
INTRODUCTION
Extracellular vesicles (EVs) are membrane-enclosed particles released from either endosomes or the cell surface [1–3]. EVs are composed of an array of proteins, nucleic acids, lipids, and other metabolites that reflect the cell of origin. They offer an intercellular route to transfer oncogenic material that change the functions of non-malignant cells, i.e. proliferation, invasion, and angiogenesis [2]. Their secretion is correlated to the cell’s ability to produce invadopodia (actin-rich cellular protrusions with proteolytic activity); i.e., inhibition of invadopodia formation decreased exosome release [3–5]. Importantly, glioblastoma (GBM)-derived EVs can cross the brain–blood-barrier and are detectable in the systemic blood circulation [6]. Profiling the composition of GBM-derived EVs may, therefore, offer a non-invasive means of assessing tumors in situ [4].
Studies have described extensive RNA expression analyses of GBM-derived EVs, however, proteomic profiles are currently limited [4, 7]. Among the vesicular proteins, one study identify the chloride intracellular channels (CLIC) carried by exosomes between GBM cells [8]. The CLIC family form a class of proteins that do not fit the paradigm set by classical ion channels (for review see; [9–11]). They can exist as both soluble globular proteins and integral membrane proteins with ion channel function. The first member of CLIC, namely CLIC1 (also known as NCC27), holds pathological implications in a variety of tumors, being involved in cell proliferation, motility, and angiogenesis [12–15]. CLIC1 is overexpressed in glioblastoma (GBM), with highest expression in patients with poor prognosis [13]. CLIC1 is also secreted in extracellular vesicles (EVs) by cancer cells [8] and is detected in biological fluids [8, 16, 17], fostering the hypothesis that secreted CLIC1 protein may increase GBM growth. Interestingly, Setti et al [8] have shown that the secretion of CLIC1 via EVs is common to all human GBM cell lines (U87MG, A172, LN405, U118MG, T98G, DBTRG-05MG and U373 MG). If the number of secreted EVs differs from one type of lineage to another, the membrane markers and biophysical properties of EVs are similar.
Using U87 GBM cell line, we have recently described that miR-5096 increases the outgrowth of filopodia in glioma cells, and promotes the extracellular release of EVs by U87 thereby promoting its own transfer to surrounding cells [18]. Here, we show that EVs also contain active CLIC1 whose amount is not significantly increased by miR-5096. The transfer of CLIC1 to human microvascular endothelial cells (HMEC) requires Ca2+ spikes and TRPM7 for their uptake, and contributes to endothelial sprouting [19, 20].
RESULTS
Extracellular vesicles from GBM cells transfer active CLIC1 to HMEC
Both U87 and HMEC expressed CLIC1 proteins, as already reported [12, 21]. Immunoblot analysis of whole cell lysates (WCL) from homotypic cultures revealed that the cell loading with miR-5096 mimic or inhibitor did not significantly change CLIC1 expression after 48h in both U87 and HMEC (Figure 1A). This is in agreement with the absence of miR-5096 effect on CLIC1 mRNA expression (not shown) and predictions from bioinformatics tools which failed to identify any target site for miR-5096 in CLIC1 gene and mRNA. However, the endothelial CLIC1 level was increased after 24h-exposure of HMEC to U87-conditioned media (Figure 1B). We next separated EVs from the effluent (soluble fraction) of culture media as described previously [18]. In all cases, EVs and effluents were adjusted to the same number of U87 (i.e. 4 x 106 cells), then applied to homotypic HMEC cultures for 24h. Cell exposure to EVs released from miR-5096-loaded U87 significantly increased CLIC1 levels in HMEC, while the effluent (EVs-free) did not (Figure 1B). The immunoblot analysis of EVs showed an enrichment in the exosome specific protein tsg101 (tumor susceptibility gene 101) [8, 18] (Figure 1C). Clearly, EVs contained CLIC1 proteins and their level seemed to be higher in EVs from miR-loaded U87 than from empty-loaded U87. A possible explanation might be that miR5096 induced an increase in EVs release [18], rather than a significant increase in CLIC1 vesicular content. To confirm the transfer of CLIC1 to HMEC, endogenous CLIC1 was silenced by using siRNA in a series of experiments (i.e. relative OD of 0.406±0.061 and 0.015±0.007, respectively before and after CLIC1 silencing; P<0.05, n=3). As shown in Figure 1C, both cellular (WCL) and vesicular (EVs lysates) CLIC1 contents were suppressed in HMEC by CLIC1 siRNA. The CLIC1 immuno-labelling showed that CLIC1 was mostly found in perinuclear areas of control HMEC (Figure 1D). No labelling was observed after silencing CLIC1 in HMEC. After 24h of cell incubations with EVs from miR-5096 loaded U87, we detected CLIC1 in both the cytosol and the plasma membrane of control and silenced HMEC. We also overexpressed a fluorescent-tagged version of human CLIC1 in U87 and collected conditioned media after 48h (see Supplementary Figure 2). Exposure of HMEC to isolated EVs resulted to the fluorescent labelling of HMEC after 24h. Thus, the increase of CLIC1 in HMEC resulted more from a vesicular transfer of CLIC1 rather than an up-regulation of its endogenous expression in recipient HMEC. Are the transferred proteins active in the recipient cells? Using a 24-hour three-dimensional in vitro angiogenesis assay, we explored the ability of vesicular CLIC1 to induce endothelial spheroid sprouting [22]. As shown in Figure 1E, EVs stimulated HMEC sprouting even more when collected from miR-loaded U87 (M) (see also Supplementary Figure 1A). This effect was partially prevented by silencing CLIC1 in U87 (Msi). To confirm the contribution of vesicular CLIC1 to angiogenesis, EVs were also tested on branching morphogenesis in vitro [22]. HMEC control and CLIC1 silenced were plated in ECM gel and exposed or not (Co) to EVs collected from the same number of U87. Quantification of average number of processes per cell was performed after 12h (Figure 1F; see also Supplementary Figure 1B). Silencing CLIC1 in HMEC decreased the branching as expected for its contribution to in vitro angiogenesis [12]. Exposure to EVs stimulated the branching even more when collected from miR-5096-loaded U87 (M). This effect was partially prevented by silencing CLIC1 in secretory U87 (Msi). Thus, the vesicular CLIC1 was active in recipient HMEC and contributed to early steps of in vitro angiogenesis [12].
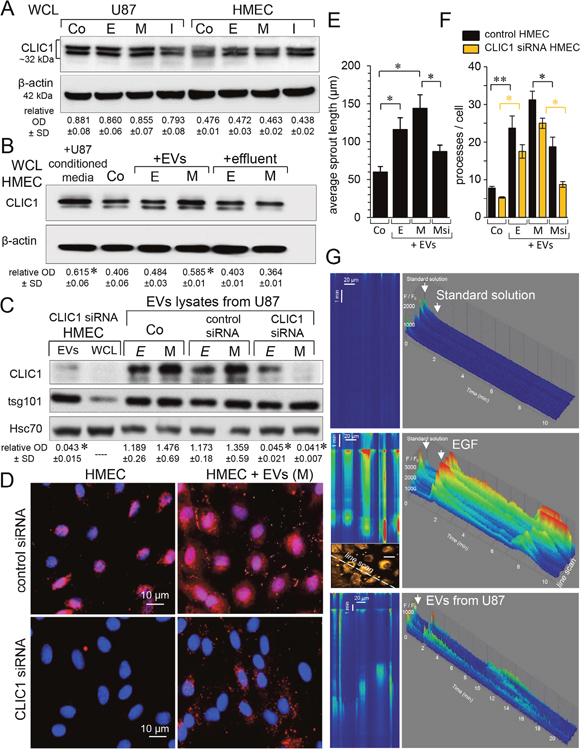
Figure 1: Active CLIC1 protein is transferred via vesicles from GBM to endothelial cells. Immunoblot analysis of CLIC1 in whole cell lysates (WCL) from homotypic cultures of U87 and HMEC, 48 h after loading. Untreated cells were used as control (Co). Cells were loaded empty (E) or with 30nM miR5096 mimic (M) or inhibitor (I). β-actin as loading control (60μg proteins/lane). Numbers indicate mean values of optical densities (OD) of CLIC1 relative to β–actin (± SD; P>0.05 vs Co; n = 3). (B) CLIC1 increased in HMEC after 24 h of incubation with EVs. Cell-conditioned media were collected from homotypic U87 miR-loaded (M) or not (E), 48h after loading. HMEC were exposed to EVs or effluent (soluble fraction) separated from U87-conditioned media. Numbers indicate mean OD values of CLIC1 relative to β-actin (± SD; * P<0.05 vs Co; n = 3). (C) EVs contained CLIC1. Lysates of EVs were immunoblotted for the marker tsg101. Homotypic U87 were loaded (M) or not (E) upon transfection of control siRNA or siRNA targeting CLIC1. Silencing CLIC1 was also tested in HMEC (WCL) and HMEC-released EVs (Hsc70 as loading control). Numbers indicate mean OD values of CLIC1 related to tsg101 for EVs lysates (± SD; * P<0.05 vs Co (E); n = 4). (D) Endothelial cell localization of CLIC1. HMEC were silenced by siRNA CLIC1 then exposed to EVs collected from homotypic U87 (M) (n = 3). CLIC1 stained with alexa Fluor 594 (red) and nuclear DNA with Dapi (blue). (E) CLIC1 effect on the length of endothelial sprouts formed from spheroïds in Matrigel for 24h, in the absence (Co) or the presence of EVs collected from homotypic U87 empty (E) or miR5096-loaded (M). When indicated by Msi, CLIC1 was silenced by siRNA in U87 (M). Data are means ± SD (* P-values<0.05 vs control; n=10) in two independent experiments. (F) Contribution of CLIC1 to the branching morphogenesis in HMEC cultured in collagen ECM gel for 12 h, in the absence (Co) or presence of EVs. Histogram shows the average number of processes per cell. Control and CLIC1 siRNA HMEC are filled black and yellow, respectively. Data are means ± SD (* P<0.05, **P<0.01 vs Co; n=10) in two experiments. (G) EVs induced Ca2+ spikes in HMEC. Representative line scan images of cytosolic [Ca2+] in Fluo-4-loaded HMEC exposed to the standard solution, to EGF (10 ng/ml) and to EVs (from U87) as indicated by arrows. Space and time ordinates are displayed in the horizontal and vertical directions, respectively (scan rate 22.3 μsec/line). Amplitudes of Ca2+ signal are expressed as the fluorescent rapport F/F0 (pseudo-colors) in a tridimensional histogram (F/F0 vs space/time). In all cases, the line scan crossed both cytosol and nuclei of 4 adjacent cells, as shown in the 1024x1024 pixel panel. Note that Ca2+ spikes were observed at the beginning of all recordings (due to the initial cell perfusion) and were not reproduced by reapplying the standard solution.
EVs-mediated CLIC1 transfer to HMEC requires TRPM7-dependent Ca2+ signaling
In order for EVs to elicit a signaling response from recipient cells, they can fuse with plasma membrane or are taken up via endocytosis or attach to the cell surface [23] (see Supplementary Figure 2). Since endocytosis, receptor internalization and trafficking are regulated by cytosolic Ca2+ level [24, 25], we determined whether the endothelial uptake of vesicular CLIC1 was associated with cytosolic Ca2+ fluctuations. Spatio-temporal Ca2+ variations were recorded in HMEC loaded with Fluo-4 by using line scanning of confocal microscopy [26]. No Ca2+ spike was observed in standard external conditions (Figure 1G; upper panel). As a positive control, HMEC were exposed to epidermal growth factor (EGF), inducing a typical pattern of Ca2+ signal [27], i.e. a rapid increase in cytosolic Ca2+ which was maintained several minutes before decaying to the resting level, followed by a second transient increase observed about 6 min later. The endothelial cell exposure to EVs from U87 elicited a similar Ca2+ signal although with a lower amplitude than did EGF (i.e. max F/F0 of 1000 ± 500 with EVs and 3000±600 with EGF; P<0.05, n=6). Of note, the lag time between successive Ca2+ waves was similar (6.61 ± 1.25 min; P>0.05; n=6). The well-known inhibitor of Ca2+ pathways, 2-APB [28–31], suppressed EVs-evoked Ca2+ signal (Figure 2A, lower panel) as well as the subsequently CLIC1 delivery to HMEC (Figure 2B). Indeed, incubations of 2-APB-treated HMEC with EVs for 24 h did not increase CLIC1 labelling, especially in CLIC1-silenced HMEC (Figure 2B). Among 2-APB-sensitive channels in plasma membranes, the melastatin-subfamily of TRP (TRPM7) is a non-specific divalent cation channel upregulated in GBM [29]. To estimate its contribution in both Ca2+ waves and CLIC1 delivery to HMEC, endogenous TRPM7 was silenced by using siRNA (Figure 2C, 2D). Cell exposure to EVs already produced cytosolic Ca2+ waves which were decreased by silencing TRPM7 (Figure 2E). Unfortunately, we could not knockdown both TRPM7 and CLIC1 since HMEC did not survive. After 24h of cell incubations with EVs, CLIC1 labeling was also decreased in cytosol or plasma membrane of TRPM7-silenced HMEC, attesting the partial contribution of TRPM7 in this process (Figure 2F). Of note, cell exposure to soluble fractions (EVs-free effluent) from U87-conditioned media did not increase TRPM7 levels in HMEC, while the miR5096 loading or Kir4.1 silencing did (Figure 2C, 2G). Thus, the vesicular transfer of CLIC1 to HMEC required Ca2+ signaling mediated, at least in part, by TRPM7.
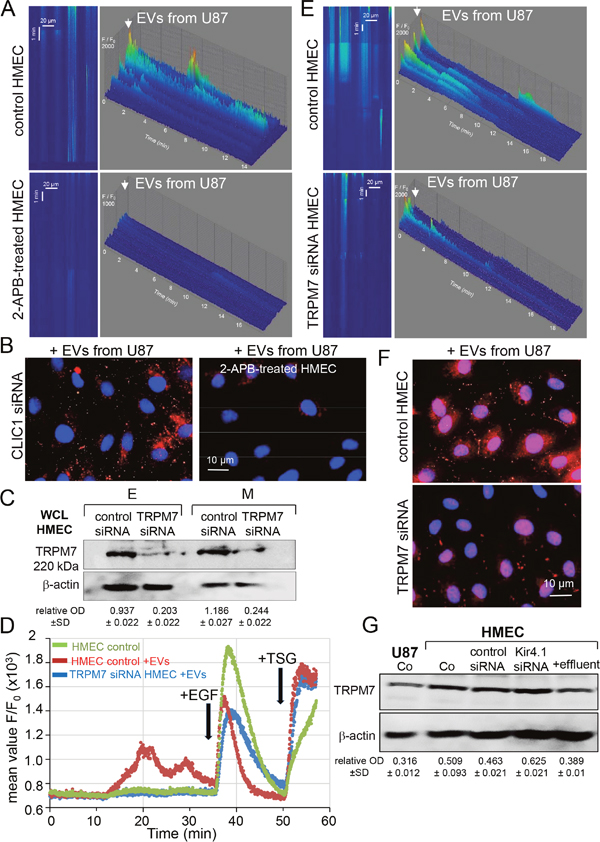
Figure 2: The vesicular transfer of CLIC1 requires cytosolic Ca2+ increases in HMEC. (A) Line scan images of cytosolic [Ca2+] increases in Fluo-4 loaded HMEC exposed to the same amount of EVs collected from the same homotypic U87 culture. HMEC were pre-treated with 2-APB (50 μM; lower panel). Tridimensional histogram (F/F0 vs space/time) are representative of 3 experiments. (B) Transfer of vesicular CLIC1 was blocked by 2-APB (50 μM). HMEC silenced by siRNA CLIC1 were exposed to EVs from U87 (n = 3). After 24h of incubation with EVs, HMEC were stained for CLIC1 (red) and nuclei (blue). (C) Expression of TRPM7 in HMEC. Homotypic HMEC were loaded (M) or not (E) with miR-5096 upon transfection of control siRNA or siRNA targeting TRPM7. Numbers indicate mean OD values of TRPM7 related to β-actin (± SD; n = 2; 80μg proteins/lane). (D) Spatially average Ca2+ profile showing the dynamic change of Ca2+ signals with time and induced by EVs (applied at the beginning of the records) then EGF (10 ng/ml) applied at the time indicated by arrow. Cytosolic Ca2+ store depletion was performed by the addition of thapsigargin (TSG, 5μM) at the end of recordings. Values are means of fluorescent ratio F/F0 ± SD; n = 3. (E) Silencing TRPM7 in HMEC reduced the Ca2+ signal induced by EVs collected from homotypic U87 for 48h. (F) Control and silenced TRPM7 HMEC were exposed to EVs and stained for CLIC1 (red) after 24 h of culture (representative of 3 experiments). (G) Expression of TRPM7 in homotypic HMEC upon transfection of control siRNA or siRNA targeting Kir4.1 [18]. HMEC were exposed to the effluent (soluble fraction) from homotypic U87. Numbers indicate mean OD values of TRPM7 related to β-actin (± SD; n = 2).
DISCUSSION
We report here that CLIC1 protein is transferred via EVs from the GBM cell line U87 to microvascular endothelial cells where it remains active, i.e. induces endothelial sprouting. When applied onto HMEC, EVs elicit Ca2+ “spikes” which can be prevented by 2-APB. Although 2-APB inhibits numerous channels including IP3 receptors [28], store-operated Ca2+ channels [30] and TRP channels [29, 31], silencing TRPM7 prevents Ca2+ spikes and the subsequent CLIC1 uptake by HMEC. Altogether, our data show that CLIC1 secreted by cancer cells via extracellular vesicles, modulates the activity of neighboring endothelial cells in a TRPM7 dependent manner, promoting tumor angiogenesis.
We previously reported that miR-5096 favors its own transfer from U87 to HMEC via an increased release of EVs, two days after its loading in U87 [18]. Here, we show that EVs also contain CLIC1 and mostly ensure its transfer to HMEC (Figure 1; see also Supplementary Figure 2). EVs are composed of an array of proteins, nucleic acids, lipids, and other metabolites that reflect the cell of origin [4]. We report that EVs, secreted by the same number of U87, produce a greater increase in active CLIC1 in the recipient HMEC when the donor U87 are previously loaded with miR-5096. The most likely explanation is that miR-5096 induces an increase in EVs release [18], rather than a significant increase in CLIC1 vesicular content. How miR-5096 exerts such an effect is currently unknown and not explored in our study. Nevertheless, evidence increasingly points to a connection between lipid metabolism and cancer, characterized by an alteration in the mechanisms that regulate cholesterol homeostasis [32]. It is known that the survival of GBM cells is dependent on uptake of cholesterol [33] in which some microRNAs are the fine tuners [34, 35]. Interestingly, cholesterol promotes the conversion of CLIC1 from cytosolic to transmembrane proteins [36], thus facilitates its docking to the membranes [37, 38]. Drawing on the data above, we propose a pure speculative model where miR-5096 increases cholesterol and CLIC1 is involved in recruiting EVs, leading to an increased secretion of EVs by U87 cells (see Supplementary Figure 3).
By overexpressing fluorescent-tagged CLIC1 proteins (CLIC-OFP) in U87, we observe CLIC1-OFP inside the invadopodia (see Supplementary Figure 2B). Upon exposure to EVs, HMEC become fluorescent and change their morphology; i.e. showing invadopodia formation. Invadopodia act as multivesicular endosome docking sites and are a site of EVs release, meaning the cell’s ability to form invadopodia determines their ability to release of EVs [4, 5]. In this process, CLIC1 would contribute to the formation of invadopodia in endothelial and tumor cells, by inducing integrin-mediated actomyosin dynamic [15]. Changes in CLIC1 location from cytosolic to transmembrane proteins are associated with malignant transformation [15]. Our study does not allow to distinguish the two forms of CLIC1 (i.e. soluble form and membrane-inserted chloride conducting pore). However, immuno-labeling of CLIC1 confirms its previously described nuclear location in steady HMEC [9]. Following exposure to EVs, CLIC1 is also detected in cytosol and weakly at the plasma membrane of HMEC within 24h. On the other hand, the endothelial sprouting in matrigel is increased by EVs within 24h. This effect is partially prevented by silencing CLIC1 in donor U87 and is not attributed to miR-5096 itself [39]. Our results are in agreement with the literature showing that a low CLIC1 expression in endothelium decreases capillary-like sprouting in matrigel [12, 14, 15]. We observe a functional difference between silencing all CLIC1 and preventing only the U87-derived CLIC1 delivery. This cannot be easily explained since EVs probably contain proangiogenic factors, miRNAs and extracellular proteases which are required by endothelial cells to proliferate, migrate, and organize into new tubular structures [4, 40]. For instance, EVs from xenografts of glioblastomas contain an oncogenic variant of the epidermal growth factor receptor (EGFRvIII), which can be transferred to endothelial cells, producing proliferation and tubulogenesis [41, 42]. A broad array of cell surface and signaling proteins is involved in tubulogenesis [43], and could explain why the perivascular invasion is more important in vascular endothelial growth factor (VEGF)-deficient glioblastoma cells and brain tumor xenografts treated with anti-VEGF blocking antibodies such as bevacizumab [44, 45].
We hypothesize that EVs bind to recipient endothelial cells (see Supplementary Figure 4). These EVs may remain at the plasma membrane [46] or may be internalized by endocytosis either mediated by clathrin [47, 48] or via caveolae and lipid rafts [49]. These mechanisms require Ca2+ increase at the submembrane level [50, 51]. While the precise mechanism of CLIC1 uptake and processing in HMEC remains unclear, we show for the first time that CLIC1 transfer requires an initial Ca2+ signaling in recipient cells within 1h. Moreover, the EVs-induced Ca2+ signal is suppressed in nominally Ca2+-free standard solution (i.e. no CaCl2 added; data not shown), attesting the involvement of an external Ca2+ entry. Among the putative Ca2+ entry pathways, the presence of functional TRPM7 channels is known in human endothelial cells [52–55]. By using specific siRNA, we identify TRPM7 as a mediator for this Ca2+ entry needed for CLIC1 uptake by HMEC. Of note, TRPM7 contributes to the EGF-induced Ca2+ signal, without affecting the Ca2+ content of internal Ca2+ stores sensitive to thapsigargin (see Supplementary Figure 1C).
High CLIC1 expression is involved in the progression of GBM and other tumors [56–58] and correlates with a poor patient outcome [13]. Our data foster the hypothesis that CLIC1 transfer to endothelial cells via EVs contributes to GBM growth by promoting capillary formation [12, 14, 15, 56]. Moreover, the pharmacological inhibition or silencing of TRPM7 inhibits adhesion or invasion in cancer cell lines [19, 59–61] as well as migration of HMEC [53]. Of note, TRPM7 expression is increased in miR-5096 loaded U87 (see Supplementary Figure 1D). Because the silencing of potassium Kir4.1 channels [18] also produces this increase, this up-regulation of TRPM7 should result more from a membrane potential variation than from a direct effect of miR-5096 on TRPM7 gene or mRNA [62]. Nevertheless, it is still not clear whether other proteins and miRNAs could be transferred via EVs to modulate channels in recipient cells [63, 64]. Further investigations are required to fully resolve the functional capabilities of EVs [65–67].
MATERIALS AND METHODS
Cells
Human primary microvascular HMEC (HMVEC-D; Lonza) and U87-MG cells (ATCC HTB-14) were grown in DMEM plus 10% FCS (5% CO2; 37°C). Cells were incubated 48 h in FCS-free media before use.
Reagents
Monoclonal anti-Tsg101, anti-Hsc70 and anti-β-actin were purchased from Santa Cruz Biotech (Clinisciences, Fr). Monoclonal anti-CLIC1 (ab77214) and anti-TRPM7 (ab109438) were from Abcam. Fluo-4 acetomethyl (AM) ester was from Invitrogen (ThermoFisher). 2-Aminoethyl diphenylborinate (2-APB), thapsigargin (TSG) and other chemicals were from Sigma-Aldrich.
Transfection
Cells were transfected by lipofectamine RNAiMAX according to the manufacturer’s protocol (Invitrogen). Human hsa-miR-5096 mimic (mirVana TM miRNA, 4464066-MC22429) and inhibitor (4464084-MH22429) were purchased from Ambion (Invitrogen; ThermoFisher) [18]. Human TRPM7 siRNA (ID 1490) and CLIC1 siRNA (ID 145733) were purchased from (Ambion, AM51331). The sequences were: siRNA CLIC1 (5’-GAGCUUGUGUUGUGCUGAAtt-3’ and 5’-UUCAGCACAACACAAGCUCtt-3’); siRNA TRPM7 (5’-GGACCCUCACAGAUGCCUUtt-3’ and 5’-AAGG CAUCUGUGAGGGUCCtt-3’). To downregulate Kir4.1, cells were transfected with human Kcnj10 siRNA SMARTpool (30 nM) purchased from Dharmacon (ThermoFisher), as we described previously [18]. To overexpress fluorescent CLIC1 proteins, we transfected U87 cells with the human CLIC1/NCC27 gene ORF cDNA clone expression plasmid, C-OFPSpark (HG15242-ACR, Sino Biological Inc.; purchased from Interchim, Montluçon, Fr). Cells were used after 48h.
Immunoblotting
Cells were lysed in RIPA buffer, and Western blots were performed as previously described [18]. EVs pellets were lysed in RIPA buffer containing protease inhibitor cocktail (Roche, Indianapolis, IN) then sonicated for 10 s. Insoluble material was pelleted by centrifugation for 15 min at 14,000 g at 4°C.
Immunofluorescence
Cells were fixed in 4% PFA and permeabilized with 0.1% Triton X-100. Images were performed using a Leica SP2 RS confocal microscope (Z-series of 0.6 μm-optical sections; 512x512 pixels).
EVs isolation
After 48h of culture in FCS-free conditions, cell-conditioned media were collected and sequentially centrifuged at 300 g for 10 min (4°C) then at 2,000 g for 10 min to remove cell debris [18, 68]. EVs were collected by ultracentrifugation at 100,000 g for 90 min. Concentrations were adjusted to the same number of cells (i.e. corresponding to the secretion from 4 x 106 cells).
Cell sprouting assay in collagen gels
Sprouting of HMEC spheroids was performed as previously described [69]. For each gel, 8 spheroids (each containing 400 – 500 cells) were seeded into 0.7 ml collagen solution in 24-well plates (PromoCell GmbH) and incubated with the tested solutions for 24h (5% CO2; 37°C). The cumulative sprout length of 8 randomly selected spheroids was measured for each tested group.
Endothelial tube formation assay in collagen gels
Control and silenced CLIC1 HMEC were plated in DMEM ECM gel, with or without EVs collected from U87, according to the manufacturer’s instructions (from Cell Biolabs, Inc). After 12 hours of incubation at 37°C, 80 single cells were scored for the number of processes per cell. Each well is duplicated for each experiment, and each experiment was repeated three times.
Calcium imaging
Spatiotemporal Ca2+ variations were recorded in HMEC cultured on uncoated glass then loaded with 4μM Fluo4/AM in FCS-free conditions for 40 min at 37°C [26]. Cells were bathed in the standard solution containing (in mM): 136 NaCl, 5 KCl, 1 MgCl2, 1.8 CaCl2, 0.3 NaH2PO4, 10 Glucose, and 10 HEPES (pH 7.4). Measurements were performed by a line scan crossing 3-4 cells in a 1024x1024 pixel panel using a confocal microscope (Nikon C1Si) with 100x objective (Nikon, Melville, NY). Excitation was at 488 nm and emission-selected at 500-570 nm. Line scan images in pseudo-colors were acquired at a sampling rate of 22.3 μsec per line (32 lines/sec). To compare cytosolic [Ca2+] fluctuations, fluorescent measurements (F) were expressed as the F/F0 ratio, where F0 refers to the basal [Ca2+] fluorescence at rest [70]. The signal amplitudes were shown in a tridimensional histogram (F/F0 as a function of time/length).
Statistical analysis
Results are expressed as mean ± SD. A Mann-Whitney U test was used to compare data groups. Statistics were also made with Tanagra software using a Kruskal-Wallis 1-way ANOVA. In all cases, * P values < 0.05 were significant.
ACKNOWLEDGMENTS
We thank greatly Dr Anne Cantereau (ImageUP, STIM CNRS ERL7003 Poitiers) and Dr André Bouchot (CellImap/IFR100, Dijon) for their technical assistance.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by Centre National de la Recherche Scientifique (CNRS) and French National Research Agency ANR-11-LABX-0021 (Lipstic Labex). Gaetan Chanteloup is a fellow of Fondation Recherche Médicale (Grant ECO20160736090).
REFERENCES
1. Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci. 2018; 75:193-208.
2. Quezada C, Torres A, Niechi I, Uribe D, Contreras-Duarte S, Toledo F, San Martin R, Gutierrez J, Sobrevia L. Role of extracellular vesicles in glioma progression. Mol Aspects Med. 2018; 60:38-51.
3. Paterson EK, Courtneidge SA. Invadosomes are coming: new insights into function and disease relevance. FEBS J. 2018; 285:8-27.
4. Mallawaaratchy DM, Hallal S, Russell B, Ly L, Ebrahimkhani S, Wei H, Christopherson RI, Buckland ME, Kaufman KL. Comprehensive proteome profiling of glioblastoma-derived extracellular vesicles identifies markers for more aggressive disease. J Neurooncol. 2017; 131:233-244.
5. Hoshino D, Kirkbride KC, Costello K, Clark ES, Sinha S, Grega-Larson N, Tyska MJ, Weaver AM. Exosome secretion is enhanced by invadopodia and drives invasive behavior. Cell Rep. 2013; 5:1159-1168.
6. García-Romero N, Carrión-Navarro J, Esteban-Rubio S, Lázaro-Ibáñez E, Peris-Celda M, Alonso MM, Guzmán-De-Villoria J, Fernández-Carballal C, de Mendivil AO, García-Duque S, Escobedo-Lucea C, Prat-Acín R, Belda-Iniesta C, Ayuso-Sacido A. DNA sequences within glioma-derived extracellular vesicles can cross the intact blood-brain barrier and be detected in peripheral blood of patients. Oncotarget. 2017; 8:1416–28. https://doi.org/10.18632/oncotarget.13635.
7. Park J, Im H, Hong S, Castro CM, Weissleder R, Lee H. Analyses of Intravesicular Exosomal Proteins Using a Nano-Plasmonic System. ACS Photonics. 2018; 5:487-494.
8. Setti M, Osti D, Richichi C, Ortensi B, Del Bene M, Fornasari L, Beznoussenko G, Mironov A, Rappa G, Cuomo A, Faretta M, Bonaldi T, Lorico A, Pelicci G. Extracellular vesicle-mediated transfer of CLIC1 protein is a novel mechanism for the regulation of glioblastoma growth. Oncotarget. 2015; 6:31413–27. https://doi.org/10.18632/oncotarget.5105
9. Edwards JC, Kahl CR. Chloride channels of intracellular membranes. FEBS Lett. 2010; 584:2102-2111.
10. Littler DR, Harrop SJ, Goodchild SC, Phang JM, Mynott AV, Jiang L, Valenzuela SM, Mazzanti M, Brown LJ, Breit SN, Curmi PM. The enigma of the CLIC proteins: Ion channels, redox proteins, enzymes, scaffolding proteins? FEBS Lett. 2010; 584:2093-2101.
11. Argenzio E, Moolenaar WH. Emerging biological roles of Cl- intracellular channel proteins. J Cell Sci. 2016; 129:4165-4174.
12. Tung JJ, Kitajewski J. Chloride intracellular channel 1 functions in endothelial cell growth and migration. J Angiogenes Res. 2010; 2:23.
13. Wang L, He S, Tu Y, Ji P, Zong J, Zhang J, Feng F, Zhao J, Zhang Y, Gao G. Elevated expression of chloride intracellular channel 1 is correlated with poor prognosis in human gliomas. J Exp Clin Cancer Res. 2012; 31:44.
14. Knowles LM, Malik G, Hood BL, Conrads TP, Pilch J. CLT1 targets angiogenic endothelium through CLIC1 and fibronectin. Angiogenesis. 2012; 15:115-129.
15. Gurski LA, Knowles LM, Basse PH, Maranchie JK, Watkins SC, Pilch J. Relocation of CLIC1 promotes tumor cell invasion and colonization of fibrin. Mol Cancer Res. 2015; 13:273-280.
16. Chang YH, Wu CC, Chang KP, Yu JS, Chang YC, Liao PC. Cell secretome analysis using hollow fiber culture system leads to the discovery of CLIC1 protein as a novel plasma marker for nasopharyngeal carcinoma. J Proteome Res. 2009; 8:5465-5474.
17. Pisitkun T, Shen RF, Knepper MA. Identification and proteomic profiling of exosomes in human urine. Proc Natl Acad Sci U S A. 2004; 101:13368-13373.
18. Thuringer D, Chanteloup G, Boucher J, Pernet N, Boudesco C, Jego G, Chatelier A, Bois P, Gobbo J, Cronier L, Solary E, Garrido C. Modulation of the inwardly rectifying potassium channel Kir4.1 by the pro-invasive miR-5096 in glioblastoma cells. Oncotarget. 2017; 8:37681–93. https://doi.org/10.18632/oncotarget.16949.
19. Wong R, Turlova E, Feng ZP, Rutka JT, Sun HS. Activation of TRPM7 by naltriben enhances migration and invasion of glioblastoma cells. Oncotarget. 2017; 8:11239–48. https://doi.org/10.18632/oncotarget.14496.
20. Gautier M, Perriere M, Monet M, Vanlaeys A, Korichneva I, Dhennin-Duthille I, Ouadid-Ahidouch H. Recent Advances in Oncogenic Roles of the TRPM7 Chanzyme. Curr Med Chem. 2016; 23:4092-4107.
21. Li H, Lei B, Xiang W, Wang H, Feng W, Liu Y, Qi S. Differences in Protein Expression between the U251 and U87 Cell Lines. Turk Neurosurg. 2017; 27:894-903.
22. Thuringer D, Jego G, Wettstein G, Terrier O, Cronier L, Yousfi N, Hebrard S, Bouchot A, Hazoume A, Joly AL, Gleave M, Rosa-Calatrava M, Solary E, Garrido C. Extracellular HSP27 mediates angiogenesis through Toll-like receptor 3. FASEB J. 2013; 27:4169-4183.
23. Edgar JR. Q&A: What are exosomes, exactly? BMC Biol. 2016; 14:46.
24. Simard M, Nedergaard M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience. 2004; 129:877-896.
25. Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev. 2001; 81:1415-1459.
26. Cheng H, Lederer WJ. Calcium sparks. Physiol Rev. 2008; 88:1491-1545.
27. Moccia F, Berra-Romani R, Tritto S, Signorelli S, Taglietti V, Tanzi F. Epidermal growth factor induces intracellular Ca2+ oscillations in microvascular endothelial cells. J Cell Physiol. 2003; 194:139-150.
28. Saleem H, Tovey SC, Molinski TF, Taylor CW. Interactions of antagonists with subtypes of inositol 1,4,5-trisphosphate (IP3) receptor. Br J Pharmacol. 2014; 171:3298-3312.
29. Leng TD, Li MH, Shen JF, Liu ML, Li XB, Sun HW, Branigan D, Zeng Z, Si HF, Li J, Chen J, Xiong ZG. Suppression of TRPM7 inhibits proliferation, migration, and invasion of malignant human glioma cells. CNS Neurosci Ther. 2015; 21:252-261.
30. Choi KJ, Kim KS, Kim SH, Kim DK, Park HS. Caffeine and 2-Aminoethoxydiphenyl Borate (2-APB) Have Different Ability to Inhibit Intracellular Calcium Mobilization in Pancreatic Acinar Cell. Korean J Physiol Pharmacol. 2010; 14:105-111.
31. Chokshi R, Fruasaha P, Kozak JA. 2-aminoethyl diphenyl borinate (2-APB) inhibits TRPM7 channels through an intracellular acidification mechanism. Channels (Austin). 2012; 6:362-369.
32. Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J. 2012; 279:2610-2623.
33. Villa GR, Hulce JJ, Zanca C, Bi J, Ikegami S, Cahill GL, Gu Y, Lum KM, Masui K, Yang H, Rong X, Hong C, Turner KM, et al. An LXR-Cholesterol Axis Creates a Metabolic Co-Dependency for Brain Cancers. Cancer Cell. 2016; 30:683-693.
34. Adlakha YK, Saini N. MicroRNA: a connecting road between apoptosis and cholesterol metabolism. Tumour Biol. 2016; 37:8529-8554.
35. Loh WP, Yang Y, Lam KP. miR-92a enhances recombinant protein productivity in CHO cells by increasing intracellular cholesterol levels. Biotechnol J. 2017; 12:12.
36. Valenzuela SM, Alkhamici H, Brown LJ, Almond OC, Goodchild SC, Carne S, Curmi PM, Holt SA, Cornell BA. Regulation of the membrane insertion and conductance activity of the metamorphic chloride intracellular channel protein CLIC1 by cholesterol. PLoS One. 2013; 8:e56948.
37. Jiang L, Phang JM, Yu J, Harrop SJ, Sokolova AV, Duff AP, Wilk KE, Alkhamici H, Breit SN, Valenzuela SM, Brown LJ, Curmi PM. CLIC proteins, ezrin, radixin, moesin and the coupling of membranes to the actin cytoskeleton: a smoking gun? Biochim Biophys Acta. 2014; 1838:643-657.
38. Hossain KR, Holt SA, Le Brun AP, Al Khamici H, Valenzuela SM. X-ray and Neutron Reflectivity Study Shows That CLIC1 Undergoes Cholesterol-Dependent Structural Reorganization in Lipid Monolayers. Langmuir. 2017; 33:12497-12509.
39. Thuringer D, Boucher J, Jego G, Pernet N, Cronier L, Hammann A, Solary E, Garrido C. Transfer of functional microRNAs between glioblastoma and microvascular endothelial cells through gap junctions. Oncotarget. 2016; 7:73925–34. https://doi.org/10.18632/oncotarget.12136.
40. Khan IN, Ullah N, Hussein D, Saini KS. Current and emerging biomarkers in tumors of the central nervous system: Possible diagnostic, prognostic and therapeutic applications. Semin Cancer Biol. 2017 Jul 31. https://doi.org/10.1016/j.semcancer.2017.07.004. [Epub ahead of print].
41. D'Asti E, Chennakrishnaiah S, Lee TH, Rak J. Extracellular Vesicles in Brain Tumor Progression. Cell Mol Neurobiol. 2016; 36:383-407.
42. Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, Rak J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008; 10:619-624.
43. Tung JJ, Tattersall IW, Kitajewski J. Tips, stalks, tubes: notch-mediated cell fate determination and mechanisms of tubulogenesis during angiogenesis. Cold Spring Harb Perspect Med. 2012; 2:a006601.
44. Carbonell WS, DeLay M, Jahangiri A, Park CC, Aghi MK. beta1 integrin targeting potentiates antiangiogenic therapy and inhibits the growth of bevacizumab-resistant glioblastoma. Cancer Res. 2013; 73:3145-3154.
45. Baker GJ, Yadav VN, Motsch S, Koschmann C, Calinescu AA, Mineharu Y, Camelo-Piragua SI, Orringer D, Bannykh S, Nichols WS, deCarvalho AC, Mikkelsen T, Castro MG, Lowenstein PR. Mechanisms of glioma formation: iterative perivascular glioma growth and invasion leads to tumor progression, VEGF-independent vascularization, and resistance to antiangiogenic therapy. Neoplasia. 2014; 16:543-561.
46. Denzer K, Kleijmeer MJ, Heijnen HF, Stoorvogel W, Geuze HJ. Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J Cell Sci. 2000; 113:3365–74.
47. Nakase I, Kobayashi NB, Takatani-Nakase T, Yoshida T. Active macropinocytosis induction by stimulation of epidermal growth factor receptor and oncogenic Ras expression potentiates cellular uptake efficacy of exosomes. Sci Rep. 2015; 5:10300.
48. Tian T, Zhu YL, Zhou YY, Liang GF, Wang YY, Hu FH, Xiao ZD. Exosome uptake through clathrin-mediated endocytosis and macropinocytosis and mediating miR-21 delivery. J Biol Chem. 2014; 289:22258-22267.
49. Laulagnier K, Javalet C, Hemming FJ, Chivet M, Lachenal G, Blot B, Chatellard C, Sadoul R. Amyloid precursor protein products concentrate in a subset of exosomes specifically endocytosed by neurons. Cell Mol Life Sci. 2018; 75:757-773.
50. Shin W, Ge L, Arpino G, Villarreal SA, Hamid E, Liu H, Zhao WD, Wen PJ, Chiang HC, Wu LG. Visualization of Membrane Pore in Live Cells Reveals a Dynamic-Pore Theory Governing Fusion and Endocytosis. Cell. 2018; 173:934–945.e12.
51. Oh HG, Chun YS, Park CS, Kim TW, Park MK, Chung S. Regulation of basal autophagy by transient receptor potential melastatin 7 (TRPM7) channel. Biochem Biophys Res Commun. 2015; 463:7–12.
52. Inoue K, Xiong ZG. Silencing TRPM7 promotes growth/proliferation and nitric oxide production of vascular endothelial cells via the ERK pathway. Cardiovasc Res. 2009; 83:547-557.
53. Baldoli E, Maier JA. Silencing TRPM7 mimics the effects of magnesium deficiency in human microvascular endothelial cells. Angiogenesis. 2012; 15:47-57.
54. Zeng Z, Inoue K, Sun H, Leng T, Feng X, Zhu L, Xiong ZG. TRPM7 regulates vascular endothelial cell adhesion and tube formation. Am J Physiol Cell Physiol. 2015; 308:C308-318.
55. Visser D, Langeslag M, Kedziora KM, Klarenbeek J, Kamermans A, Horgen FD, Fleig A, van Leeuwen FN, Jalink K. TRPM7 triggers Ca2+ sparks and invadosome formation in neuroblastoma cells. Cell Calcium. 2013; 54:404-415.
56. Wang P, Zhang C, Yu P, Tang B, Liu T, Cui H, Xu J. Regulation of colon cancer cell migration and invasion by CLIC1-mediated RVD. Mol Cell Biochem. 2012; 365:313-321.
57. Zhang S, Wang XM, Yin ZY, Zhao WX, Zhou JY, Zhao BX, Liu PG. Chloride intracellular channel 1 is overexpression in hepatic tumor and correlates with a poor prognosis. APMIS. 2013; 121:1047-1053.
58. Gritti M, Würth R, Angelini M, Barbieri F, Peretti M, Pizzi E, Pattarozzi A, Carra E, Sirito R, Daga A, Curmi PM, Mazzanti M, Florio T. Metformin repositioning as antitumoral agent: selective antiproliferative effects in human glioblastoma stem cells, via inhibition of CLIC1-mediated ion current. Oncotarget. 2014; 5:11252–68. https://doi.org/10.18632/oncotarget.2617.
59. Chen WL, Barszczyk A, Turlova E, Deurloo M, Liu B, Yang BB, Rutka JT, Feng ZP, Sun HS. Inhibition of TRPM7 by carvacrol suppresses glioblastoma cell proliferation, migration and invasion. Oncotarget. 2015; 6:16321–40. https://doi.org/10.18632/oncotarget.3872
60. Sander P, Mostafa H, Soboh A, Schneider JM, Pala A, Baron AK, Moepps B, Wirtz CR, Georgieff M, Schneider M. Vacquinol-1 inducible cell death in glioblastoma multiforme is counter regulated by TRPM7 activity induced by exogenous ATP. Oncotarget. 2017; 8:35124–37. https://doi.org/10.18632/oncotarget.16703.
61. Leng T, Lin S, Xiong Z, Lin J. Lidocaine suppresses glioma cell proliferation by inhibiting TRPM7 channels. Int J Physiol Pathophysiol Pharmacol. 2017; 9:8-15.
62. Andres-Leon E, Cases I, Alonso S, Rojas AM. Novel miRNA-mRNA interactions conserved in essential cancer pathways. Sci Rep. 2017; 7:46101.
63. Haug BH, Hald OH, Utnes P, Roth SA, Lokke C, Flaegstad T, Einvik C. Exosome-like Extracellular Vesicles from MYCN-amplified Neuroblastoma Cells Contain Oncogenic miRNAs. Anticancer Res. 2015; 35:2521-2530.
64. Yee NS. Role of TRPM7 in Cancer: Potential as Molecular Biomarker and Therapeutic Target. Pharmaceuticals (Basel). 2017; 10:39.
65. Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, Lee JJ, Kalluri R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017; 546:498-503.
66. Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr, Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008; 10:1470-1476.
67. Wang R, Gurguis CI, Gu W, Ko EA, Lim I, Bang H, Zhou T, Ko JH. Ion channel gene expression predicts survival in glioma patients. Sci Rep. 2015; 5:11593.
68. Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, Dingli F, Loew D, Tkach M, Thery C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A. 2016; 113:E968-977.
69. Korff T, Augustin HG. Integration of endothelial cells in multicellular spheroids prevents apoptosis and induces differentiation. J Cell Biol. 1998; 143:1341-1352.
70. Guatimosim S, Guatimosim C, Song LS. Imaging calcium sparks in cardiac myocytes. Methods Mol Biol. 2011; 689:205-214.