
Introduction
Gastrointestinal stromal tumors (GISTs) are the most common type of mesenchymal neoplasms in the gastrointestinal tract [1, 2]. Approximately 85% of GISTs harbor gain-of-function mutations in KIT or PDGFRA, which lead to the promotion of cell survival and escape from apoptosis. In GISTs, KIT mutations occur mainly in the exon 11 juxtamembrane domain, followed by the exon 9 extracellular domain, the exon 13/14 ATP-binding domain, and the exon 17 activation loop domain [3-5]. Imatinib mesylate (IM; Gleevec®, Novartis Pharma, Basel, Switzerland) and sunitinib malate (SU; Sutent®, Pfizer Inc., CA, USA) are the first-line and second-line drugs for metastatic/unresectable GISTs and IM-resistant GISTs, respectively [6, 7]. In a pivotal phase III trial, regorafenib yielded notable improvement in progression-free survival in IM/SU failure patients comparing to placebo control and has recently been approved as a third-line drug for IM/SU-resistant GISTs [8]. Unfortunately, TKI resistance remains an increasing issue after long-term tyrosine kinase inhibitor (TKI) treatment. HSP90AA1, a chaperone protein that assists the folding and maturation of its client proteins, is an alternative therapeutic target for cancer therapy [9-11]. Inhibition of HSP90AA1 by 17-AAG, the first HSP90AA1 inhibitor tested in clinical trials, led to KIT downregulation and cell death in both mutant KIT-expressing mast cells and GIST cell lines [12, 13].
However, 17-AAG has several pharmacological limitations, including poor bioavailability, difficulty in formulation, and hepatotoxicity to prevent its further application in clinical setting. Therefore, we evaluated the anti-proliferation effects of a next-generation HSP90AA1 inhibitor, NVP-AUY922 (AUY922), which has high affinity against HSP90AA1 in vitro, for mutant KIT expressing GIST cell in our previous study [14-17]. In that study, AUY922 effectively downregulated both total and phosphorylated KIT and induced cell apoptosis in both IM-sensitive and IM-resistant GIST cells. However, it was surprisingly to find that AUY922-induced KIT reduction as well as endogenous KIT turnover, were mediated by both autophagy and proteasome degradation pathways. These results highlight the feasibility of AUY922 in the treatment of mutant KIT-expressing GISTs and the novel role of autophagy in endogenous and AUY922-induced KIT degradation. However, despite the high antitumor activity of AUY922 against GIST cells, AUY922 therapy at dose of 70 mg/m2 weekly infusion, the maximum tolerated dose defined in phase I trial, was associated with unneglectable ocular adverse events, including night blindness, photopsia, blurred vision and visual impairment [18].
Based on our previous findings, we hypothesize that the combination of AUY922 with an autophagy inducer that may synergistically or additively enhance KIT downregulation, and thus diminish the dose of AUY922 for GIST treatment and subsequently minimize the incidence and severity of ocular adverse events. Classically, mammalian target of rapamycin (MTOR) kinase is the well-known modulator of autophagy in human cells. Inhibition of MTOR leading to autophagy activation has been demonstrated as a therapeutic mechanism for various cancer types [19-22]. Rapamycin, a MTOR inhibitor that widely used as an immunosuppressant in organ-transplanted patients, was able to induce autophagy and enhance degradation of aggregate-prone proteins, including huntingtin in several Huntington’s disease models [23-25]. Moreover, rapamycin has also been demonstrated antitumor activity through the induction of autophagy in malignant gliomas and chronic myeloid leukemia [21, 22]. Numerous clinical trials are undergoing to investigating its effects as autophagy modulators either alone or in combination with standard drug therapy for various cancer types, including pancreatic cancer, advanced solid tumor, multiple myeloma, and melanoma [26].
In this study, we investigated whether the combination of AUY922 and rapamycin would be a potential strategy to improve the therapeutic index of AUT922 in mutant KIT-expressing GISTs. We evaluated the effect of rapamycin alone and the potential synergism between AUY922 and rapamycin on induction of autophagy activation, KIT reduction and growth inhibition in IM-resistant, mutant KIT-expression GIST cells both in vitro and in vivo. These results represent a strategy toward optimizing the use of AUY922 for TKI-resistant GISTs.
RESULTS
Rapamycin induced autophagy, downregulated KIT expression, and led to cell apoptosis
Initially, we examined the inhibitory effects of rapamycin in GIST cells. The data showed that 10 µM rapamycin effectively reduced phospho-RPS6KB1, a downstream MTOR target molecule, and that 10 to 40 µM rapamycin induced clear MAP1LC3B accumulation, an index of autophagy activity, in both GIST48 and GIST430 cells. (Fig. 1A) These findings were accompanied by downregulation of phospho- and total KIT and KIT-modulated phospho-AKT in both GIST48 and GIST430 cells, but phospho-MAPK1/3 only in GIST430 cells. In the time series experiments, 40 µM rapamycin abolished phospho-RPS6KB1 level after 4 h of exposure in both GIST cell lines. (Fig. 1B) Phospho-KIT and phospho-AKT were clearly downregulated at 8 h in GIST48 cells and at 24 h in GIST430 cells, which was consistent with the time course of MAP1LC3B accumulation. In GIST430 cells, 40 µM rapamycin reduced phospho-MAPK1/3 at 24 h, which was consistent with phospho-KIT downregulation, but phospho-MAPK1/3 was not decreased after rapamycin treatment in GIST48 cells. These results imply that phospho-MAPK1/3 could be activated by other signaling cascades in GIST48 cells.
The rapamycin drug concentrations that inhibited cell viability by 50% (IC50) were 12.2 and 13.2 µM for GIST48 and GIST430 cells, respectively. (Fig. 1C) In the colony formation assay, the IC50 of rapamycin were 2.48 µM and 6.67 µM for GIST48 and GIST430 cells, respectively. (Fig. 1D) Then, we examined the effect of rapamycin on the induction of GIST cell death. Fig. 1E showed that in GIST48 cells, 40 µM rapamycin induced Annexin V-positive cells in 11.50%, 30.10%, 38.52%, and 74.09% of total cells after 0, 12, 24, and 48 h of incubation, respectively, and Annexin V-positive/PI-negative cells were 4.72%, 17.08%, 6.24%, and 5.52%, respectively. For GIST430 cells, the Annexin V-positive cells constituted 10.37%, 26.06%, 36.07%, and 67.74% of total cells, respectively, and Annexin V-positive/PI-negative cells constituted 3.86%, 11.98%, 15.19%, and 24.79% of cells after 0, 12, 24, and 48 h of 40 µM rapamycin incubation, respectively. Moreover, rapamycin induced PARP1 cleavage in the same treatment condition in both GIST cell lines. (Fig. 1F) These results demonstrate that rapamycin alone could downregulate phospho- and total KIT and induce apoptosis.
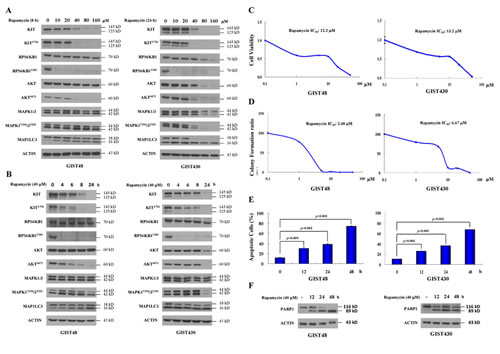
Figure 1: Rapamycin showed antitumor activity and reduced KIT expression in GIST430 and GIST48 cells. GIST430 and GIST48 cells were treated with rapamycin at the indicated doses (A) and times (B) and analyzed by immunoblotting against phospho- and total KIT, RPS6KB1, MAPK1/3, AKT, or MAP1LC3 proteins. GIST430 and GIST48 cells were incubated with rapamycin at the indicated doses, and IC50 was determined by the cell viability assay (C) and clonogenic assay (D). GIST430 and GIST48 cells were treated with 40 µM rapamycin from 12 to 48 h and then analyzed by Annexin V staining (E) or immunoblotting against PARP1 (F). All experiments were repeated at least three times. The data are expressed as the mean ± S.E. of two or more independent experiments.
Rapamycin-induced KIT reduction occurred through enhancement of autophagy
Because the inhibition of MTOR influenced protein synthesis, transcription, and autophagy activity, we investigated whether the rapamycin-induced KIT reduction is a result of autophagy activation. Cells were pretreated with the autophagy inhibitor 3-MA or bafilomycin A1 for 4 h and then incubated with 40 µM rapamycin for an additional 8 and 24 h for GIST48 and GIST430 cells, respectively. The data showed that rapamycin-induced KIT downregulation could be partially rescued by 3-MA or bafilomycin A1 in both GIST48 and GIST430 cells. (Fig. 2A) To further confirm the role of autophagy in rapamycin-induced KIT downregulation, we independently silenced the expression of two proteins essential for autophagosome formation, BECN1 and ATG5, and found that rapamycin-induced KIT reduction was diminished in BECN1- or ATG5-deficient GIST48 and GIST430 cells. (Fig. 2B and C) Using immunofluorescence staining and confocal microscopy analysis, we found no obvious nonspecific staining in normal rabbit, goat, or mouse antibody-incubated cells. After treatment of both GIST cells, rapamycin clearly led to more puncta of MAP1LC3B and more aggregates of SQSTM1, an index of autophagosome, as well as KIT condensation and aggregation. (Fig. 2D-G) Furthermore, the aggregates of SQSTM1 and accumulation of MAP1LC3B-labeled autophagosomes co-localized with KIT in the autophagosomes that were visualized as yellow foci. These results indicate that rapamycin enhanced autophagy activity and led to KIT downregulation.
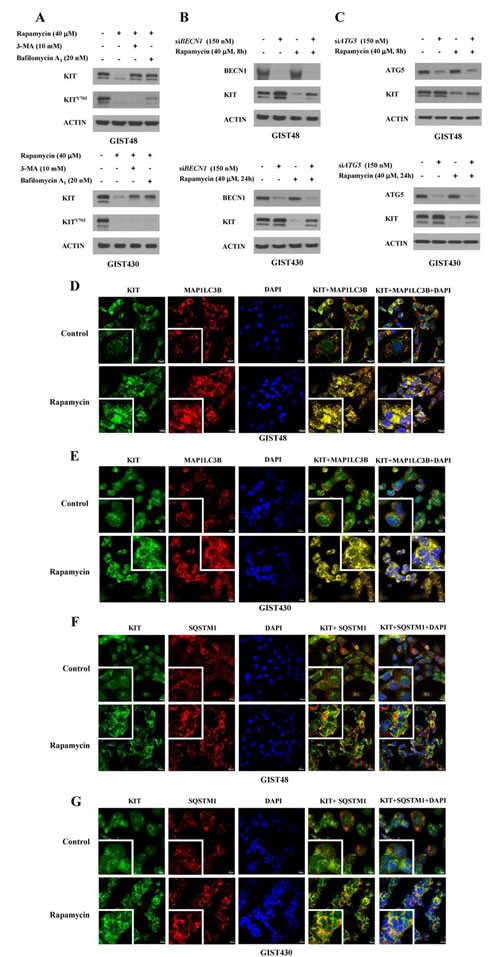
Figure 2: Rapamycin-induced KIT downregulation via autophagy activation. GIST430 and GIST48 cells were pretreated with 10 mM 3-MA and 20 nM bafilomycin A1 (A) for 4 h and then treated with 40 µM rapamycin for an additional 24 and 8 h, respectively. The cells were lysed and analyzed by immunoblotting against phospho- and total KIT. GIST430 and GIST48 cells were transfected with 150 nM siRNA targeting BECN1 (B) or ATG5 (C) for 72 h and then treated with 40 µM rapamycin for another 24 and 8 h, respectively. Cell lysates were extracted and analyzed by immunoblotting against BECN1, ATG5, and KIT. GIST430 and GIST48 cells were treated with 40 µM rapamycin for 24 and 4 h, respectively, and then stained with KIT, MAP1LC3B (D, E), or SQSTM1 (F, G). After immunostaining, cells were visualized by confocal microscopy, and images were acquired through the Cy2, Rhodamine, or DAPI channels (600 x). The inserted figure in the corner showed magnified (2400 x) and representative cells of each image. The data were representative images of 5 fields/pictures for each sample.
AUY922 downregulated phospho- and total KIT expression and induced apoptosis
In previous studies, we demonstrated that AUY922 reduces KIT expression in IM-sensitive GIST882 and IM-resistant GIST48 cells. In the present study, we first assessed whether AUY922 could reduce phospho- and total KIT in another IM-resistant GIST430 cells. The data showed that 1 µM AUY922 elevated the level of HSPA1A expression in a time-dependent manner, which is a positive indicator of HSP90AA1 inhibition. (Fig. 3A) This result suggests that AUY922 effectively inhibited HSP90AA1 activity in GIST430 cells. Moreover, AUY922 downregulated the phospho- and total KIT expression levels in GIST430 cells after incubation for 0.5 h. KIT downstream signaling was assessed by levels of phospho-MAPK and phospho-AKT and was found to be inactivated, consistent with phospho-KIT suppression. After drug exposure for 8 h, AUY922 elevated HSPA1A expression in a dose-dependent manner in a range from 0.05 to 1 µM, and it simultaneously reduced phospho- and total KIT proteins in GIST430 cells. (Fig. 3A) Consistent with phospho-KIT downregulation, the phospho-MAPK1/3 and phospho-AKT levels were also decreased in GIST430 cells.
To examine the antitumor activity of AUY922 in GIST430 cells, cells were incubated with AUY922 and then analyzed using the cell viability assay (Fig. 3B) and the clonogenic assay. (Fig. 3C) The IC50 of AUY922 was 32 nM for both the cell viability assay and clonogenic assay. Then, we treated GIST430 cells with 0.1 µM AUY922 for 12 to 48 h to determine whether AUY922 could induce apoptosis. After AUY922 treatment for 0, 12, 24, or 48 h, Annexin V-positive cells were in 10.52%, 29.05%, 50.64%, and 71.54% of total cell population, respectively, and Annexin V-positive/PI-negative cells were in 5.63%, 7.58%, 16.88%, and 26.63% of total cell population, respectively. (Fig. 3D) Furthermore, 0.1 µM AUY922 induced PARP1 cleavage with incubation times ranging from 12 to 48 h. (Fig. 3E) These findings indicate that AUY922 is highly effective in reducing KIT expression in GIST430 cells, similar to its previously reported effects in GIST48 and GIST882 cells.
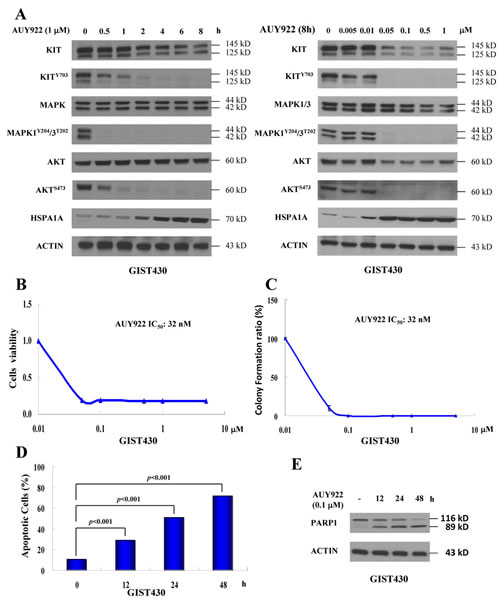
Figure 3: AUY922 reduced KIT expression and induced apoptosis in GIST430 cells. GIST430 cells were treated with AUY922 at the indicated doses and times (A) and analyzed by immunoblotting against phospho- and total KIT, MAPK1/3, AKT, or HSPA1A proteins. GIST430 cells were incubated with AUY922 at the indicated doses and IC50 was determined by the cell viability assay (B) or clonogenic assay (C). Cells were treated with 0.1 µM AUY922 for 12 to 48 h and then analyzed by Annexin V staining (D) or immunoblotting against PARP1 (E). All experiments were repeated at least three times. The data are expressed as the mean ± S.E. of two or more independent experiments.
Rapamycin enhanced AUY922-induced KIT downregulation and cell death
Rapamycin alone showed its ability to induce KIT reduction and cell death via autophagy activation. Next, we examined whether the drug combination of rapamycin and AUY922 could achieve better effects on KIT downregulation and cell death than each drug treatment alone. In GIST48 cells, 0.005 to 1 µM AUY922 combined with 40 µM rapamycin more robustly reduced KIT expression after 8 h of incubation than rapamycin or AUY922 alone. (Fig. 4A) In GIST430 cells, the drug combination of 0.005 to 1 µM AUY922/40 µM rapamycin similarly had stronger effects on KIT downregulation than each drug independently. (Fig. 4B) In GIST48 cells, 20 nM AUY922/5 µM rapamycin treatment increased the proportion of Annexin V-positive cells from 6.27% to 20.33%, 40.78%, and 52.13% after 12, 24, and 48 h of incubation, respectively, as compared with that of after either rapamycin (5.38%, 5.54%, and 6.36%) or AUY922 (15.04%, 28.75%, and 35.93%) alone. (Fig. 4C) In GIST430 cells, the Annexin V-positive cell fraction did not significantly altered after 12 or 24 h exposure to study drugs, but it increased to 39.56% after 48 h incubation with 20 nM AUY922/5 µM rapamycin as compared with that of 16.18% and 28.7% after 5 µM rapamycin alone and 20 nM AUY922 alone, respectively. To further evaluate the potential synergy between AUY922 and rapamycin, the effect of drug combination on cell viability in GIST48 and GIST430 cells was analyzed. Fig. 4D showed that the drug combination achieved a lower IC50 than either drug alone, implying that AUY922 and rapamycin had a synergistic effect on the viability of both GIST48 and GIST430 cells. In summary, rapamycin enhances AUY922-induced KIT downregulation, cell apoptosis, and cell viability in GIST cells.
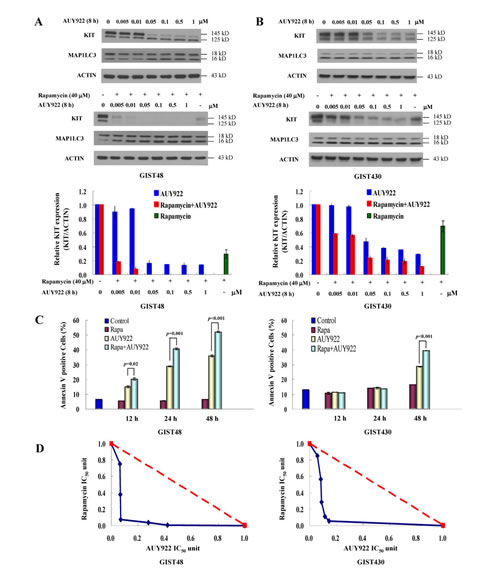
Figure 4: The combination effect of AUY922 and rapamycin on KIT protein degradation in GIST430 and GIST48 cells. GIST430 (A) and GIST48 (B) cells were treated with the indicated dose of AUY922 alone or in a combination with 40 µM rapamycin for 8 h. Cells were lysed and analyzed by immunoblotting against KIT and MAP1LC3. Relative KIT expression was normalized to ACTIN and compared with that of the untreated control. Cells were treated with 20 nM AUY922 and 5 µM rapamycin, either alone or in combination, for 12 to 48 h and analyzed by Annexin V staining (C). Cells were treated with various concentrations of AUY922 combined with rapamycin (D). Cell growth was determined by the methylene blue dye assay as described in the Materials and Methods section. The interaction between AUY922 and rapamycin at the IC50 point was analyzed using the isobologram method. The data are expressed as the mean ± S.E. of two or more independent experiments.
The combination of low-dose AUY922 and rapamycin achieved comparable tumor growth inhibition as high-dose AUY922
The synergism between AUY922 and rapamycin in inhibiting the growth of GIST cells in vitro was further evaluated in GIST430 xenograft animal model. Based on previous studies of AUY922 for cancer xenograft models, AUY922 at a dose of 25 mg/kg (high-dose) twice weekly i.p. for 4 weeks was firstly explored. Our results showed that mice with 25 mg/kg b.i.w. of AUY922 treatment had significantly lower tumor volumes as compared with the DMSO-treated control group (data not shown). To assess drug synergism, the concentration of AUY922 was reduced to 12.5 mg/kg (low-dose) in a combination with rapamycin for experimental arms. Mice were treated with DMSO (placebo control), high-dose AUY922 b.i.w. (active control), and low-dose AUY922 b.i.w., 3 mg/kg rapamycin b.i.w., or a combination of both. The mean tumor volumes in corresponding group of mice were 1232.94, 112.00, 325.71, 348.31, and 185.03 mm3, respectively. (Fig. 5A) At the end of treatment, the tumor volume of mice with high-dose AUY922 was significantly smaller than those with low-dose AUY922 (p= 0.004) or rapamycin (p= 0.002). While the mean tumor volume was comparable, there is no significantly different between mice treated with high-dose AUY922 and those with low-dose AUY922 and rapamycin combo. Immunohistochemical staining of tumor tissue sections from the GIST430 xenograft revealed that low-dose AUY922/rapamycin combo, as well as high-dose AUY922, induced more prominent tumor necrosis, as visualized by hematoxylin and eosin (H&E) staining, and achieved a more pronounced effect on KIT downregulation and MAP1LC3B accumulation than low-dose AUY922 or rapamycin alone in vivo. (Fig. 5C-E) Moreover, a marker of angiogenesis (platelet/endothelial cell adhesion molecule 1, PECAM1) and a key component of this pathway (hypoxia inducible factor 1A, HIF1A) were both more obviously reduced in the tumor tissue from the high-dose AUY922 alone and low-dose AUY922/rapamycin treated mice compared with those from each treatment alone.
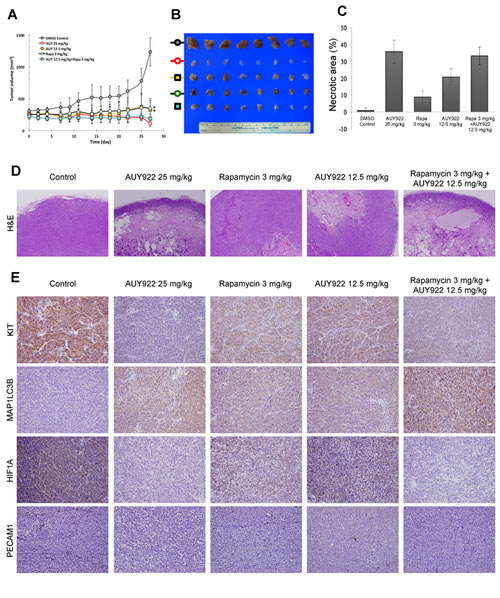
Figure 5: The effect of AUY922 either alone or in a combination with rapamycin in the GIST430 xenograft animal model. The GIST430 xenograft was established as described in Materials and Methods. Mice received an intraperitoneal injection of DMSO, AUY922 25 mg/kg twice weekly, AUY922 12.5 mg/kg twice weekly, rapamycin 3 mg/kg twice weekly, or a combination of AUY922 and rapamycin for 4 weeks. (A) The tumor volume was determined as 1/2 x length (mm) x width (mm)2. The dissected tumors were collected at the end of drug administration (B) and then analyzed by H&E staining (D) and immunohistochemical staining against KIT, MAP1LC3B, HIF1A, or PECAM1 (E). The necrotic areas of the tumor specimens were determined by quantification, as shown in (C). The data are expressed as the mean ± S.D. of two or more independent experiments. * p value is less than 0.05.
Discussion
Recently, HSP90AA1 has been shown to be a novel target for cancer therapy because its inhibition can enhance the degradation of its client proteins, typically receptor tyrosine kinases, signaling molecules and kinases, and transcription factors, to result in modulating many crucial cell functions, including survival, proliferation, metastasis, angiogenesis, cell cycle regulation, and apoptosis [27]. Although many HSP90AA1 inhibitors exhibited antitumor activity against GIST in vitro, only IPI-504, STA-9090, BIIB021, and AUY922 have been evaluated as single-agent therapies for GIST refractory to IM and SU in vivo [28-31]. IPI-504, a derivative of 17-AAG, has displayed antitumor activity against TKI-refractory GISTs, but its clinical trial was early terminated because of the occurrence of on-treatment deaths along with severe unexpected adverse events including renal and liver failure, metabolic acidosis, and cardiopulmonary arrest [28, 32]. Of the phase II study of STA-9090 in patients with metastatic GISTs showed that weekly administration of STA-9090 was not sufficient to inhibit KIT activity and its downstream pathways [33]. BIIB021 achieved a 22% overall metabolic response rate according to 18 fluoro-2-deoxyglucose positron emission tomography (FDG-PET), while the objective tumor response rate was 0% and 4% by RECIST and Choi’s criteria, respectively [34]. AUY922, one of the most potent HSP90AA1 inhibitors, is currently under clinical investigation for TKI-refractory GIST. However, the occurrence of ocular adverse events, notably night blindness, frequently leads to treatment interruption and/or dose reduction in our phase II study. Developing a novel strategy to increase the therapeutic index of HSP90AA1 inhibitor will be critical for future development of this class of compounds in patients with TKI-refractory GISTs [17]. In our previous study, we showed that AUY922-induced KIT downregulation was via both proteasome and autophagy degradation pathways. In current study, we demonstrated that rapamycin, a MTOR inhibitor and inducer of autophagy, could reduce the expression of both phospho- and total KIT in time- and dose-dependent manners, and thus enhance the apoptosis of KIT-expressing GIST cells. AUY922 and rapamycin synergistically enhanced KIT downregulation and growth inhibition as compared with individual drug alone. These findings suggest that combining AUY922 with an autophagy inducer can be a potential therapeutic strategy for TKI-refractory GISTs.
Inhibition of MTOR that leads to autophagy activation has been demonstrated as a therapeutic approach for cancers in many studies. Temsirolimus, an inhibitor of MTOR, induces autophagy and cell cycle arrest to result in proliferation inhibition of mantle cell lymphoma [19]. Another MTOR inhibitor, everolimus, also induces autophagy and cell cycle arrest and improves the median survival of mice harboring human childhood B-cell progenitor acute lymphoblastic leukemia [20]. Takeuchi et al. and Carayol et al. also showed that rapamycin exhibits an antitumor effect by inducing autophagy in malignant glioma cells and malignant chronic myeloid leukemia, respectively [21, 22]. In our study, high concentrations of rapamycin, up to 10 to 40 µM, were required to induce clear MAP1LC3B accumulation. (Fig. 1A) Typically, rapamycin is known to inhibit phospho-RPS6KB1 and cell proliferation under nM concentration [35, 36]. However, recent studies suggested that low-does (nM) and high-dose (µM) of rapamycin had different target and effects on cancer cells. Low-dose rapamycin can inhibit MTOR complex 1 (MTORC1) and lead to cell growth inhibition. On the other hand, much higher dose (at µM range) is required to suppress MTOR complex 2 (MTORC2) and induce cell apoptosis [37]. Yellen P. et al. showed that high-dose (µM) rapamycin-induced cell apoptosis was closely associated with the suppression of EIF4EBP1 phosphorylation [38]. Based on these findings, rapamycin induced autophagy and cell apoptosis at high-dose is probably through inhibiting MTORC2 and its downstream EIF4EBP1.
MTOR also plays a dominant role in nutrient sensing and the regulation of cell growth. Further studies have demonstrated that protein synthesis factors that lie downstream of MTOR affect cell proliferation and survival, cell-cycle progression, angiogenesis, energy metabolism, and metastasis [39]. Therefore, MTOR as a developing therapeutic target for cancer treatment with many rapamycin analogues (rapalogs) have been submitted to clinical trials in recently years. In 2007, temsirolimus was the first MTOR inhibitor to be approved by the FDA for the treatment of advanced-stage renal cell carcinoma. Subsequently, several MTOR inhibitors have been evaluated for the treatment of various cancers in phase III clinical trials, including Non-Hodgkin lymphoma, neuroendocrine tumors, gastric cancer, sarcoma, hepatocellular carcinoma, breast cancer, and etc. [40]. Everolimus, an oral form of MTOR inhibitor, has been shown to inhibit the proliferation of GIST cell in human GISTs xenograft model [41, 42]. In a phase I–II study, the combination of everolimus with IM, at dose of 2.5 mg and 600 mg daily, respectively, could achieve a median progression-free survival and overall survival of 3.5 months and 10.7 months, respectively, in patients with IM/SU-resistant GISTs [43].
In our study, we also found that combination of AUY922 and rapamycin could more effectively downregulate KIT expression and inhibit tumor growth in IM-resistant GIST cells in vitro and in vivo compared with individual drug alone. (Fig. 4 and 5. However, induction of autophagy activation and KIT downregulation may not be the only synergistic/additive effects exerted by the combination of AUY922 and rapamycin on GIST430 xenograft model in mice. As shown in Fig. 5, GIST 430 xenograft tumors in mice with either AUY922 or rapamycin alone, or in a combination had less PRCAM1 staining-positive blood vessels as well as HIF1A expression than the control tumors. HIF1A, the most potent pro-angiogenic protein that drives the expression of several angiogenic factors, including vascular endothelial growth factor (VEGF), is one of the client proteins of HSP90AA1. Inhibition of HSP90AA1 could enhance proteasome-mediated degradation of HIF1A followed by downregulation of HIF-mediated VEGF expression and angiogenesis [44, 45]. Furthermore, several studies have demonstrated that HSP90AA1 inhibitors decrease VEGF secretion from cancer cells, impair endothelial cell tubule formation in vitro, and reduce tumor size and vascularization in vivo [46, 47]. MTOR has also been shown to regulate the transcription and translation of HIF1A [48]. Inhibition of MTOR-induced anti-angiogenic and antitumor effects has been shown to mediate through the reduction of VEGF production and diminished vessel sprouting in tumors [49]. Moreover, Mercier et al. demonstrated that rapamycin treatment could decrease stromal content in tumor animal model [50]. Taken together, the combination of a HSP90AA1 inhibitor and a MTOR inhibitor not only activates autophagy, leading to KIT downregulation, but also likely exerts anti-angiogenic effects that to contribute to tumor growth inhibition in vivo in our study.
In conclusion, we have demonstrated that rapamycin can alone enhance autophagy and induce KIT protein downregulation and apoptosis in both IM-resistant GIST48 and GIST430 cells. Rapamycin-induced KIT degradation via the autophagy pathway was supported by co-localization of KIT with MAP1LC3B or SQSTM1 after drug incubation. In addition, blockage of autophagy by pharmacological inhibition or silencing of essential autophagy-related proteins could partially rescue rapamycin-induced KIT downregulation. The combination of AUY922 and rapamycin enhanced KIT reduction and GIST cell killing compared with each treatment alone. Furthermore, high-dose (25 mg/kg) of AUY922 could achieve significant better tumor growth inhibition than either low-dose (12.5 mg/kg) AUY922 or rapamycin 3 mg/kg monotherapy; while the low-dose AUY922/rapamycin combo had similar tumor growth inhibition as high-dose (25 mg/kg) of AUY922 did, as shown in Fig. 5. Our data highlight the combination of AUY922 and an autophagy inducer, such as rapamycin, is a potential treatment strategy for TKI-refractory, KIT-expressing GISTs and deserves further study in clinical setting.
Materials and Methods
Cell lines, chemicals, and antibodies
GIST430 and GIST48 cells encoding the exon 11V560_L576del (het)/13V654A and exon 11V560D/17D820A mutant KIT oncoprotein, respectively, were gifts from Dr. Jonathan Fletcher (Harvard Medical School, USA). AUY922 was kindly supplied by Novartis. Rapamycin, 3-MA, bafilomycin A1, CelLyticTM cell lysis reagent, and rabbit anti-MAP1LC3A/B were purchased from Sigma Aldrich. Primary antibodies against KIT and phospho-KIT (Tyr703) were obtained from DAKO and Invitrogen, respectively. Antibodies against RPS6KB1, phospho-RPS6KB1 (Thr389), MAPK1/3, phospho-MAPK1/3 (Tyr204/Thr202), AKT, phospho-AKT (Ser473), BECN1, and ATG5 were purchased from Cell Signaling Technology. Normal rabbit IgG, normal goat IgG, normal mouse IgG, goat anti-MAP1LC3B, rabbit anti-PARP1, and mouse anti-SQSTM1 were obtained from Santa Cruz Biotechnology. Antibodies against HSPA1A and ACTIN were obtained from Stressgen and Millipore, respectively.
RNA interference
Small interfering RNA (siRNA) specific to human BECN1 and negative control siRNA were purchased from Invitrogen. Specific siRNA targeted to human ATG5 was synthesized by Invitrogen using the published sequence 5-GGACGAAUUCCAACUUGUU-3 [17]. GIST430 and GIST48 cells were transfected with siRNA of BECN1, ATG5, or negative control using Lipofectamine 2000® (Invitrogen) according to the manufacturer’s protocol. Briefly, GIST430 and GIST48 cells at 90% confluence in 6-well plates were transfected with the indicated dose of annealed RNA duplexes mixed with 2 µL of Lipofectamine 2000® for 6 h, and then replaced with serum-containing complete medium for 72 h. The influence of siRNA on BECN1 or ATG5 protein expression was confirmed by immunoblotting with specific antibodies.
Cell viability assay
For this assay, 5×104 GIST430 or 4×104 GIST48 cells were seeded in each well of 24-well plates. GIST430 and GIST48 cells were exposed to various concentrations of drugs for 4 days. The methylene blue dye assay was used to evaluate the effects of each drug on the relative number of viable cells [17]. Data were measured with a SpectraMax M5 microplate reader (Molecular Device) at 595 nm and normalized to the DMSO-only control group. The IC50 was determined by plotting growth relative to that of the untreated controls. All experimental points were measured in duplicate wells for each plate and replicated in at least three plates.
To assess the interaction between AUY922 and rapamycin, the combination index (CI) was analyzed using the method of Chou and Talalay, based on the multiple drug effect equation [51]. Briefly, the constant ratio combination design was chosen to assess the combination effect of both drugs. Dose-response curves were determined with both drugs in combination at a fixed ratio equivalent to the ratio of their IC50 values. The CI value indicates that the effects of the two drugs were additive (CI≈1), synergistic (CI<1), or antagonistic (CI>1). All experimental points were measured in duplicate wells for each plate and were replicated in at least three plates.
Clonogenic assay
1×103 GIST430 or GIST48 cells in the logarithmic growth phase were seeded in each well in 6-well plates. GIST430 and GIST48 cells were incubated with the indicated doses of drugs for 1 day and then replaced with growth media for an additional 14 days. The cells were stained with 50% ethanol containing 0.5% methylene blue for 5 h. The plates were washed five times with water and allowed to air-dry. Colonies were countered manually. The IC50 value corresponding to 50% inhibition of cell growth was determined after plotting growth relative to the untreated controls. Each value represented the average of at least three independent experiments performed in triplicate.
Immunoblotting studies
Cells were lysed in CelLyticTM M Cell Lysis Reagent containing protease inhibitors and phosphatase inhibitors. The protein concentration was determined using the Bradford method (Bio-Rad). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed after loading equal amounts of protein into each lane. Proteins were transferred to polyvinylidene fluoride (PVDF) membranes for immunoblotting. After blocking with bovine serum albumin (BSA), the membranes were blotted by adding primary antibodies against specific proteins, followed by the appropriate secondary antibodies. Protein bands were detected by enhanced chemiluminescence (PerkinElmer), developed by autoradiography and quantified using 1Dscan EX gel analysis software (Scanalytics). The data are expressed as the mean ± S.E.
Immunofluorescence staining
For immunofluorescence staining, GIST430 and GIST48 cells were fixed with paraformaldehyde, permeabilized with Triton X-100, and blocked with 5% BSA in PBS at 4°C overnight. Cells were incubated with primary antibodies against KIT, MAP1LC3B, or SQSTM1. Cells used as nonspecific staining control were incubated with normal rabbit, goat, or mouse antibodies. The cells were washed three times with PBS, incubated with a fluorescently labeled secondary antibody, washed three times with PBS, and examined under a confocal microscope (FV1000, Confocal Laser Scanning Biological Microscope, Olympus).
Flow cytometry
Apoptosis was determined by staining cells with Annexin V-FITC and PI (BD Biosciences) according to the manufacturer’s protocol, followed by flow cytometry analysis. Briefly, GIST430 and GIST48 cells were incubated with drugs for the indicated times and doses and then trypsinized. Each sample containing 1×105 cells was washed with cold PBS and resuspended in 100 µl binding buffer. Then, 5 µl Annexin V-FITC and 5 µl PI were added to the cells, which were then incubated for 15 min at room temperature (RT) in the dark. An additional 400 µl binding buffer was added to the reaction prior to analysis.
GIST430 xenograft animal model and the drug treatment
NOD/SCID mice were purchased from LASCO. The animals were subcutaneously implanted with 2x107 GIST430 cells mixed with equal volume of Matrigel (BD Biosciences) in 0.1 mL in one flank per mouse. Tumor volume was estimated from caliper measurements every two days after implantation and calculated as 1/2 x length (mm) x width2 (mm). After tumors developed for approximately 4 weeks and tumor volumes reached 50 to 100 mm3, mice were intraperitoneally injected with the indicated dose of AUY922 and rapamycin dissolved in DMSO, either alone or in combination, twice a week for 4 consecutive weeks. The control group was treated with DMSO only. Tumor volumes were measured every two days after drug treatment. At the end of the experiments, animals were sacrificed and tumors were visualized and stored at -80°C for further immunohistochemical analysis.
Immunohistochemical analysis
Sections of tumor tissue blocks were cut onto adhesive-coated glass slides (Instrumedics, Hackensack) at a thickness of 3 µm. The slides were dewaxed in xylene and rehydrated through a graded alcohol series. Then, slides were pressure-cooked in 10 mM citrate buffer (pH 6) for 7 minutes for antigen retrieval and washed using TBS with 0.1% Tween 80 for 5 minutes. Endogenous peroxidase activity was blocked by 3% H2O2 treatment. After washing, the slides were incubated with primary antibodies against KIT (Epitomics), MAP1LC3B, HIF1A (Novus Biologicals), and PECAM1 (BD Biosciences) at RT for 1 hour. Primary antibodies were detected following the user’s manual of the ChemMate DAKO EnVision kit (DAKO). The slides were incubated with the secondary antibody for 30 minutes and developed with 3,3-diaminobenzidine for 5 minutes. Slides were then counterstained with hematoxylin. Incubation without the primary antibody was used as a negative control.
Statistical analysis
We analyzed the data with the Statistical Package for the Social Science (Software version 16 for Windows (SPSS, Inc.)). All data are expressed as the mean ± SEM. Comparison of means between the groups was performed by one-way analysis of variance (ANOVA) followed by the least significant difference (LSD) test. The level of significance was set at p < 0.05.
Acknowledgments
The authors are grateful for the administration and laboratory support from the National Institute of Cancer Research, National Health Research Institute, and funding support from the National Health Research Institute (CA-103-PP-15) and Ministry of Health and Welfare (CA-103-SP-01; DOH102-TD-PB-111-TM-014), Executive Yuan, Taiwan.
Author Contributions
Study concept and design: Yuan-Shuo Hsueh, Chueh-Chuan Yen, Chien-Feng Li, Li-Tzong Chen.
Experiment execution and data analysis: Yuan-Shuo Hsueh, Hui Hua Chang, Nai-Jung Chiang.
New reagents/materials: Chueh-Chuan Yen, Chien-Feng Li.
Administration and funding support: Li-Tzong Chen.
All of the investigators participated in the data interpretation and final report approval.
Manuscript preparation: Yuan-Shuo Hsueh, Hui Hua Chang, Nai-Jung Chiang, Li-Tzong Chen.
Study supervision: Li-Tzong Chen.
Conflict of interest/Disclosure
Research funding was from the National Health Research Institutes (CA-103-PP-15) and the Ministry of Health and Welfare (DOH102-TD-PB-111-TM-014 and CA-103-SP-01) of Taiwan to Dr. Li-Tzong Chen, while research compound (NVP-AUY922) was kindly provided by Novartis. Dr. Li-Tzong Chen also received honorarium and research grants for other studies from Novartis.
References
1. Nishida T, Hirota S. Biological and clinical review of stromal tumors in the gastrointestinal tract. Histol Histopathol 2000;15(4):1293-1301.
2. Rubin BP, Heinrich MC, Corless CL. Gastrointestinal stromal tumour. Lancet 2007;369(9574):1731-1741.
3. Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, Hibbard MK, Chen CJ, Xiao S, Tuveson DA, Demetri GD, Fletcher CD,Fletcher JA. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res 2001;61(22):8118-8121.
4. Corless CL, Heinrich MC. Molecular pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol 2008;3:557-586.
5. Lasota J, Miettinen M. Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology 2008;53(3):245-266.
6. Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, van Oosterom AT, Blay JY, Leyvraz S, Stul M, Casali PG, Zalcberg J, Verweij J, Van Glabbeke M, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 2006;42(8):1093-1103.
7. Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 2006;368(9544):1329-1338.
8. Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, Hohenberger P, Leahy M, von Mehren M, Joensuu H, Badalamenti G, Blackstein M, Le Cesne A, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295-302.
9. Heath EI, Gaskins M, Pitot HC, Pili R, Tan W, Marschke R, Liu G, Hillman D, Sarkar F, Sheng S, Erlichman C, Ivy P. A phase II trial of 17-allylamino-17- demethoxygeldanamycin in patients with hormone-refractory metastatic prostate cancer. Clin Prostate Cancer. 2005;4(2):138-141.
10. Ronnen EA, Kondagunta GV, Ishill N, Sweeney SM, Deluca JK, Schwartz L, Bacik J, Motzer RJ. A phase II trial of 17-(Allylamino)-17-demethoxygeldanamycin in patients with papillary and clear cell renal cell carcinoma. Invest New Drugs. 2006;24(6):543-546.
11. Solit DB, Osman I, Polsky D, Panageas KS, Daud A, Goydos JS, Teitcher J, Wolchok JD, Germino FJ, Krown SE, Coit D, Rosen N, Chapman PB. Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clin Cancer Res. 2008;14(24):8302-8307.
12. Sambol EB, Ambrosini G, Geha RC, Kennealey PT, Decarolis P, O’connor R, Wu YV, Motwani M, Chen JH, Schwartz GK, Singer S. Flavopiridol targets KIT transcription and induces apoptosis in gastrointestinal stromal tumor cells. Cancer Res. 2006;66(11):5858-5866.
13. Fumo G, Akin C, Metcalfe DD, Neckers L. 17-Allylamino-17-demethoxygeldanamycin (17-AAG) is effective in down-regulating mutated, constitutively activated KIT protein in human mast cells. Blood. 2004;103(3):1078-1084.
14. Egorin MJ, Rosen DM, Wolff JH, Callery PS, Musser SM, Eiseman JL. Metabolism of 17-(allylamino)-17-demethoxygeldanamycin (NSC 330507) by murine and human hepatic preparations. Cancer Res. 1998;58(11):2385-2396.
15. Kelland LR, Sharp SY, Rogers PM, Myers TG, Workman P. DT-Diaphorase expression and tumor cell sensitivity to 17-allylamino, 17-demethoxygeldanamycin, an inhibitor of heat shock protein 90. J Natl Cancer Inst. 1999;91(22):1940-1949.
16. Brough PA, Aherne W, Barril X, Borgognoni J, Boxall K, Cansfield JE, Cheung KM, Collins I, Davies NG, Drysdale MJ, Dymock B,Eccles SA, Finch H, et al. 4,5-diarylisoxazole Hsp90 chaperone inhibitors: potential therapeutic agents for the treatment of cancer. J Med Chem. 2008;51(2):196-218.
17. Hsueh YS, Yen CC, Shih NY, Chiang NJ, Li CF, Chen LT. Autophagy is involved in endogenous and NVP-AUY922-induced KIT degradation in gastrointestinal stromal tumors. Autophagy. 2013;9(2):220-233.
18. Sessa C, Shapiro GI, Bhalla KN, Britten C, Jacks KS, Mita M, Papadimitrakopoulou V, Pluard T, Samuel TA, Akimov M, Quadt C,Fernandez-Ibarra C, Lu H, et al. First-in-human phase I dose-escalation study of the HSP90 inhibitor AUY922 in patients with advanced solid tumors. Clin Cancer Res. 2013;19(13):3671-3680.
19. Yazbeck VY, Buglio D, Georgakis GV, Li Y, Iwado E, Romaguera JE, Kondo S, Younes A. Temsirolimus downregulates p21 without altering cyclin D1 expression and induces autophagy and synergizes with vorinostat in mantle cell lymphoma. Exp Hematol. 2008;36(4):443-450.
20. Crazzolara R, Cisterne A, Thien M, Hewson J, Baraz R, Bradstock KF, Bendall LJ. Potentiating effects of RAD001 (Everolimus) on vincristine therapy in childhood acute lymphoblastic leukemia. Blood. 2009;113(14):3297-3306.
21. Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, Kondo S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphate- dylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65(8):3336-3346.
22. Carayol N, Vakana E, Sassano A, Kaur S, Goussetis DJ, Glaser H, Druker BJ, Donato NJ, Altman JK, Barr S, Platanias LC. Critical roles for mTORC2- and rapamycin- insensitive mTORC1-complexes in growth and survival of BCR-ABL-expressing leukemic cells. Proc Natl Acad Sci U S A. 2010;107(28):12469-12474.
23. Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009;16(1):46-56.
24. Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36(6):585-595.
25. Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11(9):1107-1117.
26. Cheng Y, Ren X, Hait WN, Yang JM. Therapeutic targeting of autophagy in disease: biology and pharmacology. Pharmacol Rev. 2013;65(4):1162-1197.
27. Banerji U. Heat shock protein 90 as a drug target: some like it hot. Clin Cancer Res. 2009; 15(1):9-14.
28. Floris G, Debiec-Rychter M, Wozniak A, Stefan C, Normant E, Faa G, Machiels K, Vanleeuw U, Sciot R, Schöffski P. The heat shock protein 90 inhibitor IPI-504 induces KIT degradation, tumor shrinkage, and cell proliferation arrest in xenograft models of gastrointestinal stromal tumors. Mol Cancer Ther. 2011;10(10):1897-1908
29. Floris G, Sciot R, Wozniak A, Van Looy T, Wellens J, Faa G, Normant E, Debiec-Rychter M, Schöffski P. The Novel HSP90 inhibitor, IPI-493, is highly effective in human gastrostrointestinal stromal tumor xenografts carrying heterogeneous KIT mutations. Clin Cancer Res. 2011;17(17):5604-5614
30. Lin TY, Bear M, Du Z, Foley KP, Ying W, Barsoum J, London C. The novel HSP90 inhibitor STA-9090 exhibits activity against Kit-dependent and -independent malignant mast cell tumors. Exp Hematol. 2008;36(10):1266-1277.
31. Marcu MG, Schulte TW, Neckers L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J Natl Cancer Inst. 2000;92(3):242-248.
32. Demetri GD, Le Cesne A, von Mehren M, Chmielowski B, Bauer S, Chow WA, Rodenas E, McKee K, Grayzel DS, Kang Y, Final results from a phase III study of IPI-504 (retaspimycin hydrochloride) versus placebo in patients (pts) with gastrointestinal stromal tumors (GIST) following failure of kinase inhibitor therapies. [abstract]. In: Proceedings of the ASCO Gastrointestinal Cancers Symposium; 2010 Jan 22–24; Orlando, FL. Alexandria (VA): ASCO; 2010. Abstract nr 64.
33. Demetri GD, Heinrich MC, Chmielowski B, Morgan JA, George S, Bradley R, Blackman RK, Teofilovici F, Fletcher JA, Tap WD, von Mehren M. An open-label phase II study of the Hsp90 inhibitor ganetespib (STA-9090) in patients (pts) with metastatic and/or unresectable GIST. [abstract]. J Clin Oncol 29: 2011 (suppl; abstr 10011).
34. Dickson MA, Okuno SH, Keohan ML, Maki RG, D’Adamo DR, Akhurst TJ, Antonescu CR, Schwartz GK. Phase II study of the HSP90-inhibitor BIIB021 in gastrointestinal stromal tumors. Ann Oncol. 2013;24(1):252-257.
35. Pospelova TV, Leontieva OV, Bykova TV, Zubova SG, Pospelov VA, Blagosklonny MV. Suppression of replicative senescence by rapamycin in rodent embryonic cells. Cell Cycle. 2012;11(12):2402-2407
36. Blagosklonny MV. Increasing healthy lifespan by suppressing aging in our lifetime: preliminary proposal. Cell Cycle. 2010;9(24):4788-4794.
37. Foster DA, Toschi A. Targeting mTOR with rapamycin: one dose does not fit all. Cell Cycle. 2009;8(7):1026-1029.
38. Yellen P, Saqcena M, Salloum D, Feng J, Preda A, Xu L, Rodrik-Outmezguine V, Foster DA. High-dose rapamycin induces apoptosis in human cancer cells by dissociating mTOR complex 1 and suppressing phosphorylation of 4E-BP1. Cell Cycle. 2011;10(22):3948-3956.
39. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274-293.
40. Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest. 2011;121(4):1231-1241.
41. Pantaleo MA, Nicoletti G, Nanni C, Gnocchi C, Landuzzi L, Quarta C, Boschi S, Nannini M, Di Battista M, Castellucci P, Fanti S, Lollini PL, Bellan E, et al. Preclinical evaluation of KIT/PDGFRA and mTOR inhibitors in gastrointestinal stromal tumors using small animal FDG PET. J Exp Clin Cancer Res. 2010;29:173.
42. Rossi F, Ehlers I, Agosti V, Socci ND, Viale A, Sommer G, Yozgat Y, Manova K, Antonescu CR, Besmer P. Oncogenic Kit signaling and therapeutic intervention in a mouse model of gastrointestinal stromal tumor. Proc Natl Acad Sci U S A. 2006;103(34):12843-12848.
43. Schöffski P1, Reichardt P, Blay JY, Dumez H, Morgan JA, Ray-Coquard I, Hollaender N, Jappe A, Demetri GD. A phase I-II study of everolimus (RAD001) in combination with imatinib in patients with imatinib-resistant gastrointestinal stromal tumors. Ann Oncol. 2010;21(10):1990-1998.
44. Milkiewicz M, Doyle JL, Fudalewski T, Ispanovic E, Aghasi M, Haas TL. HIF-1alpha and HIF-2alpha play a central role in stretch-induced but not shear-stress-induced angiogenesis in rat skeletal muscle. J Physiol. 2007;583(Pt 2):753-766.
45. Alqawi O, Moghaddas M, Singh G. Effects of geldanamycin on HIF-1alpha mediated angiogenesis and invasion in prostate cancer cells. Prostate Cancer Prostatic Dis. 2006;9(2):126-135.
46. Lang SA, Klein D, Moser C, Gaumann A, Glockzin G, Dahlke MH, Dietmaier W, Bolder U, Schlitt HJ, Geissler EK, Stoeltzing O. Inhibition of heat shock protein 90 impairs epidermal growth factor-mediated signaling in gastriccancer cells and reduces tumor growth and vascularization in vivo. Mol Cancer Ther. 2007;6(6):1123-1132.
47. Lang SA, Moser C, Gaumann A, Klein D, Glockzin G, Popp FC, Dahlke MH, Piso P, Schlitt HJ, Geissler EK, Stoeltzing O. Targeting heat shock protein 90 in pancreatic cancer impairs insulin-like growth factor-I receptor signaling, disrupts an interleukin-6/signal-transducer and activator of transcription 3/hypoxia-inducible factor-1alpha autocrine loop, and reduces orthotopic tumor growth. Clin Cancer Res. 2007;13(21):6459-6468.
48. Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, Giaccia AJ, Abraham RT. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol. 2002;22(20):7004-7014.
49. Falcon BL, Barr S, Gokhale PC, Chou J, Fogarty J, Depeille P, Miglarese M, Epstein DM, McDonald DM. Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer Res. 2011;71(5):1573-1583.
50. Mercier I, Camacho J, Titchen K, Gonzales DM, Quann K, Bryant KG, Molchansky A, Milliman JN, Whitaker-Menezes D, Sotgia F, Jasmin JF, Schwarting R,Pestell RG, Blagosklonny MV, Lisanti MP. Caveolin-1 and accelerated host aging in the breast tumor microenvironment: chemoprevention with rapamycin, an mTOR inhibitor and anti-aging drug. Am J Pathol. 2012;181(1):278-293.
51. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27-55.