
INTRODUCTION
Colorectal cancer is one of the most common neoplastic diseases of the western world [1]. Carcinogenesis of the colon and other tissue is a multi-levelled process characterised by the accumulation of numerous mutations and epigenetic aberrations [2, 3]. In addition to characteristic mutations in tumour suppressors such as APC (adenomatous polyposis coli) and genes of the Wnt (wingless/lnt1) signalling pathway [4], epigenetic aberrations are known to have an important impact on the neoplastic progression of the colonic epithelia [5–11]. Aberrant, loci-specific DNA hypermethylation – one of the best-studied epigenetic hallmarks of cancer – can lead to silencing of tumour suppressors that control, e.g. cell cycle and DNA repair processes [11, 12].
Initially, DNA methyltransferase (DNMT)-mediated DNA methylation was considered a non-reversible modification in mammals. It has since become clear, however, that the group of TET (ten-eleven translocation) dioxygenases are capable of oxidizing methylated 2’-deoxycytidines (5-methyl-2’-deoxycytidine; 5-mdC) to hydroxymethylated 2’-deoxycytidines (5-hydroxymethyl-2’-deoxycytidine, 5-hmdC) [13], a known mechanism of active DNA demethylation. TETs can further oxidize 5-hmdC to 5-formyl-2’-deoxycytidine (5-fdC) and 5-carboxy-2’-deoxycytidine (5-cadC), which are eventually replaced by unmodified 2’-deoxycytidines as a result of thymine-DNA glycosylase (TDG) mediated base excision repair [14–17]. This cascade of 5-mdC oxidation by TETs might represent the active demethylation mechanism leading to a genes transcriptional reactivation.
In addition to its role as a demethylation intermediate, 5-hmdC has unique epigenetic properties that result in specific gene expression profiles [18, 19]. Indeed, a genome-wide decrease in 5-hmdC- levels is considered an epigenetic hallmark in many cancers [20, 21] and several studies have highlighted the diagnostic and prognostic value of this mechanism [22]. From a therapeutic standpoint, there are a limited number of known TET enzyme activators capable of inducing significant increases to genome-wide 5-hmdC levels. Vitamin C (ascorbate) is one of the best-known substrates for TET enzymes. Unlike other mammals, humans are incapable of synthesising ascorbate de novo, making it an essential dietary requirement. It enters the cell by the sodium-dependent vitamin C transporter SVCT1, as expressed in intestinal and renal epithelial cells, or the ubiquitously expressed SVCT2. In human blood plasma, ~50 μM ascorbate is considered optimal, < 11.4 μM is defined as a deficiency. Depending on the tissue, 1-10 mM ascorbate is found within cells [23]. It serves as an antioxidant, a radical scavenger, and an essential regenerative cofactor for Fe2+ and 2-oxoglutarate (2OG)-dependent dioxygenases (e.g. prolyl-4-hydroxylase [P4H], TET). In the latter, ascorbate reduces the inactive oxidised form of Fe3+ to the catalytically active Fe2+ in the active centre [24, 25]. Though the mode of ascorbate-induced TET dioxygenases activation is still controversial [26], it is clearly capable of re-establishing normal 5-hmdC-levels in cancer cells [21].
Many epigenetically potent compounds have been identified as potential cancer therapeutics that could play an important role in the management of neoplasias. Currently, two DNMT inhibitors – azanucleosides – have been approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the treatment of certain forms of acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS): 5-aza-cytidine (AZA; Vidaza®, Celgene, New Jersey, USA) [27–29] and 5-aza-2’-deoxycytidine (DAC; Dacogen®, Astex, Cambridge, UK) [30, 31]. As recent clinical trials suggest that azacytidine has wide therapeutic application in AML, it has been approved by the FDA for AML with 20 – 30% blasts in the bone marrow only, the EMA approved it for AML with 30% bone marrow blasts or higher [32]. Though both cytosine analogues were originally developed as cytostatic agents for leukaemia chemotherapy, their epigenetic properties have since been revealed to play an important role in their anti-cancer activity [31, 33–38]. Both act as DNMT inhibitors, DAC in particular, an action used for the treatment of myeloid malignancies such as AML. Indeed, both drugs show high efficiency against haematological cancers (MDS, AML) [39, 40]. Though AZA and DAC were once viewed as mechanistically similar working compounds, major differences in the mode of action by which they demethylate the cellular epigenome have since been revealed. During mitosis, DAC is exclusively incorporated into the newly synthesised DNA strand instead of 2’-deoxycytidine after cell division. Upon binding DAC, DNMT1 is inhibited and subsequently degraded by proteolysis. In contrast, 80-90% of AZA is incorporated into RNA and leading to mRNA and protein metabolism disruption and inhibition of malignant proliferation [38]. In addition, 10-15% of AZA is metabolised into DAC and can be introduced into DNA, as well [41, 42].
Although hypomethylating actions can be observed at concentrations as low as 30 nM DAC and 300 nM AZA, in vitro higher concentrations (3-10 μM DAC or AZA) prove to be more cytotoxic with no improvement to hypomethylation. In this regard, it is notable that such levels (3-11 μM AZA; 0.3–1.6 μM DAC) can be found in human plasma following normal administrations to cancer patients [43–45]. Higher concentrations of DAC or AZA (3-10 μM) do not lead to improved hypomethylating actions but more to cytotoxic effects which can also be found in human plasma (3-11 μM AZA) and (0.3–1.6 μM DAC) [43].
Several studies have shown that epigenetic therapies can induce re-expression of aberrantly silenced genes. An important example is the silencing of the potent tumour suppressors p16 (CDKN2A), p21 (CDKN1A) and p27 (CDKN1B) [46]. Such loss of functions allow cells to bypass the signals for cell cycle control, apoptosis induction and senescence. Accordingly, the epigenetic therapy and reactivation by the action of demethylating substances, like DAC and AZA, led to a better prognosis of cancer patients [47–49].
Although DAC and AZA appear to be beneficial in combination with chemotherapy against solid tumours [50, 51], treatment with the demethylating agents alone is less effective. As already mentioned, DAC can only be used for epigenetic modulation at concentrations less than 3-11 μM, cytotoxicity becoming a prominent issue at higher doses [52, 53]. A multitude of new compounds for the epigenetic treatment of solid tumours are presently in clinical trials [38]. Innovative combinations of epigenetically active substances have been shown to improve therapeutic actions against solid tumours. However, the combination of DNMT inhibitors and compounds capable of reactivating DNA hydroxymethylation, such as vitamin C (ascorbate) [25, 54], is yet to be investigated as a novel therapeutic avenue in colorectal neoplasia.
This newly discovered function of vitamin C prompted us to determine whether additive or synergistic effects can be achieved when sub-cytotoxic concentrations of the DNMT inhibitors DAC and AZA are used in combination with ascorbate. Investigations were conducted in HCT116 cells, a commonly used colorectal cancer cell line, to assess whether such an epigenetic therapy has therapeutic application in colorectal cancers. Herein, we demonstrate the positive combinatorial effect ascorbate has upon the demethylating agents AZA and DAC, resulting in higher 5-hmdC-levels and increased expression of the tumour suppressor levels of CDKN1A.
RESULTS
Cell viability following exposure to DAC, AZA and vitamin C
First, MTT (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) reduction assay was conducted to detect potential detrimental effects to the metabolic activity of HCT116 cells following 72 h treatment periods. Vitamin C was well tolerated up to 1 mM, though notable cytotoxicity was evident past this (Figure 1A). Well in line with previous studies [55], decreased cell viability was observed after exposure to 1 μM DAC or greater (Figure 1B), as illustrated by the white boxes with increasing DAC concentrations. However, after combined incubation of DAC and 50 μM vitamin C no additional cytotoxic effects were observed, as seen in Figure 1B. Exposure to AZA led to even greater reductions in cell viability at concentrations of 1 μM and above (Figure 1C). No additional toxic effects on cell viability were observed after exposure to indicated AZA concentrations in combination with 50 μM vitamin C (Figure 1C).
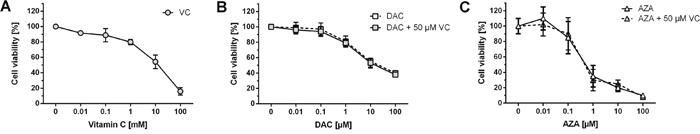
Figure 1: Effect of DAC, AZA and vitamin C on cell viability. HCT116 cells were exposed to the indicated concentrations of vitamin C (A), DAC (B) or AZA (C) alone or in combination with 50 μM vitamin C (grey boxes, DAC + VC – B; AZA + VC – C) for 72 h, and cell viabilities assessed by MTT assay. Data are expressed as a percentage of the untreated control, viable cell levels < 75% were taken to indicate cytotoxic induction (error bars = SD; n=3).
Impact of AZA, DAC and vitamin C on global DNA methylation and ALU expression
Next, we aimed to determine the genome-wide 5-mdC levels of colon cancer cells treated with vitamin C and DAC or AZA over 72 h, by isotope-dilution LC-MS/MS (Figure 2). Treatment with increasing concentrations of vitamin C alone did not lead to significant changes in the 5-mdC/dC-levels in the HCT116 cells, as illustrated by the grey bars (Figure 2A). In contrast, exposure to DAC significantly decreased the 5-mdC levels in a concentration-dependent manner; 5.5% to 2% [5-mdC/dC] from untreated to 1 μM DAC treated HCT116 cells, as shown by Figure 2B. Concentrations of DAC in 0.1 μM (p < 0.05) and 1 μM (p < 0.01) led to a significant decrease of 5-mdC/dC levels compared to the untreated control. Higher DAC doses did not further decrease 5-mdC/dC levels (data not shown). The combined treatment of HCT116 with 10 μM and 50 μM vitamin C did not alter the demethylating effect of DAC (Figure 2B). Incubation with increasing concentrations of AZA produced similar though weaker reductions; 5.5% to 3% [5-mdC/dC] from untreated to 1 μM AZA-treated cells (Figure 2C, black bars). Notably, only cytotoxic concentrations of AZA were capable of effecting large reductions to 5-mdC/dC levels in HCT116 cells not significantly. However, 0.1 μM AZA, as a non-toxic concentration, reduced the 5-mdC/dC levels in HCT116 cells not significantly. Treatment with 10 μM (bright grey bars) or 50 μM (dark grey bars) vitamin C and AZA (Figure 2C) had no apparent effect on genome-wide 5-mdC-levels. Accordingly, although treatment with DAC or AZA induced the expected dose dependent-decreases in 5-mdC levels, co-treatment with vitamin had no further influence on this effect (Figure 2B and 2C).
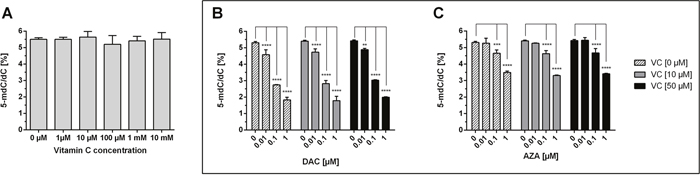
Figure 2: Impact of DAC, AZA and vitamin C on genomic 5-mdC/dC levels in human colon cancer cells. HCT116 cells were exposed to increasing concentrations of vitamin C (A) for 72h. In addition, DAC (B) and AZA (C), alone (striped bars) or in combination with 10 μM vitamin C (grey bars) and 50 μM vitamin C (black bars), were exposed to the HCT116 colon cancer cells for 72 h. The genomic levels of 5-mdC/dC were then assessed by LC-MS/MS. Statistical significance was calculated using 2-way ANOVA and Tukey post-test (****p < 0.001 – significant results; error bars = SD; n=3).
Genome-wide demethylation induced by DAC or AZA should lead to the re-expression of silenced transposable elements like ALU (athrobacter luteus). ALU elements can serve as effective markers for methylation and demethylation owing to their high abundance of CpG-sites and the high methylation levels found in their promoter region; these contribute up to 30% of genome-wide methylated DNA [56]. Concordant to the genome-wide loss of DNA methylation induced, we found DAC induced significant induction of ALU in a concentration-dependent manner (two-way ANOVA, p < 0.0001) that the addition of vitamin C did not alter (Figure 3A). Similar, though weaker dose-dependent reactivation of ALU expression was noted following AZA treatment (two-way ANOVA, p < 0.0001) (Figure 3B). Again, the addition of vitamin C at concentrations as high as 50 μM had no significant impact.
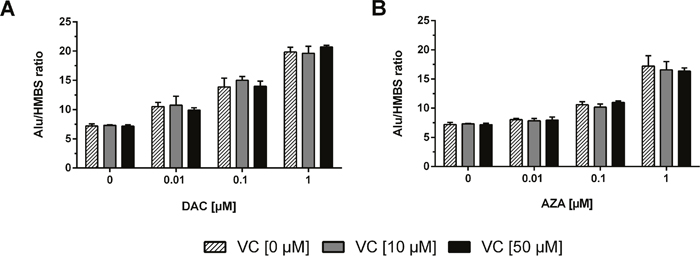
Figure 3: Reactivation of ALU transposable elements as control of epigenetic treatment and DNMT inhibition. The mRNA levels of the ALU1 transposon were measured after 72 h incubation with DAC (A) or AZA (B) alone or in combination with 10 μM (grey bars) or 50 μM (black bars) vitamin C. The relative quantitation was calculated by 2-ΔΔCT method. HMBS served as control. (Columns: mean. Error bars: SD, n=3).
Impact of AZA, DAC and vitamin C on global DNA hydroxymethylation
As vitamin C is a potent activator of the TET enzymes and leads to an increase of genome-wide 5-hmdC/dC levels, we aimed to analyse the genome-wide DNA hydroxymethylation in vitamin C and DAC or AZA-treated HCT116 cells (Figure 4). Well in line with previous work, vitamin C treated HCT116 cells showed increased 5-hmdC levels in a dose-dependent manner (Figure 4A) [50]. At 100 μM, vitamin C was able to increase 5-hmdC/dC levels 2.55 fold (p < 0.001), although no further increases were seen above this. Interestingly, although DAC caused only negligible 5-hmdC/dC increases when applied alone, in combination with vitamin C its efficacy was greatly enhanced (Figure 4B). For instance, where 0.1 μM DAC alone increases 5-hmdC/dC levels 1.05 fold relative to untreated cells (two way ANOVA with Tukey post analysis for multiple comparisons, hereafter referred to as ‘Tukey’: p > 0.05), a 1.38 fold and a 1.57 fold increase relative to matched DAC only treated cells was seen in the presence of 10 and 50 μM vitamin C respectively (Tukey: p < 0.0001 in both instances). Although this effect saturated between 0.1 and 1 μM DAC within a given vitamin C concentration, it is possible that higher vitamin C concentrations could achieve even higher 5-hmdC/dC levels. Notably, greater 5-hmdC/dC levels were achievable with DAC/vitamin C combinatorial treatments than with even the most effective vitamin C concentration alone. A similar scenario is also seen when AZA is applied in combination with vitamin C. Although 1 μM AZA alone was capable of inducing a 2.69 fold increase in 5-hmdC/dC levels (p < 0.0001), when combined with 50 μM vitamin C, 0.1 μM AZA effected a 1.42 fold increase (p < 0.01), the only non-toxic AZA concentration to induce a significant change to 5-hmdC/dC levels (Figure 4). It is again notable that combinatorial AZA/vitamin C treatments were capable of inducing notably higher genome-wide 5-hdmC/dC levels than either alone.
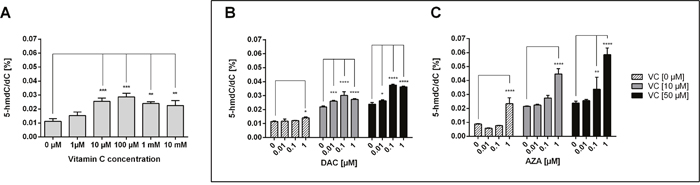
Figure 4: Impact of DAC, AZA and vitamin C on genomic 5-hmdC/dC levels in human colon cancer cells. HCT116 cells were exposed to increasing concentrations of vitamin C (A) for 72 h. In addition, DAC (B) and AZA (C), alone or in combination with vitamin C (10 μM and 50 μM), were exposed to the colon cancer cells for 72 h. The genomic levels of 5-hmdC/dC were then assessed by LC-MS/MS. Statistical significance was calculated using 2-way ANOVA and Tukey post-test (*p < 0.05, **p < 0.01, *** < 0.001, **** < 0.0001– significant results for treatment groups. Error bars = SD; n=3).
Expression of TETs and DNMTs in colon cancer cells
To further elucidate the mechanism by which vitamin C and DAC or AZA change 5-hmdC levels, we measured the gene expressions of DNMT1, 3A and 3B (Figure 5) in treated HCT116 cells. These genes are responsible for the catalysis of cytosine methylation in CG dinucleotides.
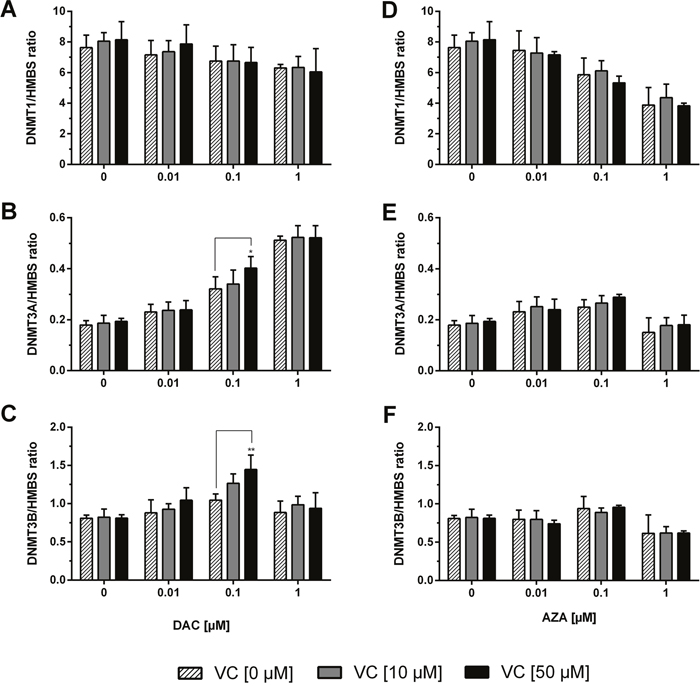
Figure 5: Expression profile of DNMTs in human colon cancer cells. The mRNA levels of the DNMTs were determined after 72 h treatment with increasing concentrations of DAC (A, B, C), AZA (D, E, F) in combination with or without vitamin C (0, 10 and 50 μM). The relative quantification was calculated by 2-ΔΔCT method. HMBS served as control. Statistical significance was calculated using 2-way ANOVA and Tukey post-test: *p < 0.05, **p < 0.01. Error bars = SD; n=3.
While application of DAC alone did result in decreased expression of DNMT1, and a significant DAC concentration-dependent effect was measured (p < 0.01), addition of vitamin C did not further alter DNMT1 expression (Figure 5A). DAC application also significantly affected DNMT3A expression (p < 0.0001) causing increases of up to 2.9 fold. Notably, the effect at 0.1 μM DAC was measurably enhanced upon co-treatment with vitamin C at 10 or 50 μM (Tukey: p < 0.05) (Figure 5B). Similarly, although treatment with vitamin C or DAC alone caused no significant change to DNMT3B expression (Figure 5C), increases of up to 1.8 fold were seen when applied together, and which positively correlated to the concentration of both compounds below 1 μM DAC (p < 0.0001).
Although stimulation of HCT116 cells with increasing concentrations of AZA significantly modulated the expressions of DNMT1, DNMT3A and DNMT3B in concentration-dependent manners (p < 0.0001 in all three instances), no significant effect was measured upon co-treatment with either 10 or 50 μM vitamin C (Figure 5D-5F). A marked decrease in the expression of the three genes can be seen at 1 μM AZA; the large decrease in cell viability seen at this same concentration is notable in this regard.
The expression of TET1, 2, and 3 – the dioxygenases responsible for the oxidation of 5-mdCs to 5-hmdC and further oxidised forms – were also assessed (Figure 6). TET1 expression was unaffected by either DAC or vitamin C alone or in combination (Figure 6A). Although DAC and vitamin C had no significant impact on TET2 applied alone, measurable increases were seen in combination, reaching as high as a 1.5 fold increase on the untreated cells, and a significant difference detected with 0.1 μM DAC and 50 μM vitamin C relative to matched DAC only treated cells (Tukey: p < 0.05) (Figure 6B). TET3 expression was increased in a DAC dependent manner (p < 0.01) that was enhanced by the co-administration of vitamin C, most pronouncedly at 0.1 μM DAC, where a 1.7 fold increase was seen relative to untreated cells (Figure 6C). Interestingly, at 1 μM DAC the addition of vitamin C induced significant decreases to TET3 expression (Tukey: p < 0.05 with 50 μM vitamin C).
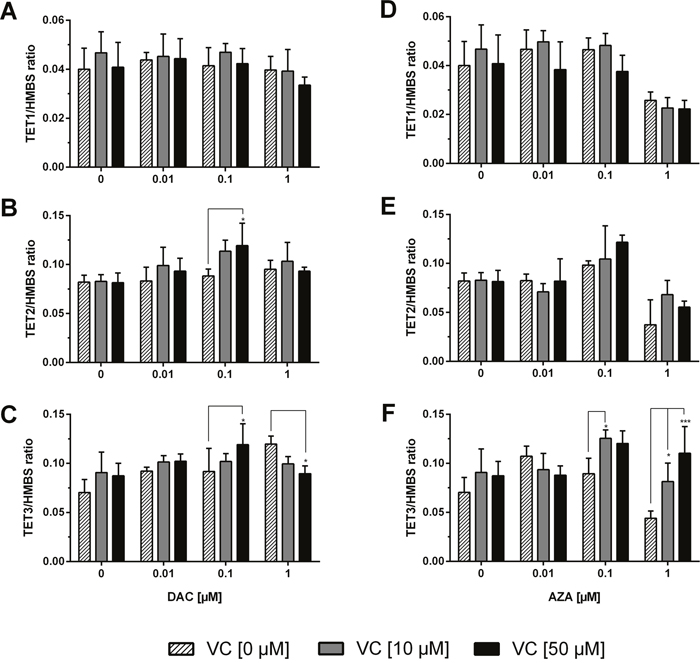
Figure 6: Expression profile of TET dioxygenases in human colon cancer cells. The mRNA levels of TETs were determined after 72 h treatment with increasing concentrations of DAC (A, B, C), AZA (D, E, F) in combination with or without vitamin C (0, 10 and 50 μM). The relative quantification was calculated by 2-ΔΔCT method. HMBS served as control. Statistical significance was calculated using 2-way ANOVA and Tukey post-test: *p < 0.05, **p < 0.01, *** < 0.001. Error bars = SD; n=3.
Upon treatment with AZA, TET1 transcript levels were not altered until 1 μM AZA, where decreases are observed, again correlating to the large decreases in cell viability at this AZA concentration (p < 0.001) (Figure 6D). Application of vitamin C did not significantly alter these values. A significant effect between TET2 expression and AZA concentration was also measured (p < 0.0001), and though no significant difference was measured upon vitamin C addition, the highest observed increases were seen with combinatorial treatments; 0.1 μM AZA + 50 μM vitamin C caused a 1.5 fold increase relative to untreated cells. TET3 expression with 0.1 μM AZA was, by contrast, significantly enhanced in the presence of 10 μM vitamin C (Tukey: p < 0.05) causing a 1.7 fold increase relative to untreated cells (Figure 6F). Interestingly, the co-administration of vitamin C was able to mitigate the decreased TET3 expression caused by treatment with 1 μM AZA significantly (Tukey: p < 0.05 and p < 0.001 for 10 and 50 μM vitamin C respectively).
Expression of vitamin C transporters
The cellular uptake of vitamin C is primarily facilitated by the sodium-dependent transporters SVCT1 and SVCT2. Expression changes to the transporters were analysed as one of the mechanisms by which vitamin C could exhibit its epigenetic action in combination with DAC or AZA. Therefore, it was of great interest to determine the expression levels of these SVCTs in cancer cells before and after exposure to DAC, AZA and vitamin C (VC). Both SVCT1 and SVCT2 gene transcripts were detectable in HCT116 cells (Figure 7), although the expression level of SVCT1 was much lower than that of SVCT2. After exposure to DAC (Figure 7A and 7B) a dose-dependent increase in SVCT1 was observed (p < 0.0001) that vitamin C did not affect. Similar increases were seen in SVCT2 (p < 0.0001), but which were affected by dual administration with vitamin C, and indeed were visible even in the absence of DAC (p < 0.001). Importantly, the application of 50 μM vitamin C significantly enhanced the SVCT2 expression induced by 0.1 (Tukey: p < 0.05) and 1 μM DAC (Tukey: p < 0.01) by, respectively, 1.3 and 1.4 fold relative to matched DAC only treatment.
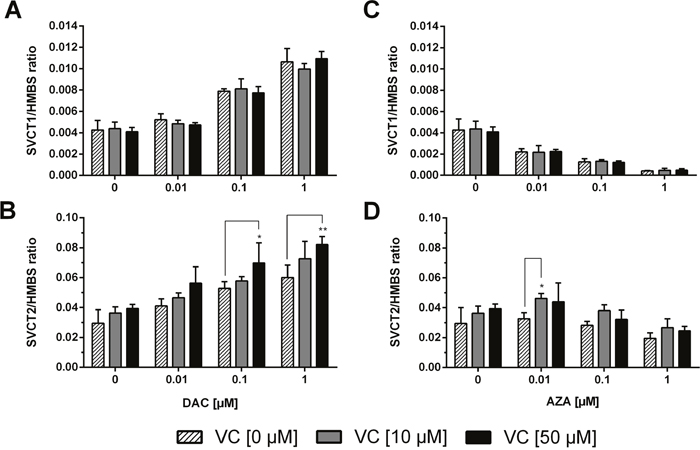
Figure 7: Gene expression profile of sodium dependent vitamin C transporters (SVCT1 and SVCT2) in human colon cancer cells. After 72 h incubation with 0.01, 0.1, or 1 μM DAC (A, B) or AZA (C, D), alone or in combination with vitamin C (0 and 50 μM), mRNA expression levels of SVCT1 (A, C) and SVCT2 (B, D) in HCT116 cells were calculated, relative to the HMBS housekeeper gene, using the 2ΔΔCT method. Statistical significance was calculated using 2-way ANOVA and Tukey post-test: *p < 0.05, **p < 0.01. Error bars = SD; n=3.
Contrastingly, treatment with increasing concentrations of AZA led to a significant dose-dependent decrease of SVCT1 expression (p < 0.0001; up to 8.8 fold) that combination with vitamin C did not alter (Figure 7C). SVCT2 expression was not notably altered by AZA treatment except at 0.01, 0.1 μM in combination with 10 or 50 μM vitamin C (Figure 7D; Tukey: p < 0.05 and p > 0.05 respectively, 1.6 fold and 1.5 fold respectively).
Reactivation of CDKN1A (p21) gene and protein expression
To explore the mechanisms of DAC and AZA dependent reactivation of tumour suppressor expression in colon cancer cells, alone and in combination with vitamin C, the expression of CDKN1A (p21) was assessed by real-time quantitative reverse transcriptase (RT)-PCR following 72 h incubations (Figure 8A, 8B). Evidence suggests that CDKN1A (p21) is an important regulator of the cell cycle that is frequently silenced by epigenetic mechanisms in cancer cells [57]. In untreated HCT116 cancer cells, significant reductions in the expressions of CDKN1A were found, as compared to the normal colon cells (HCEC; data not shown).
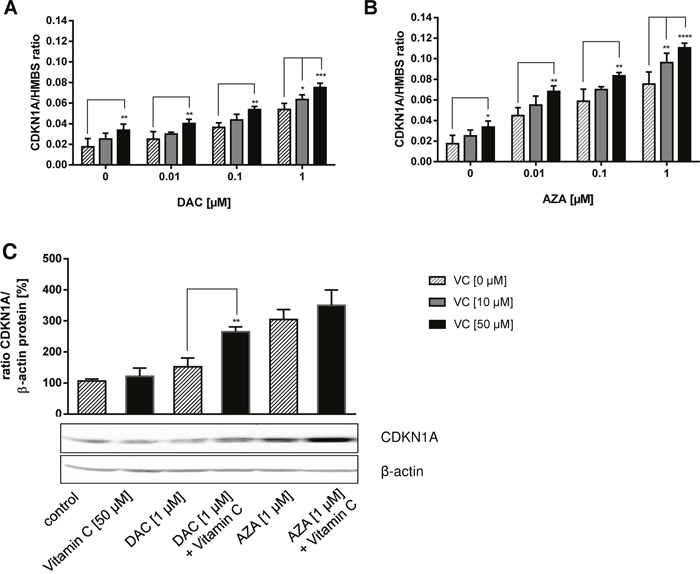
Figure 8: Reactivation of CDKN1A (p21) tumour suppressor on mRNA and protein level by DAC, AZA and vitamin C in colon cancer cells. HCT116 cells were incubated with increasing concentrations of DAC or AZA, alone or in combination with 0 μM (striped bars), 10 μM (grey bars) and 50 μM (black bars) vitamin C for 72 h. The mRNA levels of CDKN1A (p21) after treatment with DAC (A) and AZA (B) were then calculated by 2-ΔΔCT method in relation to HMBS, which served as a control. Additionally, the protein levels of CDKN1A (p21) were determined by Western Blotting and ECL detection (C). The blotting intensities were normalized to β-actin and the untreated control. The experiments were conducted in triplicate. Statistical significance of the treated groups to the untreated control was calculated using 2-way ANOVA and Tukey post-test: *p < 0.05, **p < 0.01, error bars = SD; n=3.
Clear dose-dependent increases in CDKN1A expression were achieved by treatment with DAC or vitamin C alone that were significantly enhanced by combinatorial treatment (Figure 8A). These same trends were also seen on treatment with AZA in the presence or absence of vitamin C (Figure 8B). In comparison to DAC, AZA induced stronger CDKN1A expression at matched concentration, either alone or in combination with vitamin C (Figure 8B).
Further, we examined whether the induction of CDKN1A gene expression resulted in increased CDKN1A protein levels, as well. The protein expression of CDKN1A in HCT116 cells treated with DAC or AZA in combination with vitamin C was analysed by Western blot (Figure 8C). Concordantly, increases in CDKN1A protein levels were seen following treatment with 1 μM DAC (p > 0.05, 1.4 fold) or AZA (p < 0.001, 2.9 fold) (Figure 8C). Though treatment with 50 μM vitamin C alone had no impact on CDKN1A, it measurably enhanced the effects of DAC and AZA (Tukey: p < 0.01 and p > 0.05 respectively; 1.7 fold and 1.2 fold increases relative to DAC or AZA only treated cells respectively).
Apoptotic induction by DAC and AZA in combination with vitamin C
Earlier studies stated that DAC and AZA have pro-apoptotic activities in myeloid leukaemia cells [58, 59], neoplastic mast cells [60] and have the ability sensitize colorectal cancer cells to apoptosis induction by chemotherapy or immunotherapy [61, 62]. To elucidate whether the pro-apoptotic activity of DAC and AZA could be enhanced by vitamin C, combined treatments in HCT116 cells were assessed by Annexin V/propidium iodide staining and flow cytometric analysis (Figure 9). The median fluorescent intensities of cells positive for Annexin V were used to quantify apoptotic cells, of which, those positive for both Annexin V and propidium iodide were defined as late apoptotic, and those positive for Annexin V only as early apoptotic.
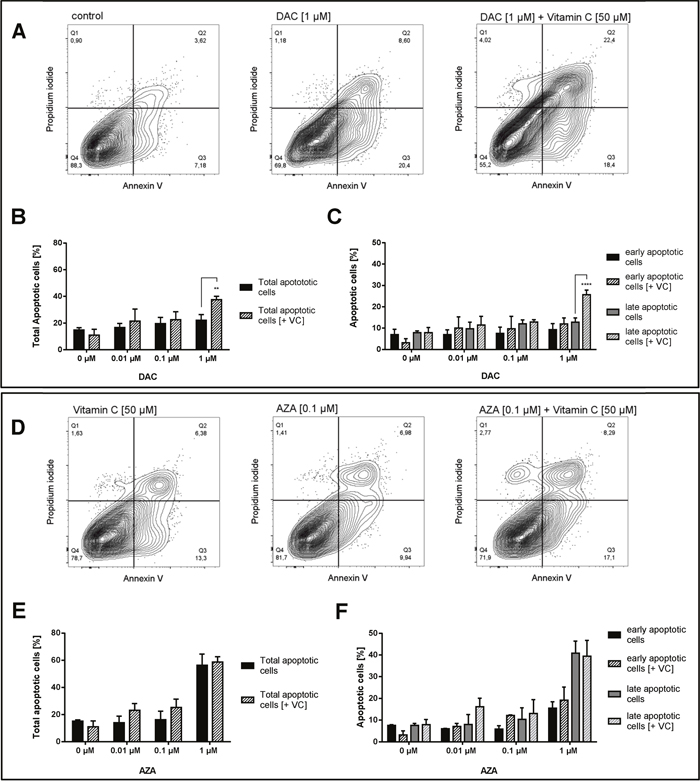
Figure 9: Increase of apoptotic cells in human colon cancer cells by vitamin C and DAC/AZA incubation. Representative contour plots of Annexin V/propidium iodide staining in HCT116 colon cancer cells treated for 72 h with DAC (A) and vitamin C, alone or in combinations, as indicated. Q1 gates for necrotic cells, Q2 for late apoptosis, Q3 for early apoptosis and Q4 for healthy cells. TNFα (20 ng/mL) and cycloheximide (20 μg/mL) were used as positive controls for the induction of apoptosis. Quantitative representation of total apoptotic cells (B) and early + late apoptotic cells separately (C) after treatment with DAC with or without vitamin C, with indicated concentrations. Representative contour plots of Annexin V/propidium iodide staining in HCT116 colon cancer cells treated for 72 h with AZA in indicated concentrations and vitamin C [50 μM] (D). Quantitative representation of total apoptotic cells (E) and early + late apoptotic cells separately (F) after treatment with AZA with or without vitamin C, with indicated concentrations (error bars = SD; n=3; Statistical significance of the treated groups to the untreated control was calculated using 2-way ANOVA and Tukey post-test **** = p< 0.0001, *** = p<0.001, ** = p<0.01, * = p<0.05).
Following 72 h incubation with DAC, significant induction of HCT116 cell apoptosis relative to untreated cells was only seen upon treatment with 1 μM DAC and 50 μM vitamin C (Tukey: p < 0.0001; 2.5 fold increase), where the addition of vitamin C also significantly enhanced the effect of DAC alone (Tukey: p < 0.01; 1.7 fold increase) (Figure 9A and 9B). This effect appeared to derive primarily from an increase in late apoptosis, where the addition of vitamin C increased DAC induced late apoptosis 3.3 fold (Tukey: p < 0.0001) (Figure 9C). Though significant differences were also seen in early apoptosis, these derived from the apparent reduction in early apoptosis induced by vitamin C alone, and did not significantly increase the apoptosis seen in untreated cells.
Following 72 h incubation with AZA, significant dose-dependent increases in HCT116 cell apoptosis were seen (p < 0.0001), reaching up to a 3.8 fold increase relative to untreated cells (Tukey: p < 0.0001) (Figure 9D and 9E). Though slight increases in total apoptosis were seen at 0.01 and 0.1 μM AZA on addition of 50 μM vitamin C, no significant differences were detected in total apoptosis, late apoptosis or early apoptosis (Figure 9E and 9F). However, a significant increase relative to 50 μM vitamin C only treated cells was seen on co-treatment with 0.1 μM AZA (p < 0.05), though this again relates to the decrease in early apoptosis induced by vitamin C alone. Similar to DAC, it is interesting to note the emergence of a necrotic population on addition of 1 μM AZA and 50 μM vitamin C not seen with AZA alone (Figure 9D).
DISCUSSION
Although epigenetic events are a well-studied hallmark of cancer development, very few epigenetic therapies against cancer are approved. The cytosine-analogues DAC and AZA, the most frequently used epigenetic drugs, are highly effective against haematological malignancies such as AML in older individuals [38]. Previous efforts to introduce cytosine analogues to the treatment of solid tumour malignancies have concentrated on the use of DAC and AZA in high doses in cancer cells, which led to more cytotoxic induction without epigenetic modulation [50, 51, 63, 64].
Colorectal cancer is one of the most common solid tumour cancers and epigenetic aberrations are very common [65–69]. However, as for most solid tumour cancers, an epigenetic therapy is still lacking. Therefore, we studied here the efficacy of an innovative combinatorial therapy with hypomethylating agents DAC and AZA and physiological relevant doses of vitamin C on colon cancer cells. Vitamin C has attracted attention as a potential cancer therapy through induction of oxidative stress that selectively kills cancer cells [70, 71]. However, vitamin C has also been shown to act as a potent activator and possible co-factor of the TET dioxygenases, which mediate the oxidation of 5-mdC to 5-hmdC in genomic DNA, a possible therapeutic avenue for active demethylation in aberrantly hypermethylated cancer loci [72, 73]. Notably, there is an underestimated prevalence of vitamin C deficiencies in cancer patients [74–76]. Moreover, sufficient supplementations with physiologically relevant concentrations of vitamin C have been shown to significantly improve the epigenetic outcomes of the epigenetic therapies such as DAC and AZA [74].
Here, we investigated whether vitamin C can enhance the demethylating actions of DAC or AZA, and whether such combinatorial treatments could be used as an innovative therapy in colorectal cancer. The human colon cancer cell line HCT116 was selected as an in vitro model of colorectal cancer. Firstly, cytotoxic assessments of DAC and AZA in HCT116 cells were performed. From these, the sub-cytotoxic concentrations of DAC (0.001–1 μM) and AZA (0.001–0.1 μM) were identified, as well as one AZA concentration of moderate toxicity (1 μM). These were selected to reflect a realistic pharmacological scenario in which efficient demethylating activity, rather than cytotoxicity, might be seen [43]. In addition, concentrations of 10 and 50 μM vitamin C were chosen to reflect plasma levels achieved by deficient and normal dietary intakes, rather than by pharmacological supplementation. Although several studies have demonstrated that vitamin C inhibits proliferation and induces apoptotic effects at very high concentrations [55, 70], our MTT viability studies confirmed our selected ascorbate doses did not induce significant toxicity to HCT116 cells. Importantly, the addition of 50 μM vitamin C did not cause further AZA or DAC induced toxicity at any of the tested concentrations.
Next, the impact of vitamin C of DAC or AZA on the state of HCT116 cell apoptosis was investigated by flow cytometry. While supplementation with physiologically relevant vitamin C concentrations did not alter the cytotoxic potential of either agent, a shift in the number of cells undergoing late apoptosis was seen, particularly for DAC treated cells where combinations with vitamin C induced the highest measured levels of apoptosis. Indeed, vitamin C resulted in significant increases to apoptotic induction with 1 μM DAC, both sub-cytotoxic concentrations alone or in combination with vitamin C. Notably, little cell necrosis was seen at even the highest AZA and DAC concentrations alone or in combination with vitamin C. These data indicate that a combination of demethylating and hydroxymethylating substances could have significant impacts on cell cycle normalization and apoptosis induction in colon cancer cells, not related to direct cytotoxic potential.
To elucidate whether epigenetic activity was increased by the combinatorial treatments of AZA or DAC with vitamin C, genome-wide changes in DNA-methylation and DNA-hydroxymethylation states were assessed by a highly sensitive LC-MS/MS approach. Previous studies have shown that global DNA methylation status is a poor marker of cell division changes within cancer cells after DAC treatment [77, 78]. This has led to the assumption that 5-hmdC/dC status is a more accurate reflection of gene re-expression for at least certain gene subsets, e.g. the tumour suppressor CDKN1A (p21) [79–81].
Reductions to 5-mdC levels occurred in a concentration-dependent manner for both AZA and DAC, and were not affected by the addition of vitamin C. By contrast, the demethylating agents alone mostly failed to elicit any change to 5-hmdC-levels, with only the moderately toxic dose of AZA showing any kind of effect. This is in contrast to a previous study that reported DAC could induce an increase of 5-hmdC-levels in leukaemia cell lines HL-60 (AML) and TK6 (CML) [82]. Instead, treatment with vitamin C induced concentration-dependent increases to 5-hmdC/dC levels that plateaued between 50 and 100 μM. Remarkably, combinatorial treatments with AZA or DAC resulted in significant enhancements to this effect, suggesting some form of synergism or additive effect was taking place between sub-toxic concentrations of vitamin C and AZA or DAC.
Interestingly, the combination of the moderately toxic AZA dose (1 μM) and vitamin C induced the most prominent increase in 5-hmdC levels. The results of the LC-MS/MS analysis illustrate that VC is not involved in the passive DNA hypomethylation mediated by inhibition of the DNMTs, but is involved in the active global 5-hmdC increase. Concordant gene expression data re-affirmed the role of vitamin C in AZA or DAC mediated epigenetic modulation. The expression of ALU, the promoter regions of which are highly CpG rich and so particularly prone to DNA methylation, were increased by AZA or DAC mediated demethylation in a concentration-dependent manner that was not affected by addition of vitamin C. Expression of CDK1NA was also increased in an AZA or DAC concentration-dependent manner, however here vitamin C was shown to significantly enhance this effect at the mRNA and protein level. Indeed vitamin C was capable of moderately increasing CDK1NA expression when applied alone. CDKN1A (p21) is a tumour suppressor that plays an important role in preventing tumour development by inhibiting cell cycle progression and inducing apoptotic signals within the cell [83]. Its increased expression may in part explain the increased prevalence of apoptosis in HCT116 cells following the combination of vitamin C to AZA or DAC treatments. However, the treatment failed to reactivate other epigenetically silenced tumour suppressors like CDKN1B (p27) or CDKN2A (p16) (data not shown).
To unravel the mechanism by which vitamin C and AZA or DAC may interact to enhance genome-wide hydroxymethylation states, the expression levels of DNMT and TET family genes were assessed. As already noted, AZA and DAC act to inhibit the actions of DNMTs, thereby preventing DNA methylation, and vitamin C enhanced TET activities resulting in increased DNA hydroxymethylation. Interestingly, while vitamin C alone had no impact on DNMT expression, significantly enhanced expression of DNMT3A and DNMT3B were seen when applied in combination with 0.1 μM DAC. Though the enhanced expression of DNMTs on addition of DAC is perhaps counterintuitive, this perhaps reflects a feedback loop in response to the role of DAC in depleting DNMT. Similarly, though vitamin C alone did not impact on TET expression, measurable enhancements to TET2 and TET3 expression were seen in combination with 0.1 μM AZA or DAC, an action that could well improve the hydroxymethylating activity of vitamin C. It should be noted that these effects were seen almost solely at 0.1 μM AZA and DAC, correlating only partly with hydroxymethylation states, suggesting further mechanisms are at play, including the vitamin C induced increase to the enzymatic activity of the TETs.
It was also of interest to examine the expression of the sodium-dependent transporters that bring vitamin C into the cell. Exposing HCT116 cells to 50 μM vitamin C and DAC could increase SVCT1 and SVCT2 expression. This led us to the hypothesis that an increase in SVCT1 and SVCT2 could enhance the vitamin C uptake into the cell and therefore potentiate the vitamin C related apoptosis induction in the cancer cells, possibly explaining the co-operative effect seen between vitamin C and DAC on genomic 5-hmdC/dC levels [71, 74]. In this regard, a local vitamin C deficiency, along with a decreased level of TETs, could cause loss of SVCTs expression that may contribute to the global loss of 5-hmdC/dC in colon adenocarcinoma cells [74]. In our study, treatment with AZA failed to induce SVCT expression, probably based on their different mode of action [38].
A recent study stated that the combinational therapy of elderly AML patients with DAC and vitamin C led to improvements in overall survival and remission rates compared to patients treated with DAC alone [84]. In accordance to our data, it was found that low doses of vitamin C (300 μM) could enhance the effects of 2.5 μM DAC on the leukaemia cell lines HL60 and NB4, reducing proliferation, increasing apoptosis and enhancing TET2 activity. In our study, we were able to achieve similar in vitro results in colorectal cancer cells with even lower concentrations of the compounds. In contrast, though increased TET expression was identified, this only occurred within a limited DAC concentration range with apparent decreases at lower doses than used by Zhao (2018) and co-workers [84]. However, a significant increase of 5-hmdC/dC levels was demonstrated after combinatorial treatments with vitamin C compared to AZA or DAC treatment alone. This led us to suggest that not an increased TET-expression but an increased TET activity may lead to the higher levels of 5-hmdC/dC.
The present study shows an important impact on the epigenetic landscape of colorectal cancer cells through the combined actions of functionally distinct substances, leading to significant improvements in the apoptotic induction of HCT116 cells. Furthermore, the combination of sub- or low-micromolar doses of demethylating agents like DAC and AZA with moderate, physiological levels of vitamin C was shown to result in enhanced epigenetic actions, increased expression of suppressed tumour suppressors, and a positive shift in the apoptotic state without significant cytotoxic induction. Although this is not the first time a counter-intuitive link between the application of hypomethylating agents and increased genomic hmdC levels has been identified [82], this is the first time a direct link between this and vitamin C levels has been observed. In conclusion, our results indicate epigenetic therapies mediated by sub-micromolar concentrations of DAC or AZA are capable of enhanced anti-cancerous activities when administered in combination with vitamin C at physiologically relevant doses. Vitamin C deficiencies are common among many cancer patients [76]. As such, our data suggest that therapeutic responses to low, non-toxic doses of DNMT inhibitors can be improved by additional vitamin C supplementation.
MATERIALS AND METHODS
Cell culture and treatment
HCT116 (ATCC no. CCL-247), a human colorectal carcinoma cell line, was purchased from the American Type Culture Collection (ATCC; https://www.atcc.org/). Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 2 mM L-glutamine supplemented with 10% fetal bovine serum (FBS), 45 IU/ml penicillin, and 45 IU/ml streptomycin. The cell lines were confirmed as negative for mycoplasma infection within six months prior to use. HCT116 cells were treated daily with DAC or AZA (0, 10, 100 or 1000 nM) for three consecutive days, alone or in combination with vitamin C (0, 10 or 50 μM). DAC, AZA and vitamin C were administered at the same time points.
Cell viability assay
Potential cytotoxic effects induced by vitamin C, AZA and DAC were assessed by 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) reduction assay, as previously described [85]. HCT116 cells were seeded into 96-well plates (TPP, Trasadingen, Switzerland). After 24 h, the indicated concentrations were added for 24, 48 and 72 h. Subsequently, the cells were incubated with 100 μL MTT solution (0.5 mg/mL in PBS) for 4 h. After removing the supernatants, 50 μL dimethyl sulfoxide was added to dissolve the formazan salt and its optical density (OD) was measured using a microplate reader (Tecan, Crailsheim, Germany) at 540 nm. The positive controls were treated with 0.002% SDS. A cell viability < 75% predicts cytotoxic effects.
Determination of genome-wide DNA methylation and hydroxymethylation using isotope-dilution liquid chromatography tandem-mass spectrometry (LC-MS/MS)
Samples of genomic DNA (20 μg) were hydrolyzed to 2’-deoxynucleosides using micrococcal nuclease from Staphylococcus aureus, bovine spleen phosphodiesterase, and calf intestinal alkaline phosphatase (all from Sigma-Aldrich, Taufkirchen, Germany) as described previously [86] with slight modifications. 10 μL of 50 nM 5-hmdC-d3 (Toronto Research Chemicals, Toronto, Canada) was added to the DNA digestion mixture as an internal standard, and the incubation times of the two-step hydrolysis were 1 h each. Afterwards, DNA hydrolysates were centrifuged (5 min; 16,000 x g) and 10 μL of their supernatants taken for quantification of dC and 5-mdC by means of the stable isotopically labelled reference compounds [15N2,13C1]dC and 5-mdC-d3 (both from Toronto Research Chemicals, Toronto, Canada) as recently described [87]. The remaining DNA hydrolysates (~ 310 μL) were evaporated to dryness under reduced pressure using a Savant SpeedVac Concentrator (Thermo Fisher Scientific, Dreieich, Germany). The dried residues were then mixed with 100 μL methanol, briefly vortexed, and stored overnight (-20°C). The next day, samples were thoroughly vortexed (1,400 rpm) for 10 min followed by centrifugation at 16,000 x g for 10 min. Supernatants were transferred to new sample tubes. Extraction of the protein pellets was repeated by addition of a further 100 μL methanol and vortexing (1,400 rpm) for 5 min. After centrifugation at 16,000 x g for 10 min, both methanolic fractions were combined and evaporated to dryness under reduced pressure. The dried residues were reconstituted in 50 μL water containing 0.0075% formic acid, ultra-sonicated for 10 min, vortexed (1,400 rpm) for 5 min, and centrifuged for 5 min at 16,000 x g. LC-MS/MS analyzes of the supernatants were conducted with an Agilent 1260 Infinity LC system coupled to an Agilent 6490 triple quadrupole mass spectrometer (both from Waldbronn, Germany) interfaced with an electrospray ion source operating in the positive ion mode (ESI+). Chromatographic conditions and settings of the ESI source were as described recently for quantification of dC and 5-mdC [87] with the following exceptions: a longer separation column (Agilent Poroshell 120 EC-C18, 2.7 μm, 3.0 x 150 mm) was used and the injection volume was 5 μL. Quantification of 5-hmdC in relation to its stable isotope labelled standard, both eluted at 4.9 min from the LC column (retention times of dC and 5-mdC were 4.7 and 6.0 min, respectively), was carried out using the multiple reaction monitoring (MRM) approach. The following mass transitions (loss of 2’-deoxyribose) were used as quantifiers (optimized collision energies in parentheses): 5-hmdC: m/z 258.1 → 142.0 (8 eV) and 5-hmdC-d3: m/z 261.1 → 145.0 (8 eV). Additional mass transitions were recorded for unambiguous identification. The dwell time for each of the four mass transitions analyzed was 50 ms.
Apoptosis assay
Levels of apoptotic and dead cells were determined by flow cytometry using eBioscience™ Annexin V Apoptosis Detection Kit APC (Thermo Fisher, Darmstadt, Germany). Briefly, 2 x 105 HCT116 cells/well were seeded into 6-well plates (TPP, Trasadingen, Switzerland). After 24 h, the cells were incubated with the substances at the indicated concentrations for 72 h. Incubation with TNFα (20 ng/mL) and cycloheximide (20 μg/mL) for 72 h served as positive control for apoptosis induction. Subsequently, the cells were washed and stained with Annexin V antibody and propidium iodide (PI) according to the manufacturer’s instructions. The cells were distinguished between viable cells (Annexin V− / PI−), early apoptotic cells (Annexin V+ / PI−), late apoptotic/necrotic cells (Annexin V+ / PI+) and late necrotic cells (Annexin V− / PI+). Per run 10,000 events were counted and analyzed on a FACSCanto II (BD Biosciences, Heidelberg, Germany). FlowJo software (Treestar, Ashland, USA) was used for data analysis.
RNA extraction and quantitative real-time PCR
RNA extraction was performed with the RNA High Pure RNA Kit (Roche, Mannheim, Germany) according to the manufacturer’s instructions. The cDNA synthesis was then conducted using the RevertAid reverse transcriptase (Thermo Fisher, Darmstadt, Germany) and 0.5-5 μg (ideally 3 μg) RNA. The qRT-PCR was performed using the Maxima SYBR Green qPCR Mix (ThermoFisher, Darmstadt, Germany) on a LightCycler 480 II Real-Time PCR system (Roche, Mannheim, Germany). Quantification was performed using the ΔΔ Ct method with hHMBS expression as an internal reference. Melt curve analysis confirmed that all the qRT-PCR products were generated in the form of double-stranded DNA. The primers used are listed in Table 1.
Table 1: Primer sequences and fragment sizes for qPCR experiments
Target gene |
Sequence |
Fragment size (bp) |
|---|---|---|
hHMBS |
fw: ACCAAGGAGCTTGAACATGC |
143 |
hDNMT1 |
fw: ACCTGGCTAAAGTCAAATCC |
80 |
hDNMT3a |
fw: ACTACATCAGCAAGCGCAAG |
359 |
hDNMT3b |
fw: CCAGCTCTTACCTTACCATC |
285 |
hTET1 |
fw: GCTGCTGTCAGGGAAATCAT |
209 |
hTET2 |
fw: CCAATAGGACATGATCCAGG |
232 |
hTET3 |
fw: TCGGAGACACCCTCTACCAG |
179 |
CDKN2A (p16) |
fw: GAGCAGCATGGAGCCTTC |
124 |
CDKN1A (p21) |
fw: AGTGGACAGCGAGCAGCTGA |
381 |
CDKN1B (p27) |
fw: AAACGTGCGAGTGTCTAACGGGA |
456 |
SVCT1 |
fw: TCATCCTCCTCTCCCAGTACCT |
141 |
SVCT2 |
fw: TCTTTGTGCTTGGATTTTCGAT |
106 |
Protein extraction and western blot
Briefly, cell pellets were lysed in 200 μL RIPA buffer (50 mM Tris/HCl, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS in PBS) and incubated for 30 min at 4 °C. Subsequently, cell lysates were centrifuged at 10,000 x g for 30 min, and the supernatant used for protein analysis.
Western blotting was performed according to standard procedure. Protein samples (50 μg per lane) were separated by SDS-PAGE, and then transferred to PVDF membrane. Following blocking (5% milk powder in TBST buffer) membranes were probed with antibodies against CDKN1A (Abcam, Cambridgeshire, UK) and anti-β-actin (Abcam, Cambridgeshire, UK), followed by reaction with the corresponding horseradish peroxidase-conjugated secondary antibody (Cell Signaling, Boston, USA). Proteins were visualized using the ECL detection system (Thermo Scientific) and ChemiDoc™ system (Bio-Rad Laboratories, Munich, Germany).
Statistical analysis
Statistically significant differences were determined two-way ANOVA followed Tukey post hoc test, where p-values ≤ 0.05 were considered as significant. Differences in apoptosis induction were calculated by 2 way ANOVA analysis, where p-values ≤ 0.05 were considered as significant (* = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = P < 0.0001). All statistical analyses were conducted using the software GraphPad Prism (Graphpad Software, Inc., La Jolla, USA).
Abbreviations
5-mdC: 5-methyl-2’-deoxycytidine; 5-hmdC: 5-hydroxymethyl-2’-deoxycytidine; 5-fdC: 5-formyl-2’-deoxycytidine; 5-cadC: 5-carboxy-2’-deoxycytidine; ALU: athrobacter luteus; AML: acute myeloid leukemia; APC: adenomatous polyposis coli; ATCC: American type culture collection; AZA: 5-aza-cytidine; CDKN1A: cyclin-dependent kinase inhibitor 1; CDKN2A: cyclin-dependent kinase inhibitor 2A; CDKN2B: cyclin-dependent kinase inhibitor 2B; DAC: 5-aza-2’-deoxycytidine; DNMTs: DNA methyltransferases; FDA: US Food and Drug Administration; HMBS: hydroxymethylbilane synthase; ITGA4: integrin a subunit 4; LC-MS/MS: liquid chromatography tandem-mass spectrometry; MTT: 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide; P4H: prolyl-4-hydroxylase; SVCT1: sodium dependent vitamin C transporter 1; SVCT2: sodium dependent vitamin C transporter 2; TET: ten eleven translocation; TDG: thymine-DNA glycosylase; TFPI2: tissue factor pathway inhibitor 2; VC: vitamin C; Wnt: wingless/Int1.
Author contributions
CG and BK designed experiments; CG, FS, AE, AW and LKN performed experiments; CG, LKN, FS, AE, GY and BS analyzed data; CG, FS, AE, AW, TH, BS, GY and BK wrote the manuscript, CG, BK and BS advised on experimental design and provided critical feedback, all authors reviewed the manuscript.
ACKNOWLEDGMENTS
We would like to thank Monika Haseloff and Dennis Thurack for the excellent technical assistance in conducting the experiments.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest to disclose.
FUNDING
C.G. and A.E. were financially supported by the German Research Foundation (Collaborative Research Center 1112, project Z01). The study was supported by the NutriAct – Competence Cluster Nutrition Research Berlin-Potsdam funded by the Federal Ministry of Education and Research (FKZ: 01EA1408A-B).
REFERENCES
1. Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017; 66:683–91. https://doi.org/10.1136/gutjnl-2015-310912.
2. Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011; 6:479–507. https://doi.org/10.1146/annurev-pathol-011110-130235.
3. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990; 61:759–67. https://doi.org/10.1016/0092-8674(90)90186-I.
4. Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature. 1992; 359:235–37. https://doi.org/10.1038/359235a0.
5. Issa JP, Shen L, Toyota M. CIMP, at last. Gastroenterology. 2005; 129:1121–4. https://doi.org/10.1053/j.gastro.2005.07.040.
6. Chirieac LR, Shen L, Catalano PJ, Issa JP, Hamilton SR. Phenotype of microsatellite-stable colorectal carcinomas with CpG island methylation. Am J Surg Pathol. 2005; 29:429–36. https://doi.org/10.1097/01.pas.0000155144.53047.7d.
7. Kondo Y, Issa JP. Epigenetic changes in colorectal cancer. Cancer Metastasis Rev. 2004; 23:29–39. https://doi.org/10.1023/A:1025806911782.
8. Frazier ML, Xi L, Zong J, Viscofsky N, Rashid A, Wu EF, Lynch PM, Amos CI, Issa JP. Association of the CpG island methylator phenotype with family history of cancer in patients with colorectal cancer. Cancer Res. 2003; 63:4805–08.
9. Jones PA. DNA methylation errors and cancer. Cancer Res. 1996; 56:2463–67.
10. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002; 3:415–28. https://doi.org/10.1038/nrg816.
11. Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007; 128:683–92. https://doi.org/10.1016/j.cell.2007.01.029.
12. Taberlay PC, Jones PA. DNA methylation and cancer. Prog Drug Res. 2011; 67:1–23. https://doi.org/10.1007/978-3-7643-8989-5_1.
13. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009; 324:930–35. https://doi.org/10.1126/science.1170116.
14. Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010; 466:1129–33. https://doi.org/10.1038/nature09303.
15. Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011; 333:1300–03. https://doi.org/10.1126/science.1210597.
16. He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011; 333:1303–07. https://doi.org/10.1126/science.1210944.
17. Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem. 2011; 286:35334-8. https://doi.org/10.1074/jbc.C111.284620.
18. Hu L, Li Z, Cheng J, Rao Q, Gong W, Liu M, Shi YG, Zhu J, Wang P, Xu Y. Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell. 2013; 155:1545–55. https://doi.org/10.1016/j.cell.2013.11.020.
19. Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013; 14:341–56. https://doi.org/10.1038/nrm3589.
20. Lian CG, Xu Y, Ceol C, Wu F, Larson A, Dresser K, Xu W, Tan L, Hu Y, Zhan Q, Lee CW, Hu D, Lian BQ, et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell. 2012; 150:1135–46. https://doi.org/10.1016/j.cell.2012.07.033.
21. Delatte B, Deplus R, Fuks F. Playing TETris with DNA modifications. EMBO J. 2014; 33:1198–211. https://doi.org/10.15252/embj.201488290.
22. Koivunen P, Laukka T. The TET enzymes. Cell Mol Life Sci. 2018; 75:1339–48. https://doi.org/10.1007/s00018-017-2721-8.
23. Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim Biophys Acta. 2012; 1826:443–57. https://doi.org/10.1016/j.bbcan.2012.06.003.
24. Young JI, Züchner S, Wang G. Regulation of the Epigenome by Vitamin C. Annu Rev Nutr. 2015; 35:545–64. https://doi.org/10.1146/annurev-nutr-071714-034228.
25. Yin R, Mao SQ, Zhao B, Chong Z, Yang Y, Zhao C, Zhang D, Huang H, Gao J, Li Z, Jiao Y, Li C, Liu S, et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J Am Chem Soc. 2013; 135:10396–403. https://doi.org/10.1021/ja4028346.
26. Hore TA, von Meyenn F, Ravichandran M, Bachman M, Ficz G, Oxley D, Santos F, Balasubramanian S, Jurkowski TP, Reik W. Retinol and ascorbate drive erasure of epigenetic memory and enhance reprogramming to naïve pluripotency by complementary mechanisms. Proc Natl Acad Sci U S A. 2016; 113:12202–07. https://doi.org/10.1073/pnas.1608679113.
27. Raj K, Mufti GJ. Azacytidine (Vidaza(R)) in the treatment of myelodysplastic syndromes. Ther Clin Risk Manag. 2006; 2:377–88. https://doi.org/10.2147/tcrm.2006.2.4.377.
28. Kaminskas E, Farrell AT, Wang YC, Sridhara R, Pazdur R. FDA drug approval summary: azacitidine (5-azacytidine, Vidaza) for injectable suspension. Oncologist. 2005; 10:176–82. https://doi.org/10.1634/theoncologist.10-3-176.
29. Azacitine (vidaza) for myelodysplastic syndrome. Med Lett Drugs Ther. 2005; 47:11.
30. Decitabine (Dacogen) for myelodysplastic syndromes. Med Lett Drugs Ther. 2006; 48:91–92.
31. Saba HI. Decitabine in the treatment of myelodysplastic syndromes. Ther Clin Risk Manag. 2007; 3:807–17.
32. Schuh AC, Döhner H, Pleyer L, Seymour JF, Fenaux P, Dombret H. Azacitidine in adult patients with acute myeloid leukemia. Crit Rev Oncol Hematol. 2017; 116:159–77. https://doi.org/10.1016/j.critrevonc.2017.05.010.
33. Malik P, Cashen AF. Decitabine in the treatment of acute myeloid leukemia in elderly patients. Cancer Manag Res. 2014; 6:53–61. https://doi.org/10.2147/CMAR.S40600.
34. Wijermans PW, Rüter B, Baer MR, Slack JL, Saba HI, Lübbert M. Efficacy of decitabine in the treatment of patients with chronic myelomonocytic leukemia (CMML). Leuk Res. 2008; 32:587–91. https://doi.org/10.1016/j.leukres.2007.08.004.
35. Kantarjian H, Oki Y, Garcia-Manero G, Huang X, O’Brien S, Cortes J, Faderl S, Bueso-Ramos C, Ravandi F, Estrov Z, Ferrajoli A, Wierda W, Shan J, et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. 2007; 109:52–57. https://doi.org/10.1182/blood-2006-05-021162.
36. Sorm F, Pískala A, Cihák A, Veselý J. 5-Azacytidine, a new, highly effective cancerostatic. Experientia. 1964; 20:202–03. https://doi.org/10.1007/BF02135399.
37. Sorm F, Vesely J. The Activity of a New Antimetabolite, 5-Azacytidine, against Lymphoid Leukaemia in Ak Mice. Neoplasma. 1964; 11:123–30.
38. Pleyer L, Greil R. Digging deep into “dirty” drugs - modulation of the methylation machinery. Drug Metab Rev. 2015; 47:252–79. https://doi.org/10.3109/03602532.2014.995379.
39. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, Schoch R, Gattermann N, Sanz G, List A, Gore SD, Seymour JF, Bennett JM, et al, and International Vidaza High-Risk MDS Survival Study Group. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009; 10:223–32. https://doi.org/10.1016/S1470-2045(09)70003-8.
40. Pleyer L, Stauder R, Burgstaller S, Schreder M, Tinchon C, Pfeilstocker M, Steinkirchner S, Melchardt T, Mitrovic M, Girschikofsky M, Lang A, Krippl P, Sliwa T, et al. Azacitidine in patients with WHO-defined AML - results of 155 patients from the Austrian Azacitidine Registry of the AGMT-Study Group. J Hematol Oncol. 2013; 6:32. https://doi.org/10.1186/1756-8722-6-32.
41. Aimiuwu J, Wang H, Chen P, Xie Z, Wang J, Liu S, Klisovic R, Mims A, Blum W, Marcucci G, Chan KK. RNA-dependent inhibition of ribonucleotide reductase is a major pathway for 5-azacytidine activity in acute myeloid leukemia. Blood. 2012; 119:5229–38. https://doi.org/10.1182/blood-2011-11-382226.
42. Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008; 123:8–13. https://doi.org/10.1002/ijc.23607.
43. Hollenbach PW, Nguyen AN, Brady H, Williams M, Ning Y, Richard N, Krushel L, Aukerman SL, Heise C, MacBeth KJ. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS One. 2010; 5:e9001. https://doi.org/10.1371/journal.pone.0009001.
44. Marcucci G, Silverman L, Eller M, Lintz L, Beach CL. Bioavailability of azacitidine subcutaneous versus intravenous in patients with the myelodysplastic syndromes. J Clin Pharmacol. 2005; 45:597–602. https://doi.org/10.1177/0091270004271947.
45. Cashen AF, Shah AK, Todt L, Fisher N, DiPersio J. Pharmacokinetics of decitabine administered as a 3-h infusion to patients with acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS). Cancer Chemother Pharmacol. 2008; 61:759–66. https://doi.org/10.1007/s00280-007-0531-7.
46. Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, Shin JJ, Harbom KM, Beaty R, Pappou E, Harris J, Yen RW, Ahuja N, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012; 21:430–46. https://doi.org/10.1016/j.ccr.2011.12.029.
47. Otterson GA, Khleif SN, Chen W, Coxon AB, Kaye FJ. CDKN2 gene silencing in lung cancer by DNA hypermethylation and kinetics of p16INK4 protein induction by 5-aza 2'deoxycytidine. Oncogene. 1995; 11:1211–16.
48. Boldt DH. p21(CIP1/WAF1/SDI1) hypermethylation: an exciting new lead in ALL biology. Blood. 2002; 99:2283–84. https://doi.org/10.1182/blood.V99.7.2283.
49. Xu XL, Yu J, Zhang HY, Sun MH, Gu J, Du X, Shi DR, Wang P, Yang ZH, Zhu JD. Methylation profile of the promoter CpG islands of 31 genes that may contribute to colorectal carcinogenesis. World J Gastroenterol. 2004; 10:3441–54. https://doi.org/10.3748/wjg.v10.i23.3441.
50. Schwartsmann G, Schunemann H, Gorini CN, Filho AF, Garbino C, Sabini G, Muse I, DiLeone L, Mans DR. A phase I trial of cisplatin plus decitabine, a new DNA-hypomethylating agent, in patients with advanced solid tumors and a follow-up early phase II evaluation in patients with inoperable non-small cell lung cancer. Invest New Drugs. 2000; 18:83–91. https://doi.org/10.1023/A:1006388031954.
51. Pohlmann P, DiLeone LP, Cancella AI, Caldas AP, Dal Lago L, Campos O Jr, Monego E, Rivoire W, Schwartsmann G. Phase II trial of cisplatin plus decitabine, a new DNA hypomethylating agent, in patients with advanced squamous cell carcinoma of the cervix. Am J Clin Oncol. 2002; 25:496–501. https://doi.org/10.1097/00000421-200210000-00015.
52. Flis S, Gnyszka A, Misiewicz-Krzemińska I, Spławiński J. Decytabine enhances cytotoxicity induced by oxaliplatin and 5-fluorouracil in the colorectal cancer cell line Colo-205. Cancer Cell Int. 2009; 9:10. https://doi.org/10.1186/1475-2867-9-10.
53. Vijayaraghavalu S, Dermawan JK, Cheriyath V, Labhasetwar V. Highly synergistic effect of sequential treatment with epigenetic and anticancer drugs to overcome drug resistance in breast cancer cells is mediated via activation of p21 gene expression leading to G2/M cycle arrest. Mol Pharm. 2013; 10:337–52. https://doi.org/10.1021/mp3004622.
54. Minor EA, Court BL, Young JI, Wang G. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J Biol Chem. 2013; 288:13669–74. https://doi.org/10.1074/jbc.C113.464800.
55. Sajadian SO, Tripura C, Samani FS, Ruoss M, Dooley S, Baharvand H, Nussler AK. Vitamin C enhances epigenetic modifications induced by 5-azacytidine and cell cycle arrest in the hepatocellular carcinoma cell lines HLE and Huh7. Clin Epigenetics. 2016; 8:46. https://doi.org/10.1186/s13148-016-0213-6.
56. Rodriguez J, Vives L, Jordà M, Morales C, Muñoz M, Vendrell E, Peinado MA. Genome-wide tracking of unmethylated DNA Alu repeats in normal and cancer cells. Nucleic Acids Res. 2008; 36:770–84. https://doi.org/10.1093/nar/gkm1105.
57. Lao VV, Grady WM. Epigenetics and colorectal cancer. Nat Rev Gastroenterol Hepatol. 2011; 8:686–700. https://doi.org/10.1038/nrgastro.2011.173.
58. Flotho C, Claus R, Batz C, Schneider M, Sandrock I, Ihde S, Plass C, Niemeyer CM, Lübbert M. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia. 2009; 23:1019–28. https://doi.org/10.1038/leu.2008.397.
59. Bogenberger JM, Kornblau SM, Pierceall WE, Lena R, Chow D, Shi CX, Mantei J, Ahmann G, Gonzales IM, Choudhary A, Valdez R, Camoriano J, Fauble V, et al. BCL-2 family proteins as 5-Azacytidine-sensitizing targets and determinants of response in myeloid malignancies. Leukemia. 2014; 28:1657–65. https://doi.org/10.1038/leu.2014.44.
60. Ghanim V, Herrmann H, Heller G, Peter B, Hadzijusufovic E, Blatt K, Schuch K, Cerny-Reiterer S, Mirkina I, Karlic H, Pickl WF, Zöchbauer-Müller S, Valent P. 5-azacytidine and decitabine exert proapoptotic effects on neoplastic mast cells: role of FAS-demethylation and FAS re-expression, and synergism with FAS-ligand. Blood. 2012; 119:4242–52. https://doi.org/10.1182/blood-2011-09-382770.
61. Sharma A, Vatapalli R, Abdelfatah E, Wyatt McMahon K, Kerner Z, A Guzzetta A, Singh J, Zahnow C, B Baylin S, Yerram S, Hu Y, Azad N, Ahuja N. Hypomethylating agents synergize with irinotecan to improve response to chemotherapy in colorectal cancer cells. PLoS One. 2017; 12:e0176139. https://doi.org/10.1371/journal.pone.0176139.
62. Yu G, Wu Y, Wang W, Xu J, Lv X, Cao X, Wan T. Low-dose decitabine enhances the effect of PD-1 blockade in colorectal cancer with microsatellite stability by re-modulating the tumor microenvironment. Cell Mol Immunol. 2018 Apr 5. https://doi.org/10.1038/s41423-018-0026-y. [Epub ahead of print].
63. Seymour JF, Fenaux P, Silverman LR, Mufti GJ, Hellström-Lindberg E, Santini V, List AF, Gore SD, Backstrom J, McKenzie D, Beach CL. Effects of azacitidine compared with conventional care regimens in elderly (≥ 75 years) patients with higher-risk myelodysplastic syndromes. Crit Rev Oncol Hematol. 2010; 76:218–27. https://doi.org/10.1016/j.critrevonc.2010.04.005.
64. Fenaux P, Mufti GJ, Hellström-Lindberg E, Santini V, Gattermann N, Germing U, Sanz G, List AF, Gore S, Seymour JF, Dombret H, Backstrom J, Zimmerman L, et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol. 2010; 28:562–69. https://doi.org/10.1200/JCO.2009.23.8329.
65. Baylin SB, Jones PA. Epigenetic Determinants of Cancer. Cold Spring Harb Perspect Biol. 2016; 8:a019505. https://doi.org/10.1101/cshperspect.a019505.
66. Sweeney C, Boucher KM, Samowitz WS, Wolff RK, Albertsen H, Curtin K, Caan BJ, Slattery ML. Oncogenetic tree model of somatic mutations and DNA methylation in colon tumors. Genes Chromosomes Cancer. 2009; 48:1–9. https://doi.org/10.1002/gcc.20614.
67. Steine EJ, Ehrich M, Bell GW, Raj A, Reddy S, van Oudenaarden A, Jaenisch R, Linhart HG. Genes methylated by DNA methyltransferase 3b are similar in mouse intestine and human colon cancer. J Clin Invest. 2011; 121:1748–52. https://doi.org/10.1172/JCI43169.
68. Shen L, Toyota M, Kondo Y, Lin E, Zhang L, Guo Y, Hernandez NS, Chen X, Ahmed S, Konishi K, Hamilton SR, Issa JP. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci U S A. 2007; 104:18654–59. https://doi.org/10.1073/pnas.0704652104.
69. Ahuja N, Li Q, Mohan AL, Baylin SB, Issa JP. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res. 1998; 58:5489–94.
70. Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K, Roper J, Chio II, Giannopoulou EG, Rago C, Muley A, Asara JM, Paik J, et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015; 350:1391–96. https://doi.org/10.1126/science.aaa5004.
71. Sant DW, Mustafi S, Gustafson CB, Chen J, Slingerland JM, Wang G. Vitamin C promotes apoptosis in breast cancer cells by increasing TRAIL expression. Sci Rep. 2018; 8:5306. https://doi.org/10.1038/s41598-018-23714-7.
72. Blaschke K, Ebata KT, Karimi MM, Zepeda-Martínez JA, Goyal P, Mahapatra S, Tam A, Laird DJ, Hirst M, Rao A, Lorincz MC, Ramalho-Santos M. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. 2013; 500:222–26. https://doi.org/10.1038/nature12362.
73. Chen J, Guo L, Zhang L, Wu H, Yang J, Liu H, Wang X, Hu X, Gu T, Zhou Z, Liu J, Liu J, Wu H, et al. Vitamin C modulates TET1 function during somatic cell reprogramming. Nat Genet. 2013; 45:1504–09. https://doi.org/10.1038/ng.2807.
74. Liu M, Ohtani H, Zhou W, Ørskov AD, Charlet J, Zhang YW, Shen H, Baylin SB, Liang G, Grønbæk K, Jones PA. Vitamin C increases viral mimicry induced by 5-aza-2′-deoxycytidine. Proc Natl Acad Sci U S A. 2016; 113:10238–44. https://doi.org/10.1073/pnas.1612262113.
75. Schleicher RL, Carroll MD, Ford ES, Lacher DA. Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003-2004 National Health and Nutrition Examination Survey (NHANES). Am J Clin Nutr. 2009; 90:1252–63. https://doi.org/10.3945/ajcn.2008.27016.
76. Mayland CR, Bennett MI, Allan K. Vitamin C deficiency in cancer patients. Palliat Med. 2005; 19:17–20. https://doi.org/10.1191/0269216305pm970oa.
77. Daskalakis M, Nguyen TT, Nguyen C, Guldberg P, Köhler G, Wijermans P, Jones PA, Lübbert M. Demethylation of a hypermethylated P15/INK4B gene in patients with myelodysplastic syndrome by 5-Aza-2′-deoxycytidine (decitabine) treatment. Blood. 2002; 100:2957–64. https://doi.org/10.1182/blood.V100.8.2957.
78. Fandy TE, Herman JG, Kerns P, Jiemjit A, Sugar EA, Choi SH, Yang AS, Aucott T, Dauses T, Odchimar-Reissig R, Licht J, McConnell MJ, Nasrallah C, et al. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood. 2009; 114:2764–73. https://doi.org/10.1182/blood-2009-02-203547.
79. Vasanthakumar A, Godley LA. 5-hydroxymethylcytosine in cancer: significance in diagnosis and therapy. Cancer Genet. 2015; 208:167–77. https://doi.org/10.1016/j.cancergen.2015.02.009. Erratum in: Erratum to "5-hydroxymethylcytosine in cancer: Significance in diagnosis and therapy" [Cancer Genet 208 (2015):167-177]. [Cancer Genet. 2016].
80. Rodger EJ, Chatterjee A, Morison IM. 5-hydroxymethylcytosine: a potential therapeutic target in cancer. Epigenomics. 2014; 6:503–14. https://doi.org/10.2217/epi.14.39.
81. Ye C, Li L. 5-hydroxymethylcytosine: a new insight into epigenetics in cancer. Cancer Biol Ther. 2014; 15:10–15. https://doi.org/10.4161/cbt.27144.
82. Chowdhury B, McGovern A, Cui Y, Choudhury SR, Cho IH, Cooper B, Chevassut T, Lossie AC, Irudayaraj J. The hypomethylating agent Decitabine causes a paradoxical increase in 5-hydroxymethylcytosine in human leukemia cells. Sci Rep. 2015; 5:9281. https://doi.org/10.1038/srep09281.
83. Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002; 1:639–49.
84. Zhao H, Zhu H, Huang J, Zhu Y, Hong M, Zhu H, Zhang J, Li S, Yang L, Lian Y, Wang S, Mao J, Chen Y, et al. The synergy of Vitamin C with decitabine activates TET2 in leukemic cells and significantly improves overall survival in elderly patients with acute myeloid leukemia. Leuk Res. 2018; 66:1–7. https://doi.org/10.1016/j.leukres.2017.12.009.
85. Kumar S, Alnasif N, Fleige E, Kurniasih I, Kral V, Haase A, Luch A, Weindl G, Haag R, Schäfer-Korting M, Hedtrich S. Impact of structural differences in hyperbranched polyglycerol–polyethylene glycol nanoparticles on dermal drug delivery and biocompatibility. Eur J Pharm Biopharm. 2014; 88:625–34. https://doi.org/10.1016/j.ejpb.2014.10.014.
86. Schumacher F, Herrmann K, Florian S, Engst W, Glatt H. Optimized enzymatic hydrolysis of DNA for LC-MS/MS analyses of adducts of 1-methoxy-3-indolylmethyl glucosinolate and methyleugenol. Anal Biochem. 2013; 434:4–11. https://doi.org/10.1016/j.ab.2012.11.001.
87. Speckmann B, Schulz S, Hiller F, Hesse D, Schumacher F, Kleuser B, Geisel J, Obeid R, Grune T, Kipp AP. Selenium increases hepatic DNA methylation and modulates one-carbon metabolism in the liver of mice. J Nutr Biochem. 2017; 48:112–19. https://doi.org/10.1016/j.jnutbio.2017.07.002.