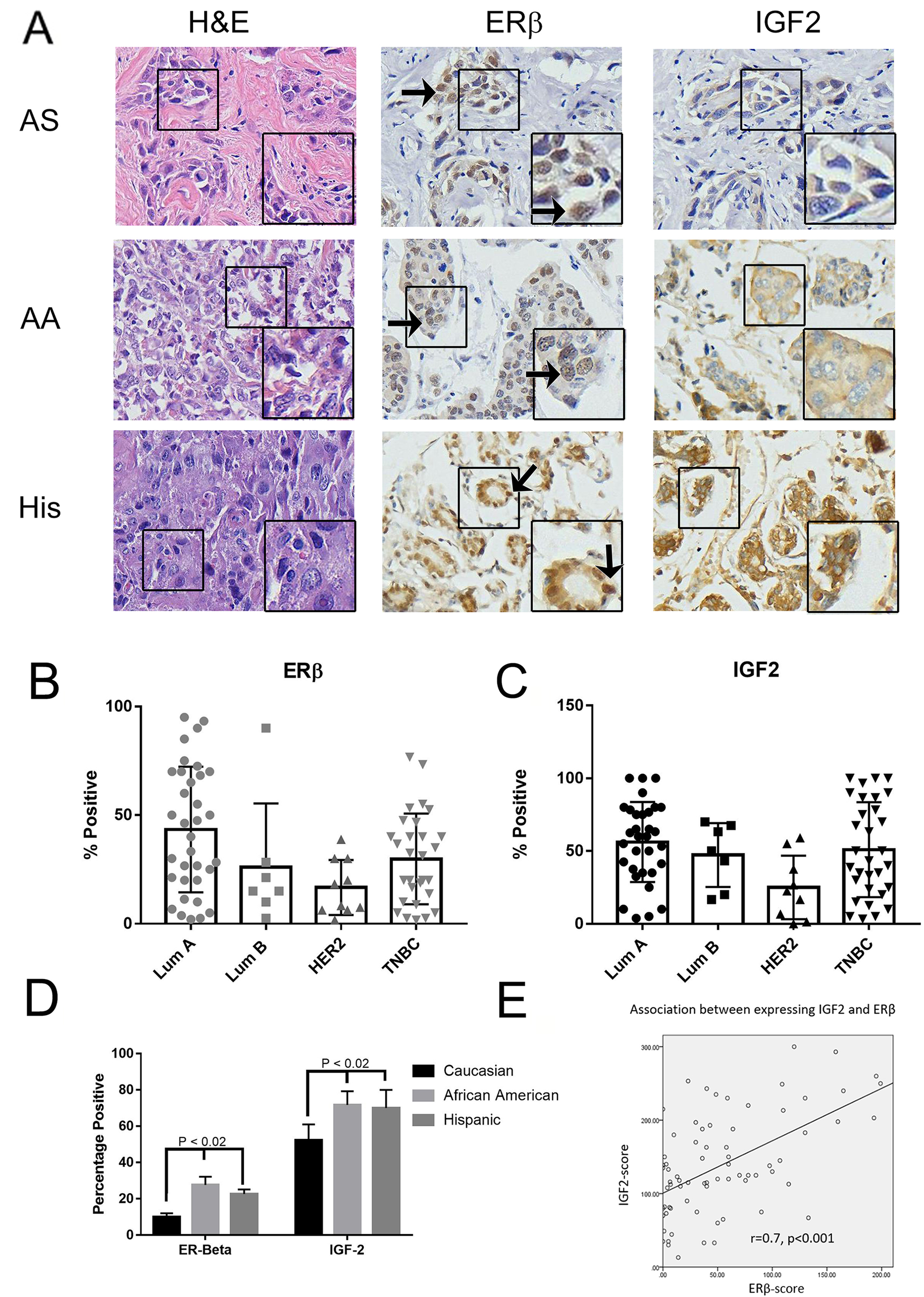
Figure 8: ERβ is expressed in breast cancer patients’ tissue microarray. (A) Immunohistochemical (IHC) analysis was used to determine ERβ1 and IGF2 expression in breast cancer primary tissues as described in methods. Represented images from IHC of H&E, ERβ1 and IGF2 in TNBC from Asian (AS), African American (AA), and Hispanic/Latina (His) patients were shown. (B and C) Bar graphs showed quantified IHC data of ERβ1 (B) and IGF2 (C) expression in different subtypes of breast cancer. The bars indicated percentage of positive staining for ERβ1 or IGF2 (mean ±SD) in the indicated subtype of breast cancers. (D) IHC data of ERβ1 was quantified and the bars indicated percentage positive expression of ERβ1 in the indicated racial ethnicities. (E) The correlation between expressing ERβ and IGF2 in patients was evaluated by Bivariate Correlations with two tailed test, performed with SPSS v.22.
Table 1: Subjects in CDU TMA
N (%) |
|
|---|---|
Total |
204 |
Ethnicity |
|
African American |
50 (24.5) |
Hispanic |
28 (13.7) |
Asian |
126 (61.8) |
Age (yr) |
|
<50 |
90 (44.1) |
≥50 |
114 (55.9) |
ERα/PR status |
|
Positive |
130 (63.7) |
Negative |
74 (36.3) |
HER2 status |
|
Positive (IHC 3+/FISH amplified) |
52 (25.5) |
Negative |
152 (74.5) |
Subtype (by receptors) |
|
ERα/PR+/HER2- |
104 (51.0) |
HER2+ |
52 (25.5) |
TNBC |
48 (23.5) |
Tumor size |
|
≤ 2cm |
45 (22.1) |
2cm – 5cm |
115 (56.4) |
> 5cm |
32 (15.7) |
Unknown |
12 (5.9) |
Node |
|
Negative |
68 (33.3) |
Positive |
127 (62.3) |
Unknown |
9 (4.4) |
Stage |
|
0-II |
134 (65.7) |
III-IV |
66 (32.4) |
unknown |
4 (2.0) |
ERβ (nuclear) |
|
Positive |
121 (59.3) |
Negative |
83 (40.7) |
IGF2 |
|
IGF2-positive |
41 (19.7) |
IGF2-negative |
159 (76.4) |
missing |
8 (3.8) |
Table 2: ERβ and IGF2 status by ethnicity, age, tumor pathology and stage
N (%) |
|||||||
|---|---|---|---|---|---|---|---|
Total |
204 |
ERβ-positive |
ERβ-negative |
p-Value |
IGF2-Positive |
IGF2-negative |
P-Value |
Ethnicity |
|||||||
African American |
50 (24.5) |
36 (72.0) |
14 (28.0) |
26 (55.3) |
21 (44.7) |
||
Hispanic |
28 (13.7) |
18 (64.3) |
10 (35.7) |
11 (40.7) |
16 (59.3) |
||
Asian |
126 (61.8) |
67 (53.2) |
59 (46.8) |
0.019 |
4 (3.2) |
122 (96.8) |
<0.001 |
Age (yr) |
|||||||
<50 |
90 (44.1) |
52 (57.8) |
38 (42.2) |
21 (23.9) |
67 (76.1) |
||
≥50 |
114 (55.9) |
69 (60.5) |
45 (39.5) |
0.774 |
20 (17.9) |
92 (82.1) |
0.378 |
ERα/PR status |
|||||||
Positive |
130 (63.7) |
81 (62.3) |
49 (37.7) |
26 (20.3) |
102 (79.7) |
||
Negative |
74 (36.3) |
40 (54.1) |
34 (45.9) |
0.3 |
15 (20.8) |
57 (79.2) |
1.0 |
HER2 status |
|||||||
Positive |
52 (25.5) |
26 (50.0) |
26 (50.0) |
7 (14.0) |
43 (86.0) |
||
Negative |
152 (74.5) |
95 (62.5) |
57 (37.5) |
0.141 |
34 (22.7) |
116 (77.3) |
0.228 |
Subtype |
|||||||
ERα/PR+/HER2- |
104 (51.0) |
69 (66.3) |
35 (33.7) |
23 (22.3) |
80 (77.7) |
||
HER2+ |
52 (25.5) |
26 (50.0) |
26 (50.0) |
7 (14.0) |
43 (86.0) |
||
TNBC |
48 (23.5) |
26 (54.2) |
22 (45.8) |
0.089 |
11 (23.4) |
36 (76.6) |
0.912 |
Tumor size |
|||||||
≤ 2cm |
45 (22.1) |
29 (64.4) |
16 (35.6) |
12 (27.3) |
32 (72.7) |
||
2cm – 5cm |
115 (56.4) |
63 (54.8) |
52 (45.2) |
11 (9.7) |
102 (90.3) |
||
> 5cm |
32 (15.7) |
23 (71.9) |
9 (28.1) |
0.677 |
14 (43.8) |
18 (56.3) |
0.208 |
Node |
|||||||
Negative |
68 (33.3) |
44 (64.7) |
24 (35.3) |
15 (22.4) |
52 (77.6) |
||
Positive |
127 (62.3) |
74 (58.3) |
53 (41.7) |
0.443 |
23 (18.4) |
102 (81.6) |
0.570 |
Stage |
|||||||
0-II |
134 (65.7) |
79 (59.0) |
55 (41.0) |
27 (20.6) |
104 (79.4) |
||
III-IV |
66 (32.4) |
41 (62.1) |
25 (37.9) |
0.759 |
13 (19.7) |
53 (80.3) |
1.0 |
Table 3: Association between expressing IGF2 and ERβ
IGF2 Status |
P-Value |
||
|---|---|---|---|
High |
Low |
||
ERβ-positive |
32 (78%) |
87 (54.7) |
|
ERβ-negative |
9 (22.0) |
72 (45.3) |
0.07 |
DISCUSSION
Breast cancer patients who are presented with TNBC are frequently diagnosed at an advance stage of the disease, with high histologic grade, and are associated with a significantly higher probability of relapse and metastasis. This clinical diagnosis is as a health disparity, and currently, the most common treatment is systemic chemotherapy [3-11]. Discovery and higher expression of ERβ made it a logical therapeutic target because of its expression in TNBC and its structural similarity to ERα [13-15], however there have been conflicting results, some suggesting that ERβ is of favorable prognostic value [20, 55, 56], no prognostic value [57], or worse prognosis [16, 30, 58]. Some of the discrepancies can also be due to the fact that TNBC is a heterogeneous disease, and most studies that look at TNBC used the more accessible TNBC cell lines (MB231 and Hs578T) and did not account for the heterogeneity of TNBC. There are four subclasses of TNBCs [59] such as basal-like (BL) [7], mesenchymal (MS) [60], immune-enriched (IM) [7, 61], and the luminal AR subtype (LAR) [61]. Others have suggested that the discrepancy may be due to a lack of standardized detection methods, poorly validated antibodies, inconsistent cutoffs for defining ERβ positive cancers via IHC, and variable tissue preparation and processing methods [62, 63]. To address these discrepancies we used validated TNBC specimens (Negative for ERα. PR, and HER2 overexpression). We used validated ERβ antibodies [30, 63, 64], TNBC subtypes (BL1/2, MS, and MSL), and different ethnicities (AA: MB468 HCC-70/1806 CA: MCF7 T-47D, MB-231, BT-549).
Here we examined the role ERβ in disease progression, proliferation, invasion, and migration of TNBC. Our findings show that contrary to previous studies, ERβ activation is pro-tumorigenic in vitro. The relationship between the expression of ERβ and breast cancer clinical progression has been ambiguous, in the publically available data. TNBCs reviewed for our study have shown that high ERβ expression results in significantly lower RFS with or without chemotherapy. We have also shown that in TNBC cell lines that activation of ERβ results in increased secretion of IGF2 which can bind to IR/IGF1R, and activate growth promoting capabilities. Recent studies have shown that targeting the IGF1R pathway could be a therapeutic target [65, 66]. These results and the previous data suggest that endocrine therapy may be beneficial for improving TNBC outcomes.
ERβ has been suggested to control a myriad of functions in BC. Here we have shown that in most TNBC cells, activation of ERβ by DPN significantly increased cell invasion and migration, especially in cells that aren’t highly invasive (MB468, HCC-70/1806). However, in MB-231 cells, DPN caused significant decrease in cellular invasion. This discrepancy could be due to the that fact that a recent study [67] had shown that MB-231 cells express androgen receptor (AR) and that in AR positive TNBC cells, ERβ decreases cellular invasion and migration. Another discrepancy could be due to the fact that these authors used an over-expressing ERβ construct, used FBS and phenol red containing media to determine invasion instead of using hormonal free FBS/phenol red free media. In our study we used a specific agonist to activate ERβ expression, DPN. Even though our results corroborated on the decreased invasion by the activation of ERβ in MB-231 cells, the inhibition of migration (wound closure) due to ERβ activation didn’t agree. Also, we saw in another MS subtype, BT-549, that ERβ activation increased cellular invasion. This supports the notion that in specific subtypes of TNBC ERβ could have different effects.
In this study we also wanted to look at what pathways are activated by ERβ. In our ERα+ cell lines (MCF7, T47D) ERβ activation results in the increase expression of p21WAF/CIP1 and p27kip indicating that in ERα+ cell lines, ERβ provides and anti-proliferative role and agrees with overall BC RFS and other studies showing the anti-proliferative role of ERβ in ERα+ tumors [14-16, 20, 68, 69]. Cyclin–CdK activity is regulated by phosphorylation events and cyclin kinase inhibitors, inhibit their activity. Our results showed that the upregulation of p21/27 might be one of the possible mechanisms of proliferation inhibition by ERβ in ERα+ tumors and the lack of activation of these inhibitors in TNBC. In TNBC, the activation of phospho-ERK1/2, p38, SAPK/JNK, and PI3K which are involved in the MAPK and PI3K signaling pathways. This supports previous studies that activation of ERβ resulted in the activation of MAPK/PI3K signaling [70, 71]. Knocked down (siRNA)/inhibited ERβ by a specific ERβ antagonist (PHTPP) saw a down regulation in Cyclin D1, EGFR, and MAPK/PI3K signaling, indicating ERβ roles in cell cycle and MAPK/PI3K signaling. We have also shown that down regulation of ERβ increase apoptosis in TNBC cell lines. Further studies are warranted to identify the precise mechanism by which ERβ overexpression can repress apoptosis in TNBC tumors.
In conclusion, the results of this study indicates that expression ERβ in TNBC patients is significantly associated with decreased RFS and RFS with/without chemotherapy. ERβ and IGF2 expression is positively correlated with each other, ERβ/IGF2 expression is increased in AA/HS women when compared to CA and AS. ERβ activation significantly increases invasion, migration, proliferation, and IGF2 secretion. When silencing ERβ or using a specific ERβ antagonist, we demonstrated that ERβ suppression resulted in decreased IGF2 secretion and proliferation. This suppression could possibly be due to the suppression of the MAPK/PI3K/AKT pathways and IGF2 activation on IR/IGF-1R. This work further highlights the roles ERβ plays in the heterogeneity of TNBC and can lead us to more promising therapeutics to specifically target ERβ or use combinational targets for ERβ, IR/IGF-1R, MAPK/PI3K/AKT pathway signaling.
MATERIALS AND METHODS
Patients
Patients were selected from an ongoing breast cancer study conducted in the Division of Cancer Research and Training at Charles R. Drew University of Medicine and Science in South Los Angeles. Women were informed and consented from Martin Luther King Ambulatory Care Center (MACC) between 1995 and 2007. This study was approved by the Charles R. Drew University of Science and Medicine Institutional Review Board and written informed consent was obtained from all participants (Approval 00-06-041-13). A total 1400 participants have been consented into the study, and 370 subjects have breast cancer confirmed by surgical biopsy/pathology and follow-up data. Characteristics of tissue microarray are found in Table 1. Charles R. Drew University breast cancer patient tissue microarray contains African-American (AA) and Hispanic (His) breast cancer tissue. Asian (AS) tissue microarray came from US Biomax.
Definition of breast cancer subtypes
The receptor subtypes were categorized in the following manner: (a) Luminal A (LumA) (ER+ and/or PR+) and HER2-, (b) LumB (ER+ and/or PR+) and HER2+, (c) and HER2+, and (d) triple negative (TNBC) (ER-/PR-/HER2-) based on immunohistochemistry (IHC) analysis. The ER/PR, HER2, and Ki67 status were obtained from the patient’s pathology reports. HR+ was defined as >5% nuclear positive for ER and/or PR in tumor cells. HER2+ was defined as HER2 3+ by IHC and/or more than 2.2 HER2 genes counted for every copy of chromosome 17 (HER2/CEP17 ratio) by FISH analysis. Ki67 Low was defined as <= 20% nuclear positive, and Ki67 High was defined as > 20% nuclear positive. Asian patient tissue microarray was obtained from US Bio Max, Inc (Rockville, MD). Staining and receptor status are located on manufactures web page, sample: HBre-Duc140Sur-01 (https://www.biomax.us/tissue-arrays/Breast/HBre-Duc140Sur-01).
Cells, reagents, and antibodies
Triple negative breast cancer cells (TNBC; MDA-MB-231/468, BT-649,) and ER/PR positive breast cancer cells (MCF7 and T-47D) were cultured in DMEM/F12 (Gibco, Grand Island, NY), 10% fetal bovine serum (Gibco), and 1% penicillin/streptomycin (Gibco). Triple negative cell lines HCC 70/1806 were cultured in RPMI 1640 (Gibco), 10% FBS (Gibco), or 5% dextran-coated charcoal-stripped FBS (CSFBS) (GE Life Sciences Pittsburgh, PA), and 1% penicillin/streptomycin (Gibco). Primary antibodies against ERβ1 (clone PPG5/10) were purchased from BioRad(AbDSerotec) (Valencia, CA), Insulin Receptor (Abcam) IGF2 from ThermoFisher (Rockford, IL), CNND1, EGFR, ERα, Beta-Actin from Santa Cruz Biotechnology (Santa Cruz, CA), p-ERK1/2, p-p38, p27, p21 from Cell signaling (Beverly, MA). ERβ agonist, Diarylpropionitrile (DPN) and antagonist, 4-[2-phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]-pyrimidin-3-yl]phenol (PHTPP) were purchased from Tocris.
Cell migration test
TNBC cells were seeded in 8.5cm3 dishes and cultured in culture-insert 2 wells from Ibidi (Munich, Germany) to give a uniform scratch at 500μM. A line was created by removing the culture insert. Cells were then treated with DPN or PHTPP for 24hrs and pictures were taken at 0, 2, 4, 8, 12, and 24hr. The wound area was marked and cell migration into the scratched area was daily photographed using an inverted microscope.
Cell invasion assay
One day prior to beginning the assay, TNBC cells were starved in a serum-free medium in top chamber of Corning BioCoat matrigel invasion chamber with 8.0mm PET membrane (Corning Corning, NY). Invasion assay was performed using the matrigel invasion assay kit. Briefly, top chamber was plated with 5 × 104 cells in serum-free, phenol red free medium with either 20mM DPN or 100mM PHTPP. Bottom chamber was filled with medium containing CSFBS and incubated at 37°C with 5% CO2 for 24 hours. After the fixation of cells to the bottom chamber, the upper chamber was removed of all cells, and then coated with crystal violet for 15mins. Chamber was then wash and analyzed for cell invasion by the detection of crystal violet positive cells on the bottom chamber. The total numbers of invaded cells were counted by hand in 4 independent sections. All experiments were independently repeated at least three times.
Apoptosis assay
Measurement of apoptosis was done by using Mitochondrial Membrane Potential Apoptosis Kit, with Mitotracker™ Red & Annexin V Alexa Fluor™ 488, for flow cytometry per manufacture protocol by Thermofisher. Cells were stained and then analyze the cells by flow cytometry, measuring the fluorescence emission at 530 nm and 585 nm. The cells should resolve into two principal populations: live cells with a low level of green fluorescence and high red fluorescence, and apoptotic cells with moderate level of green fluorescence and low red fluorescence by our Attune NxT Flow Cytometer.
Transfection assay
ERβ si/shRNA was purchased from Santa Cruz Biotechnology. The transfection was performed on the Lonza 4D-Nucleofector™ System per manufacture protocol.
Cell viability assay
Cell viability assay was performed using CellTiter-Glo (Promega) according to manufacturer’s instructions. TNBC and control cells were seeded at 2,000 cells/well in a 96-well plate, grown over 3 and 5 days in the presence and absence of DMSO, DPN, and PHTPP. CellTiter-Glo measurements were taken at several time points to track cell survival.
Quantitative-real-time PCR, western blot and IHC
For quantitative real-time PCR (qPCR) RNA was extracted with Trizol (Ambion) from 46 Surgical BPH and 53 Incidental BPH specimens. Subsequently, 500 ng RNA was reverse transcribed into cDNA using iScript cDNA Synthesis Kit (BioRad, Valencia, CA). qPCR was performed using IQ SYBR Green Supermix (BioRad, Hercules, CA) and results were analyzed using BioRad CFX manager software. All results were calculated using ΔΔCt analysis and normalized to GAPDH expression.
For Western blotting, protein was extracted with 2% SDS buffer and 25 μg protein was run on pre-made 4-12% polyacrylamide gels (Life Technologies). Primary antibodies were incubated in 5% BSA in TBST for 1 hour or overnight or at 4°C followed by incubation in secondary antibodies and development using ECL. Membranes were stripped and re-probed with an antibody against β-actin (Sigma).
Immunohistochemistry was performed as previously described [72] Briefly, 5 μm sections were de-waxed, rehydrated and endogenous peroxidases were blocked with hydrogen peroxide. Sections were then boiled in citrate and blocked in 5% serum for 1 hr. Primary antibodies were incubated overnight at 4°C at the following concentrations: ERb (1:100) and IGF2 (1:200). Biotinylated anti-mouse or -rabbit secondary antibodies (DAKO Carpentaria, CA) were incubated for 60 min at room temperature after slides were washed for 1 hr in PBS. Slides were incubated in ABC-HRP complex (Vector Laboratories Burlingame, CA) for 30 min. Bound antibodies were then visualized by incubation with 3,3′ diaminobenzidine tetrahydrochloride (liquid DAB, DAKO). Slides were then rinsed in tap water, counterstained with hematoxylin, and mounted.
Statistical analysis
Log-rank (Mantel-Cox) test was used to determine overall survival and relapse free survival between High and low ER-β gene expression. Mann–Whitney tests were used for univariate comparisons of study characteristics between two groups. A two-sided P-value of 0.05 or less was considered statistically significant. A two-way ANOVA test was used for comparisons of characteristics. Correlation between IGF2 and ER-β was evaluated by the determination of the Spearman correlation coefficient (GraphPad Prism).
Abbreviations
AA: African-American women, AR: androgen receptor, BC: Breast Cancer, CA: Caucasian American women, DPN: Diarylpropionitrile, ELISA: enzyme-linked immunosorbent assay, ER: Estrogen Receptor, ERα: ER-alpha, ERβ: ER-beta, HER2: Human Epidermal Growth Factor Receptor 2, HS: Hispanic women, IGF: insulin-like growth factor (IGF-1 and IGF-2), IGF-1R: IGF-1 receptor, IGF2R: IGF-2 receptor, IHC: immunohistochemical, LAR: luminal AR subtype, mIGF2: mature IGF2, PHTPP: 4-[2-phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]-pyrimidin-3-yl]phenol, pIGF2: precursor IGF2, PR: Progesterone Receptor, qPCR: quantitative-real-time PCR, TNBC: Triple negative breast cancer.
CONFLICTS OF INTEREST
The authors have declared that no competing interests exist.
FUNDING
This work was supported in part by NIH/NCI Grants: 1U54CA14393, R25DK067015, BC043180, U54MD007598 to Jaydutt V. Vadgama. Diversity supplement from U54MD007598/NIMHD to David Austin, and NIH/NIMHD CRECD R25 MD007610, and NIMHD 5S21MD 000103-Faculty Retention Award to Yanyuan Wu.
REFERENCES
1. Hudis CA, Gianni L. Triple-negative breast cancer: an unmet medical need. Oncologist. 2011; 16:1–11. https://doi.org/10.1634/theoncologist.2011-S1-01.
2. Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010; 363:1938–48. https://doi.org/10.1056/NEJMra1001389.
3. Morris GJ, Naidu S, Topham AK, Guiles F, Xu Y, McCue P, Schwartz GF, Park PK, Rosenberg AL, Brill K, Mitchell EP. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: a single-institution compilation compared with the National Cancer Institute’s Surveillance, Epidemiology, and End Results database. Cancer. 2007; 110:876–84. https://doi.org/10.1002/cncr.22836.
4. Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007; 13:4429–34. https://doi.org/10.1158/1078-0432.CCR-06-3045.
5. Early Breast Cancer Trialists' Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005; 365:1687–717. https://doi.org/10.1016/S0140-6736(05)66544-0.
6. Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, Li Q, Juul N, Leong CO, Calogrias D, Buraimoh A, Fatima A, Gelman RS, Ryan PD, et al. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J Clin Oncol. 2010; 28:1145–53. https://doi.org/10.1200/JCO.2009.22.4725.
7. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011; 121:2750–67. https://doi.org/10.1172/JCI45014.
8. Haffty BG, Yang Q, Reiss M, Kearney T, Higgins SA, Weidhaas J, Harris L, Hait W, Toppmeyer D. Locoregional relapse and distant metastasis in conservatively managed triple negative early-stage breast cancer. J Clin Oncol. 2006; 24:5652–57. https://doi.org/10.1200/JCO.2006.06.5664.
9. Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S, Deming SL, Geradts J, Cheang MC, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006; 295:2492–502. https://doi.org/10.1001/jama.295.21.2492.
10. Amirikia KC, Mills P, Bush J, Newman LA. Higher population-based incidence rates of triple-negative breast cancer among young African-American women: implications for breast cancer screening recommendations. Cancer. 2011; 117:2747–53. https://doi.org/10.1002/cncr.25862.
11. Reddy GM, Suresh PK, Pai RR. Clinicopathological Features of Triple Negative Breast Carcinoma. J Clin Diagn Res. 2017; 11:EC05–08.
12. Madak-Erdogan Z, Kieser KJ, Kim SH, Komm B, Katzenellenbogen JA, Katzenellenbogen BS. Nuclear and extranuclear pathway inputs in the regulation of global gene expression by estrogen receptors. Mol Endocrinol. 2008; 22:2116–27. https://doi.org/10.1210/me.2008-0059.
13. Tamoxifen for early breast cancer: an overview of the randomised trials. Early Breast Cancer Trialists' Collaborative Group. Lancet. 1998; 351:1451–67. https://doi.org/10.1016/S0140-6736(97)11423-4.
14. Skliris GP, Leygue E, Curtis-Snell L, Watson PH, Murphy LC. Expression of oestrogen receptor-beta in oestrogen receptor-alpha negative human breast tumours. Br J Cancer. 2006; 95:616–26. https://doi.org/10.1038/sj.bjc.6603295.
15. Novelli F, Milella M, Melucci E, Di Benedetto A, Sperduti I, Perrone-Donnorso R, Perracchio L, Venturo I, Nisticò C, Fabi A, Buglioni S, Natali PG, Mottolese M. A divergent role for estrogen receptor-beta in node-positive and node-negative breast cancer classified according to molecular subtypes: an observational prospective study. Breast Cancer Res. 2008; 10:R74. https://doi.org/10.1186/bcr2139.
16. Jensen EV, Cheng G, Palmieri C, Saji S, Mäkelä S, Van Noorden S, Wahlström T, Warner M, Coombes RC, Gustafsson JA. Estrogen receptors and proliferation markers in primary and recurrent breast cancer. Proc Natl Acad Sci U S A. 2001; 98:15197–202. https://doi.org/10.1073/pnas.211556298.
17. Hartman J, Ström A, Gustafsson JA. Estrogen receptor beta in breast cancer—diagnostic and therapeutic implications. Steroids. 2009; 74:635–41. https://doi.org/10.1016/j.steroids.2009.02.005.
18. Chen JQ, Russo J. ERalpha-negative and triple negative breast cancer: molecular features and potential therapeutic approaches. Biochim Biophys Acta. 2009; 1796:162–75.
19. Phipps AI, Chlebowski RT, Prentice R, McTiernan A, Wactawski-Wende J, Kuller LH, Adams-Campbell LL, Lane D, Stefanick ML, Vitolins M, Kabat GC, Rohan TE, Li CI. Reproductive history and oral contraceptive use in relation to risk of triple-negative breast cancer. J Natl Cancer Inst. 2011; 103:470–77. https://doi.org/10.1093/jnci/djr030.
20. Honma N, Horii R, Iwase T, Saji S, Younes M, Takubo K, Matsuura M, Ito Y, Akiyama F, Sakamoto G. Clinical importance of estrogen receptor-beta evaluation in breast cancer patients treated with adjuvant tamoxifen therapy. J Clin Oncol. 2008; 26:3727–34. https://doi.org/10.1200/JCO.2007.14.2968.
21. Yan Y, Li X, Blanchard A, Bramwell VH, Pritchard KI, Tu D, Shepherd L, Myal Y, Penner C, Watson PH, Leygue E, Murphy LC. Expression of both estrogen receptor-beta 1 (ER-β1) and its co-regulator steroid receptor RNA activator protein (SRAP) are predictive for benefit from tamoxifen therapy in patients with estrogen receptor-alpha (ER-α)-negative early breast cancer (EBC). Ann Oncol. 2013; 24:1986–93. https://doi.org/10.1093/annonc/mdt132.
22. Shaaban AM, Green AR, Karthik S, Alizadeh Y, Hughes TA, Harkins L, Ellis IO, Robertson JF, Paish EC, Saunders PT, Groome NP, Speirs V. Nuclear and cytoplasmic expression of ERbeta1, ERbeta2, and ERbeta5 identifies distinct prognostic outcome for breast cancer patients. Clin Cancer Res. 2008; 14:5228–35. https://doi.org/10.1158/1078-0432.CCR-07-4528.
23. Murphy LC, Leygue E. The role of estrogen receptor-β in breast cancer. Semin Reprod Med. 2012; 30:5–13. https://doi.org/10.1055/s-0031-1299592.
24. Pietras RJ, Márquez-Garbán DC. Membrane-associated estrogen receptor signaling pathways in human cancers. Clin Cancer Res. 2007; 13:4672–76. https://doi.org/10.1158/1078-0432.CCR-07-1373.
25. Hammes SR, Levin ER. Extranuclear steroid receptors: nature and actions. Endocr Rev. 2007; 28:726–41. https://doi.org/10.1210/er.2007-0022.
26. Tonetti DA, Rubenstein R, DeLeon M, Zhao H, Pappas SG, Bentrem DJ, Chen B, Constantinou A, Craig Jordan V. Stable transfection of an estrogen receptor beta cDNA isoform into MDA-MB-231 breast cancer cells. J Steroid Biochem Mol Biol. 2003; 87:47–55. https://doi.org/10.1016/j.jsbmb.2003.07.003.
27. Shanle EK, Zhao Z, Hawse J, Wisinski K, Keles S, Yuan M, Xu W. Research resource: global identification of estrogen receptor β target genes in triple negative breast cancer cells. Mol Endocrinol. 2013; 27:1762–75. https://doi.org/10.1210/me.2013-1164.
28. Galluzzo P, Caiazza F, Moreno S, Marino M. Role of ERbeta palmitoylation in the inhibition of human colon cancer cell proliferation. Endocr Relat Cancer. 2007; 14:153–67.
29. Caiazza F, Galluzzo P, Lorenzetti S, Marino M. 17Beta-estradiol induces ERbeta up-regulation via p38/MAPK activation in colon cancer cells. Biochem Biophys Res Commun. 2007; 359:102–07. https://doi.org/10.1016/j.bbrc.2007.05.059.
30. Hamilton N, Márquez-Garbán D, Mah V, Fernando G, Elshimali Y, Garbán H, Elashoff D, Vadgama J, Goodglick L, Pietras R. Biologic roles of estrogen receptor-β and insulin-like growth factor-2 in triple-negative breast cancer. Biomed Res Int. 2015; 2015:925703. https://doi.org/10.1155/2015/925703.
31. Belardi V, Gallagher EJ, Novosyadlyy R, LeRoith D. Insulin and IGFs in obesity-related breast cancer. J Mammary Gland Biol Neoplasia. 2013; 18:277–89. https://doi.org/10.1007/s10911-013-9303-7.
32. Westley BR, May FE. Role of insulin-like growth factors in steroid modulated proliferation. J Steroid Biochem Mol Biol. 1994; 51:1–9. https://doi.org/10.1016/0960-0760(94)90109-0.
33. Osborne CK, Clemmons DR, Arteaga CL. Regulation of breast cancer growth by insulin-like growth factors. J Steroid Biochem Mol Biol. 1990; 37:805–09. https://doi.org/10.1016/0960-0760(90)90423-I.
34. Richardson AE, Hamilton N, Davis W, Brito C, De León D. Insulin-like growth factor-2 (IGF-2) activates estrogen receptor-α and -β via the IGF-1 and the insulin receptors in breast cancer cells. Growth Factors. 2011; 29:82–93. https://doi.org/10.3109/08977194.2011.565003.
35. Mancini M, Gariboldi MB, Taiana E, Bonzi MC, Craparotta I, Pagin M, Monti E. Co-targeting the IGF system and HIF-1 inhibits migration and invasion by (triple-negative) breast cancer cells. Br J Cancer. 2014; 110:2865–73. https://doi.org/10.1038/bjc.2014.269.
36. Pierobon M, Frankenfeld CL. Obesity as a risk factor for triple-negative breast cancers: a systematic review and meta-analysis. Breast Cancer Res Treat. 2013; 137:307–14. https://doi.org/10.1007/s10549-012-2339-3.
37. Sturtz LA, Melley J, Mamula K, Shriver CD, Ellsworth RE. Outcome disparities in African American women with triple negative breast cancer: a comparison of epidemiological and molecular factors between African American and Caucasian women with triple negative breast cancer. BMC Cancer. 2014; 14:62. https://doi.org/10.1186/1471-2407-14-62.
38. LeRoith D, Baserga R, Helman L, Roberts CT Jr. Insulin-like growth factors and cancer. Ann Intern Med. 1995; 122:54–59. https://doi.org/10.7326/0003-4819-122-1-199501010-00009.
39. Yang XF, Beamer WG, Huynh H, Pollak M. Reduced growth of human breast cancer xenografts in hosts homozygous for the lit mutation. Cancer Res. 1996; 56:1509–11.
40. Sciacca L, Costantino A, Pandini G, Mineo R, Frasca F, Scalia P, Sbraccia P, Goldfine ID, Vigneri R, Belfiore A. Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene. 1999; 18:2471–79. https://doi.org/10.1038/sj.onc.1202600.
41. Klotz DM, Hewitt SC, Ciana P, Raviscioni M, Lindzey JK, Foley J, Maggi A, DiAugustine RP, Korach KS. Requirement of estrogen receptor-alpha in insulin-like growth factor-1 (IGF-1)-induced uterine responses and in vivo evidence for IGF-1/estrogen receptor cross-talk. J Biol Chem. 2002; 277:8531–37. https://doi.org/10.1074/jbc.M109592200.
42. Singh SK, Moretta D, Almaguel F, Wall NR, De León M, De León D. Differential effect of proIGF-II and IGF-II on resveratrol induced cell death by regulating survivin cellular localization and mitochondrial depolarization in breast cancer cells. Growth Factors. 2007; 25:363–72. https://doi.org/10.1080/08977190801886905.
43. Pacher M, Seewald MJ, Mikula M, Oehler S, Mogg M, Vinatzer U, Eger A, Schweifer N, Varecka R, Sommergruber W, Mikulits W, Schreiber M. Impact of constitutive IGF1/IGF2 stimulation on the transcriptional program of human breast cancer cells. Carcinogenesis. 2007; 28:49–59. https://doi.org/10.1093/carcin/bgl091.
44. Singer CF, Mogg M, Koestler W, Pacher M, Marton E, Kubista E, Schreiber M. Insulin-like growth factor (IGF)-I and IGF-II serum concentrations in patients with benign and malignant breast lesions: free IGF-II is correlated with breast cancer size. Clin Cancer Res. 2004; 10:4003–09. https://doi.org/10.1158/1078-0432.CCR-03-0093.
45. Livingstone C. IGF2 and cancer. Endocr Relat Cancer. 2013; 20:R321–39.
46. Kalla Singh S, Tan QW, Brito C, De León M, De León D. Insulin-like growth factors I and II receptors in the breast cancer survival disparity among African-American women. Growth Horm IGF Res. 2010; 20:245–54. https://doi.org/10.1016/j.ghir.2010.03.001.
47. Mathieu M, Rochefort H, Barenton B, Prebois C, Vignon F. Interactions of cathepsin-D and insulin-like growth factor-II (IGF-II) on the IGF-II/mannose-6-phosphate receptor in human breast cancer cells and possible consequences on mitogenic activity of IGF-II. Mol Endocrinol. 1990; 4:1327–35. https://doi.org/10.1210/mend-4-9-1327.
48. Cullen KJ, Allison A, Martire I, Ellis M, Singer C. Insulin-like growth factor expression in breast cancer epithelium and stroma. Breast Cancer Res Treat. 1992; 22:21–29. https://doi.org/10.1007/BF01833330.
49. De Leon DD, Wilson DM, Powers M, Rosenfeld RG. Effects of insulin-like growth factors (IGFs) and IGF receptor antibodies on the proliferation of human breast cancer cells. Growth Factors. 1992; 6:327–36. https://doi.org/10.3109/08977199209021544.
50. Giani C, Campani D, Rasmussen A, Fierabracci P, Miccoli P, Bevilacqua G, Pinchera A, Cullen KJ. Insulin-like growth factor II (IGF-II) immunohistochemistry in breast cancer: relationship with the most important morphological and biochemical prognostic parameters. Int J Biol Markers. 2002; 17:90–95.
51. Györffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010; 123:725–31. https://doi.org/10.1007/s10549-009-0674-9.
52. Lánczky A, Nagy Á, Bottai G, Munkácsy G, Szabó A, Santarpia L, Győrffy B. miRpower: a web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res Treat. 2016; 160:439–46. https://doi.org/10.1007/s10549-016-4013-7.
53. Harrington WR, Sheng S, Barnett DH, Petz LN, Katzenellenbogen JA, Katzenellenbogen BS. Activities of estrogen receptor alpha- and beta-selective ligands at diverse estrogen responsive gene sites mediating transactivation or transrepression. Mol Cell Endocrinol. 2003; 206:13–22. https://doi.org/10.1016/S0303-7207(03)00255-7.
54. Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor-beta potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J Med Chem. 2001; 44:4230–51. https://doi.org/10.1021/jm010254a.
55. Mann S, Laucirica R, Carlson N, Younes PS, Ali N, Younes A, Li Y, Younes M. Estrogen receptor beta expression in invasive breast cancer. Hum Pathol. 2001; 32:113–18. https://doi.org/10.1053/hupa.2001.21506.
56. Sugiura H, Toyama T, Hara Y, Zhang Z, Kobayashi S, Fujii Y, Iwase H, Yamashita H. Expression of estrogen receptor beta wild-type and its variant ERbetacx/beta2 is correlated with better prognosis in breast cancer. Jpn J Clin Oncol. 2007; 37:820–28. https://doi.org/10.1093/jjco/hym114.
57. Miller WR, Anderson TJ, Dixon JM, Saunders PT. Oestrogen receptor beta and neoadjuvant therapy with tamoxifen: prediction of response and effects of treatment. Br J Cancer. 2006; 94:1333–38. https://doi.org/10.1038/sj.bjc.6603082.
58. Bialesova L, Xu L, Gustafsson JÅ, Haldosen LA, Zhao C, Dahlman-Wright K. Estrogen receptor β2 induces proliferation and invasiveness of triple negative breast cancer cells: association with regulation of PHD3 and HIF-1α. Oncotarget. 2017; 8:76622–33. https://doi.org/10.18632/oncotarget.20635.
59. Ahn SG, Kim SJ, Kim C, Jeong J. Molecular Classification of Triple-Negative Breast Cancer. J Breast Cancer. 2016; 19:223–30. https://doi.org/10.4048/jbc.2016.19.3.223.
60. Weigelt B, Ng CK, Shen R, Popova T, Schizas M, Natrajan R, Mariani O, Stern MH, Norton L, Vincent-Salomon A, Reis-Filho JS. Metaplastic breast carcinomas display genomic and transcriptomic heterogeneity [corrected]. Mod Pathol. 2015; 28:340–51. https://doi.org/10.1038/modpathol.2014.142. Erratum in: Corrigendum: metastatic breast carcinomas display genomic and transcriptomic heterogeneity. [Mod Pathol. 2015].
61. Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua SA, Savage MI, Osborne CK, Hilsenbeck SG, Chang JC, Mills GB, Lau CC, Brown PH. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res. 2015; 21:1688–98. https://doi.org/10.1158/1078-0432.CCR-14-0432.
62. Carder PJ, Murphy CE, Dervan P, Kennedy M, McCann A, Saunders PT, Shaaban AM, Foster CS, Witton CJ, Bartlett JM, Walker RA, Speirs V. A multi-centre investigation towards reaching a consensus on the immunohistochemical detection of ERbeta in archival formalin-fixed paraffin embedded human breast tissue. Breast Cancer Res Treat. 2005; 92:287–93. https://doi.org/10.1007/s10549-004-4262-8.
63. Wu X, Subramaniam M, Negron V, Cicek M, Reynolds C, Lingle WL, Goetz MP, Ingle JN, Spelsberg TC, Hawse JR. Development, characterization, and applications of a novel estrogen receptor beta monoclonal antibody. J Cell Biochem. 2012; 113:711–23. https://doi.org/10.1002/jcb.23443.
64. Mah V, Marquez D, Alavi M, Maresh EL, Zhang L, Yoon N, Horvath S, Bagryanova L, Fishbein MC, Chia D, Pietras R, Goodglick L. Expression levels of estrogen receptor beta in conjunction with aromatase predict survival in non-small cell lung cancer. Lung Cancer. 2011; 74:318–25. https://doi.org/10.1016/j.lungcan.2011.03.009.
65. Wu W, Ma J, Shao N, Shi Y, Liu R, Li W, Lin Y, Wang S. Co-Targeting IGF-1R and Autophagy Enhances the Effects of Cell Growth Suppression and Apoptosis Induced by the IGF-1R Inhibitor NVP-AEW541 in Triple-Negative Breast Cancer Cells. PLoS One. 2017; 12:e0169229. https://doi.org/10.1371/journal.pone.0169229.
66. Hamilton N, Austin D, Márquez-Garbán D, Sanchez R, Chau B, Foos K, Wu Y, Vadgama J, Pietras R. Receptors for Insulin-Like Growth Factor-2 and Androgens as Therapeutic Targets in Triple-Negative Breast Cancer. Int J Mol Sci. 2017; 18:E2305. https://doi.org/10.3390/ijms18112305.
67. Song W, Tang L, Xu Y, Sun Q, Yang F, Guan X. ERβ1 inhibits metastasis of androgen receptor-positive triple-negative breast cancer by suppressing ZEB1. J Exp Clin Cancer Res. 2017; 36:75. https://doi.org/10.1186/s13046-017-0545-x.
68. Charn TH, Liu ET, Chang EC, Lee YK, Katzenellenbogen JA, Katzenellenbogen BS. Genome-wide dynamics of chromatin binding of estrogen receptors alpha and beta: mutual restriction and competitive site selection. Mol Endocrinol. 2010; 24:47–59. https://doi.org/10.1210/me.2009-0252.
69. Järvinen TA, Pelto-Huikko M, Holli K, Isola J. Estrogen receptor beta is coexpressed with ERalpha and PR and associated with nodal status, grade, and proliferation rate in breast cancer. Am J Pathol. 2000; 156:29–35. https://doi.org/10.1016/S0002-9440(10)64702-5.
70. Marty B, Maire V, Gravier E, Rigaill G, Vincent-Salomon A, Kappler M, Lebigot I, Djelti F, Tourdès A, Gestraud P, Hupé P, Barillot E, Cruzalegui F, et al. Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase pathway in basal-like breast cancer cells. Breast Cancer Res. 2008; 10:R101. https://doi.org/10.1186/bcr2204.
71. Wang J, Zhang C, Chen K, Tang H, Tang J, Song C, Xie X. ERβ1 inversely correlates with PTEN/PI3K/AKT pathway and predicts a favorable prognosis in triple-negative breast cancer. Breast Cancer Res Treat. 2015; 152:255–69. https://doi.org/10.1007/s10549-015-3467-3.
72. Strand DW, Jiang M, Murphy TA, Yi Y, Konvinse KC, Franco OE, Wang Y, Young JD, Hayward SW. PPARγ isoforms differentially regulate metabolic networks to mediate mouse prostatic epithelial differentiation. Cell Death Dis. 2012; 3:e361. https://doi.org/10.1038/cddis.2012.99.