
INTRODUCTION
“Double-hit” lymphoma (DHL) represents a subset of B-cell malignancies characterized by the presence of MYC (8q24) rearrangement and concurrent BCL2 (18q21) or BCL6 (3q27) rearrangements [1]. In recognition of its unique biology and clinical behavior, DHL has been included in the 2016 revision of the World Health Organization (WHO) classification of lymphoid neoplasms as a new category of “high-grade B-cell lymphoma (HGBL) with MYC and BCL2 or BCL6 rearrangements” [2, 3]. Based on reviews in the literature [1, 4, 5], cases of HGBL with MYC and BCL2 rearrangements (MYC/BCL2 DHL) form the great majority of DHLs (60–85%), whereas cases of HGBL with MYC and BCL6 rearrangements (MYC/BCL6 DHL) are relatively rare (5–8%) and even less common than “triple-hit” lymphoma (THL) that involves MYC, BCL2, and BCL6 simultaneously (16%). This is because most of what we know about DHLs is based on cases with MYC/BCL2 DHL, which has an inferior prognosis when treated with regimens for diffuse large B-cell lymphoma (DLBCL) and has a very high recurrence rate with a reported median survival of only 0.2 to 1.5 years [1, 6, 7]. In contrast, there are far fewer data available for MYC/BCL6 DHL. Some studies have suggested that the clinicopathologic features of MYC/BCL6 DHL are distinct from those of MYC/BCL2 DHL [8–11]. Cases of MYC/BCL6 DHL more often involve extranodal sites and have less complex karyotypes [9, 10]. In addition, gene expression profiling of MYC+BCL2–BCL6+ lymphoma cells has shown them to be different from MYC+BCL2+BCL6– lymphoma cells [11]. Thus, MYC/BCL6 DHL is likely a different disease biologically from MYC/BCL2 DHL and remains an incompletely characterized disease entity.
One of the major limitations in understanding the pathogenesis of MYC/BCL6 DHL is the lack of in vitro and in vivo models by which unlimited supplies of lymphoma cells with concurrent MYC and BCL6 rearrangements can be studied repeatedly and extensively. So far, there have been various lymphoma cell lines that appear to have both MYC and BCL2 rearrangements [12–14]. Most of these cell lines were reported primarily before sufficient recognition of the clinical importance of DHL and have contributed to the study of lymphomas bearing alterations of both MYC and BCL2. However, they have not been well authenticated genetically against primary lymphoma cells. In this context, generation of MYC/BCL6 DHL cell lines is a prerequisite for increasing our knowledge of the less common forms of DHL and for the identification of valid therapeutic targets.
Herein, we describe a fully characterized lymphoma cell line harboring simultaneous MYC and BCL6 rearrangements, designated DH-My6, that is proved to be immunophenotypically and genetically consistent with a primary DHL tumor. DH-My6 is a new validated MYC/BCL6 DHL cell line carrying both fusion genes of MYC with the immunoglobulin heavy-chain (IGH) locus (14q32) and BCL6–IGH. Using this model, we evaluated the potential of MYC- and BCL6-targeted strategies in combination with agents targeting molecules associated with cell proliferation, such as polo-like kinase 1 (PLK1) and histone deacetylase (HDAC) inhibitors, as possible therapeutic approaches to aggressive MYC/BCL6 DHL.
RESULTS
Generation and characteristics of the DH-My6 cell line
The DH-My6 cell line was generated from tumor tissue of a patient with MYC/BCL6 DHL. The cells began to proliferate 2 weeks after the initiation of culture and then could be regularly passaged in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS). The cells could be frozen under standard conditions using medium containing 10% FCS and 10% dimethylsulfoxide (DMSO), and could be revived after storage in liquid nitrogen. DH-My6 cells grew in single-cell suspensions with a doubling time of 20 h (Figure 1A). The cell line was composed of medium-to-large-sized cells (Figure 1B). The nuclei were round or slightly irregular with slightly coarse chromatin and had one or more nucleoli. The cytoplasm was basophilic and occasionally contained small vacuoles. The morphology of DH-My6 cells closely resembled the primary lymphoma cells. The cells were shown to be negative for Epstein–Barr virus by polymerase chain reaction (PCR) analysis.
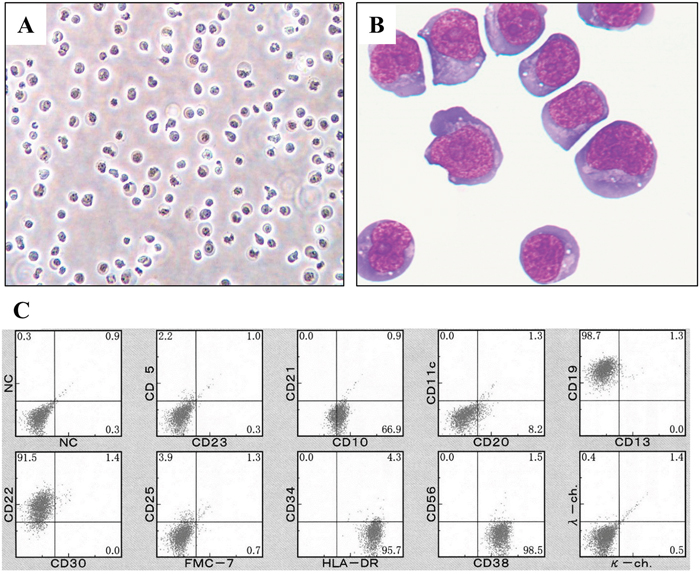
Figure 1: Appearance and surface immunophenotype of DH-My6 cells. (A) Phase-contrast microphotograph of growing DH-My6 cells. (B) Cytospin preparation of DH-My6 cells closely resembling the primary lymphoma cells (May-Giemsa staining). (C) Representative flow cytometric histograms of DH-My6 cells.
The immunophenotypes of DH-My6 cells were virtually identical to the primary tumor cells. DH-My6 cells were positive for CD10, CD19, and CD22, and negative for CD5, CD11c, CD13, CD21, CD23, CD25, CD30, CD34, CD56, FMC-7, and surface Ig kappa- and lambda-light chains (Figure 1C). The cells had a germinal center B-cell like (GCB) phenotype. Notably, DH-My6 cells express a high level of CD38, and a fraction of weakly CD20-positive or -negative cells was consistently detected during cell passages.
G-banding chromosomal analysis of DH-My6 cells 2 months after cell line establishment showed a complex karyotype, including der(3)t(3;14)(q27;q32) and der(14)t(8;14)(q24;q32) (Figure 2A). The karyotype showed a close resemblance to that of the primary lymphoma cells, indicating that that the DH-My6 cells were indeed derived from the primary lymphoma cells. Fluorescence in situ hybridization (FISH) analyses revealed the occurrence of the IGH–MYC fusion gene and the BCL6 gene rearrangement in all DH-My6 cells analyzed (Figure 2B–2C). Spectral karyotyping (SKY) analysis of the metaphase cells showed the presence of der(14)t(8;14)(q24;q32) (Figure 2D). In addition, it revealed a cryptic translocation on der(8)t(3;8)(q27;q24). Combined G-banding and SKY analysis yielded the following karyotype: 47, XY, der(1)(pter→q21::1?::q21→qter), der(3)del(3)(p25p26)t(3;14)(q27;q32), der(8)t(3;8)(q27;q24), der(9)t(9;13)(p13;q14), inv(9)(p21q11), der(12)(pter→q24.1::12?), der(14)t(8;14)(q24;q32), der(19)t(7;19)(?;p13).
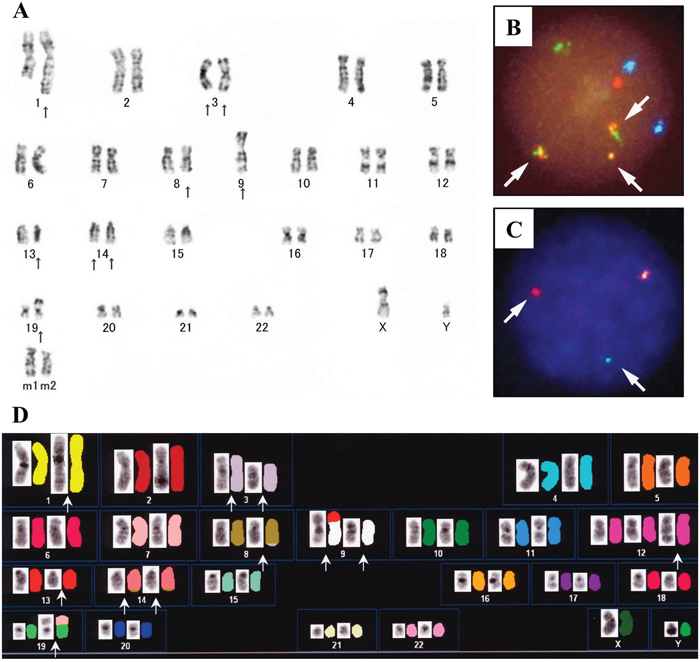
Figure 2: Cytogenetic analyses of DH-My6 cells. (A) Giemsa-banding karyotype, showing the following karyotype: 47, XY, ins(1;?)(q21:?), add(3)(p25), der(3)del(3)(p?)t(3;14)(q27;q32), add(8)(q24), -9, add(9)(p11), del(13)(q?), der(14)t(8;14)(q4;q32), add(19)(p13), +mar1, +mar2. (B) FISH analysis with the IGH/MYC dual-color probes, showing fusion signals for the IGH-MYC gene rearrangement (arrows). The green signal corresponds to the normal IGH allele, red to the normal MYC allele, yellow to the IGH-MYC fusion gene, and blue to the centromeric region of chromosome 8. (C) FISH analysis for BCL6 gene rearrangement with the break-apart probes, showing split signals for BCL6 (arrows). Yellow signal corresponds to the intact, nonrearranged BCL6 locus, while separate red (5' BCL6 FISH DNA probe) and green (3' BCL6 FISH DNA probe) signals indicate the BCL6 rearrangement. (D) SKY karyotype (left side, reverse 4′,6-diamidino-2-phenylindole staining; right side, SKY), showing the following karyotype: 47, XY, der(1)(pter→q21::1?::q21→qter), del(3)(p25p26), del(3)(q27), der(8)t(3;8)(q27;q24), der(9)t(9;13)(p13;q14), inv(9)(p21q11), der(12)(pter→q24.1::12?), der(14)t(8;14)(q24;q32), der(19)t(7;19)(?;p13).
Presence of both BCL6–IGH and MYC–IGH linkages
Although the SKY assay did not visualize the der(3)t(3;14)(q27;q32) karyotype, we determined successfully the exact fusion positions of BCL6–IGH linkage by long–distance PCR (LD–PCR). We selected primers targeting the sequences of BCL6 intron 1 and the IGH switch region, because BCL6/IGH translocations frequently occur at these regions in B-cell lymphomas [15, 16]. The primer BCL6/09 combined with primer JXI yielded a PCR product sized approximately 2.5 kilobase (kb), and subsequent sequence analysis confirmed fusion between the first intron of the BCL6 gene (205 base pairs apart from the 3' end of exon 1) and the IGH switch region Sμ (Figure 3A). Likewise, MYC–IGH linkage as a result of der(14)t(8;14)(q24;q32) karyotype was demonstrated by LD–PCR using primers recognizing the MYC exon 2 (MYC/M6) and the IGH switch region Sɑ (ɑR2), which produced a PCR product sized approximately 3.8 kb. We found breakpoints within MYC intron 1 (1412 base pairs apart from the 3' end of exon 1) and the IGH region Sα (Figure 3B). Sequences around fusion regions on the MYC–IGH fusion gene and the BCL6–IGH fusion gene are also shown in Figure 3.
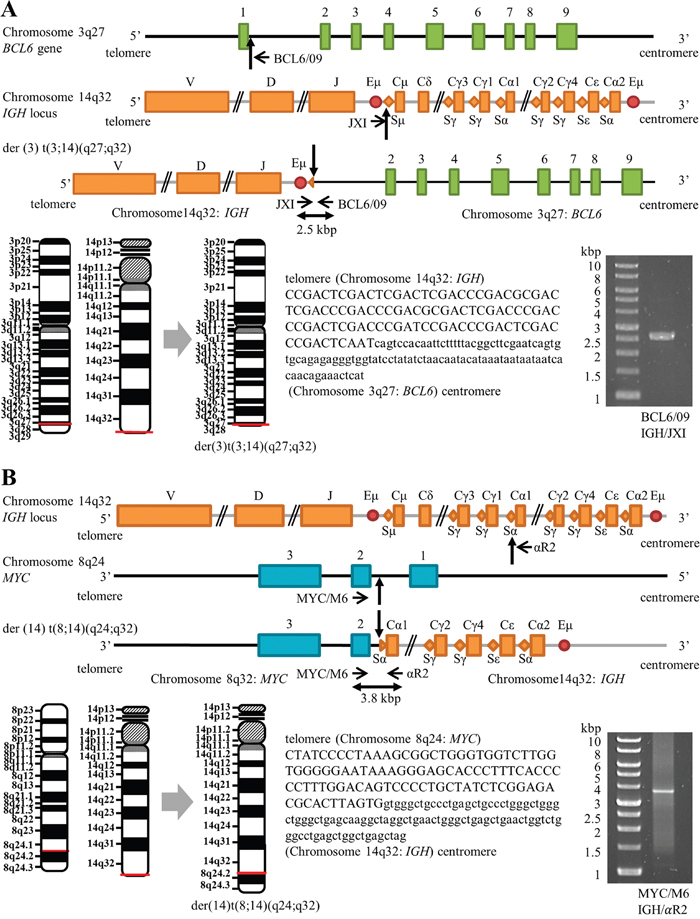
Figure 3: Schematic positions of BCL6–IGH and MYC–IGH breakpoints (not to scale). (A) Maps of the BCL6 on chromosome 3, the IGH locus on chromosome 14, and the BCL6–IGH fusion gene on der(3) t(3;14)(q27;q32), showing the positions of breakpoints (vertical arrows) and positions of forward (JX1) and reverse (BCL6/09) primers (horizontal arrows). Sequence alignments of the der(3) t(3;14)(q27;q32) junction are shown with capital letters representing the IGH 14q32 sequence and small letters representing the BCL6 3q27 sequence. (B) Maps of the MYC on chromosome 8, the IGH locus on chromosome 14, and the MYC–IGH fusion gene on der(14)t(8;14)(q24;q32), showing the positions of breakpoints (vertical arrows) and positions of forward (MYC/M6) and reverse (αR2) primers (horizontal arrows). Sequence alignments of the der(14)t(8;14)(q24;q32) junction are shown with capital letters representing the MYC 8q24 sequence and small letters representing the IGH 14q32 sequence. Horizontal red lines shown on the schematic chromosomes indicate the breakage or fusion points for the chromosomal translocations. Images of ethidium bromide-stained gel electrophoresis separation of LD–PCR products are also shown. The sizes of the products are 2.5 kb and 3.8 kb for the BCL6–IGH and MYC–IGH fusion genes, respectively. Molecular weight markers are shown on the left.
Short tandem repeat (STR) DNA fingerprinting analysis of primary cells and DH-My6 cells
The relatedness of the primary cells and DH-My6 cells was determined by comparing their STR loci profiles. STR DNA fingerprinting analysis based on genotyping of 10 loci showed that the primary lymphoma cells and DH-My6 cells shared 100% identity (Table 1). These results demonstrated that the DH-My6 cells were indeed derived from the patient’s tumor cells.
Table 1: STR DNA fingerprinting of primary cells and DH-My6 cells
Sample |
STR Loci |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
TH01 |
D21S11 |
D5S818 |
D13S317 |
D7S820 |
D16S539 |
CSF1PO |
AMEL |
vWA |
TPOX |
|
Primary cells |
6, 7 |
30 |
10, 12 |
8 |
10, 12 |
12, 13 |
12 |
X, Y |
14, 17 |
8 |
DH-My6 cells |
6, 7 |
30 |
10, 12 |
8 |
10, 12 |
12, 13 |
12 |
X, Y |
14, 17 |
8 |
Abbreviation: STR, short tandem repeat.
Xenotransplantation of DH-My6 cells into nude mice
DH-My6 cells gave rise to tumors in all nude mice tested after simultaneous inoculation. The palpable subcutaneous nodules could be detected within 3 weeks and grew larger with no sign of regression. Histology of the tumors showed diffuse infiltrations of medium-to-large-sized lymphoma cells with round or slightly irregular nuclei and one or more prominent nucleoli (Figure 4A). Immunohistochemistry showed the expressions of MYC and BCL6 (Figure 4B–4C). LD–PCR confirmed that the tumor cells carried both the IGH–MYC and IGH–BCL6 fusion genes. These results indicated that the same features were maintained in this in vivo model.
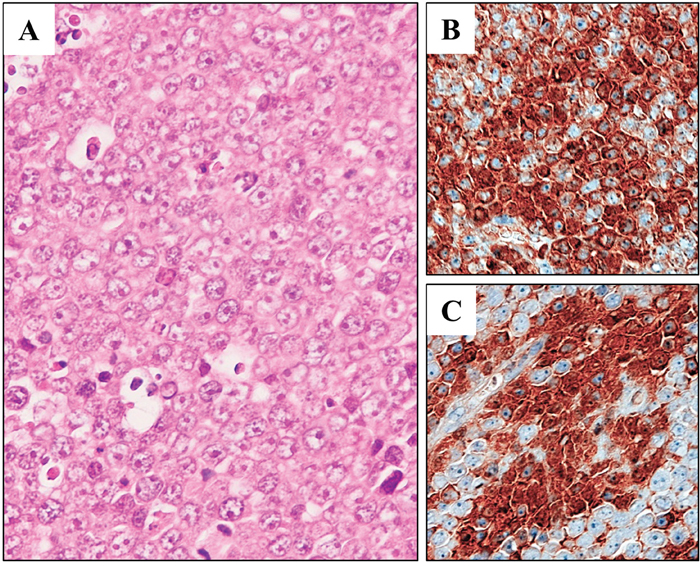
Figure 4: Xenotransplantation of DH-My6 cells into nude mice. (A) Hematoxylin and eosin staining of a tumor, showing lymphoma cells with an indistinct cell border and prominent nucleoli. (B) Immunohistochemistry showing positive staining for MYC in a significant proportion of lymphoma cells. (C) Immunohistochemistry showing positive staining for BCL6 in a significant proportion of lymphoma cells.
Antiproliferative effects of MYC and BCL6 inhibitions evaluated using small interfering RNAs (siRNAs)
The MYC and BCL6 gene expression levels were assayed by real-time quantitative reverse-transcription PCR (RT–qPCR) on the DH-My6 cell line and a panel of seven DLBCL cell lines classified as the GCB subtype. Both MYC and BCL6 genes were expressed at relatively higher levels in the DH-My6 cell line (Supplementary Figure 1). Given these results, we assessed the effects of MYC and BCL6 inhibitions on cell proliferation by applying gene-specific siRNAs. Transduction of each MYC and BCL6 siRNA resulted in a significant decrease in cell proliferation compared with the control siRNA and caused significant increases in the proportion of cell population at the G0/G1 phase of the cell cycle, whereas they did not cause significant apoptosis (Figure 5). These results implied that the antiproliferative effects of blocking MYC and BCL6 were mediated mainly through cell cycle inhibition.
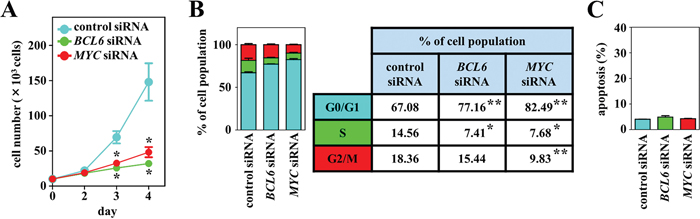
Figure 5: Effects of MYC and BCL6 knockdown using siRNAs on cell growth, cell cycle, and apoptosis in DH-My6 cells. (A) Cell growth assay. After transfection with MYC, BCL6 or control siRNAs, viable cells were counted every 24 h. (B) Cell cycle analysis conducted at 48 h after transfection. Percentages of the cell population in each stage of the cell cycle are presented outside the graph. (C) Apoptosis assay conducted at 48 h after transfection. The graph shows the percentage of apoptotic cells in the total cell population. All experiments were repeated independently three times, and data are expressed as the mean ± SEM. Significant expression differences are shown as *P < 0.05; **P <0.01.
Antiproliferative effects of MYC and BCL6 inhibitors
We next evaluated the effects of chemical inhibitors targeting MYC and BCL6 on proliferation by exposing the DH-My6 cells to them at various concentrations. In parallel with experiments on DH-My6 cells, four B-cell lymphoma cell lines with a GCB type, including two DLBCL cell lines (Su-DHL-5 and HT) and two MYC/BCL2 DHL cell lines (Su-DHL-10 and Nu-DHL-1), were also tested. First, we employed 10058-F4, a direct MYC inhibitor that targets the interaction between MYC and MAX [17]. 10058-F4 significantly suppressed DH-My6 cell growth by induction of cell cycle arrest at the G0/G1 phase (Supplementary Figure 2A). Alternatively, because 10058-F4 has limited clinical utility because of its rapid degradation [17], we also treated DH-My6 cells with JQ1. This is an inhibitor targeting the bromodomain and extraterminal domain (BET) family of bromodomain (BRD) proteins, with the highest affinity for BRD4, and suppresses oncogenic transcription factors mainly through inhibiting the function of MYC [18, 19]. Like 10058-F4, JQ1 treatment significantly attenuated cell growth in a dose-responsive manner (Figure 6A). The estimated half maximal inhibitory concentration (IC50) at 72 h treatment in DH-My6 cells was 141 nM, which indicates a sensitivity to BET inhibition [19, 20], but was higher than the IC50 values in cell lines tested (Table 2). Cell cycle analysis showed an increase in the G0/G1 phase and reduction in S and G2/M phases following JQ1 (250 nM) treatment (Figure 6B). However, JQ1 treatment with concentrations as high as 2,000 nM induced cell apoptosis only weakly or not at all (Figure 6C).
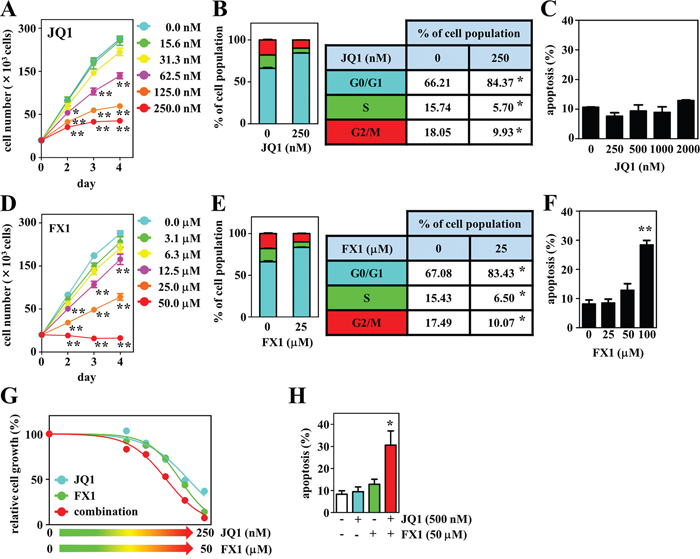
Figure 6: Effects of the BET inhibitor JQ1, the BCL6 inhibitor FX1, and their combination on cell growth, cell cycle, and apoptosis in DH-My6 cells. (A) Cell growth assay following treatment with the indicated doses of JQ1. (B) Cell cycle analysis conducted at 48 h after treatment with JQ1 (250 nM). (C) Apoptosis assay conducted at 48 h after treatment with the indicated doses of JQ1. (D) Cell growth assay following treatment with the indicated doses of FX1. (E) Cell cycle analysis conducted at 48 h after treatment with FX1 (25 μM). (F) Apoptosis assay conducted at 48 h after treatment with the indicated doses of FX1. (G) Cell growth assay following treatment with the various doses of JQ1, FX1, and their combinations for 72 h. The numbers of viable cells are normalized as a percentage of the viable cell numbers of DMSO-treated controls. (H) Apoptosis assay conducted at 48 h after a single treatment with JQ1 (500 nM), FX1 (50 μM), and their combination at each concentration that gave a limited effect on apoptosis when treated with a single agent. Data are shown as the mean ± SEM of three independent experiments. Significant expression differences are shown as *P < 0.05; **P <0.01.
Table 2: Estimated IC50 values in the lymphoma cell lines treated with inhibitors for 72 h
cell line |
origin |
IC50 |
|||
|---|---|---|---|---|---|
JQ1 (nM) |
FX1 (μM) |
Volasertib (nM) |
Vorinostat (nM) |
||
HT |
GCB-type |
55.7 |
21.6 |
7.4 |
630.9 |
Su-DHL-5 |
GCB-type |
48.5 |
8.3 |
5.4 |
468.8 |
Su-DHL-10 |
MYC/BCL2 |
34.6 |
15.9 |
8.5 |
322.8 |
Nu-DHL-1 |
MYC/BCL2 |
65.7 |
21.6 |
>10.0 |
608.1 |
DH-My6 |
MYC/BCL6 |
140.6 |
20.9 |
4.6 |
346.7 |
Abbreviation: IC50, half maximal inhibitory concentration.
We also explored the antitumor effects of two BCL6 inhibitors, 79-6 and FX1, both of which bind to the groove of the BCL6 BTB domain and disrupt BCL6 transcriptional complexes [21, 22]. These agents led to significant suppression of DH-My6 cell growth in a dose-dependent manner (Supplementary Figure 2B and Figure 6D). FX1 showed a stronger antiproliferative effect (FX1, IC50 21 μM vs. 79-6, IC50 >100 μM), consistent with previous findings that FX1 exhibits 10-fold greater inhibitory activity against BCL6 BTB domain than 79-6 [22, 23]. FX1 treatment (25 μM) induced cell cycle arrest at the G0/G1 phase (Figure 6E), but significant apoptosis was not elicited with concentrations as high as 50 μM (Figure 6F). Taken together, consistent with genetic inhibition, single chemical inhibition targeting MYC and BCL6 induced cell cycle arrest consistently but led to only limited apoptosis.
Next, we assessed the effects of combination treatments with the BET and BCL6 inhibitors. DH-My6 cells were treated with JQ1 plus FX1, and the efficacy was compared with that of the single agent. Cotreatment showed an additive inhibitory effect on DH-My6 cell growth with a combination index (CI) value of 1.0 (Figure 6G). Notably, apoptosis was strikingly induced in combination with JQ1 (500 nM) and FX1 (50 μM) at each concentration that gave a limited effect on apoptosis when treated with single agents (Figure 6H). These data imply that dual inhibition of MYC and BCL6 produced better antigrowth efficacy than the single agent.
The estimated IC50 values of these agents for other cell lines tested in this study are shown in Table 2.
Agents leading to the effective inhibition of tumor cell growth
We explored the inhibitory effects of a chemotherapeutic agent that targets cell cycle regulation in combination with JQ1 or FX1. Because PLK1 is a master regulator of several cell cycle events [24, 25], we investigated its antitumor effects, using the selective PLK1 inhibitor volasertib [26]. PLK1 expression was detected in DH-My6 cells with expression levels similar to the DLBCL cell lines tested, except for Nu-DHL-1 in which lower levels of PLK1 were detected (Supplementary Figure 1). Treatment with volasertib at concentrations of 5.0 nM or higher resulted in a drastic reduction of DH-My6 cell proliferation (Figure 7A). The estimated IC50 was 4.6 nM, lower than in the MYC/BCL2 DHL cell lines tested in this study (Su-DHL-10, 8.5 nM; Nu-DHL-1, >10.0 nM; Table 2) and than in a panel of various cancer cell lines reported previously (11–37 nM) [27]. Exposure of DH-My6 cells to volasertib caused a significant increase in the proportion of cells at the G2/M phase and also induced a marked increase in apoptosis (Figure 7B–7C). Genetic inhibition of PLK1 through siRNA knockdown also diminished cell proliferation significantly over time relative to control siRNA and produced G2/M cell cycle arrest and significant apoptosis in DH-My6 cells (Figure 7D–7F). Thus, PLK1 interruption by the chemical inhibitor and by siRNA knockdown approaches both showed efficient antitumor activity.
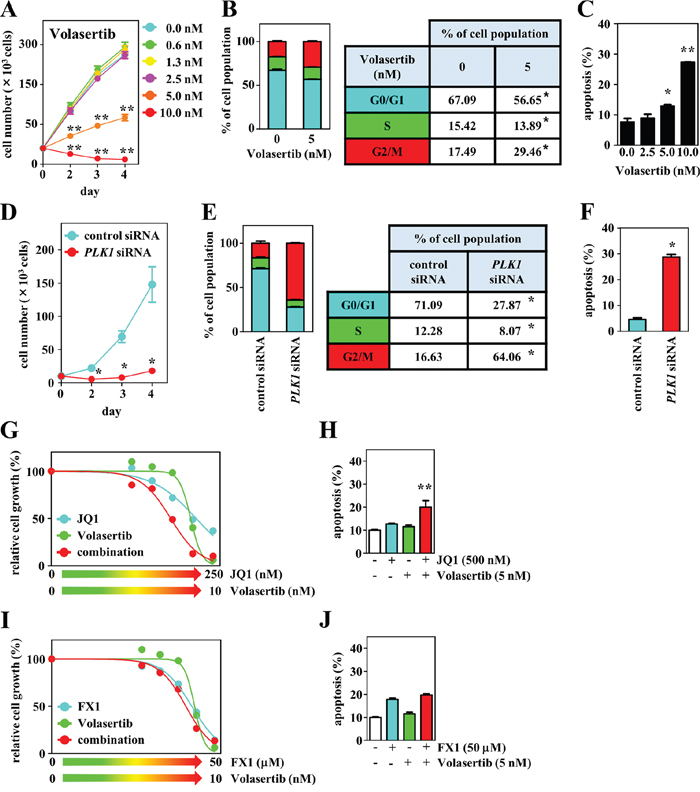
Figure 7: Effects of PLK1 interruption and its combination with BET or BCL6 inhibition on cell growth, cell cycle, and apoptosis in DH-My6 cells. (A) Cell growth assay following treatment with the indicated doses of volasertib. (B) Cell cycle analysis conducted at 48 h after treatment with volasertib (5 nM). (C) Apoptosis assay conducted at 48 h after treatment with the indicated doses of volasertib. (D) Cell growth assay following transfection with PLK1 or control siRNAs. (E) Cell cycle analysis conducted at 48 h after siRNA transfection. (F) Apoptosis assay conducted at 48 h after siRNA transfection. (G) Cell growth assay following treatment with the various doses of volasertib, JQ1, and their combinations for 72 h. (H) Apoptosis assay conducted at 48 h after a single treatment with volasertib (5 nM), JQ1 (500 nM), and their combination at doses that did not induce significant apoptosis individually. (I) Cell growth assay following treatment with the various doses of volasertib, FX1, and their combinations for 72 h. (J) Apoptosis assay conducted at 48 h after a single treatment with volasertib (5 nM), FX1 (50 μM), and their combination at doses that did not induce significant apoptosis individually. Data are shown as the mean ± SEM of three independent experiments. Significant expression differences are shown as *P < 0.05; **P <0.01.
When volasertib was combined with JQ1 or FX1, a synergistic antiproliferative effect was given only with the combination of volasertib with JQ1 with a CI value of 0.9 (Figure 7G–7J). This combination also led to a greater level of apoptosis than the use of single agents. These results suggest that coinhibition of PLK1 and BET exerted greater antitumor effects through blocking the cell cycle at both G0/G1 and G2/M phases and through the induction of apoptosis.
In addition, we evaluated the antitumor effect of HDAC inhibition using the selective HDAC inhibitor vorinostat as a single agent and in combination with BET or BCL6 inhibitors. Vorinostat significantly suppressed DH-My6 cell growth in a dose-responsive manner (Figure 8A). Vorinostat blocked cell cycle progression at the G0/G1 phase (Figure 8B) and induced pronounced apoptosis (Figure 8C). The estimated IC50 was 350 nM. Treatment of DH-My6 cells with vorinostat in combination with JQ1 resulted in synergistic inhibition of cell growth with a CI value of 0.7 (Figure 8D) and led to more efficient apoptosis than treatment with the single agents (Figure 8E). The combination of vorinostat (500 nM) with FX1 (25 μM) also resulted in greater apoptosis (Figure 8F–8G). This effect could be achieved at doses which did not induce significant apoptosis with individual agents.
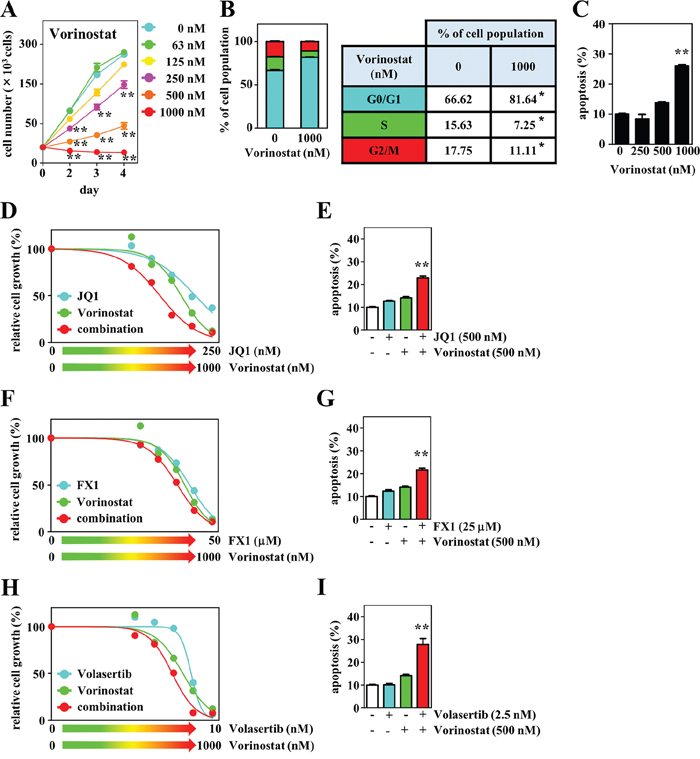
Figure 8: Effects of the HDAC inhibitor vorinostat and its combination with JQ1, FX1, or volasertib on cell growth, cell cycle, and apoptosis in DH-My6 cells. (A) Cell growth assay following treatment with the indicated doses of vorinostat. (B) Cell cycle analysis conducted at 48 h after treatment with vorinostat (1 μM). (C) Apoptosis assay conducted at 48 h after treatment with the indicated doses of vorinostat. (D) Cell growth assay following treatment with the various doses of vorinostat, JQ1, and their combinations for 72 h. (E) Apoptosis assay conducted at 48 h after a single treatment with vorinostat (500 nM), JQ1 (500 nM), and their combination at doses that did not induce significant apoptosis individually. (F) Cell growth assay following treatment with the various doses of vorinostat, FX1, and their combinations for 72 h. (G) Apoptosis assay conducted at 48 h after single treatment with vorinostat (500 nM), FX1 (25 μM), and their combination at doses that did not induce significant apoptosis individually. (H) Cell growth assay following treatment with the various doses of vorinostat, volasertib, and their combinations for 72 h. (I) Apoptosis assay conducted at 48 h after a single treatment with vorinostat (500 nM), volasertib (2.5 nM), and their combination at doses that did not induce significant apoptosis individually. Data are shown as the mean ± SEM of three independent experiments. Significant expression differences are shown as *P < 0.05; **P <0.01.
HDAC and PLK1 inhibitors exhibit complementary mechanisms of actions [28, 29]. Therefore, we also studied the antitumor effect of a combination of vorinostat with volasertib. This showed a better growth inhibitory effect than the single agents at lower concentrations (Figure 8H). Exposure of DH-My6 cells to a low concentration of volasertib (2.5 nM) in conjunction with vorinostat (500 nM) induced a dramatic increase in apoptosis, although each agent individually had no significant apoptotic effect (Figure 8I).
DISCUSSION
Features of the DH-My6 cell line are summarized in Table 3. This well-characterized DH-My6 cell line has several strengths. MYC/BCL6 DHL cases make up only 8% of all DHL/THL cases [1, 4]. Moreover, there is concern that many of the reported MYC/BCL6 DHL cases may actually represent THL [1]. Clinicopathologically, THL cases resemble MYC/BCL2 DHL cases more closely than MYC/BCL6 DHL cases [30]. Consequently, establishment of DHL cell lines that only have MYC and BCL6 translocations is meaningful in that they offer the possibility of analyzing the effects of such unique rearrangements in more detail. The DH-My6 cell line presented here is a new one derived from a “pure MYC/BCL6 DHL” case with MYC and BCL6 translocations. Its derivation from the patient was clearly authenticated by our STR analysis. DH-My6 cells with morphologic features of DLBCL are of the GCB type, which is an immunophenotype observed in 50–80% of published MYC/BCL6 DHL cases [9, 10, 31, 32]. DH-My6 cells showed only faint CD20 expression, as with the pretreated primary lymphoma cells, eliminating the effect of rituximab as a cause of this unique finding. Based on reviews in the literature [14, 33, 34], most of the MYC/BCL2 DHL cases showed decreased expression of CD20. Although mechanisms resulting in decreased expression of CD20 after rituximab treatment have been studied using rituximab-resistant lymphoma cell lines and the results have suggested the association of epigenetic or genetic changes with downregulation of CD20 [35, 36], the underlying mechanism for diminishing CD20 expression in de novo DHL cells has not been investigated. Clinically, the decreased expression of CD20 results in the loss of efficiency of anti-CD20 antibody therapies. The DH-My6 cell line will also be a useful tool for the elucidation of the mechanism of CD20 downregulation in DHL. Moreover, the DH-My6 cells show strong CD38 expression, which appears to be a characteristic phenotype of lymphomas with MYC rearrangement [34, 37]. Because a satisfactory screening for rare cases of MYC/BCL6 DHL has not been defined so far [5], these flow cytometric findings might be helpful in triaging lymphomas for confirmatory targeted FISH analysis.
Table 3: Synopsis of data on the DH-My6 cell line
Parameter |
Features |
|---|---|
Clinical data |
|
Patient |
54-year-old man |
Diagnosis |
HGBL with MYC and BCL6 rearrangements |
Treatment status |
At diagnosis |
Specimen Year of establishment |
Bone marrow |
Cell line |
|
Culture medium |
RPMI1640 + 10% FCS |
Growth pattern |
Single cells in suspension |
Doubling time |
20 h |
Optimal cell density |
5 × 105 cells/ml |
Optimal split |
1:3 every 3–4 days |
Cryopreservation |
In 80% medium, 10% FCS, 10% DMSO |
Morphology |
medium-to-large-sized cells with round or slightly irregular nuclei |
Viral status |
Negative for Epstein–Barr virus |
Immunoprofiles |
Strongly positive for CD10, CD19, CD22, CD38; |
Karyotypic analysis in conjunction with SKY |
47, XY, der(1)(pter→q21::1?::q21→qter), der(3)del(3)(p25p26)t(3;14)(q27;q32), der(8)t(3;8)(q27;q24), der(9)t(9;13)(p13;q14), inv(9)(p21q11), der(12)(pter→q24.1::12?), der(14)t(8;14)(q24;q32), der(19)t(7;19)(?;p13) |
Tumorigenic capacity |
Subcutaneous growth in BALB/c nude mice |
Authentication |
Yes (by STR DNA fingerprinting and cytogenetic characteristics) |
Abbreviations: HGBL, high-grade B-cell lymphoma; STR, short tandem repeat.
Another important characteristic of DH-My6 is that the cells carry gene rearrangements with concurrent t(8;14)(q24;q32)/MYC–IGH and t(3;14)(q27;q32)/BCL6–IGH linkages. Our SKY analysis detected der(14)t(8;14)(q24;q32) and cryptic der(8)t(3;8)(q27;q24) but not der(3)t(3;14)(q27;q32). It is probable that the chromosome 3 with deletion q27 detected by SKY was actually a der(3)t(3;14)(q27;q32), because SKY usually cannot visualize the small segment 14q32→14qter on a der(3)t(3;14)(q27;q32) background [38]. In fact, LD–PCR assays readily detected both the 3' BCL6–5' IGH fusion sequence in the BCL6 intron 1 on chromosome 3 and the 3' MYC–3' IGH fusion sequence in the MYC intron 1 on chromosome 14.
DH-My6 cells had higher expression levels of both MYC and BCL6. Ueda et al. [39] showed that BCL6 is significantly upregulated when its translocation partner is an IG gene in DLBCL. Wilda et al. [40] demonstrated that the level of MYC overexpression is dependent on breakpoint location within the MYC locus and that significant higher MYC expression is associated with breakpoint from exon 1 to intron 1. These events, as observed in DH-My6 cells, are likely to lead to higher levels of both MYC and BCL6 in this cell line. Several studies have shown that most patients with MYC/BCL6 DHL have an aggressive clinical course [9, 10, 32]. In contrast, in other studies, patients with MYC and BCL6 rearrangements were not necessarily associated with an inferior prognosis [11, 41]. A possible explanation for the discrepancy is patient selection. These previous reports on the prognostic value of MYC/BCL6 DHL included MYC/BCL6 DHL cases of a non-GCB type, which have a tendency to a better prognosis than the GCB type that is observed almost exclusively in MYC/BCL2 DHL cases [41]. In addition, such studies did not assess whether the specific BCL6 partner is an IG or a non-IG gene and might include t(3;8)(q27;q24) with simultaneous rearrangement of BCL6 and MYC in the context of a MYC/BC6 double hit. Indeed, the most common non-IG/BCL6 partner is MYC [9], and the t(3;8)(q27;q24) karyotype is frequently observed in cases of “multiple-hit” lymphoma [42–44], as shown in DH-My6 cells. It should be noted, however, that t(3;8)(q27;q24) represents a MYC/non-IG single hit activating solely MYC [44, 45]. Thus, MYC/BCL6 DHL appears to be heterogeneous group of tumors consisting of cases with both favorable and poor prognoses. The latter cases are resistant to standard frontline therapy for DLBCL and might require specific therapeutic approaches. We believe that the DH-My6 cell line carrying concurrent t(8;14)(q24;q32) and t(3;14)(q27;q32) and the higher expression of both MYC and BCL6 will also prove to be a valuable experimental tool to find potential therapeutic targets against aggressive MYC/BCL6 DHLs.
In this study, we demonstrated that targeted inhibitions of MYC by 10058-F4 and BCL6 by its inhibitors (79-6 and FX1) resulted in a significant attenuation of DH-My6 cell growth through induction of G0/G1 cell cycle arrest. Similar results were obtained when the antiproliferative effect was monitored by genetic inhibition using MYC and BCL6 gene-specific siRNAs. Although the importance of MYC in carcinogenesis is well documented, therapies targeting MYC directly are not available currently. Accordingly, the BET inhibitor JQ1 has been used as a therapeutic strategy to target MYC [18]. In one study, a panel of seven GCB-type DLBCL cell lines, including two MYC/BCL2 DHL cell lines (Su-DHL-10 and OCI-Ly18), were treated with JQ1, and all showed decreased cell growth [20]. Another study has demonstrated that the BCL2 inhibitor venetoclax enhanced the antiproliferative activity when it was combined with JQ1 in two MYC/BCL2 DHL cell lines (Sc-1 and OCI-Ly18) [46]. Because BCL6 is required for the survival of certain types of lymphoma, it is also conceivable that BCL6 inhibitors might prove to be useful for MYC/BCL6 DHL [21]. In the present study, a combination of JQ1 and FX1 showed a better inhibitory effect on DH-My6 cell growth. Notably, pronounced apoptosis was induced when these compounds were combined. These results suggest that cotargeting of MYC and BCL6 is likely beneficial for antitumor activity in MYC/BCL6 DHL cells with high expression levels of MYC and BCL6. Indeed, aggressive MYC/BCL6 DHL tumors showed a trend toward higher MYC mRNA expression compared with MYC/BCL2 DHL [31]. Thus, as a proof-of-principle, our findings provide a rationale for preclinical trials on the efficacy of BRD4 inhibitors such as JQ1 in combination with a BCL6 inhibitor for the development of valid treatment modalities for patients with aggressive MYC/BCL6 DHL.
In an effort to identify other therapeutic targets, we also demonstrated that the PLK1 inhibitor volasertib alone significantly impaired the viability of DH-My6 cells. Notably, we found synergistic antitumor effects when volasertib was combined with JQ1 by blocking the cell cycle at both G2/M and G0/G1 phases. Murga-Zamalloa et al. [47] showed that MYC and PLK1 expression levels were strongly correlated in cases of aggressive DLBCLs and that PLK1 expression was most prevalent in “double hit” HGBC with a MYC translocation. Because PLK1 has been implicated in molecular cross talk with MYC [48] and volasertib possibly has an inhibitory activity on the BRD4 protein [26], it is plausible that therapeutic targeting of PLK1 in combination with JQ1 might yield a more favorable therapeutic index in MYC-associated lymphomas. Moreover, cotreatment with the HDAC inhibitor vorinostat and JQ1 synergistically reduced tumor growth in DH-My6 cells. Bhadury et al. [49] indicated that BET and HDAC inhibitors have similar target genes and biological effects, thus synergizing to kill MYC-induced murine lymphomas. BET and HDAC coinhibition have been also proved to be effective against human acute myelogenous leukemia cells [50]. In the present study, we also found that the combination of vorinostat and volasertib in DH-My6 cells resulted in marked apoptosis at doses that exerted little antiproliferative effect or only induced cell cycle arrest when used individually. This combination treatment appears to be effective in HGBL irrespective of BCL2 or BCL6 rearrangements because exposure of MYC/BCL2 DHL cells (OCI-Ly18 and Carnaval) to volasertib in combination with the HDAC inhibitor belinostat increased cell death [29]. Furthermore, combination of vorinostat with FX1 significantly induced apoptosis in DH-My6 cells. Cotargeting of BCL6 and HDAC seems to be another attractive approach for efficient eradication of MYC/BCL6 DHL cells. This combination has yielded more potent antilymphoma effects in BCL6-dependent DLBCL cell lines [51].
In summary, we present a novel “double hit” high-grade B-cell lymphoma cell line, designated DH-My6, with concurrent MYC/IGH and BCL6/IGH rearrangements. This well-characterized MYC/BCL6 DHL cell line will provide useful models for biological studies of the less common DHL tumors. We also provide here a basis for a rational combination therapeutic strategy targeting MYC and BCL6, and in combination with PLK1 or HDAC inhibitors against aggressive MYC/BCL6 DHL tumors, which will promote further in vivo testing in a preclinical model.
MATERIALS AND METHODS
Case history and cell culture
The DH-My6 cell line was established from a 54-year-old man who was referred to our hospital for treatment of a bulky retroperitoneal tumor. Pathology of a bone marrow aspiration showed diffuse infiltration of medium-to-large-sized lymphoma cells with expression of both MYC and BCL6 proteins. Chromosomal analysis showed a complex karyotype, including der(3)t(3;14)(q27;q32) and der(14)t(8;14)(q24;q32). FISH analysis revealed concurrent MYC and BCL6 rearrangements. These pathology findings are shown in Supplementary Figure 3. The cells were strongly positive for CD10, CD19, CD22, and CD38, and weakly positive for CD20, and exhibited high positivity for Ki-67 staining (85–90%). The cells had a GCB immunophenotype. The patient was diagnosed as having an HGBL with MYC and BCL6 rearrangements. He was treated initially with one cycle of hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (hyper CVAD) and two cycles of cyclophosphamide, vincristine, doxorubicin, methotrexate, ifosfamide, etoposide, cytarabine, and rituximab (R-CODOX-M/R-IVAC). However, the disease relapsed immediately, and he died of disease progression 10 months after the initial presentation. Both chromosomal translocations t(3;14)(q27;q32) and t(8;14)(q24;q32) were consistently detected three times during the disease’s progression.
A heparinized bone marrow sample was obtained when his bone marrow aspirate contained 92% lymphoma cells. The cells were cultured in RPMI 1640 medium supplemented with 20% heat-inactivated FCS without any external stimulation. The cultures were incubated at 37°C in a humidified atmosphere of 5% CO2 in air and fed every 3 days by partial medium change.
This study was approved by the Ethics Committee of Kochi Medical School, Kochi University. Written informed consent was obtained from the patient before the patient died, and all experiments were performed in accordance with the regulations of the institutional review board.
Immunophenotypic, immunohistochemical, cytogenetic, and FISH analyses
Expression of cell surface antigens was studied with fluorescein isothiocyanate-conjugated monoclonal antibodies using flow cytometry. To detect MYC and BCL6 proteins, immunohistochemistry was performed in formalin-fixed paraffin wax-embedded tissue sections using mouse monoclonal antibodies 9E10 (Santa Cruz Biotechnology, Dallas, TX, USA) and D-8 (Santa Cruz Biotechnology), respectively, based on an indirect biotin-avidin system using a biotinylated universal secondary antibody and diaminobenzidine substrate with hematoxylin counterstaining. The specificity of staining with the primary antibodies was controlled by testing the isotype-matched control antibodies in parallel. Metaphase chromosome spreads were G-banded according to standard procedures. Karyotypes have been described according to the International System for Cytogenetic Nomenclature. For SKY analysis, a probe cocktail containing 24 uniquely labeled chromosome-specific painting probes was hybridized to metaphase chromosomes according to described methods [52]. FISH analysis was performed on interphase nuclei using commercially available probes, as described previously [53, 54]. The IGH–MYC rearrangement [t(8:14)] probe is a dual-color, dual-fusion probe that detects the juxtaposition of the IGH locus at 14q32 to the MYC sequence at 8q24. To detect the BCL6 rearrangement, a dual-color, break-apart probe flanking the major breakpoint region of BCL6 was used.
LD–PCR
LD–PCR was carried out as described previously with modifications [55]. Genomic DNA was extracted by the phenol–chloroform method. The LD–PCR reaction mixture contained 200 ng of extracted DNA, reaction buffer, a dNTP mixture, 0.2 μM of each primer, and 1.25 units of PrimeSTAR GXL DNA Polymerase (TaKaRa Bio, Shiga, Japan). To determine the fusion position of the MYC–IGH [t(8;14)] sequence, we used primers targeting the MYC exon 2 region, named MYC/M6 and targeting the IGH switch region, named αR2 [55, 56]. To determine the fusion position of the BCL6–IGH [t(3:14)] sequence, we used primers targeting the BCL6 exon 1 region, named BCL6/09 and the IGH switch region, named JXI [56, 57]. The primer sequences are listed in Supplementary Table 1. The LD–PCR conditions were 2 min at 98°C, followed by 35 cycles of 10 s at 98°C, 15 s at 60°C, and 5 min at 68°C, with a final extension for 5 min at 68°C. The PCR products were analyzed by 0.8% agarose gel electrophoresis and visualized under an ultraviolet transilluminator after staining with ethidium bromide. The purified PCR products were sequenced directly, as described previously [58].
STR DNA fingerprinting
Genomic DNA was isolated from the primary lymphoma cells and DH-My6 cells, using NucleoSpin Tissue kits (TaKaRa Bio). STR DNA fingerprinting was carried out using a GenePrint 10 System (Promega, Madison, WI, USA), which allows coamplification and detection of 10 human loci (TH01, D21S11, D5S818, D13S317, D7S820, D16S539, CSF1PO, Amelogenin, vWA, and TPOX).
In vivo studies in mice
Five-week-old male BALB/c Slc-nu/nu mice (Japan SLC, Hamamatsu, Japan) were engrafted subcutaneously with 2 × 107 cells in the flanks. The mice were maintained in a temperature-controlled (23 ± 2°C) and humidity-controlled (55 ± 10%) room under a constant day-night photoperiod and observed regularly for appearance of tumors. After tumor-bearing mice had been euthanized, tumors were resected surgically and subjected to pathological studies. All experimental protocols were approved by our Institutional Animal Care and Use Committee in compliance with our institutional guidelines on the care and use of animals for scientific purposes.
Real-time RT–qPCR
Real-time RT–qPCR was used to study expression levels of the BCL6 and MYC genes. Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) and RNeasy Mini kits (QIAGEN, Tokyo, Japan). Total RNA aliquots were treated with DNase to avoid any amplification of genomic DNA and were reverse-transcribed, using SuperScript VILO cDNA Synthesis kits (Thermo Fisher Scientific). An aliquot of each cDNA was subjected to qPCR analysis. The reaction was conducted on a StepOnePlus thermocycler (Thermo Fisher Scientific) with THUNDRBIRD SYBR qPCR mix (TOYOBO, Osaka, Japan) containing 0.3 μM of each primer. PLK1 expression in DH-My6 cells was also examined as described previously [35]. The primer sequences used to determine the gene expressions are listed in Supplementary Table 1. The PCR conditions were 1 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 62°C. Relative gene expression levels were calculated using 2–ΔCt values, with the β-actin gene (ACTB) gene used as a housekeeping control. These experiments were performed in triplicate.
Drug exposure
Cells were treated with a single drug or a combination of two drugs for 4 days. The MYC inhibitor 10058-F4 was purchased from Cayman Chemical (Ann Arbor, MI, USA). The BCL6 inhibitors 79-6 and FX1 were from Merck KGaA (Darmstadt, Germany) and Selleck Chemical (Houston, TX, USA), respectively. The BET domain inhibitor JQ1 was obtained from Merck KGaA, the HDAC inhibitor vorinostat was from Cayman Chemical, and the PLK1 inhibitor volasertib was from ChemScene (Monmouth Junction, NJ, USA). All reagents were dissolved in DMSO. DMSO concentration in the control medium was the same as that used to make up the highest concentration of drugs in growth medium for the same set of experiments.
Genetic inhibition through siRNA
Cells were transfected with siRNAs on Nucleofector (Lonza, Basel, Switzerland), using C solution and the D-23 program. Stealth RNAi siRNAs directed against MYC (HSS181389), BCL6 (HSS100968), and PLK1 (HSS108120) and nontargeting control siRNA (12935-145) were obtained from Thermo Fisher Scientific. These experiments were performed in triplicate.
Cell proliferation, apoptosis, and cell cycle analyses
For cell proliferation assays, cells were seeded in 96-well plates (2 × 104 cells/well), and viable cells were counted every 24 h on a FACSCalibur flow cytometer (Becton Dickinson, Mountain View, CA, USA) by gating out cells stained with propidium iodide. To compare the antiproliferation effect of single-agent treatments with combination treatments, synergy levels were determined from CI ranges, using the Chou–Talalay method [59]: no synergism, CI > 1.1; additive effect, CI = 1.0–1.1; and synergism, CI < 1.0. For apoptosis analysis, cells were stained with annexin V-phycoerythrin and 7-amino-actinomycin D as described [35]. For cell cycle analysis, cells were fixed in cold 70% ethanol, treated with RNase, and stained with propidium iodide. Cells were analyzed using a FACSCalibur, and all flow cytometry data were analyzed using CellQuest Pro software (Becton Dickinson). These experiments were performed in triplicate.
Statistical analysis
Data are presented as the mean ± the standard error of the mean (SEM). Differences between two groups were analyzed using the Mann–Whitney nonparametric U test when only two groups were compared, or by one-way analysis of variance when more than two groups were compared. A statistically significant difference was defined as P < 0.05.
Abbreviations
DHL: double-hit lymphoma; HGBL: high-grade B-cell lymphoma; THL: triple-hit lymphoma; DLBCL: diffuse large B-cell lymphoma; IGH: immunoglobulin heavy chain; PLK1: polo-like kinase 1; HDAC: histone deacetylase; FCS: fetal bovine serum; DMSO: dimethylsulfoxide; GCB: germinal center B-cell like; FISH: fluorescence in situ hybridization; SKY: spectral karyotyping; LD–PCR: long-distance polymerase chain reaction; STR: short tandem repeat; RT–qPCR: quantitative reverse-transcription PCR; siRNA: small interfering RNA; BET: bromodomain and extraterminal domain; BRD: bromodomain; IC50: half maximal inhibitory concentration; CI: combination index; SEM: standard error of the mean.
Author contributions
HK, TH, YH, MK, and TT performed the experiments; AT engaged in clinical work; IM advised on immunohistochemistry; AY and MF provided valuable comments on this study; MD designed the study, established the cell line, contributed to the acquisition of funding, and wrote the paper.
ACKNOWLEDGMENTS
The authors thank Dr. Takako Ujihara (Kochi Medical School, Kochi University) and Dr. Tadashi Asagiri (Kochi University Hospital) for technical assistance.
CONFLICTS OF INTEREST
The authors declare no conflicts of interests.
FUNDING
This work was supported by the Japan Society for the Promotion of Science (grant 17K09927) to MD.
REFERENCES
1. Aukema SM, Siebert R, Schuuring E, van Imhoff GW, Kluin-Nelemans HC, Boerma EJ, Kluin PM. Double-hit B-cell lymphomas. Blood. 2011; 117:2319–31. https://doi.org/10.1182/blood-2010-09-297879.
2. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD, Jaffe ES. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016; 127: 2375–90. https://doi.org/10.1182/blood-2016-01-643569.
3. Kluin PM, Harris NL, Stein H, Leoncini L, Campo E, Jaffe ES, Gascoyne RD, Swerdlow SH. High-grade B-cell lymphoma. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Arber DA, Hasserjian RP, Le Beau MM, Orazi A, Siebert R, eds. WHO classification of tumours of haematopoietic and lymphoid tissues. Internat Agency for Research on Cancer; 2017. p. 335–41.
4. Sarkozy C, Traverse-Glehen A, Coiffier B. Double-hit and double-protein-expression lymphomas: Aggressive and refractory lymphomas. Lancet Oncol. 2015; 16:e555–67. https://doi.org/10.1016/S1470-2045(15)00005-4.
5. Scott DW, King RL, Staiger AM, Ben-Neriah S, Jiang A, Horn H, Mottok A, Farinha P, Slack GW, Ennishi D, Schmitz N, Pfreundschuh M, Nowakowski GS, et al. High grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements with diffuse large B-cell lymphoma morphology. Blood. 2018; 131: 2060–4. https://doi.org/10.1182/blood-2017-12-820605.
6. Snuderl M, Kolman OK, Chen YB, Hsu JJ, Ackerman AM, Dal Cin P, Ferry JA, Harris NL, Hasserjian RP, Zukerberg LR, Abramson JS, Hochberg EP, Lee H, et al. B-cell lymphomas with concurrent IGH-BCL2 and MYC rearrangements are aggressive neoplasms with clinical and pathologic features distinct from Burkitt lymphoma and diffuse large B-cell lymphoma. Am J Surg Pathol. 2010; 34:327–40. https://doi.org/10.1097/PAS.0b013e3181cd3aeb.
7. Li S, Lin P, Young KH, Kanagal-Shamanna R, Yin CC, Medeiros LJ. MYC/BCL2 double-hit high-grade B-cell lymphoma. Adv Anat Pathol. 2013; 20:315–26. https://doi.org/10.1097/PAP.0b013e3182a289f2.
8. Horn H, Ziepert M, Becher C, Barth TF, Bernd HW, Feller AC, Klapper W, Hummel M, Stein H, Hansmann ML, Schmelter C, Möller P, Cogliatti S, et al, and German High-Grade Non-Hodgkin Lymphoma Study Group. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood. 2013; 121:2253–63. https://doi.org/10.1182/blood-2012-06-435842.
9. Pillai RK, Sathanoori M, Van Oss SB, Swerdlow SH. Double-hit B-cell lymphomas with BCL6 and MYC translocations are aggressive, frequently extranodal lymphomas distinct from BCL2 double-hit B-cell lymphomas. Am J Surg Pathol. 2013; 37:323–32. https://doi.org/10.1097/PAS.0b013e31826cebad.
10. Turakhia SK, Hill BT, Dufresne SD, Nakashima MO, Cotta CV. Aggressive B-cell lymphomas with translocations involving BCL6 and MYC have distinct clinical-pathologic characteristics. Am J Clin Pathol. 2014; 142:339–46. https://doi.org/10.1309/AJCPBWVHTF7RRSA4.
11. Ye Q, Xu-Monette ZY, Tzankov A, Deng L, Wang X, Manyam GC, Visco C, Montes-Moreno S, Zhang L, Dybkær K, Chiu A, Orazi A, Zu Y, et al. Prognostic impact of concurrent MYC and BCL6 rearrangements and expression in de novo diffuse large B-cell lymphoma. Oncotarget. 2016; 7:2401–16. https://doi.org/10.18632/oncotarget.6262.
12. Drexler HG, Eberth S, Nagel S, MacLeod RA. Malignant hematopoietic cell lines: in vitro models for double-hit B-cell lymphomas. Leuk Lymphoma. 2016; 57:1015–20. https://doi.org/10.3109/10428194.2015.1108414.
13. Drexler HG. Guide to leukemia-lymphoma cell lines. 2nd edition, Braunschweig, 2010.
14. Pham LV, Lu G, Tamayo AT, Chen J, Challagundla P, Jorgensen JL, Medeiros LJ, Ford RJ. Establishment and characterization of a novel MYC/BCL2 “double-hit” diffuse large B cell lymphoma cell line, RC. J Hematol Oncol. 2015; 8:121. https://doi.org/10.1186/s13045-015-0218-1.
15. Akasaka H, Akasaka T, Kurata M, Ueda C, Shimizu A, Uchiyama T, Ohno H. Molecular anatomy of BCL6 translocations revealed by long-distance polymerase chain reaction-based assays. Cancer Res. 2000; 60:2335–41.
16. Lu Z, Tsai AG, Akasaka T, Ohno H, Jiang Y, Melnick AM, Greisman HA, Lieber MR. BCL6 breaks occur at different AID sequence motifs in Ig-BCL6 and non-Ig-BCL6 rearrangements. Blood. 2013; 121:4551–4. https://doi.org/10.1182/blood-2012-10-464958.
17. Prochownik EV, Vogt PK. Therapeutic targeting of Myc. Genes Cancer. 2010; 1:650–9. https://doi.org/10.1177/1947601910377494.
18. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011; 146:904–17, 2011. https://doi.org/10.1016/j.cell.2011.08.017.
19. Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, Reichert E, Kung AL, Rodig SJ, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013; 24:777–90. https://doi.org/10.1016/j.ccr.2013.11.003.
20. Trabucco SE, Gerstein RM, Evens AM, Bradner JE, Shultz LD, Greiner DL, Zhang H. Inhibition of bromodomain proteins for the treatment of human diffuse large B-cell lymphoma. Clin Cancer Res. 2015; 21:113–22. https://doi.org/10.1158/1078-0432.CCR-13-3346.
21. Cerchietti LC, Ghetu AF, Zhu X, Da Silva GF, Zhong S, Matthews M, Bunting KL, Polo JM, Farès C, Arrowsmith CH, Yang SN, Garcia M, Coop A, et al. A small-molecule inhibitor of BCL6 kills DLBCL cells in vitro and in vivo. Cancer Cell. 2010; 17:400–11. https://doi.org/10.1016/j.ccr.2009.12.050.
22. Cardenas MG, Yu W, Beguelin W, Teater MR, Geng H, Goldstein RL, Oswald E, Hatzi K, Yang SN, Cohen J, Shaknovich R, Vanommeslaeghe K, Cheng H, et al. Rationally designed BCL6 inhibitors target activated B cell diffuse large B cell lymphoma. J Clin Invest. 2016; 126:3351–62. https://doi.org/10.1172/JCI85795.
23. Cardenas MG, Oswald E, Yu W, Xue F, MacKerell AD Jr, Melnick AM. The expanding role of the BCL6 oncoprotein as a cancer therapeutic target. Clin Cancer Res. 2017; 23:885–93. https://doi.org/10.1158/1078-0432.CCR-16-2071.
24. Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer. 2006; 6:321–30. https://doi.org/10.1038/nrc1841.
25. Zitouni S, Nabais C, Jana SC, Guerrero A, Bettencourt-Dias M. Polo-like kinases: structural variations lead to multiple functions. Nat Rev Mol Cell Biol. 2014; 15:433–52. https://doi.org/10.1038/nrm3819.
26. Van den Bossche J, Lardon F, Deschoolmeester V, De Pauw I, Vermorken JB, Specenier P, Pauwels P, Peeters M, Wouters A. Spotlight on volasertib: Preclinical and clinical evaluation of a promising Plk1 Inhibitor. Med Res Rev. 2016; 36:749–86. https://doi.org/10.1002/med.21392.
27. Rudolph D, Steegmaier M, Hoffmann M, Grauert M, Baum A, Quant J, Haslinger C, Garin-Chesa P, Adolf GR. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin Cancer Res. 2009; 15:3094–102. https://doi.org/10.1158/1078-0432.CCR-08-2445.
28. Stevens FE, Beamish H, Warrener R, Gabrielli B. Histone deacetylase inhibitors induce mitotic slippage. Oncogene. 2008; 27:1345–54. https://doi.org/10.1038/sj.onc.1210779.
29. Nguyen T, Parker R, Hawkins E, Holkova B, Yazbeck V, Kolluri A, Kmieciak M, Rahmani M, Grant S. Synergistic interactions between PLK1 and HDAC inhibitors in non-Hodgkin's lymphoma cells occur in vitro and in vivo and proceed through multiple mechanisms. Oncotarget. 2017; 8:31478–93. https://doi.org/10.18632/oncotarget.15649.
30. Wang W, Hu S, Lu X, Young KH, Medeiros LJ. Triple-hit B-cell lymphoma with MYC, BCL2, and BCL6 translocations/rearrangements: Clinicopathologic features of 11 cases. Am J Surg Pathol. 2015; 39:1132–9. https://doi.org/10.1097/PAS.0000000000000434.
31. Aukema SM, Kreuz M, Kohler CW, Rosolowski M, Hasenclever D, Hummel M, Küppers R, Lenze D, Ott G, Pott C, Richter J, Rosenwald A, Szczepanowski M, et al, and Molecular Mechanisms in Malignant Lymphomas Network Project. Biological characterization of adult MYCtranslocation-positive mature B-cell lymphomas other than molecular Burkitt lymphoma. Haematologica. 2014; 99:726–35. https://doi.org/10.3324/haematol.2013.091827.
32. Li S, Desai P, Lin P, Yin CC, Tang G, Wang XJ, Konoplev SN, Khoury JD, Bueso-Ramos CE, Medeiros LJ. MYC/BCL6 double-hit lymphoma (DHL): A tumour associated with an aggressive clinical course and poor prognosis. Histopathology. 2016; 68:1090–8. https://doi.org/10.1111/his.12884.
33. Wu D, Wood BL, Dorer R, Fromm JR. “Double-Hit” mature B-cell lymphomas show a common immunophenotype by flow cytometry that includes decreased CD20 expression. Am J Clin Pathol. 2010; 134:258–65. https://doi.org/10.1309/AJCP7YLDTJPLCE5F.
34. Harrington AM, Olteanu H, Kroft SH, Eshoa C. The unique immunophenotype of double-hit lymphomas. Am J Clin Pathol. 2011; 135:649–50. https://doi.org/10.1309/AJCPL11MAHISIJBQ.
35. Higuchi T, Hashida Y, Taniguchi A, Kamioka M, Daibata M. Differential gene expression profiling linked to tumor progression of splenic marginal zone lymphoma. Sci Rep. 2017; 7:11026. https://doi.org/10.1038/s41598-017-11389-5.
36. Czuczman MS, Olejniczak S, Gowda A, Kotowski A, Binder A, Kaur H, Knight J, Starostik P, Deans J, Hernandez-Ilizaliturri FJ. Acquirement of rituximab resistance in lymphoma cell lines is associated with both global CD20 gene and protein down-regulation regulated at the pretranscriptional and posttranscriptional levels. Clin Cancer Res. 2008; 14:1561-70. https://doi.org/10.1158/1078-0432.CCR-07-1254.
37. Maleki A, Seegmiller AC, Uddin N, Karandikar NJ, Chen W. Bright CD38 expression is an indicator of MYC rearrangement. Leuk Lymphoma. 2009; 50:1054–7. https://doi.org/10.1080/10428190902930470.
38. Yamamoto K, Okamura A, Inui Y, Yakushijin K, Kawakami F, Itoh T, Matsuoka H, Minami H. IGH@/BCL6 rearrangement on the der(3)t(3;14)(q27;q32) in primary mediastinal large B-cell lymphoma. Leuk Res. 2012; 36:e218–21. https://doi.org/10.1016/j.leukres.2012.07.011.
39. Ueda C, Uchiyama T, Ohno H. Immunoglobulin (Ig)/BCL6 versus non-Ig/BCL6 gene fusion in diffuse large B-cell lymphoma corresponds to a high- versus low-level expression of BCL6 mRNA. Blood. 2002; 99:2624–5. https://doi.org/10.1182/blood-2001-11-0117.
40. Wilda M, Busch K, Klose I, Keller T, Woessmann W, Kreuder J, Harbott J, Borkhardt A. Level of MYC overexpression in pediatric Burkitt's lymphoma is strongly dependent on genomic breakpoint location within the MYC locus. Genes Chromosomes Cancer. 2004; 41:178–82. https://doi.org/10.1002/gcc.20063.
41. Copie-Bergman C, Cuillière-Dartigues P, Baia M, Briere J, Delarue R, Canioni D, Salles G, Parrens M, Belhadj K, Fabiani B, Recher C, Petrella T, Ketterer N, et al. MYC-IG rearrangements are negative predictors of survival in DLBCL patients treated with immunochemotherapy: a GELA/LYSA study. Blood. 2015; 126:2466–74. https://doi.org/10.1182/blood-2015-05-647602.
42. Wang HY, Bossler AD, Schaffer A, Tomczak E, DiPatri D, Frank DM, Nowell PC, Bagg A. A novel t(3;8)(q27;q24.1) simultaneously involving both the BCL6 and MYC genes in a diffuse large B-cell lymphoma. Cancer Genet Cytogenet. 2007; 172:45–53. https://doi.org/10.1016/j.cancergencyto.2006.07.016.
43. Geiersbach K, Salama M, Sennett MD, Shetty S. Multi-hit lymphoma with intermediate features and novel t(3;8)(q27;q24) not involving MYC. Leuk Lymphoma. 2011; 52:922–9. https://doi.org/10.3109/10428194.2011.555894.
44. Ohno H, Nakagawa M, Kishimori C, Fukutsuka K, Honjo G. Cryptic t(3;8)(q27;q24) and/or MYC-BCL6 linkage associated with MYC expression by immunohistochemistry is frequent in multiple-hit B-cell lymphomas. Blood Cancer J. 2017; 7:e578. https://doi.org/10.1038/bcj.2017.59.
45. Ryan RJ, Drier Y, Whitton H, Cotton MJ, Kaur J, Issner R, Gillespie S, Epstein CB, Nardi V, Sohani AR, Hochberg EP, Bernstein BE. Detection of enhancer-associated rearrangements reveals mechanisms of oncogene dysregulation in B-cell lymphoma. Cancer Discov. 2015; 5:1058–71. https://doi.org/10.1158/2159-8290.CD-15-0370.
46. Johnson-Farley N, Veliz J, Bhagavathi S, Bertino JR. ABT-199, a BH3 mimetic that specifically targets Bcl-2, enhances the antitumor activity of chemotherapy, bortezomib and JQ1 in “double hit” lymphoma cells. Leuk Lymphoma. 2015; 56:2146–52. https://doi.org/10.3109/10428194.2014.981172.
47. Murga-Z amalloa C, Polk A, Hanel W, Chowdhury P, Brown N, Hristov AC, Bailey NG, Wang T, Phillips T, Devata S, Poonnen P, Gomez-Gelvez J, Inamdar KV, Wilcox RA. Polo-like-kinase 1 (PLK-1) and c-myc inhibition with the dual kinase-bromodomain inhibitor volasertib in aggressive lymphomas. Oncotarget. 2017; 8:114474–80. https://doi.org/10.18632/oncotarget.22967.
48. Tan J, Li Z, Lee PL, Guan P, Aau MY, Lee ST, Feng M, Lim CZ, Lee EY, Wee ZN, Lim YC, Karuturi RK, Yu Q. PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mTOR-targeted therapy. Cancer Discov. 2013; 3:1156–71. https://doi.org/10.1158/2159-8290.CD-12-0595.
49. Bhadury J, Nilsson LM, Muralidharan SV, Green LC, Li Z, Gesner EM, Hansen HC, Keller UB, McLure KG, Nilsson JA. BET and HDAC inhibitors induce similar genes and biological effects and synergize to kill in Myc-induced murine lymphoma. Proc Natl Acad Sci USA. 2014; 111:E2721–30. https://doi.org/10.1073/pnas.1406722111.
50. Fiskus W, Sharma S, Qi J, Valenta JA, Schaub LJ, Shah B, Peth K, Portier BP, Rodriguez M, Devaraj SG, Zhan M, Sheng J, Iyer SP, et al. Highly active combination of BRD4 antagonist and histone deacetylase inhibitor against human acute myelogenous leukemia cells. Mol Cancer Ther. 2014; 13:1142–54. https://doi.org/10.1158/1535-7163.MCT-13-0770.
51. Cerchietti LC, Hatzi K, Caldas-Lopes E, Yang SN, Figueroa ME, Morin RD, Hirst M, Mendez L, Shaknovich R, Cole PA, Bhalla K, Gascoyne RD, Marra M, et al. BCL6 repression of EP300 in human diffuse large B cell lymphoma cells provides a basis for rational combinatorial therapy. J Clin Invest. 2010; 120:4569–82. https://doi.org/10.1172/JCI42869.
52. Schröck E, du Manoir S, Veldman T, Schoell B, Wienberg J, Ferguson-Smith MA, Ning Y, Ledbetter DH, Bar-Am I, Soenksen D, Garini Y, Ried T. Multicolor spectral karyotyping of human chromosomes. Science. 1996; 273:494–7. https://doi.org/10.1126/science.273.5274.494.
53. Siebert R, Matthiesen P, Harder S, Zhang Y, Borowski A, Zühlke-Jenisch R, Metzke S, Joos S, Weber-Matthiesen K, Grote W, Schlegelberger B. Application of interphase fluorescence in situ hybridization for the detection of the Burkitt translocation t(8;14)(q24;q32) in B-cell lymphomas. Blood. 1998; 91:984–90.
54. Tamura A, Miura I, Iida S, Yokota S, Horiike S, Nishida K, Fujii H, Nakamura S, Seto M, Ueda R, Taniwaki M. Interphase detection of immunoglobulin heavy chain gene translocations with specific oncogene loci in 173 patients with B-cell lymphoma. Cancer Genet Cytogenet. 2001; 129:1–9. https://doi.org/10.1016/S0165-4608(01)00436-8.
55. Kiaei A, Onsori H, Alijani A, Andalib S, Ghorbian S, Sakhinia E. Detection of t(8;14) c-myc/IgH gene rearrangement by long-distance polymerase chain reaction in patients with diffuse large B-cell lymphoma. Hematol Oncol Stem Cell Ther. 2016; 9:141–6. https://doi.org/10.1016/j.hemonc.2016.05.006.
56. Sonoki T, Willis TG, Oscier DG, Karran EL, Siebert R, Dyer MJ. Rapid amplification of immunoglobulin heavy chain switch (IGHS) translocation breakpoints using long-distance inverse PCR. Leukemia. 2004; 18:2026–31. https://doi.org/10.1038/sj.leu.2403500.
57. Akasaka T, Lossos IS, Levy R. BCL6 gene translocation in follicular lymphoma: a harbinger of eventual transformation to diffuse aggressive lymphoma. Blood. 2003; 102:1443–8. https://doi.org/10.1182/blood-2002-08-2482.
58. Hashida Y, Imajoh M, Nemoto Y, Kamioka M, Taniguchi A, Taguchi T, Kume M, Orihashi K, Daibata M. Detection of Merkel cell polyomavirus with a tumour-specific signature in non-small cell lung cancer. Br J Cancer. 2013; 108:629–37. https://doi.org/10.1038/bjc.2012.567.
59. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010; 70:440–6. https://doi.org/10.1158/0008-5472.CAN-09-1947.