
INTRODUCTION
Radiotherapy is an essential component of cancer therapy [1, 2]. In the field of radiotherapy, clinical management aimed at improving treatment precision has generally focused on delivering radiations to a defined tumor target [3]. On the other hand, in the biological context, the factors used clinically for optimization of treatment planning include tumor size, imaging features, and histopathological typing [3, 4]. The response of tumors to radiotherapy varies even among tumors for which these factors are similar, highlighting the need for additional indices to improve prediction of tumor radiosensitivity.
Precision cancer medicine, which uses the genetic information of individual tumors to guide treatment, is currently emerging in the field of clinical oncology [5]. Advances in next-generation sequencing technologies have enabled the identification of multiple genetic alterations that make tumor cells responsive to molecularly targeted drugs [6–9]. However, the mutation signatures of tumors resistant to radiotherapy have not been fully elucidated. Such mutation signatures would contribute to facilitation of precision radiotherapy, enabling clinicians to stratify patients with radioresistant tumors into treatments with high-intensity radiation modalities, e.g., carbon ion radiotherapy [4].
To address this issue, we investigated the mutation signatures of a unique set of clinical specimens from a uterine cervical cancer that repeatedly locally recurred after multiple rounds of radiotherapy. Exon sequencing of 409 cancer-related genes in treatment-naïve and multiply-recurrent tumors revealed rare simultaneous mutations in KRAS and SMAD4. In addition, we validated the association between this mutation signature and radioresistance by performing meta-analysis of published in vitro data and isogenic cell-based experiments.
RESULTS
Mutation profiles of radioresistant tumors
To investigate mutation signatures associated with radioresistance, we analyzed a series of tumors collected from a patient with uterine cervical cancer who experienced repeated local recurrence after multiple rounds of curative radiotherapy (Table 1). Given that the 5-year local control rate for uterine cervical cancer treated with curative radiotherapy is as high as >90%, this case is considered extremely radioresistant [10]. At the time of diagnosis, the patient was 34 years old and the FIGO stage was IB2. The tumor was positive for HPV-16. The pathological diagnosis (according to the UICC pTNM, 6th edition) was a mucinous adenocarcinoma of the uterine cervix, intestinal plus endocervical type, pT1b2N1MX, G2 (R0, Stage IIIB). Surgery comprised radical hysterectomy and bilateral salpingo-oophorectomy. Lymphatic invasion, vascular invasion, and metastatic foci were observed in the right ovary. Adjuvant chemotherapy comprised six courses of mitomycin C (10 mg/body on Day 1), etoposide (80 mg/body on Days 1, 3, and 5), and cisplatin (45 mg/body on Day 1). Radiotherapy for the first recurrence comprised external body radiotherapy (50 Gy in 25 fractions delivered to the whole pelvis, using the center-seal technique for the latter 20 Gy) and intracavitary brachytherapy (23 Gy delivered in 4 fractions). Radiotherapy for the second recurrence comprised interstitial brachytherapy (30 Gy delivered in 5 fractions). Radiotherapy for the third recurrence comprised interstitial brachytherapy (30 Gy delivered in 5 fractions).
Table 1: Clinical course of the patient and timing of sample collection
Event |
Sample |
Treatment |
Months |
|---|---|---|---|
Diagnosis |
T1 |
0 |
|
Normal |
Surgery |
1 |
|
Adjuvant chemotherapy |
3 |
||
Recurrence at vaginal stump |
11 |
||
Radiotherapy (EBRT+ICBT) |
13 |
||
Recurrence at vaginal stump |
T2 |
31 |
|
Radiotherapy (ISBT) |
32 |
||
Recurrence at vaginal stump |
T3 |
46 |
|
Radiotherapy (ISBT) |
47 |
||
Deceased |
71 |
Abbreviations: EBRT, external body radiotherapy; ICBT, intracavitary brachytherapy; ISBT, interstitial brachytherapy. Treatment details are described in Supplementary Text.
Using the Ion AmpliSeq Comprehensive Cancer Panel (Thermo Fisher Scientific), we performed semiconductor-based next-generation sequencing of exons of 409 cancer-related genes in the treatment-naïve tumor (T1), recurrent tumor after initial radiotherapy (T2), recurrent tumor after secondary radioherapy (T3), and normal tissue of the uterine cervix (as a Control) (Table 1 and Supplementary Figure 1). T2 and T3 were diagnosed as local recurrence based on the fact that pathological findings for T2 and T3 were consistent with T1.
After quality filtering, the number of sequencing reads per sample was 10.6 million (Supplementary Table 1). Average coverage depth was 616 reads per base (Supplementary Table 1). Somatic mutations and copy number variations (CNVs) were determined using the analytical workflow described in Identification of somatic mutations and CNVs in MATERIALS AND METHODS. In total, we identified 91 somatic non-synonymous mutations and 394 CNV regions.
T1 harbored the PIK3CA E545K mutation (Figure 1A and Supplementary Table 2). PIK3CA is the gene most frequently mutated in uterine cervical cancer (14−26% of cases), and E545K is one of three hotspots for activating mutations in this gene [11−13]. T1 also had a high frequency (i.e., 60%; 3/5) of single-nucleotide substitutions in cytosines preceded by thymines, a characteristic of APOBEC mutagenesis, which is the predominant source of mutations in uterine cervical cancer (Figures 1B and 2) [11, 12, 14]. These observations suggested that the genetic characteristics of the treatment-naïve tumor were typical of uterine cervical cancer.
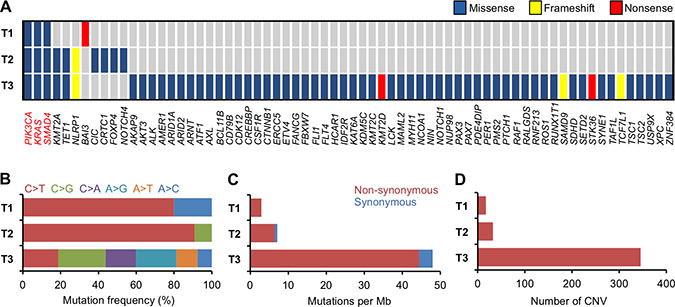
Figure 1: Summary of somatic alterations in treatment-naïve and recurrent tumors. (A) Mutated genes, according to mutation type. Genes found to be mutated in T1−T3 are indicated in red. (B) Mutation spectrum of single-nucleotide substitutions. (C) Number of mutations. (D) Number of CNV regions.
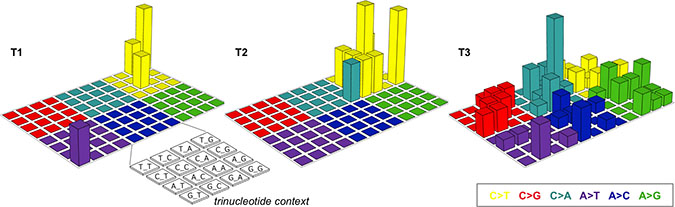
Figure 2: Lego plots of mutational patterns in a three-base context. The somatic single-nucleotide variants identified in T1–3 are categorized into six groups based on the base substitution type and are shown in different colors. Each group is further classified into 16 subgroups based on the 5’ and 3’ neighboring bases [12, 14].
Interestingly, T1−T3 had mutations in KRAS and SMAD4 in common (Figure 1A and Supplementary Table 2). KRAS G12D is a well-studied oncogenic driver, whereas the co-occurrence of R361C and R361H in SMAD4 indicated biallelic inactivation of this tumor suppressor [15−18]. The persistence of these mutations in T1−T3 indicates that they played pivotal roles in the radioresistance that enabled survival of the tumor through repeated rounds of radiotherapy.
Mutations in KMT2A, TET1, and NLRP1 were present in both T2 and T3, but not in T1 (Figure 1A and Supplementary Table 2). In addition, amplification of PIK3CA and FGFR2 genes was observed in both T2 and T3, but not in T1. These observations suggest that the gene alterations are generated, or accumulated, as the tumor survives through the initial round of radiotherapy. Therefore, these gene alterations may be involved in acquisition of radioresistance.
The number of somatic mutations and CNVs in T1 and T2 was comparable, but was higher in T3 than in T2 (Figure 1A, 1C, 1D and Supplementary Table 1). Interestingly, T3 exhibited an unbiased spectrum of single-nucleotide substitutions in contrast with T1 and T2 (Figures 1B and 2).
Association of simultaneous mutations in KRAS and SMAD4 with radioresistance
Among the identified somatic alterations, the simultaneous mutations in KRAS and SMAD4 caught our attention because (i) they have not been reported in uterine cervical cancers, and (ii) their persistence in T1−T3 indicates that this mutation signature contributed to the extreme radioresistance of the tumor. Hence, we investigated the association of this mutation signature with radiosensitivity in published in vitro data. Despite an intensive literature search, we were unable to identify uterine cervical cancer cell lines with simultaneous mutations in KRAS and SMAD4, supporting the idea that this mutation signature is rare among uterine cervical cancers. We then expanded our search to 1039 cell lines derived from various cancers registered in the Cancer Cell Line Encyclopedia (CCLE). This comprehensive search identified 12 cell lines simultaneously carrying (i) gene amplification and/or putative driver mutations in KRAS and (ii) homozygous deletion or truncation mutations in SMAD4 (hereafter referred to as KRASmt/SMAD4mt cell lines) for which in vitro radiosensitivity has been assessed in clonogenic assays (Table 2). As a control, we randomly selected 12 cancer type–matched cell lines without simultaneous mutations in KRAS and SMAD4 (hereafter referred to as Control cell lines) for which published in vitro radiosensitivity data were available (Table 2). The prevalence of KRAS and SMAD4 mutations in the Control cell lines was 50% and 17%, respectively, whereas that in cancer type– and sample size–matched clinical tumor populations was 48% and 2.5%, respectively. This indicates that the randomly selected Control cell lines reflect the profiles of clinical tumors in terms of KRAS mutations, with a relatively high prevalence of SMAD4 mutations (for details, see Identification of KRASmt/SMAD4mt and Control cell lines in CCLE in MATERIALS AND METHODS) (Table 2 and Supplementary Table 3). The surviving fraction after exposure to 2-Gy irradiation, as assessed in clonogenic assays (SF2), is relevant with respect to the tumor response to radiotherapy; the precision of SF2 is sufficient for inter-assay comparison [19, 20]. Based on this evidence, we obtained all available SF2 data for the KRASmt/SMAD4mt and Control cell lines by performing a comprehensive literature search (for details, see Acquisition of SF2 data from the literature in MATERIALS AND METHODS) and compared SF2 data between the two groups (Table 2 and Supplementary Table 4). We found that SF2 in the 12 KRASmt/SMAD4mt cell lines was significantly higher than in the 12 Control cell lines (P = 0.010) (Figure 3A), indicating that simultaneous mutations in KRAS and SMAD4 are associated with radioresistance.
Table 2: KRASmt/SMAD4mt and Control cell lines
Cell line |
Group |
Cancer type |
KRAS |
SMAD4 |
SF2 |
|---|---|---|---|---|---|
AsPC-1 |
KRASmt/SMAD4mt |
pancreatic |
G12D |
HD |
0.66 ± 0.13 |
PK-1 |
KRASmt/SMAD4mt |
pancreatic |
G12D |
HD |
0.50 |
SW1990 |
KRASmt/SMAD4mt |
pancreatic |
G12D |
HD |
0.56 ± 0.19 |
Capan-1 |
KRASmt/SMAD4mt |
pancreatic |
G12V |
S343* |
0.52 ± 0.24 |
CFPAC-1 |
KRASmt/SMAD4mt |
pancreatic |
AMP, G12V |
HD |
0.58 |
PSN1 |
KRASmt/SMAD4mt |
pancreatic |
AMP, G12R |
HD |
0.48 ± 0.00 |
T84 |
KRASmt/SMAD4mt |
colorectal |
G12D |
HD |
0.74 ± 0.28 |
SW403 |
KRASmt/SMAD4mt |
colorectal |
G13V |
HD |
0.59 |
SW480 |
KRASmt/SMAD4mt |
colorectal |
AMP, G12V |
HD |
0.67 ± 0.07 |
SW620 |
KRASmt/SMAD4mt |
colorectal |
AMP, G12V |
HD |
0.59 ± 0.16 |
AGS |
KRASmt/SMAD4mt |
stomach |
G12D |
HD |
0.45 ± 0.06 |
SW1573 |
KRASmt/SMAD4mt |
lung |
G12C |
HD |
0.60 ± 0.08 |
Capan-2 |
Control |
pancreatic |
G12V |
WT |
0.49 ± 0.04 |
MIAPaCa-2 |
Control |
pancreatic |
G12C |
WT |
0.49 ± 0.10 |
PANC-1 |
Control |
pancreatic |
AMP |
WT |
0.73 ± 0.20 |
HCT 116 |
Control |
colorectal |
G13D |
WT |
0.42 ± 0.10 |
LoVo |
Control |
colorectal |
G13D |
WT |
0.38 ± 0.10 |
LS180 |
Control |
colorectal |
G12D |
WT |
0.50 |
RKO |
Control |
colorectal |
WT |
WT |
0.33 ± 0.13 |
SW48 |
Control |
colorectal |
WT |
WT |
0.22 ± 0.06 |
NCI-N87 |
Control |
stomach |
WT |
HD |
0.39 ± 0.15 |
MKN-45 |
Control |
stomach |
WT |
HD |
0.65 ± 0.25 |
NCI-H1437 |
Control |
lung |
WT |
WT |
0.34 |
NCI-H1648 |
Control |
lung |
WT |
WT |
0.16 |
Abbreviations: AMP, amplification; HD, homozygous deletion; WT, wild type. SF2 data were obtained from previous publications, and the average ± standard deviation (if available) value for each cell line is shown (for details, see Acquisition of SF2 data from the literature in MATERIALS AND METHODS).
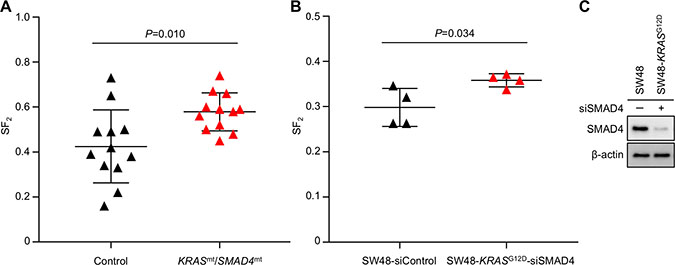
Figure 3: Higher SF2 is associated with simultaneous mutations in KRAS and SMAD4. (A, B) SF2 values (assessed in a clonogenic assay) for 12 KRASmt/SMAD4mt and 12 Control cell lines (obtained from the literature) (A) and those for siControl-treated SW48 and siSMAD4-treated SW48-KRASG12D cells (experiments were performed in quadruplicate) (B). Bars denote the mean ± standard deviation (for details, see Acquisition of SF2 data from the literature in MATERIALS AND METHODS and Supplementary Table 6). (C) Immunoblots showing expression of SMAD4 and β-actin (as a loading Control) in siControl-treated SW48 and siSMAD4-treated SW48-KRASG12D cells.
Finally, we examined the association between the KRASmt/SMAD4mt signature and radioresistance measured in isogenic cell-based experimental systems. For these experiments, we used KRAS G12D-knock-in cells (SW48-KRASG12D) treated with SMAD4-siRNA (siSMAD4) which mimics loss-of-function mutations in SMAD4, and the parental SW48 cell line (as the Control) (Table 2). We found that the SF2 values for siSMAD4-treated SW48-KRASG12D cells were significantly higher than those for SW48 cells (P = 0.034) (Figure 3B and 3C). These data support an association between radioresistance and simultaneous mutations in KRAS and SMAD4.
DISCUSSION
The profiles of mutations that occur over the clinical course of cancer have been studied extensively in the context of recurrence after chemotherapy. For example, EGFR T790M is enriched after treatment with EGFR tyrosine kinase inhibitors, and this mutation contributes to resistance to this type of therapy [21]. Recently, diverse genetic branching was reported in spatiotemporally different tumors in a single patient [22]. From this perspective, the present study is the first to compare the mutation profiles of treatment-naïve tumors with those of multiply-recurrent tumors collected from the same disease site following multiple rounds of radiotherapy. Our results highlighted (i) activating mutations in PIK3CA and KRAS, and putative inactivating mutations in SMAD4, as trunk mutation signatures that persisted over the clinical course; and (ii) mutations in KMT2A, TET1, and NLRP1, and gene amplification of PIK3CA and FGFR2, as acquired mutation signatures observed only in recurrent tumors after radiotherapy. Comprehensive mining of published in vitro data revealed that simultaneous mutations in KRAS and SMAD4 are associated with cancer cell radioresistance. Although this was a pilot study that analyzed only one case, these data provide proof-of-principle for the analytical pipeline that we employed, which explores clinically relevant mutation signatures for radioresistance, indicating that this approach is worth pursuing in larger cohorts in the future.
Several studies have demonstrated predictive biomarkers for radiosensitivity [23]. In many cases, radiosensitivity is predicted based on gene expression profiles of frozen tumor tissues, which are generally difficult to collect in routine clinical practice. Importantly in this regard, the KRASmt/SMAD4mt signature can be assessed using formalin-fixed paraffin-embedded (FFPE) tissues, which are routinely used for pathological diagnosis, indicating that this approach is suitable for application in the clinic. The predictive value of the KRASmt/SMAD4mt signature for radioresistance should be verified further in clinical studies.
To date, two large-scale whole-exome sequencing studies of uterine cervical cancer have been conducted to identify its genomic landscape [8, 9]. According to these studies, the genomic profiles of uterine cervical cancer are characterized by recurrent alterations in PIK3CA, EP300, FBXW7, PTEN, HLA-B, and KRAS (in 14−26%, 11−12%, 10−11%, 7-8%, 6-8%, and 3–9% of cases, respectively), along with a predominant APOBEC mutagenesis pattern. Mutations in PIK3CA and KRAS have been studied using targeted sequencing approaches, which identified mutation frequencies comparable with those observed in whole-exome sequencing studies [24−28]; while alterations in SMAD4 was identified in only one of the whole-exome sequencing study at 5% of cases [11]. Concomitant alterations in KRAS and SMAD4 have not been reported for uterine cervical cancer; indeed, data combined from different types of tumors suggest that such alterations are rare (i.e., 1.2%; 279/22928) (Supplementary Table 5 and Supplementary Figure 2). Nevertheless, it is noteworthy that pancreatic cancer, which is clinically radioresistant [29], shows the highest frequency of alterations (24%). Interestingly, a previous study demonstrates that treatment of hTERT-immortalized human pancreatic duct cells in which KRAS is overexpressed and SMAD4 is suppressed upregulates NF-kB [30]. Since NF-kB regulates multiple anti-apoptotic genes and is considered to be a major contributor to radioresistance [31], it is possible that simultaneous mutations in KRAS and SMAD4 (as observed in the present study) contributed to the radioresistant nature of the tumor via this mechanism.
KMT2A is a member of the KMT2 complex, which methylates lysine 4 on histone H3 (H3K4) to promote genome accessibility and transcription [32]. A recent study reports broad trimethylation of H3K4 in pan-cancer tumor suppressor genes, including TP53, in normal cells, and shortage of the trimethylation pattern in cancer cells in opposite [33]. In cancer, common mutations of KMT2A are nonsense, frameshift, and missense mutations; this suggests that KMT2A mutations observed in cancer contribute to loss-of-function of KMT2A [32], whereas suppression of TP53 contributes to resistance of cancer cells to X-rays [34]. Taken together, the evidence suggests that the KMT2A mutations observed herein might play roles in acquisition of radioresistance by tumor cells through loss-of-function of KMT2A, which contributes to suppression of TP53 and other tumor suppressor genes, transcription of which is regulated by methylation on H3K4.
TET1 is an enzyme that catalyzes successive oxidation of 5-methylcytosine to 5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxylcytosine to promote DNA demethylation [35]. Mutations in TET1 occur in a wide variety of solid tumors, albeit at relatively low frequency (0.1−10% of cases) [35]. However, we do not have solid evidence on how TET1 mutations contribute to onset and maintenance of tumors, and it is unclear whether TET1 mutations confer a selective advantage, or whether they represent inconsequential passenger mutations; this warrants further research [35].
Studies suggest that FGF2 upregulates expression of anti-apoptotic proteins such as BCL-2 by activating S6 kinase within the PI3K-AKT signaling pathway, thereby promoting resistance to chemotherapy [36]. Amplification of PIK3CA and FGFR2 might contribute to acquisition of radioresistance by the tumor in the present study. In addition, amplification of PIK3CA and FGFR2 in T2 and T3, combined with activating mutations in KRAS and PIK3CA in T1−3, indicates a critical role for the RAS-PI3K axis in tumor survival through multiple rounds of radiotherapy [37].
Several mutations detected in T1 or T2 were not detected in T3. It is possible that radiotherapy induces nucleotide modifications in these genes in some cancer cells, which was followed by clonal selection. Another possibility is that differences in the mutational profiles observed in these samples are based on genetic heterogeneity within the tumor, which was retained over the clinical course. Further studies are warranted.
The present study has the following limitations. First, we used a commercially available gene panel that contains only 409 cancer-related genes. Therefore, we were not able to investigate the mutational status of genes not listed in the panel. It should be noted that a number of mutations in genes not listed in the panel do occur in uterine cervical cancers [38]. Further studies based on whole-exome sequencing will help to identify the mutational signature associated with radioresistance in uterine cervical cancer. Second, we analyzed only one case, due to the rarity of patients who receive multiple rounds of radiotherapy at the same biopsy-accessible disease site. Third, we did not investigate the mechanisms underlying the radioresistance associated with the KRASmt/SMAD4mt signature; we considered such an effort to be outside the scope of the present study, which primarily aimed to discover clinically applicable mutation signatures associated with radioresistance. Fourth, we diagnosed T2 and T3 as local recurrence based on the fact that pathological findings for T2 and T3 are consistent with those of T1. The fact that T1−T3 shared rare simultaneous mutations in KRAS and SMAD4 support the idea that T2 and T3 recurred from T1. Nevertheless, the possibility that T2 and T3 are secondary cancers distinct from T1 cannot be ruled out completely because there is no evidence so far, on the difference in mutational signatures between locally recurrent tumors and secondary tumors. Fifth, we did not analyze samples at first recurrence because our aim was to identify mutational signatures associated with radioresistance by comparing mutational profiles between treatment-naïve tumors and tumors that recurred after radiotherapy; therefore, analyzing samples from the first recurrence, which is a tumor that recurs after chemotherapy, was beyond the scope of the present study. Nevertheless, we confirmed the presence of mutations commonly detected in T1−T3, i.e., PIK3CA (E545K), KRAS (G12D), and SMAD4 (R361C and R361H), by Sanger sequencing of tumor samples from the first recurrence, indicating that this tumor also recurred from T1 and shares the trunk mutational characteristics with T2 and T3 (Supplementary Figure 3). Sixth, Sanger sequencing results for PIK3CA (c.1633G>A) and TET1 (c.242G>A), in which mutations in T2 were not detected, should have been validated using other methods, including real-time polymerase chain reaction (PCR) and droplet digital PCR. However, we were not able to conduct these experiments due to a shortage of sample. Lastly, the T3 specimen was smaller than the other specimens, which might affect smaller coverage for T3 compared to that for the others.
In summary, we identified rare simultaneous mutations in KRAS and SMAD4 in a series of tumors that survived multiple rounds of radiotherapy and validated the association between the mutation signature and radioresistance by conducting meta-analysis of published in vitro data and isogenic cell-based experiments. Based on this pilot study, we propose that our analytical pipeline would be useful for identification of clinically relevant mutation signatures of radioresistance, and that this approach warrants validation in larger cohorts. The predictive value of the KRASmt/SMAD4mt signature for radioresistance should also be evaluated in the clinic.
MATERIALS AND METHODS
Ethics
This study was approved by the Institutional Review Boards of Gunma University Hospital (approved number: 1109). Written informed consent was obtained from the patient. The study was conducted in accordance with the ethical principles of the Declaration of Helsinki.
Tissue sample collection
Tumor tissues were collected by punch biopsy. Normal tissues of the uterine cervix were collected by surgery. The presence of malignant cells was pathologically confirmed in all tumor specimens, and the absence of malignant cells was likewise confirmed in the normal tissue specimen.
DNA preparation
DNA was extracted from the FFPE tissues using the QIAamp DNA FFPE Tissue kit (Qiagen). The TaqMan RNase P Detection Reagents kit (Thermo Fisher Scientific) was used to quantify purified DNA.
Semiconductor-based next-generation sequencing
DNA (40 ng) was subjected to multiplex PCR amplification using an Ion AmpliSeq Comprehensive Cancer Panel that covers 95.4% of the exons of the 409 cancer-related genes. Library preparation and sequencing with the Ion Torrent sequencer were performed as previously described [39–41]. The templates were sequenced after emulsion PCR with 6–8 samples per Ion PI chip using the Ion PI HI-Q Chef kit (Thermo Fisher Scientific).
Identification of somatic mutations and CNVs
Somatic mutations and CNVs were determined as previously described, with minor modifications [42]. Briefly, the sequenced data were aligned to the Genome Reference Consortium Human Build 37 (hg19), and reads were counted using Torrent Suite version 5.0 (Thermo Fisher Scientific). Single-nucleotide variants, insertions, and deletions were determined using the Ion Reporter software 5.0 tumor-normal workflow (Thermo Fisher Scientific), in which germline variants were subtracted from the tumor variants. The following criteria were used as cutoffs: total coverage >20, variant coverage >10, variant frequency >5%, and minor allele frequency <0.1%. All identified single-nucleotide variants, insertions, and deletions were visually inspected using the Integrative Genomics Viewer software to filter out possible strand-specific errors, such as a mutation detected only in either the forward or reverse DNA strand [43]. The dbSNP database was used to exclude SNPs from the called variants. SIFT, Polyphen-2, and Grantham scores were used to estimate evolutionary conservation and the effects of amino-acid substitution on the structure and function of the protein. CNVs were determined with the Ion Reporter software using an algorithm based on a Hidden Markov Model. To verify the robustness of next-generation sequencing, Sanger sequencing was performed for the somatic non-synonymous mutations in PIK3CA, KRAS, and SMAD4 that were commonly identified in T1−T3, as well as for those in KMT2A, TET1, and NLRP1 that were commonly identified in T2 and T3 but not in T1. As a result, the mutational status for 93.7% (30/32) of the samples analyzed was consistent with that observed after next-generation sequencing, demonstrating the robustness of the analytical workflow (Supplementary Figures 4 and 5).
Identification of KRASmt/SMAD4mt and Control cell lines in CCLE
The KRASmt/SMAD4mt and Control cell lines were identified based on genetic information in cBioportal for Cancer Genomics (Table 2) [44, 45]. The KRASmt/SMAD4mt cell lines were derived from pancreatic, colorectal, stomach, and lung cancers. Control cell lines were selected randomly from CCLE, matching the cancer type to the KRASmt/SMAD4mt cell lines. Differences in the distribution of cancer types between the KRASmt/SMAD4mt and Control cell lines were not statistically significant (P = 0.68, as assessed by Fisher’s exact test). The prevalence of KRAS and SMAD4 mutations in clinical tumor specimens of pancreatic, colorectal, stomach, and lung cancer was confirmed using cBioportal for Cancer Genomics, and the combined prevalence of the mutations in the four cancer types was calculated by matching the relative proportion for each cancer type to those of the Control cell lines (Supplementary Table 3).
Acquisition of SF2 data from the literature
All published SF2 data for the KRASmt/SMAD4mt and Control cell lines were obtained as previously described [20]. Briefly, a PubMed search was performed for each cell line using the terms “[cell line name] AND (X-rays OR gamma rays OR radiation)”. Two radiation oncologists (EN and TBMP) examined all manuscripts identified by the search in their entirety, and identified publications reporting data obtained from clonogenic assays after treatment with X-rays or γ-rays alone. This examination identified 95 relevant papers, from which SF2 data were obtained (Table 2 and Supplementary Table 4). For each cell line, the average SF2 was calculated and used for analysis.
Cell culture
SW48 and SW48-KRASG12D cells were purchased from ATCC and cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum (Sigma).
siRNA knockdown
Transfection of siRNA was performed using HiPerFect (Qiagen), as previously described [46]. Briefly, siSMAD4 (Thermo Fisher Scientific, 4390824) or Control-siRNA (siControl, GGGAUACCUAGACGUUCUAdTdT; Sigma) was added to suspended cells after trypsinization. After 24 h, cells were trypsinized, suspended, and re-transfected with the siRNAs. Cells were incubated for 24 h after the second transfection and then subjected to clonogenic assay and immunoblotting (in parallel).
Clonogenic assay
The clonogenic assay was performed as previously described [47]. Briefly, cells were seeded in 6 well plates at the specified numbers and then exposed to X-ray irradiation using a Faxitron RX-650 apparatus (100 kVp, 1.14 Gy/min; Faxitron Bioptics). After incubating for 10 days, cells were fixed with methanol and stained with crystal violet. Colonies comprising at least 50 cells were counted. The surviving fraction of irradiated cells was normalized to that of the corresponding unirradiated Controls.
Immunoblotting
Immunoblotting was performed as previously described [48]. The following antibodies were purchased from Cell Signaling Technology: SMAD4 (38454) and β-actin (3700). Uncropped versions of the immunoblots are shown in Supplementary Figure 6.
Statistical analysis
Differences in SF2 between two groups were examined as follows: First, normality was confirmed using the Shapiro-Wilk test. Next, variance was examined using an F test. Differences between two groups with equal variance were examined using Student’s t-test. Differences between groups without equal variance were examined using Welch’s t-test. Differences in the distribution of cancer type between the KRASmt/SMAD4mt cell lines and Control cell lines were examined using Fisher’s exact test. All statistical analyses were performed using Prism 6 (GraphPad) or EZR (Saitama Medical Center), which is a graphical user interface for R ver. 3.3.2 (The R Foundation for Statistical Computing) [49]. A P value < 0.05 was considered significant.
Abbreviations
CCLE: Cancer Cell Line Encyclopedia; CNVs: copy number variations; FFPE: formalin-fixed paraffin-embedded; PCR: polymerase chain reaction; SF2: the surviving fraction after 2-Gy irradiation as assessed by clonogenic assays; siControl: Control-siRNA; siSMAD4: SMAD4-siRNA.
Author contributions
EN, YS, HS, SK, YY, TBMP, Y. Hagiwara, and Y. Hirota conducted experiments. EN, YS, TBMP, and Y. Hagiwara drafted the manuscript. KM, KA, NK, NO, YT, AA, MI, KT, SN, and T. Tamaki acquired clinical samples. AS, T. Ohno, T. Tokino, and TN contributed to study design. T. Oike contributed to data interpretation and takes full responsibility for the manuscript and all data presented. All authors read and approved the manuscript.
ACKNOWLEDGMENTS
We thank Dr. Yasuyuki Kanke of the National Cancer Institute for technical assistance.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
FUNDING
This work was supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (programs for Leading Graduate Schools, Cultivating Global Leaders in Heavy Ion Therapeutics and Engineering). This work was also supported by Grants-in-Aid from the Japan Society for the Promotion of Science for KAKENHI, Grant Number 16H06279.
REFERENCES
1. Thariat J, Hannoun-Levi JM, Sun Myint A, Vuong T, Gérard JP. Past, present, and future of radiotherapy for the benefit of patients. Nat Rev Clin Oncol. 2013; 10:52–60. https://doi.org/10.1038/nrclinonc.2012.203.
2. Durante M, Orecchia R, Loeffler JS. Charged-particle therapy in cancer: clinical uses and future perspectives. Nat Rev Clin Oncol. 2017; 14:483–95. https://doi.org/10.1038/nrclinonc.2017.30.
3. Scott JG, Berglund A, Schell MJ, Mihaylov I, Fulp WJ, Yue B, Welsh E, Caudell JJ, Ahmed K, Strom TS, Mellon E, Venkat P, Johnstone P, et al. A genome-based model for adjusting radiotherapy dose (GARD): a retrospective, cohort-based study. Lancet Oncol. 2017; 18:202–11. https://doi.org/10.1016/S1470-2045(16)30648-9.
4. Oike T, Sato H, Noda SE, Nakano T. Translational research to improve the efficacy of carbon ion radiotherapy: experience of Gunma University. Front Oncol. 2016; 6:139. https://doi.org/10.3389/fonc.2016.00139.
5. Abrams J, Conley B, Mooney M, Zwiebel J, Chen A, Welch JJ, Takebe N, Malik S, McShane L, Korn E, Williams M, Staudt L, Doroshow J. National Cancer Institute’s Precision Medicine Initiatives for the new National Clinical Trials Network. Am Soc Clin Oncol Educ Book. 2014; 34:71–76. https://doi.org/10.14694/EdBook_AM.2014.34.71.
6. Jordan EJ, Kim HR, Arcila ME, Barron D, Chakravarty D, Gao J, Chang MT, Ni A, Kundra R, Jonsson P, Jayakumaran G, Gao SP, Johnsen HC, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. 2017; 7:596–609. https://doi.org/10.1158/2159-8290.CD-16-1337.
7. Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A, Wu W, Goessl C, Runswick S, Conte P. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med. 2017; 377:523–33. https://doi.org/10.1056/NEJMoa1706450.
8. Lamba JK, Chauhan L, Shin M, Loken MR, Pollard JA, Wang YC, Ries RE, Aplenc R, Hirsch BA, Raimondi SC, Walter RB, Bernstein ID, Gamis AS, et al. CD33 Splicing Polymorphism Determines Gemtuzumab Ozogamicin Response in De Novo Acute Myeloid Leukemia: Report From Randomized Phase III Children’s Oncology Group Trial AAML0531. J Clin Oncol. 2017; 35:2674–82. https://doi.org/10.1200/JCO.2016.71.2513.
9. Harris MH, DuBois SG, Glade Bender JL, Kim A, Crompton BD, Parker E, Dumont IP, Hong AL, Guo D, Church A, Stegmaier K, Roberts CW, Shusterman S, et al. Multicenter Feasibility Study of Tumor Molecular Profiling to Inform Therapeutic Decisions in Advanced Pediatric Solid Tumors: The Individualized Cancer Therapy (iCat) Study. JAMA Oncol. 2016; 2:608. https://doi.org/10.1001/jamaoncol.2015.5689.
10. Ohno T, Noda SE, Okonogi N, Murata K, Shibuya K, Kiyohara H, Tamaki T, Ando K, Oike T, Ohkubo Y, Wakatsuki M, Saitoh JI, Nakano T. In-room computed tomography-based brachytherapy for uterine cervical cancer: results of a 5-year retrospective study. J Radiat Res. 2017; 58:543–51.
11. Cancer Genome Atlas Research Network; Albert Einstein College of Medicine; Analytical Biological Services; Barretos Cancer Hospital; Baylor College of Medicine; Beckman Research Institute of City of Hope; Buck Institute for Research on Aging; Canada’s Michael Smith Genome Sciences Centre; Harvard Medical School; Helen F. Graham Cancer Center &Research Institute at Christiana Care Health Services; HudsonAlpha Institute for Biotechnology; ILSbio, LLC; Indiana University School of Medicine, et al. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017; 543:378–84. https://doi.org/10.1038/nature21386.
12. Ojesina AI, Lichtenstein L, Freeman SS, Pedamallu CS, Imaz-Rosshandler I, Pugh TJ, Cherniack AD, Ambrogio L, Cibulskis K, Bertelsen B, Romero-Cordoba S, Treviño V, Vazquez-Santillan K, et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014; 506:371–75. https://doi.org/10.1038/nature12881.
13. Karakas B, Bachman KE, Park BH. Mutation of the PIK3CA oncogene in human cancers. Br J Cancer. 2006; 94:455–59. https://doi.org/10.1038/sj.bjc.6602970.
14. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013; 499:214–18. https://doi.org/10.1038/nature12213.
15. Haigis KM. KRAS Alleles: The Devil Is in the Detail. Trends Cancer. 2017; 3:686–97. https://doi.org/10.1016/j.trecan.2017.08.006.
16. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012; 72:2457–67. https://doi.org/10.1158/0008-5472.CAN-11-2612.
17. Fleming NI, Jorissen RN, Mouradov D, Christie M, Sakthianandeswaren A, Palmieri M, Day F, Li S, Tsui C, Lipton L, Desai J, Jones IT, McLaughlin S, et al. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res. 2013; 73:725–35. https://doi.org/10.1158/0008-5472.CAN-12-2706.
18. Sonkin D, Hassan M, Murphy DJ, Tatarinova TV. Tumor suppressors status in cancer cell line Encyclopedia. Mol Oncol. 2013; 7:791–98. https://doi.org/10.1016/j.molonc.2013.04.001.
19. Torres-Roca JF. A molecular assay of tumor radiosensitivity: a roadmap towards biology-based personalized radiation therapy. Per Med. 2012; 9:547–57. https://doi.org/10.2217/pme.12.55.
20. Nuryadi E, Mayang Permata TB, Komatsu S, Oike T, Nakano T. Inter-assay precision of clonogenic assays for radiosensitivity in cancer cell line A549. Oncotarget. 2018; 9:13706–12. https://doi.org/10.18632/oncotarget.24448.
21. Suda K, Onozato R, Yatabe Y, Mitsudomi T. EGFR T790M mutation: a double role in lung cancer cell survival? J Thorac Oncol. 2009; 4:1–4. https://doi.org/10.1097/JTO.0b013e3181913c9f.
22. Ma P, Fu Y, Cai MC, Yan Y, Jing Y, Zhang S, Chen M, Wu J, Shen Y, Zhu L, Chen HZ, Gao WQ, Wang M, et al. Simultaneous evolutionary expansion and constraint of genomic heterogeneity in multifocal lung cancer. Nat Commun. 2017; 8:823. https://doi.org/10.1038/s41467-017-00963-0.
23. Hall WA, Bergom C, Thompson RF, Baschnagel AM, Vijayakumar S, Willers H, Li XA, Schultz CJ, Wilson GD, West CML, Capala J, Coleman CN, Torres-Roca JF, et al. Precision Oncology and Genomically Guided Radiation Therapy: A Report From the American Society for Radiation Oncology/American Association of Physicists in Medicine/National Cancer Institute Precision Medicine Conference. Int J Radiat Oncol Biol Phys. 2017; S0360–3016:30982–3.
24. Wright AA, Howitt BE, Myers AP, Dahlberg SE, Palescandolo E, Van Hummelen P, MacConaill LE, Shoni M, Wagle N, Jones RT, Quick CM, Laury A, Katz IT, et al. Oncogenic mutations in cervical cancer: genomic differences between adenocarcinomas and squamous cell carcinomas of the cervix. Cancer. 2013; 119:3776–83. https://doi.org/10.1002/cncr.28288.
25. McIntyre JB, Wu JS, Craighead PS, Phan T, Köbel M, Lees-Miller SP, Ghatage P, Magliocco AM, Doll CM. PIK3CA mutational status and overall survival in patients with cervical cancer treated with radical chemoradiotherapy. Gynecol Oncol. 2013; 128:409–14. https://doi.org/10.1016/j.ygyno.2012.12.019.
26. Wegman P, Ahlin C, Sorbe B. Genetic alterations in the K-Ras gene influence the prognosis in patients with cervical cancer treated by radiotherapy. Int J Gynecol Cancer. 2011; 21:86–91. https://doi.org/10.1097/IGC.0b013e3182049924.
27. Kang S, Kim HS, Seo SS, Park SY, Sidransky D, Dong SM. Inverse correlation between RASSF1A hypermethylation, KRAS and BRAF mutations in cervical adenocarcinoma. Gynecol Oncol. 2007; 105:662–66. https://doi.org/10.1016/j.ygyno.2007.01.045.
28. Pappa KI, Choleza M, Markaki S, Giannikaki E, Kyroudi A, Vlachos G, Voulgaris Z, Anagnou NP. Consistent absence of BRAF mutations in cervical and endometrial cancer despite KRAS mutation status. Gynecol Oncol. 2006; 100:596–600. https://doi.org/10.1016/j.ygyno.2005.09.029.
29. Seshacharyulu P, Baine MJ, Souchek JJ, Menning M, Kaur S, Yan Y, Ouellette MM, Jain M, Lin C, Batra SK. Biological determinants of radioresistance and their remediation in pancreatic cancer. Biochim Biophys Acta. 2017; 1868:69–92.
30. Bera A, Zhao S, Cao L, Chiao PJ, Freeman JW. Oncogenic K-Ras and loss of Smad4 mediate invasion by activating an EGFR/NF-κB Axis that induces expression of MMP9 and uPA in human pancreas progenitor cells. PLoS One. 2013; 8:e82282. https://doi.org/10.1371/journal.pone.0082282.
31. Magné N, Toillon RA, Bottero V, Didelot C, Houtte PV, Gérard JP, Peyron JF. NF-kappaB modulation and ionizing radiation: mechanisms and future directions for cancer treatment. Cancer Lett. 2006; 231:158–68. https://doi.org/10.1016/j.canlet.2005.01.022.
32. Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. 2015; 15:334–46. https://doi.org/10.1038/nrc3929.
33. Chen K, Chen Z, Wu D, Zhang L, Lin X, Su J, Rodriguez B, Xi Y, Xia Z, Chen X, Shi X, Wang Q, Li W. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat Genet. 2015; 47:1149–57. https://doi.org/10.1038/ng.3385.
34. Amornwichet N, Oike T, Shibata A, Ogiwara H, Tsuchiya N, Yamauchi M, Saitoh Y, Sekine R, Isono M, Yoshida Y, Ohno T, Kohno T, Nakano T. Carbon-ion beam irradiation kills X-ray-resistant p53-null cancer cells by inducing mitotic catastrophe. PLoS One. 2014; 9:e115121. https://doi.org/10.1371/journal.pone.0115121.
35. Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016; 30:733–50. https://doi.org/10.1101/gad.276568.115.
36. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010; 10:116–29. https://doi.org/10.1038/nrc2780.
37. Castellano E, Downward J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer. 2011; 2:261–74. https://doi.org/10.1177/1947601911408079.
38. Agarwal SM, Raghav D, Singh H, Raghava GP. CCDB: a curated database of genes involved in cervix cancer. Nucleic Acids Res. 2011; 39:D975–79. https://doi.org/10.1093/nar/gkq1024.
39. Grasso C, Butler T, Rhodes K, Quist M, Neff TL, Moore S, Tomlins SA, Reinig E, Beadling C, Andersen M, Corless CL. Assessing copy number alterations in targeted, amplicon-based next-generation sequencing data. J Mol Diagn. 2015; 17:53–63. https://doi.org/10.1016/j.jmoldx.2014.09.008.
40. Ikeda H, Ishiguro K, Igarashi T, Aoki Y, Hayashi T, Ishida T, Sasaki Y, Tokino T, Shinomura Y. Molecular diagnostics of a single drug-resistant multiple myeloma case using targeted next-generation sequencing. Onco Targets Ther. 2015; 8:2805–15. https://doi.org/10.2147/OTT.S86515.
41. Singh RR, Patel KP, Routbort MJ, Reddy NG, Barkoh BA, Handal B, Kanagal-Shamanna R, Greaves WO, Medeiros LJ, Aldape KD, Luthra R. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J Mol Diagn. 2013; 15:607–22. https://doi.org/10.1016/j.jmoldx.2013.05.003.
42. Nakagaki T, Tamura M, Kobashi K, Koyama R, Fukushima H, Ohashi T, Idogawa M, Ogi K, Hiratsuka H, Tokino T, Sasaki Y. Profiling cancer-related gene mutations in oral squamous cell carcinoma from Japanese patients by targeted amplicon sequencing. Oncotarget. 2017; 8:59113–22. https://doi.org/10.18632/oncotarget.19262.
43. Website for Integrative Genomics Viewer. http://www.broadinstitute.org/igv.
44. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012; 483:603–07. https://doi.org/10.1038/nature11003.
45. Website for cBioportal for Cancer Genomics. http://www.cbioportal.org.
46. Sato H, Niimi A, Yasuhara T, Permata TB, Hagiwara Y, Isono M, Nuryadi E, Sekine R, Oike T, Kakoti S, Yoshimoto Y, Held KD, Suzuki Y, et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun. 2017; 8:1751. https://doi.org/10.1038/s41467-017-01883-9.
47. Kobayashi D, Oike T, Shibata A, Niimi A, Kubota Y, Sakai M, Amornwhichet N, Yoshimoto Y, Hagiwara Y, Kimura Y, Hirota Y, Sato H, Isono M, et al. Mitotic catastrophe is a putative mechanism underlying the weak correlation between sensitivity to carbon ions and cisplatin. Sci Rep. 2017; 7:40588. https://doi.org/10.1038/srep40588.
48. Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, van Rossum-Fikkert S, Kertokalio A, Romoli F, Ismail A, Ismalaj E, Petricci E, et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell. 2014; 53:7–18. https://doi.org/10.1016/j.molcel.2013.11.003.
49. Kanda Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013; 48:452–58. https://doi.org/10.1038/bmt.2012.244.