
INTRODUCTION
APL, a blood cancer characterized by a translocation mutation between chromosome 15 and chromosome 17, strikes about 1,500 patients in the United States annually [1, 2]. This mutation leads to the formation of two fusion genes (oncogenes), promyelocytic leukemia– retinoic acid receptor alpha (PML–RARα) and RARα–PML. PML–RARα fusion transcript is involved in pathogenesis of APL whereas RARα–PML fusion transcript, an important molecular marker for the diagnosis and monitoring of APL [2, 3]. Current treatment options for APL patients include: all - trans retinoic acid (ATRA), combination of ATRA and anthracyclines, ATRA and arsenic trioxide (TX) combination, and TX alone. ATRA acts through induction of differentiation of leukemic promyelocytes into mature granulocytes in APL patients [4–7]. It also degrades promyelocytic leukemia (PML)-retinoic acid receptor-α (PML-RARA) fusion protein and eradicates leukemia–initiating cells in mouse model of APL [8]. The advent of ATRA in APL pathophysiology has made revolutionary changes in APL patient’s treatment strategies and almost ended previous life-threatening chemotherapy treatment with 6-mercatopurine (6-MP), methyl-glyoxal guanyl hydrazine, daunorubicin (DNR), and cytarabine [9]. It has been reported to induce complete remission (CR) rates of 85% from investigations conducted by the Shanghai group [7], and of 72% from studies done by the North American Intergroup [4]. However, APL patients often relapse from ATRA treatment due to excessive clearance and lower blood plasma level in course of treatment. Also, ATRA is good mainly for low risk APL patients [9, 10]. It may cause life-threatening complication of ATRA syndrome [11]. Moreover, ATRA works only PML-RARAα containing APL patients, and not in RA-resistant promyelocytic leukemia zinc finger (PLZF)-RARα patients [8]. Combination of ATRA and anthracyclines have successfully improved the relapse rate and lowered the incidence of ATRA syndrome in APL patients [9, 11]. These combinations also enhance the anti-leukemic efficacy of APL patients by raising CR rates to more than 90%, leading to a virtual absence of resistance, and a survival rate of nearly 80% [12]. They are considered the standard induction treatment for newly diagnosed APL patients [13]. Although the ombination of ATRA and anthracyclines is used prominently for low risk APL patients in induction therapy, it not able to cure relapsed or refractory patients properly, and also it is not effective in the treatment of APL patients all age groups for longer survival [9, 12, 14]. ATRA and arsenic trioxide (ATO) combination is a targeted therapy without inclusion of chemotherapy. This combination acts through induction of maturation of promyelocytes, apoptosis, and eradication of APL-initiating cells through degradation of PML-RARAα [15–18]. In consolidation therapy APL patients undergoing ATRA-ATO combination treatment show improved CR rate between around 90-95% with a decrease in relapse rate [19]. ATRA-ATO combination has also emerged as a standard care for both low and intermediate risk APL patients, with possible superior outcomes. It is also best suited for older and cardiovascular disease-complicated APL patients [20]. ATRA-ATO combination is not recommended to high risk refractory APL patients. It also does not work in RA-resistant promyelocytic leukemia zinc finger (PLZF)-RARα patients and may causes some sort of cardiovascular problems [8, 9, 12, 14]. TX is the preferred drug of choice for relapsed or refractory APL patients of all age groups. It is used in both induction and consolidation therapy and also high-risk APL patients with combination of ATRA and idarubicin, providing a maximum CR (~ 99 %) with longer survival rate [9, 12, 14, 21]. TX acts inside APL cells by various mechanisms, producing reactive oxygen species (ROS), oxidative stress, DNA damage, and p53 activation leading to cell cycle regulation/ arrest and apoptosis [9, 12, 14, 21–23]. However, few cases of TX drug resistance have been reported in some APL patients with different fusion gene/oncogene such as PLZF-RARα responsible for pathogenesis of APL [24, 25]. Hence, there exists a need to find new targets of TX action that would help in designing new drugs to cure APL patients quickly and overcome drug resistance. P53 induces cell cycle arrest and apoptosis in response of genotoxic and other stresses in several cancer cells [26, 27]. Its expression level is kept normal in cells by several ubiquitin ligases (E3), predominantly mouse double minute 2(Mdm2) and Mdm4/MDMX, through ubiquitination and proteasomal degradation. MDM2 is a negative regulator of p53 that interacts with DAXX and HAUSP to form tertiary complex (MDM2-DAXX-HAUSP) [28]. Tertiary complex reduces self-ubiquitination of MDM2, maintaining MDM2 ligase activity toward p53 inside the cell. During cellular stress, complex is disrupted, increasing MDM2 self-ubiquitination and degradation, which leads to accumulation of p53 [28–30]. Accumulating evidence suggests that TX inhibits proliferation through activation of p53, p21, cell cycle arrest, and apoptosis in fibroblast cells [23], human gastric cancer cells [31], human myeloma cells [32], HL-60 cells [21, 33], lymphoid malignant cells [34], and NB4 cells [35]. TX promotes accumulation of DAXX, which modulates transcription of death-related genes in apoptosis [36]. Berberine, doxorubicin, and VP-16 kill cancer cells through disruption of complex, self-ubiquitination and degradation of MDM2, which leads to accumulation of p53 [28, 37]. Promyelocytic leukemia (PML) gene co-localizes with p53 and is involved in pro-apoptotic events [38]. Promyelocytic leukemia zinc finger-retinoic acid receptor α (PLZF-RARα) promotes cell proliferation in APL patients by repression of p53 and p21 proteins expression [39]. PML-transformation related protein (Trp53) is required to control leukemia –initiating cells in mouse model of APL [8]. Pseudokinase Tribble 3 (TRIB3) promotes APL progression by inhibition of p53 mediated senescence. Arsenic trioxide interacts with TRIB3/ PML–RARα and eradicates APL [40]. Nutlin-3, an antagonist of MDM2 induces p53 dependent apoptosis in acute lymphoblastic leukemia cells [41, 42]. In the present research, we discovered a new target of action of TX through transmission of stress signal with protein kinases, and their downstream phosphorylation targets, disruption of MDM2-DAXX-HAUSP complex, and activation of p53 leading to cell cycle regulation and apoptosis in APL cell lines.
RESULTS
Trisenox inhibits APL cells proliferation
To investigate the effect of TX on APL cells growth, we conducted cell proliferation assay using methyl thymidine incorporation protocol. Cells were treated to various concentrations (0, 0.5, 1,2, 4, 6 and 8 μg/ml) of TX for 24 hours and then further incubated with tritium labeled methyl thymidine for 21 hours. Study results indicated that TX stimulated cell proliferation up to one μg/ml and inhibited cells growth significantly (p < 0.01) at high doses (4, 6 and 8 μg/ml) of TX in a dose - dependent manner, in all three APL cell lines (Figure 1A–1C) tested.
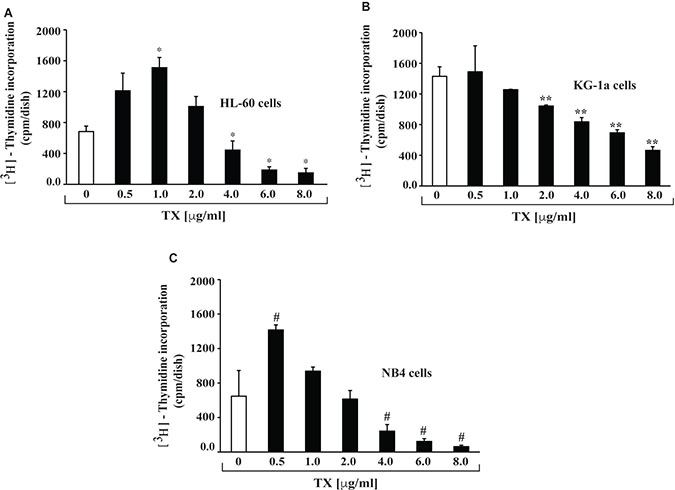
Figure 1: TX inhibits proliferation of APL cells. APL cell lines were treated with different concentrations of TX for 24 hours and 21 hours with tritium labeled thymidine. After incubation, cells were harvested, counted, and 3H-methyl thymidine incorporation expressed as cpm/dish. Highly statistically significant decreases (p < 0.01) in cell proliferation were observed in HL-60 (A), KG-1a (B) and KG-1a (C).
Trisenox activates p53 expression and cell cycle arrest
To study TX –induced p53 accumulation and cell cycle arrest of APL cells, we treated Hl-60, KG1a, NB4 and U937 with various concentrations (2, 4, 6 and 8 μg/ml) of TX for 24 hours and analyzed cell cycle regulatory proteins expression by Western blotting. Interestingly, p53 and p21 expressions were stimulated significantly in TX treated APL cells compared to untreated cells (Figure 2A, 2C–2E). Whereas cyclins (cyclin D1&3) and cyclin dependent kinases (CDK2, 4 &6) expressions were down regulated concentration - dependently in APL cells treated with TX (Figure 2A, 2C–2D). Our immunocytochemistry experiment also showed a significant reduction in ki67 expression TX treated APL cells (Figure 2B (i-v)). Overall, TX induced accumulation of p53, p21 leading to cell cycle arrest, which was also confirmed by a significant decrease in the expression of ki67 and increase in the percentage of cell cycle arrest at S & G2 phase (Figure 2F).
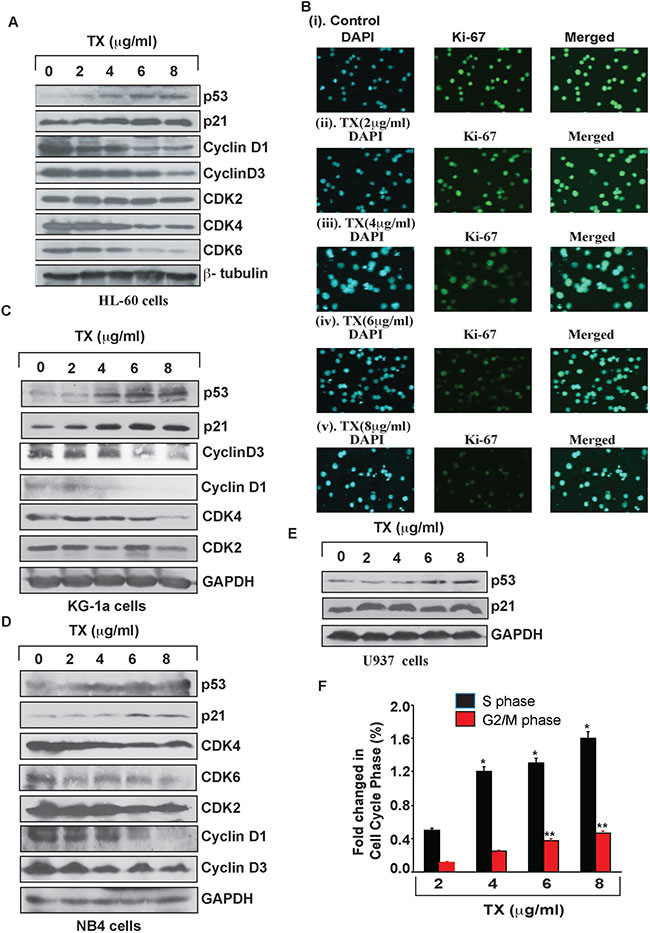
Figure 2: TX activates p53 leading to cell cycle arrest. TX activated p53 and p21 through down regulation of expression of cyclins, cdks, and ki67 in HL-60 (A), KG-1a (C), NB4 (D), and U937 (E). TX induced cell cycle arrest at S and G2/M phase prominently (F) in APL cells.
Trisenox induces intrinsic pathway of apoptosis
We investigated TX-induced p53 mediated apoptosis in APL cells by Western blot analysis of the expression levels of proapoptotic proteins and Bcl-2 by Western blotting both in all samples. Our finding showed that p53 stimulated the expression levels of pro-apoptotic proteins and also down regulated the expression levels of anti-apoptotic proteins (Figure 3A–3C). Our experimental results showed that TX –induced p53 significantly decreased mitochondrial membrane potential (Figure 3D–3F) in APL cells.
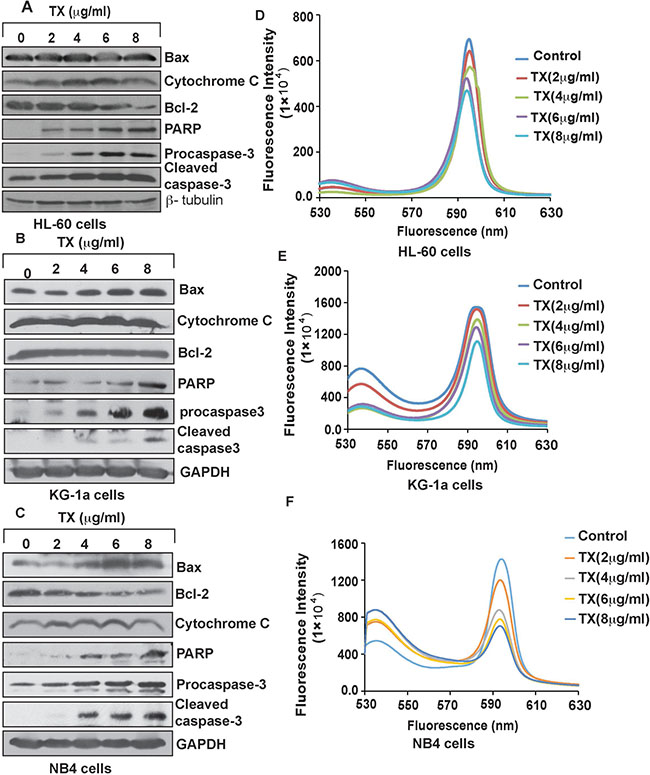
Figure 3: TX induces intrinsic pathway of apoptosis. TX upregulated the expression levels of Bax, Cytochrome C, PARP, and caspase 3 through down regulation of Bcl-2 expression in HL-60 (A), KG-1a (B), and NB4 (C). Mitochondria were isolated from samples and mitochondrial membrane potentials [MMPs] were measured by spectrofluoremetry. TX was reduced the MMPs in HL-60 (D), KG-1a (E) and NB4 (F) cells.
Trisenox disrupts MDM2- DAXX-HAUSP complex
To investigate TX effect on complex disruption with interaction of DAXX and degradation of MDM2 in APL cells, we treated HL-60 and NB4 cells with different concentrations of TX and analyzed complex disruption and interaction through immunoprecipitation (IP) and Western blotting. Our experimental results revealed that TX down regulated the expression level of complex molecules in both cells (Figure 4A, 4B). Also, IP results indicated that all complex molecules were associated with each other in APL cells (Figure 4C, 4D).
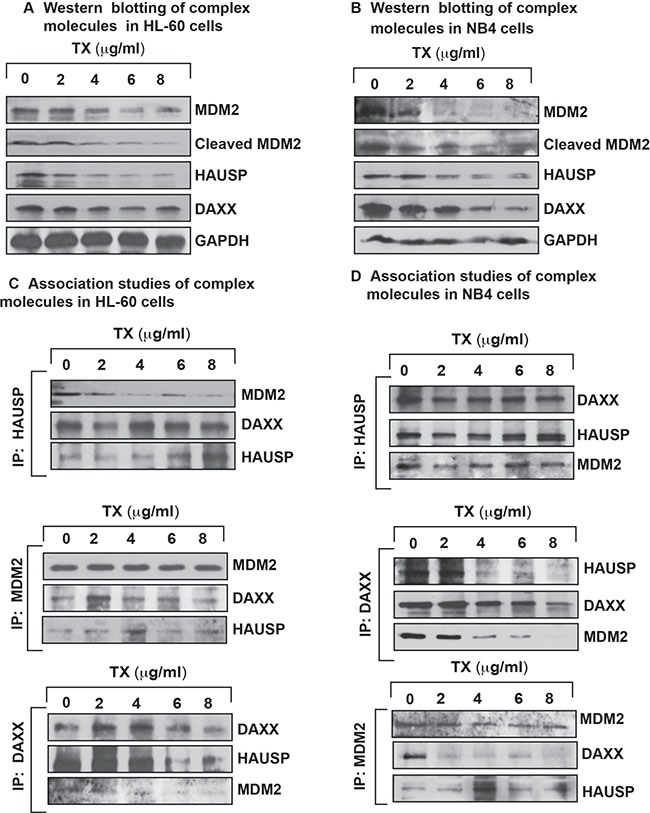
Figure 4: TX disrupts complex molecules expression and association. TX effect on both expression and association of complex molecules in APL cell lines was analyzed by Western blotting and immunoprecipitation (IP) method. TX disrupted/changed the expression level of complex molecules in a concentration dependent manner and changed association level of complex molecules in HL-60 (A and C) and NB4 (B and D) cells.
Functional studies of Trisenox in complex disruption
TX–induced stress signal involved in complex disruption is transmitted though stimulation of phosphorylation of CHK1 at Ser 345 residue, CHK2 at Thr 68 residue ATM at Ser 1981 residue and ATR at Ser428 residue in both KG1a and HL-60 cells (Figure 5A, 5B). We evaluated the functional significance of p53 in MDM2-DAXX-HAUSP complex molecules expression by Western blotting. TX reduced the expression level of complex molecules in normal NB4 cells, but in p53 knock-down cells, the expression was not significantly reduced (Figure 5D). We also assessed the role of nutlin-3 in overcoming TX effect in complex disruption. We found that nutrilin-3 neutralized TX-induced down-regulation of complex molecules in KG1a cells (Figure 5C).
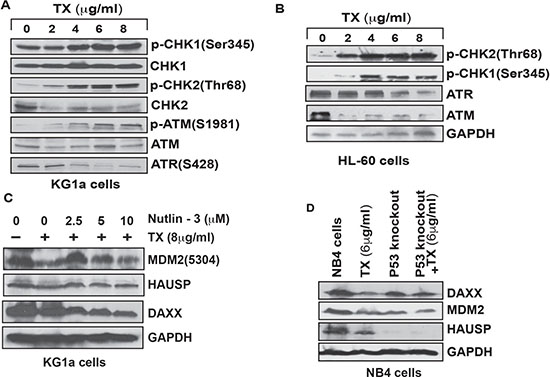
Figure 5: Functional mechanism of TX in complex disruption. TX induced the phosphorylation of CHK1, CHK2, ATM, and ATR at different residues and changed the expression levels of ATM and ATR in both KG1a cells and HL-60 cells (A, B). Nutilin-3, an antagonist of MDM2 neutralized TX repression of MDM2 without effecting DAXX and HAUSP expression (C). TX treatment did not significantly reduce the expression of complex molecules in p53-knockout NB4 cells (D).
DISCUSSION
Generally, TX enters into APL cells through diffusion and acts by various mechanisms in different pathways [21, 23, 43, 44]. Previous studies have reported that it induces differentiation at lower doses, but at higher doses, produces cell cycle arrest and apoptosis in APL cells [44, 45]. Earlier studies suggested that it interacts with p53 and co-localizes together into PML nuclear body (PML-NB) leading to apoptosis [38, 46, 47]. In most of human cancers, p53 is mutated or remains functionally inactive by MDM2 and MDMX through E3 ligase activity. Reactivation of p53 could be an attractive and effective cancer therapy [48]. TX-induced p53 mediated cell cycle arrest and apoptosis in APL cells/ tissues mostly remains unknown. We used four APL cell lines to investigate TX-induced molecular mechanisms of action involving p53 activation, MDM2-DAXX-HAUSP complex disruption, cell cycle regulation, and apoptosis. Some of our gene expression research data are not consistent across all cell lines. This may be due to the source origin, genotype, and/or culture conditions of each specific APL cell line. Our findings indicate that both HL-60 and NB4 cells underwent proliferation at lower doses of TX, but this proliferation was significantly inhibited at higher doses of TX (Figure 1A–1C). Existing evidence suggests that TX induced cell cycle arrest at G1 or G2/M phase in several cell types [22, 23, 31, 49] by down regulation of cyclins and cdks expression [32]. Our finding provides additional evidence that TX activated the expression of p53 and p21 involved in cell cycle arrest prominently at S and G2/M phase, through down regulation of expression of ki67, cyclins and cdks in APL cell lines (Figure 2A–2F). Accumulating evidence also suggests that TX-induced p53 up-regulates the intrinsic pathway of apoptosis in several cell types [21, 23, 31]. Our data show that TX induced p53 was linked to the intrinsic pathway of apoptosis (Figure 3A–3F).
Several research groups have reported that p53 is accumulated in response of cellular stress and DNA damage due to DAXX interaction, MDM2-DAXX-HAUSP complex disruption and MDM2 degradation inside the stressed cells [28, 29, 30, 37]. Our new findings provide evidence that TX disrupted complex through reduced expression of complex molecules, associations, and degraded MDM2 in both APL cells (Figure 4A–4D). Accumulating evidence also suggests that p53 expression level and activity are controlled by MDM2 E3 ubiquitin-ligase through recruitment of E2 ubiquitin-conjugating enzymes to transfer ubiquitins onto p53 [50, 51]. Our findings show that TX-induced stress signal is transmitted through modulation of significantly phosphorylation of CHK1 at Ser345 residue, CHK2 at residue Thr68, and ATM at residue Ser 1981 as well as changed expression of ATM & ATR in both KG1a and HL-60 cells (Figure 5A, 5B). This resulted in complex disruption, MDM2 degradation, and accumulation of p53 in both APL cells. (Figure 2A, 2C–2E). Our functional studies data show that TX treatment did not significantly reduce the expression of complex molecules in p53 knock-down NB4 cells (Figure 5D). We also found that nutlin-3 overcame/neutralized TX–induced down-regulation of complex molecules in kG1a cells (Figure 5C).
In summary, we present here comprehensive molecular mechanisms of TX induced p53 activation via transmission of stress signaling by protein kinases [ATM, ATR] and their downstream targets, CHK1 and CHK2 residues phosphorylation leading to disruption of complex in APL cells (Figure 6). By elucidating the p53 role in TX-induced cell cycle arrest and apoptosis in APL cells, we have provided new insights into the molecular targets.
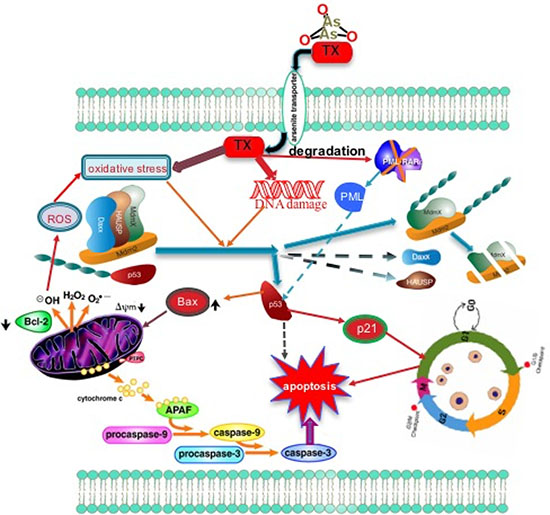
Figure 6: Summary of TX new target of action in APL cells. TX enters in APL cells, and induces oxidative stress and DNA damage, leading to a reduced expression of complex molecules, MDM2 degradation through stress signal coordinated by protein kinases (ATM & ATR) and their downstream targets CHK1 and CHK2 residues, and subsequent activation of p53. TX-activated p53 induces cell cycle arrest in APL cells and forces them to undergo apoptosis.
MATERIALS AND METHODS
Cell line and culture
HL-60, NB4, KG-1a and U937 cells, were used in this study. The cells were purchased from the American Type Culture Collection–ATCC (Manassas, VA), and maintained according to standard procedures. These four cell lines were chosen based on ATCC fact sheets, and on the fact that many previously published articles have used them as test models for APL research.
Reagents
Trisenox (arsenic trioxide) was purchased from Fisher scientific (Waltham, MA) and thymidine (methyl-3H) was obtained from MP Biomedical (Santa Ana, CA). Nutlin – 3, Mitochondrial isolation kit, caspase assay kit, and protease inhibitor were obtained from Sigma-Aldrich (St. Louis, MO). anti-DAXX, anti-HAUSP, Anti-cytochrome C, anti-Bax and anti-Bcl2 were purchased by Cell Signaling Technology (Danvers, MA). Anti-MDM2 was purchased from Santa Cruz Biotechnology Inc.(Santa Cruz, CA). Hoechst 33342, Alexa fluor 568 and Alexa fluor 568 were purchased from Life Technologies (Grand Island, NY).
Cell proliferation assay
APL cells were treated with different doses of TX in triplicate and labeled with thymidine methyl- 3H. Cells were harvested and the radioactivity measurement was performed using a liquid scintillation analyzer (Tri-carb 2700TR, Perkin Elmer) as previously described [2].
Immunoprecipitation and Western blotting
After treatment with different doses of TX, APL cells were collected and protein lysates were prepared in RIPA buffer by sonication and centrifugation. We used 500ug protein lysate of APL cells for each sample. Western blotting and immunoprecipitation (IP) were performed as previously described [28].
Measurement of mitochondrial membrane potential (Δѱm)
Mitochondria were isolated from all samples using mitochondria isolation kit (Sigma, St. Louis, MO, USA). Isolated mitochondria were incubated with 2 μl JC1 stain (from stock 1mg/ml) and 950 μl JC1 assay buffer for 10 min in dark at 25° C. The fluorescence of each sample was recorded using a Perkin Elmer LS50B spectrofluorometric (excitation 490 nm, slit, 5 nm; emission 590 nm, slit, 7.2 nm) [52].
Cell cycle analysis
APL cells (1 × 107 cells/ml) were treated with different doses (0, 2, 4, 6 and 8 μg/ml) of TX for 24 hours. After incubation, cells were harvested by centrifugation and the cell pellets were fixed in 500 μl ice cold ethanol for 15 minutes. Fixed cell pellets were mixed with 150 μl propidium iodide solution and incubated at 37° C for 40 minutes. After incubation, cell cycle analysis was done by Cellometer (Nexcelom Bioscience LLC, MA, USA) as described previously [53].
Immunocytochemistry and confocal microscopy imaging
APL cells (1 × 105) were cultured in presence or absence of TX and attached on poly-L-lysine coated slides. Immunocytochemistry of attached cells were performed using Ki67 antibody (dilution, 1:100) (cat# 33-4711) [54] or p53 antibody (cat # 9282) and PML-RARα (cat# ab43152) from Life Technology, Cell Signaling or Abcam company and imaged by confocal microscopy (Olympus Company, Center valley, PA) as described earlier [2].
Knockdown of p53 in NB4 cells
We made p53 knockdown NB4 cells using lentivirus shRNA (Dharmacon Inc; Lafayette, CO) method as described earlier [55]. In brief, we seeded 10,000 NB4 cells in 25 μl of transduction medium (RPMI 1640) without serum in each well with polybrene (8 μg/ml). Then, we added 40 MOI SMART choice lentiviral p53 shRNA particles (105 TU/ml) to each well and incubated for 20 hours at 37° C. After incubation, we added 75 ml of 20% serum containing culture medium in each well and further cultured for 2 days at 37° C. We performed microscopic examination and cell viability test and further incubated with puromycin (8 μg/ml) for a week. Puromycin-selected NB4 cells were further checked through Western blotting and fluorescence imagining. We used for our experiment more than 90% p53 knock-down NB4 cells.
Statistical analysis
Experiments were performed in triplicates. Data were presented as means ± SDs and one-way ANOVA or student paired t-test using SAS Software available in the Biostatistics Core Laboratory at Jackson State University. P-values less than 0.05 were considered statistically significant.
Abbreviations
APL: Acute promyelocytic leukemia; TX: Trisenox; P53: Tumor suppressor protein; DAXX: Death-associated protein 6; HAUSP: Herpesvirus-associated ubiquitin-specific protease; MDM2: Mouse double minute 2 homolog; JC1: 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide; DAPI- 4’,6-diamidino-2-phenylindole; PARP: poly ADP ribose polymerase; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; ATM: Ataxia-telangiectasia mutated; ATR: Ataxia telangiectasia and Rad3 related; CHK1: Checkpoint kinase 1; CHK2: Checkpoint kinase 2; PML-RARα: Promyelocytic leukemia- retinoic acid receptor alpha; ATRA: All trans retinoic acid.
Author contributions
Conception and Design: Sanjay Kumar and Paul B. Tchounwou; Research implementation: Sanjay Kumar and Andrea Brown; Analysis and interpretation of data: Sanjay Kumar; Writing, review and /or revision of the manuscript: Sanjay Kumar and Paul B. Tchounwou.
ACKNOWLEDGMENTS
We thank Dr. Ibrahim Farah for the support and advice for maintaining and breeding APL mice.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest.
FUNDING
This research was supported by National Institutes of Health NIMHD [grant# G12MD007581].
REFERENCES
1. Powell BL. Arsenic trioxide in acute promyelocytic leukemia: potion not poison. Expert Rev Anticancer Ther. 2011; 11:1317–19.
2. Kumar S, Tchounwou PB. Molecular mechanisms of cisplatin cytotoxicity in acute promyelocytic leukemia cells. Oncotarget. 2015; 6:40734–46. https://doi.org/10.18632/oncotarget.5754.
3. Grignani F, Fagioli M, Alcalay M, Longo L, Pandolfi PP, Donti E, Biondi A, Lo Coco F, Grignani F, Pelicci PG. Acute promyelocytic leukemia: from genetics to treatment. Blood. 1994; 83:10–25.
4. Tallman MS, Andersen JW, Schiffer CA, Appelbaum FR, Feusner JH, Ogden A, Shepherd L, Willman C, Bloomfield CD, Rowe JM, Wiernik PH. All-trans-retinoic acid in acute promyelocytic leukemia. N Engl J Med. 1997; 337:1021–28.
5. Chen ZX, Xue YQ, Zhang R, Tao RF, Xia XM, Li C, Wang W, Zu WY, Yao XZ, Ling BJ. A clinical and experimental study on all-trans retinoic acid-treated acute promyelocytic leukemia patients. Blood. 1991; 78:1413–19.
6. Castaigne S, Chomienne C, Daniel MT, Ballerini P, Berger R, Fenaux P, Degos L. All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I. Clinical results. Blood. 1990; 76:1704–09.
7. Huang ME, Ye YC, Chen SR, Chai JR, Lu JX, Zhoa L, Gu LJ, Wang ZY. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 1988; 72:567–72.
8. Ablain J, Rice K, Soilihi H, de Reynies A, Minucci S, de Thé H. Activation of a promyelocytic leukemia-tumor protein 53 axis underlies acute promyelocytic leukemia cure. Nat Med. 2014; 20:167–74.
9. Coombs CC, Tavakkoli M, Tallman MS. Acute promyelocytic leukemia: where did we start, where are we now, and the future. Blood Cancer J. 2015; 5:e304.
10. Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008; 111:2505–15.
11. de Botton S, Chevret S, Coiteux V, Dombret H, Sanz M, San Miguel J, Caillot D, Vekhoff A, Gardembas M, Stamatoulas A, Conde E, Guerci A, Gardin C, et al. Early onset of chemotherapy can reduce the incidence of ATRA syndrome in newly diagnosed acute promyelocytic leukemia (APL) with low white blood cell counts: results from APL 93 trial. Leukemia. 2003; 17:339–42.
12. Sanz MA, Lo-Coco F. Modern approaches to treating acute promyelocytic leukemia. J Clin Oncol. 2011; 29:495–03.
13. Sanz MA, Grimwade D, Tallman MS, Lowenberg B, Fenaux P, Estey EH, Naoe T, Lengfelder E, Büchner T, Döhner H, Burnett AK, Lo-Coco F. Management of acute promyelocytic leukemia: Recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2009; 113:1875–91.
14. Zhou GB, Zhang J, Wang ZY, Chen SJ, Chen Z. Treatment of acute promyelocytic leukemia with all-trans retinoic acid and arsenic trioxide: a paradigm of synergistic molecular targeting therapy. Philos Trans R Soc Lond B Biol Sci. 2007; 362:959–71.
15. Zheng PZ, Wang KK, Zhang QY, Huang QH, Du YZ, Zhang QH, Xiao DK, Shen SH, Imbeaud S, Eveno E, Zhao CJ, Chen YL, Fan HY, et al. Systems analysis of transcriptome and proteome in retinoic acid/arsenic trioxide-induced cell differentiation/apoptosis of promyelocytic leukemia. Proc Natl Acad Sci U S A. 2005; 102:7653–58.
16. Gianni M, Koken MH, Chelbi-Alix MK, Benoit G, Lanotte M, Chen Z, de Thé H. Combined arsenic and retinoic acid treatment enhances differentiation and apoptosis in arsenic-resistant NB4 cells. Blood. 1998; 91:4300–10.
17. Dos Santos GA, Kats L, Pandolfi PP. Synergy against PML-RARa: targeting transcription, proteolysis, differentiation, and self-renewal in acute promyelocytic leukemia. J Exp Med. 2013; 210:2793–02.
18. Nasr R, Guillemin MC, Ferhi O, Soilihi H, Peres L, Berthier C, Rousselot P, Robledo-Sarmiento M, Lallemand-Breitenbach V, Gourmel B, Vitoux D, Pandolfi PP, Rochette-Egly C, et al. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nat Med. 2008; 14:1333–42.
19. Shen ZX, Shi ZZ, Fang J, Gu BW, Li JM, Zhu YM, Shi JY, Zheng PZ, Yan H, Liu YF, Chen Y, Shen Y, Wu W, et al. All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 2004; 101:5328–35.
20. Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S, Ferrara F, Fazi P, Cicconi L, Di Bona E, Specchia G, Sica S, Divona M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013; 369:111–21.
21. Kumar S, Yedjou CG, Tchounwou PB. Arsenic trioxide induces oxidative stress, DNA damage, and mitochondrial pathway of apoptosis in human leukemia (HL-60) cells. J Exp Clin Cancer Res. 2014; 33:42.
22. Yedjou CG, Tchounwou PB. Modulation of p53, c-fos, RARE, cyclin A, and cyclin D1 expression in human leukemia (HL-60) cells exposed to arsenic trioxide. Mol Cell Biochem. 2009; 331:207–14.
23. Miller WH Jr, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002; 62:3893–03.
24. Tomita A, Kiyoi H, Naoe T. Mechanisms of action and resistance to all-trans retinoic acid (ATRA) and arsenic trioxide (As2O 3) in acute promyelocytic leukemia. Int J Hematol. 2013; 97:717–25.
25. Lou Y, Ma Y, Sun J, Ye X, Pan H, Wang Y, Qian W, Meng H, Mai W, He J, Tong H, Jin J. Evaluating frequency of PML-RARA mutations and conferring resistance to arsenic trioxide-based therapy in relapsed acute promyelocytic leukemia patients. Ann Hematol. 2015; 94:1829–37.
26. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000; 408:307–10.
27. Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002; 2:594–04.
28. Zhang X, Gu L, Li J, Shah N, He J, Yang L, Hu Q, Zhou M. Degradation of MDM2 by the interaction between berberine and DAXX leads to potent apoptosis in MDM2-overexpressing cancer cells. Cancer Res. 2010; 70:9895–04.
29. Meek DW. Tumour suppression by p53: a role for the DNA damage response? Nat Rev Cancer. 2009; 9:714–23.
30. Tang J, Qu LK, Zhang J, Wang W, Michaelson JS, Degenhardt YY, El-Deiry WS, Yang X. Critical role for Daxx in regulating Mdm2. Nat Cell Biol. 2006; 8:855–62.
31. Jiang XH, Wong BC, Yuen ST, Jiang SH, Cho CH, Lai KC, Lin MC, Kung HF, Lam SK. Arsenic trioxide induces apoptosis in human gastric cancer cells through up-regulation of p53 and activation of caspase-3. Int J Cancer. 2001; 91:173–79.
32. Park WH, Seol JG, Kim ES, Hyun JM, Jung CW, Lee CC, Kim BK, Lee YY. Arsenic trioxide-mediated growth inhibition in MC/CAR myeloma cells via cell cycle arrest in association with induction of cyclin-dependent kinase inhibitor, p21, and apoptosis. Cancer Res. 2000; 60:3065–71.
33. Yedjou C, Tchounwou P, Jenkins J, McMurray R. Basic mechanisms of arsenic trioxide (ATO)-induced apoptosis in human leukemia (HL-60) cells. J Hematol Oncol. 2010; 3:28–35.
34. Zhang W, Ohnishi K, Shigeno K, Fujisawa S, Naito K, Nakamura S, Takeshita K, Takeshita A, Ohno R. The induction of apoptosis and cell cycle arrest by arsenic trioxide in lymphoid neoplasms. Leukemia. 1998; 12:1383–91.
35. Ma DC, Sun YH, Chang KZ, Ma XF, Huang SL, Bai YH, Kang J, Liu YG, Chu JJ. Selective induction of apoptosis of NB4 cells from G2 + M phase by sodium arsenite at lower doses. Eur J Haematol. 1998; 61:27–35.
36. Torii S, Egan DA, Evans RA, Reed JC. Human Daxx regulates Fas-induced apoptosis from nuclear PML oncogenic domains (PODs). EMBO J. 1999; 18:6037–49.
37. Song MS, Song SJ, Kim SY, Oh HJ, Lim DS. The tumour suppressor RASSF1A promotes MDM-2 self-ubiquitination by disrupting the MDM2-DAXX-HAUSP complex. EMBO J. 2008; 27:1863–74.
38. Guo A, Salomoni P, Luo J, Shih A, Zhong S, Gu W, Pandolfi PP. The function of PML in p53-dependent apoptosis. Nat Cell Biol. 2000; 2:730–36.
39. Choi WI, Yoon JH, Kim MY, Koh DI, Licht JD, Kim K, Hur MW. Promyelocytic leukemia zinc finger-retinoic acid receptor α (PLZF-RARα), an oncogenic transcriptional repressor of cyclin-dependent kinase inhibitor 1A (p21WAF/CDKN1A) and tumor protein p53 (TP53) genes. J Biol Chem. 2014; 289:18641–56.
40. Li K, Wang F, Cao WB, Lv XX, Hua F, Cui B, Yu JJ, Zhang XW, Shang S, Liu SS, Yu JM, Han MZ, Huang B, et al. TRIB3 Promotes APL Progression through Stabilization of the Oncoprotein PML-RARα and Inhibition of p53-Mediated Senescence. Cancer Cell. 2017; 31:697–10.
41. Gu L, Zhu N, Findley HW, Zhou M. MDM2 antagonist nutlin-3 is a potent inducer of apoptosis in pediatric acute lymphoblastic leukemia cells with wild-type p53 and overexpression of MDM2. Leukemia. 2008; 22:730–39.
42. Trino S, Iacobucci I, Erriquez D, Laurenzana I, De Luca L, Ferrari A, Ghelli Luserna Di Rorà A, Papayannidis C, Derenzini E, Simonetti G, Lonetti A, Venturi C, Cattina F, et al. Targeting the p53-MDM2 interaction by the small-molecule MDM2 antagonist Nutlin-3a: a new challenged target therapy in adult Philadelphia positive acute lymphoblastic leukemia patients. Oncotarget. 2016; 7:12951–61. https://doi.org/10.18632/oncotarget.7339.
43. Lengfelder E, Hofmann WK, Nowak D. Impact of arsenic trioxide in the treatment of acute promyelocytic leukemia. Leukemia. 2012; 26:433–42.
44. Emadi A, Gore SD. Arsenic trioxide—An old drug rediscovered. Blood Rev. 2010; 24:191–99.
45. Chen GQ, Shi XG, Tang W, Xiong SM, Zhu J, Cai X, Han ZG, Ni JH, Shi GY, Jia PM, Liu MM, He KL, Niu C, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): I. As2O3 exerts dose-dependent dual effects on APL cells. Blood. 1997; 89:3345–53.
46. Rego EM, He LZ, Warrel RP Jr, Wang ZG, Pandolfi PP. Retinoic acid (RA) and As2O3 treatment in transgenic models of acute promyelocytic leukemia (APL) unravel the distinct nature of the leukemogenic process induced by the PML-RARalpha and PLZF-RARalpha oncoproteins. Proc Natl Acad Sci U S A. 2000; 97:10173–78.
47. Müller S, Miller WH Jr, Dejean A. Trivalent antimonials induce degradation of the PML-RAR oncoprotein and reorganization of the promyelocytic leukemia nuclear bodies in acute promyelocytic leukemia NB4 cells. Blood. 1998; 92:4308–16.
48. Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013; 13:83–96.
49. Liu Q, Hilsenbeck S, Gazitt Y. Arsenic trioxide-induced apoptosis in myeloma cells: p53-dependent G1 or G2/M cell cycle arrest, activation of caspase-8 or caspase-9, and synergy with APO2/TRAIL. Blood. 2003; 101:4078–87.
50. Wade M, Wang YV, Wahl GM. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 2010; 20:299–09.
51. Li YC, Wahl GM. What a difference a phosphate makes: life or death decided by a single amino acid in MDM2. Cancer Cell. 2012; 21:595–96.
52. Cossarizza A, Baccarani-Contri M, Kalashnikova G, Franceschi C. A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the Jaggregate forming lipophilic cation 5,5_,6,6_-tetrachloro-1,1_,3,3_-tetraethylbenzimidazolcarbocyanine iodide (JC-1). Biochem Biophys Res Commun. 1993; 197:40–45.
53. Chan LL, Zhong X, Qiu J, Li PY, Lin B. Cellometer vision as an alternative to flow cytometry for cell cycle analysis, mitochondrial potential, and immunophenotyping. Cytometry A. 2011; 79:507–17.
54. Sun X, Bizhanova A, Matheson TD, Yu J, Zhu LJ, Kaufman PD. Ki-67 Contributes to Normal Cell Cycle Progression and Inactive X Heterochromatin in p21 Checkpoint-Proficient Human Cells. Mol Cell Biol. 2017; 37. https://doi.org/10.1128/MCB.00569-16.
55. Sims JN, Graham B, Pacurari M, Leggett SS, Tchounwou PB, Ndebele K. Di-ethylhexylphthalate (DEHP) modulates cell invasion, migration and anchorage independent growth through targeting S100P in LN-229 glioblastoma cells. Int J Environ Res Public Health. 2014; 11:5006–19.