
INTRODUCTION
Breast cancer (BC) is a heterogeneous disease that develops and progresses from alterations in the genes that govern cell growth, proliferation and differentiation [1, 2]. At the same time, these molecular alterations are responsible for primary and secondary treatment resistance and may represent a major limitation to cancer treatment efficacy. In the last two decades, increasing knowledge of genomic abnormalities associated with gain of function or downstream signal activation involved in BC evolution allowed for new therapeutic approaches “tailored” on identified molecular alterations. The revolutionary era of targeted therapy shifted the classic paradigm of BC treatment from an approach based on pathological and clinical characteristics [3] to personalized medicine. This is based on the match between molecular alteration conferring a survival advantage to cancer cells, and the targeted drug [4]. An increasing number of molecularly targeted drugs are currently available for clinical practice or in the context of clinical trials. Nowadays, the main challenge remains the identification of predictive biomarkers for the selection of optimal treatment, in order to spare patients from treatment-associated side effects and to minimize the overall cost [5]. On this basis, parallel to the development of new therapeutic strategies, researches are looking for molecular biomarkers able to predict response to those treatments. For some of these targeted therapies, predictive biomarkers have already been identified in clinical trials [6]. In particular, PIK3CA mutations have already been shown to predict sensitivity to Everolimus [7], Buparlisib [8] and Taselisib [9] as well as resistance to Lapatinib [10]. AKT1 mutations can predict sensitivity to Everolimus [7], while ESR1 mutations can predict sensitivity to Fulvestrant and resistance to Exemestane [11]. Furthermore, HER2-negative patients with an ERBB2 somatic mutation are potentially good candidates for HER2-targeted therapy [12, 13], as already shown by Neratinib in two recently published clinical trials [14, 15].
The main purpose of our study was to investigate the mechanisms of treatment resistance in a sample of metastatic BC patients. Since BC behaves as an evolving entity, with metastases acquiring different biological profiles as compared to their matched primary tumors [16, 17], we evaluated a panel of 25 genes involved in the mechanisms of endocrine and targeted treatment resistance in paired BC samples (primary and recurrence) of patients with metastatic BC.
RESULTS
Patient and sample characteristics
The tumor characteristics of the 16 patients enrolled in the study are described in Table 1. The median age at BC diagnosis was 57 years (range 35–81). Following the classification proposed in the 13th St Gallen International Breast Cancer Conference in 2013, seven patients (43.75%) were diagnosed with Luminal A-like BC. This means estrogen-receptor positive, progesterone-receptor ≥20%, HER2 negative, and low levels of MIB1 (<20%). The other 9 patients (56.25%) had Luminal B-like BC (estrogen-receptor positive and progesterone-receptor <20% or HER2 positive or levels of MIB1 ≥20%). Of these patients, 4 were Luminal B-like HER2 positive.
Table 1: Patients and sample characteristics
Discordant ER, PR and HER2 reported in red.
At presentation, 3 patients (18.75%) had Stage IA BC, 4 patients (25%) had Stage IIA, 2 patients (12.5%) had Stage IIB, 3 (18.75%) had Stage IIIA, 1 (6.25%) had Stage IIIB, 1 (6.25%) had Stage IIIC and 2 (12.5%) had Stage IV disease. The most common sites of relapse were lymph-nodes (9 patients, 56.25%), bones (7 patients, 43.75%) and skin (6 patients, 37.5%). As regards the biopsy of the relapse/metastasis, 7 patients (43.75%) underwent skin or soft tissues biopsy, 5 patients (31.25%) had lymph-node biopsy, 2 patients (12.5%) had liver biopsy, one patient (6.25%) underwent single lung metastasis resection, and lastly one patient (6.25%) underwent ipsilateral mastectomy for a local recurrence. In 8 cases (50%), the relapse/metastasis maintained the same immunohistochemical characteristics [i.e. the same hormone receptors (HR) and HER2 status] of the primary tumor, whereas 8 (50%) changed during progression. More specifically, estrogen receptor (ER) and progesteron receptor (PR) were considered positive when ≥1%, according to the last ASCO/CAPS Guidelines [18]. Rates of discordance were 18.75%, 31.25% and 6.25% for ER, PR and HER2, respectively.
Predictive value of primary tumor mutations
Two patients (12.5%) received neoadjuvant chemotherapy (anthracycline and taxane-based), whereas 8 patients (50%) received adjuvant chemotherapy (1 anthracycline and taxane-based, 3 anthracycline-based, 2 taxane-based and 2 patients with cyclophosphamide, methotrexate and fluorouracil). One patient (Patient 12) received endocrine treatment (letrozole and fulvestrant) with neoadjuvant purposes, but she achieved no response and underwent radical mastectomy with axillary lymph node dissection. All primary tumor tissues analyzed were taken before initiation of systemic treatment. Six patients (37.5%) received adjuvant tamoxifen alone or with LHRH analogues, 3 patients (18.75%) received adjuvant aromatase inhibitors alone or with LHRH analogues, whereas 4 patients (25%) received an adjuvant sequence of tamoxifen and aromatase inhibitors with or without LHRH analogues. None of the patients, including those with HER2-positive tumor, received HER2-targeted therapy in the neoadjuvant or adjuvant setting.
Two patients (12.5%) were diagnosed with de novo Stage IV BC, whereas the other 14 patients (87.5%) developed loco-regional or distant recurrence after receiving treatment for primary BC and particularly during neo/adjuvant endocrine therapies. The median disease-free survival (DFS) time for these patients was 35.38 months (range 3–58) from surgery or from the end of adjuvant chemotherapy. In more details, 4 patients (25%) had primary resistance to endocrine therapy, i.e. they relapsed while on the first 2 years of adjuvant endocrine therapy, or progressed within the first 6 months of first-line endocrine therapy. On the other hand, the other 12 patients (75%) developed secondary endocrine resistance.
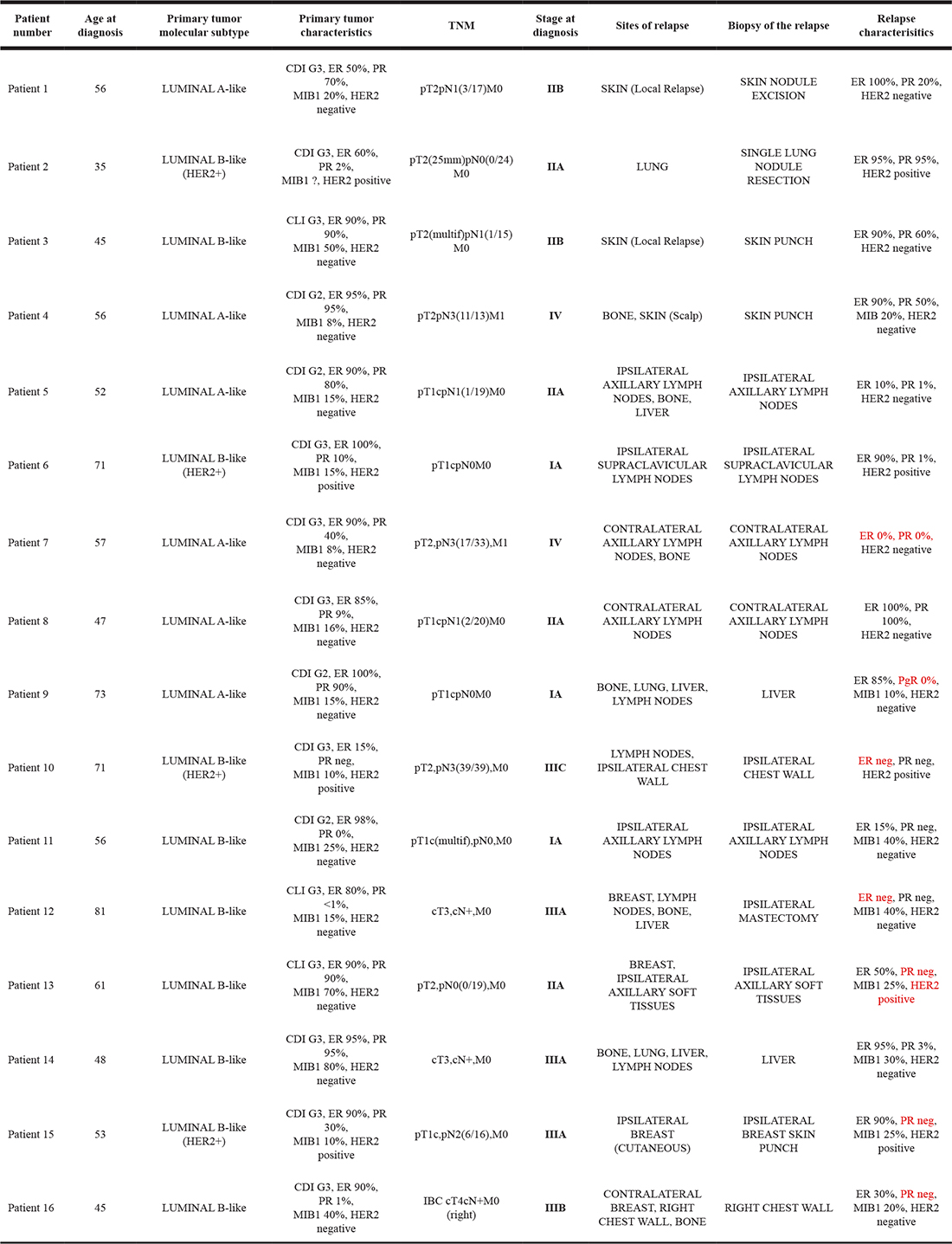
Among the 16 patients, 32 breast tumors were profiled, including 16 primary breast tumors, 10 loco-regional recurrences (ipsilateral breast, chest wall or supraclavicular/axillary lymph nodes) and 6 distant metastases (skin, contralateral axillary lymph nodes, liver and lung). Fourteen patients (87.5%) showed at least one mutation in one of the 25 genes involved in the mechanisms of targeted treatment resistance. Five patients (31.25%) had only one mutated gene, 2 patients (12.5%) had two mutated genes, whereas the other 7 patients (50%) had three or more mutated genes. Overall, we found 64 mutated genes in 16 primary tumors (Figure 1). The most common mutation detected in primary tumor tissues was PIK3CA (6 patients, 37.5%), followed by ERBB2, mTOR and INPP4B (5 patients for each one, 31.25%) (Figure 1).
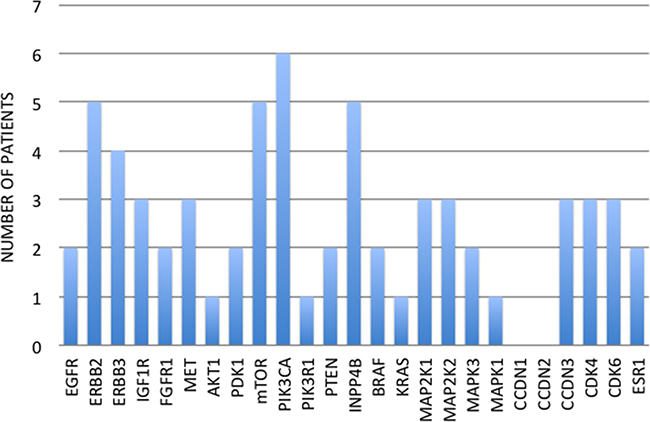
Figure 1: Number of patients for each primary tumor mutation.
Predictive value of relapse mutations
Among the 16 patients, 10 developed loco-regional recurrences in the previously operated breast or chest wall or in the ipsilateral supraclavicular/axillary lymph nodes, whereas 10 patients developed distant metastases (bone, skin, lymph nodes, liver and lung). As far as tissue samples are concerned, 10 loco-regional recurrences and 6 distant metastases have been profiled. More precisely, 10 patients have been biopsied on the site of the first relapse during adjuvant treatments, while 5 patients were biopsied on a metastatic site in progression under treatment in the metastatic setting.
Two patients (12.5%) received only one line of therapy in the metastatic setting, while 3 patients (18.75%) received two lines of therapy, and the other 11 patients (68.75%) received three or more lines of therapy in the metastatic setting. Thirteen patients (81.25%) showed at least one mutation in one of the 25 genes involved in the mechanisms of targeted treatment resistance. Five patients (31.25%) had only one mutated gene and 2 patients (12.5%) had two mutated genes, whereas the other 6 patients (46.15%) had three or more mutated genes. Overall, we found 46 mutated genes in 16 local or distant relapses (Figure 2). The most common mutation detected in relapsed tissues was PIK3CA (6 patients, 37.5%), followed by ERBB2 and MAP2K2 (5 patients for each, 31.25%) (Figure 2).
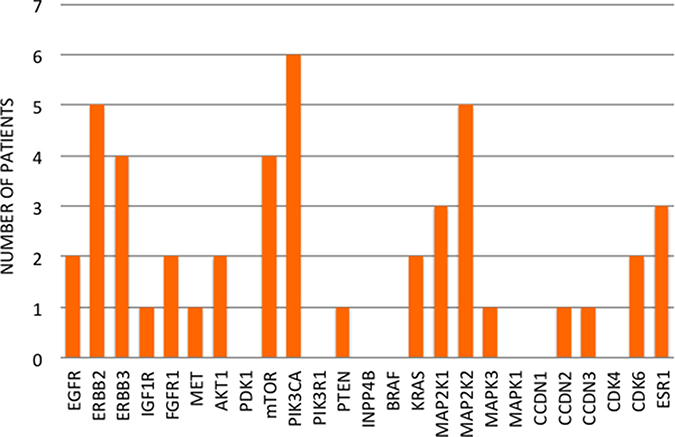
Figure 2: Number of patients for each relapse mutation.
Comparison between primary tumor and relapse mutations
In four cases (25%), primary tumors and relapsed tissues maintained the same mutational profile, whereas 12 (75%) patients changed pathogenic variants during progression. In particular, 9 patients (56.25%) acquired new mutations in the metastatic sites, while we find no primary tumor mutations in the relapsed tissues of 10 patients (62.5%). Overall, 5 patients (31.25%) showed more mutated genes (Figure 3) and more pathogenic variants (Figure 4) in the metastatic sites than in the primary tumor tissues. On the other hand, 5 patients (31.25%) had more mutated genes (Figure 3) and 6 (37.5%) had more pathogenic variants (Figure 4) in the primary tumor tissues.
Figure 3: Number of mutated genes in primary tumors and relapsed tissues.
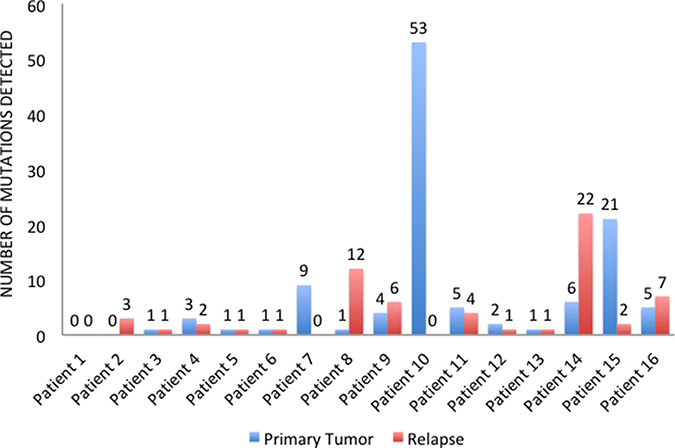
Figure 4: Number of pathogenic variants detected in primary tumors and relapsed tissues.
Finally, we analyzed the mutations detected in the genes that had already shown predictive value in clinical trials: PIK3CA, ERBB2, ESR1 and AKT1 genes (Table 2). Five out of 6 patients with a PIK3CA mutation in either primary tumor or relapse tissues exhibited one of the 3 “Hotspot” mutations shown to cause gains in protein enzymatic function and induce oncogenic transformation (E542 and E545 in the helical domain and H1047 in the kinase domain) [19, 20]. The other variants were N1044H, N345K, A1035V and V344M (Figure 5). With regard to the other genes, we found 11 different pathogenic or likely pathogenic variants for ERBB2 (Figure 5), 5 different variants for ESR1 and 4 different variants for AKT1 (Table 2).
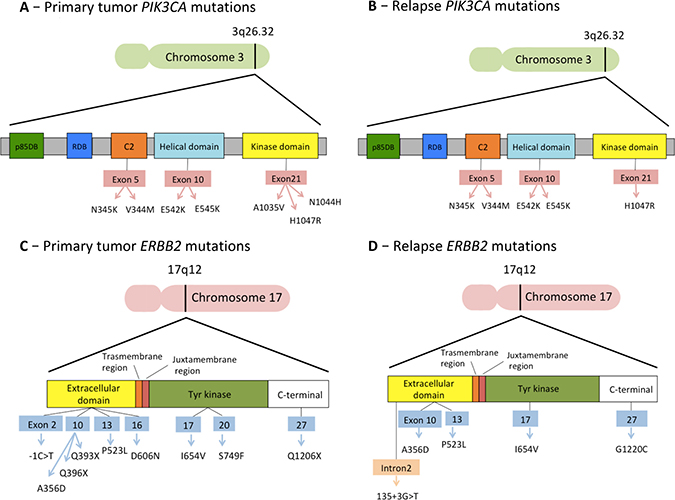
Figure 5: Mutations detected in the two most frequently mutated genes, PIK3CA and ERBB2.
Table 2: Mutations detected in PIK3CA, ERBB2, ESR1 and AKT1 genes

PIK3CA “Hotspot” mutations reported in red.
DISCUSSION
Cancer is a dynamic and heterogeneous entity following the principles of clonal evolution, with different areas of the same primary tumor showing different genomic profiles and with metastases acquiring new molecular aberrations compared to their primary tumors [21–23]. Moreover, clonal heterogeneity, exacerbated by the selective pressures imposed by treatment during the life cycle of the disease, may confer greater resistance on anti-cancer treatments and radiation therapy [24]. Acquired drug resistance is common during the course of the disease. Therefore, there is an urgent need to monitor tumor evolution and ideally predict the onset of resistance to targeted therapies. In this context, emerging evidence from clinical trials assessing targeted therapies demonstrates that the genetic landscape of any given tumor will dictate its sensitivity or resistance profile to anticancer agents [25].
Against this backdrop, we evaluated a panel of 25 genes involved in targeted treatment resistance, comparing cancers sampled at first diagnosis (primary tumors) with their matched relapsed sites. Proper understanding of the mutational landscape of breast tumors through progression and metastatization should open new avenues for the assessment of resistance to therapy and the development of better targeted treatments, with a potential for improved clinical outcomes. The study allowed for the identification of the affected genes that were more prevalent in recurrences, compared with primary tumors, and that might be involved in the resistance to therapies.
In our study, we found an overall detection rate of mutations higher than that described in the literature. The most frequently mutated genes among the 25 analyzed were PIK3CA and ERBB2 for both primary and relapsed tissues (Figure 5). Mutations of PIK3CA were present in 37.5% of primary tumors and relapsed tissues, while ERBB2 mutations were present in 31.25% of our samples. These percentages are higher than those reported in previous research (30% for PIK3CA and 2–3% for ERBB2) [26, 27], indicating that our patients have been highly selected. It should be noted that only tumors with proved targeted treatment resistance, such as those relapsing or progressing under the targeted treatment, have been included in the study. This process increases the chance of finding the mutations responsible for that resistance. Other genes mutating with high frequency in our study were mTOR and INPP4B, which mutated in 31.25% of primary tumors, and MAP2K2, which mutated in 31.25% of relapsed sites. Overall, the most frequently mutated genes among those investigated are involved in the PI3K/AKT/mTOR and the MAPK signaling pathways. Conversely, mutations in genes involved in the cell-cycle regulation are not as crucial to the mechanism of treatment resistance.
In 75% of the patients, furthermore, the number and type of mutations changed with disease progression, as the immunohistochemical profile does in 50% of cases. In contrast with the previous literature, however, most of the time (68.75% of cases) the mutations detected did not increase in number throughout the natural history of the tumor. As already described, we included patients with targeted treatment resistant tumors. As a result, this might have contributed to selecting primary tumors with a high rate of mutations. Interestingly, 12 out of 14 patients (85.71%) with early-stage or locally advanced primary tumors showed a mutation in one of the genes analyzed. The high rate of mutation detected in primary BC tissues might explain the subsequent relapse and thus, it might justify a more targeted approach in neoadjuvant or adjuvant settings for patients exhibiting these mutations at diagnosis. It is noteworthy that previous studies indicated better prognoses for early BC patients harboring a PIK3CA mutation [28–30]. Along with the results shown by m-TOR and PI3K inhibitors in metastatic settings [31, 32], these data highlight a possible dual role of PIK3CA mutations in early versus advanced settings. In early HR-positive BC, PIK3CA may represent a predictive marker for benefit of endocrine therapies. In advanced HR-positive BC selected by primary endocrine therapy, PIK3CA contribute to endocrine resistance requiring combined endocrine and targeted therapy (i.e. everolimus or buparlisib). This is a research area in which more studies are clearly needed.
According to the results reported in recent clinical trials [6, 14, 15], mutations in PIK3CA, ERBB2, AKT1 and ESR1 genes are able to predict the response to some targeted therapies, and future research may find further associations. On these grounds, the mutations detected in the relapsed tissues/metastatic sites of 11 patients (68.75%) enrolled in our study could have guided subsequent treatment choices in a different way. More specifically, these patients underwent the standard treatments approved in that period for the metastatic setting. Nevertheless, 6 patients with PIK3CA mutations could have benefited from treatment with Everolimus, Buparlisib or Taselisib in their capacity as approved agents or in the context of clinical trials. Moreover, the patient with AKT1 mutations could have benefited from Everolimus, while 3 patients with ESR1 mutations could have benefited from Fulvestrant. Finally, 5 patients with ERBB2 mutations could have benefited from treatment with Neratinib (Table 2).
In spite of such evidence, our study presents several limitations and must be considered hypothesis-generating. First of all, our study population is still small in sample size and the relapsed sites were biopsied at various time points throughout tumor course. Secondly, as patients were chosen based on sample availability, there may be inherent biases in patient selection. Furthermore, due to the retrospective nature of our analysis, samples were fixed and processed for storage in different periods and by different technicians, with no purpose of genomic analysis. This variability might have reduced the quality and preservation of some tissues, increasing the rate of variants detected in some of the samples. Overall, we described mutations with an allelic burden ≥3%. This was the case because, for research purposes, we wanted to describe the highest possible number of mutations present in our samples. Nevertheless, in some samples with a lower DNA quality we found a high number of variants, therefore we decided to use the cut-off of 5%. This is the detection limit of Sanger sequencing, the technique used in clinical practice for the confirmation of NGS variant calls. Future research should be conducted to clarify the predictive role of each mutation for each targeted therapy and define the cut-off level for test positivity, in order to select which patients would benefit most from that targeted treatment.
The re-characterization of recurrences was provided through invasive tumor biopsies, as the standard of practice. Obtaining samples of metastatic tissue is notably impractical and complicated by spatial heterogeneity and sampling bias. An attractive alternative to overcome the limitation of repeated tissue sampling is provided by peripheral blood samples as a ‘liquid biopsy’ through the analyses of CTCs, ctDNA or exosomes. Nevertheless, tumor heterogeneity, different sequencing techniques, spatial as well as temporal factors, and potential germline DNA contamination may cause low rates of concordance between liquid biopsies and tumor tissue. So far, therefore, both tissue and blood-based NGS are necessary to describe the complex biology of BC [33]. Finally, our study only focused on gene mutations, but the recent development of additional high information content assays focused on abnormalities in DNA methylation, microRNA expression and protein expression can provide further opportunities to better characterize the molecular architecture of BC.
The ability to perform NGS on small FFPE samples, such as core biopsies, creates the opportunity to comprehensively characterize cancer-relevant genes and personalize therapies. Overall, our study leads to the following conclusions:
1. Almost all patients had at least one alteration potentially targetable with approved or investigational therapeutics. This finding indicates that routine genomic profiling may be instrumental in individualized pathway-directed therapies.
2. Our study reveals substantial discordances between primary tumors and metastases, which stresses the need for analysis of metastatic tissue at recurrences. The analysis of recurrent tumors prior to selecting treatment may provide additional insights, as both gains and losses of targets are observed. Furthermore, the analysis could contribute to treatment selection. In this setting, the role of liquid biopsy instead of biopsy of the metastatic site should be further investigated.
3. Overall, the most frequently mutated genes among those investigated were involved in the PI3K/AKT/mTOR and the MAPK signaling pathway. It is likely that the deregulation of these pathways plays a crucial role in the mechanisms of targeted treatment resistance. Future research should be conducted to identify patients with a molecular profile predictive of treatment resistance, which makes them eligible for more intensive and personalized adjuvant treatments.
4. The extreme complexity of cancer biology necessitates analysis not only on a single level, as the novel targets for molecularly targeted therapy can be revealed at a DNA, RNA and protein level, or epigenetically.
Whether these alterations drive disease progression as well as survival, and therefore represent suitable therapeutic targets for the treatment of metastatic BC or for adjuvant therapy aimed at preventing a relapse warrants further study. However, our study demonstrates that mutation events may be present at diagnosis or arise during cancer treatment. Thus, profiling primary and metastatic tumor tissues may be a major step in defining optimal treatments for BC patients.
MATERIALS AND METHODS
Study design
Patients affected by metastatic hormone receptor (HR) positive and/or HER2 positive BC were eligible for the study. In order to be enrolled, patients had to have tissue samples taken from the primary tumor and at least one relapsed site stored in the archives of our Pathology Department. Using a next-generation sequencing (NGS) technology, the Ion Torrent Personalized Genome Machine (PGM) (Life Technologies, Guilford, CT, USA), we evaluated a panel of 25 genes involved in the mechanisms of endocrine and targeted treatment resistance. We analyzed formalin-fixed and paraffin-embedded (FFPE) tissues of primary BCs and the matched tissues taken from local or distant relapsed sites. Therefore, we compared the cancer genes found in cancers sampled at first diagnosis (primary tumors) with those found in relapsed cancers, after progression during treatment. The study was approved by our local research ethics committee.
Patients and samples
We retrospectively identified 143 patients affected by metastatic hormone positive and/or HER2 positive BC, treated at the Modena Cancer Center in the last 20 years (Figure 6). Among these patients, 65 relapsed or developed disease progression during a targeted treatment, including endocrine therapy. Thirty of these patients had formalin-fixed and paraffin-embedded (FFPE) tissues taken from the primary tumor and from the local or distant recurrence stored in the archives of our Pathology Department. The FFPE samples were evaluated by a pathologist for specimen suitability, tumor quality and quantity, and selection of tumor areas for microdissection. Finally, only 21 patients gave consent to the use of their stored tissues for the study and were therefore included in the analyses.
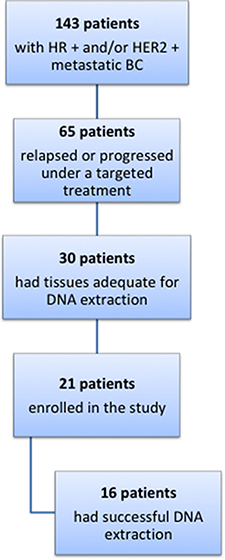
Figure 6: Flow diagram of patients evaluated for the study.
DNA was purified from 6 to 8 5 μm-thin sections of each FFPE tissue using the Maxwell® 16 FFPE Plus LEV DNA Purification Kit on the Maxwell® 16 System (Promega). DNA sample quality and quantity were assessed through a Qubit 2.0 fluorometer and the Qubit™ dsDNA HS Assay Kit (ThermoFisher), in order not to overestimate the concentration of stressed FFPE-purified DNAs. In 16 out of 21 patients, the extraction was successful and the purified DNA was adequate for the sequencing.
Finally, DNA was successfully extracted from 32 FFPE blocks stored between 2000 and 2016. According to data reported by previous research, all of these FFPE blocks were considered suitable for DNA extraction [34]. In particular, 5 primary BC tissues were stored between 2000 and 2005, 8 specimens were stored between 2006 and 2010 and 3 specimens were stored between 2011 and 2016. Of secondary cancer tissues, only one was stored in 2004, 3 FFPE blocks were stored between 2006 and 2010, while 12 were stored between 2011 and 2016.
The multi-gene panel
Twenty-five genes involved in the mechanisms of targeted treatment resistance were selected for the analyses. The list of genes and their role in treatment resistance are reported in Table 3 and described below:
Table 3: The gene panel
EGFR |
ERBB2 |
ERBB3 |
IGF1R |
FGFR1 |
MET |
AKT1 |
PDK1 |
mTOR |
PIK3CA |
PIK3R1 |
PTEN |
INPP4B |
BRAF |
KRAS |
MEK1 |
MEK2 |
ERK1 |
ERK2 |
CCDN1 |
CCDN2 |
CCDN3 |
CDK4 |
CDK6 |
ESR1 |
• EGFR, ERBB2 and ERBB3: this family of tyrosine kinase receptors is involved in cell proliferation control, differentiation and survival [35]. The mutation rate of ERBB2 in BC is about 2–3% [27].
• IGF1R, FGFR1, MET: in HR and/or HER2 positive BC, one of the possible mechanisms of resistance to endocrine and anti-HER2 agents is alternative signaling from other receptor tyrosine kinases [6].
• AKT1, PDK1, mTOR, PIK3CA, PIK3R1, PTEN, INPP4B: the PI3K/AKT/mTOR pathway is one of the main downstream pathways involved in cancer cell proliferation. Mutations of PIK3CA have been found in almost 30% of all sporadic BC [26] with a wide frequency range in BC subtypes [36, 37]. The majority of the mutations, “Hotspot” mutation, occurred in three sites: E542 and E545 in the helical domain, and H1047 in the kinase domain. These mutations cause gains in protein enzymatic function and induce oncogenic transformation [19, 20]. The somatic intragenic PTEN mutation frequency is <5% [38].
• BRAF, KRAS, MEK1 (MAP2K1), MEK2 (MAP2K2), ERK1 (MAPK3), ERK2 (MAPK1): the MAPK signaling pathway may lead to uncontrolled cell cycle, resistance to apoptosis and to chemotherapy, targeted therapies and radiotherapy [6].
• CCDN1, CCDN2, CCDN3, CDK4, CDK6: dysregulation of the cell-cycle machinery and activation of cyclin-dependent kinases (CDKs) represents one of the mechanisms of endocrine resistance. Following the literature, however, the mutation rate of these genes seems to be very low.
• ESR1: in HR positive BC, multiple mechanisms of endocrine resistance have been described, including mutations in the ESR1 gene. Mutations in ESR1 appear to be rare in treatment naive settings (up to 3% of primary tumors [39]) and more frequent in advanced BC (12.1% of a large cohort of advanced BC [40]).
Study procedures
The Ion AmpliSeq™ technology (Thermo Fisher Scientific, Waltham, MA USA 02451) was used to design the gene panel described above. An optimized primer design for the FFPE samples encompassing the CDS and the UTR regions of the selected genes was created, in order to generate amplicons up to 175 bp. Libraries were prepared using the Ion AmpliSeq™ Library Kit 2.0 starting from 15 ng of gDNA/pool, according to the manufacturer’s instructions. Template preparation was performed using an Ion OneTouch™ 2 System following the latest version of the manufacturer’s manuals. The template positive Ion Sphere Particles (ISPs+) were sequenced on an Ion Torrent™ Personal Genome Machine® (PGM™) System (Life Technologies Ltd, Paisley, UK) using the Ion 318™ Chip kit v2 following the Ion PGM™ Sequencing 200 Kit v2 manual. Run conditions were optimized to have at least 1000X coverage depth for 90% of target regions. In these conditions, LoD for mutation was about 1%. For the purpose of quality run assessment, we considered suitable for bioinformatics analysis only data coming from sequencing runs with the following run metrics: on-target % of reads >85%, amplicons with at least 1000 reads >90% and amplicon with no strand bias >95%. NGS sequence data were analyzed, processed and annotated as already described [41].
Statistical analysis
Patient and tumor characteristics were retrieved from the electronic medical files of all patients. Data were summarized by frequency and percentage for categorical variables, and by median and range for continuous variables. The median disease-free survival (DFS) time for these patients was determined as the interval from surgery, or from the end of adjuvant chemotherapy, to any BC event including local, regional or distant recurrence or contralateral disease.
The bioinformatics analyses identified recurrently mutated genes in primary tumors and relapsed tissues. Using our NGS run conditions, we considered known pathogenic and likely pathogenic variants as reported in the literature so far, with an allelic burden ≥3%. For samples with lower DNA quality, we only considered pathogenic or likely pathogenic variants with an allelic burden (mutation frequency) ≥5%.
CONFLICTS OF INTEREST
The authors declare no potential conflicts of interest.
GRANT SUPPORT
This work was supported by the “Angela Serra” Association for Cancer Research.
REFERENCES
1. Sledge GW Jr, Miller KD. Exploiting the hallmarks of cancer: the future conquest of breast cancer. Eur J Cancer. 2003; 39:1668–75.
2. Ingvarsson S. Genetics of breast cancer. Drugs Today (Barc). 2004; 40:991–1002.
3. Cardoso F, Harbeck N, Fallowfield L, Kyriakides S, Senkus E; ESMO Guidelines Working Group. Locally recurrent or metastatic breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012; 23:vii11–9.
4. Sonnenblick A, Pondé N, Piccart M. Metastatic breast cancer: The Odyssey of personalization. Mol Oncol. 2016; 10:1147–59.
5. Toss A, Cristofanilli M. Molecular characterization and targeted therapeutic approaches in breast cancer. Breast Cancer Res. 2015; 17:60.
6. Toss A, Venturelli M, Peterle C, Piacentini F, Cascinu S, Cortesi L. Molecular Biomarkers for Prediction of Targeted Therapy Response in Metastatic Breast Cancer: Trick or Treat? Int J Mol Sci. 2017; 18:18.
7. André F, Hurvitz S, Fasolo A, Tseng LM, Jerusalem G, Wilks S, O’Regan R, Isaacs C, Toi M, Burris H, He W, Robinson D, Riester M, et al. Molecular Alterations and Everolimus Efficacy in Human Epidermal Growth Factor Receptor 2-Overexpressing Metastatic Breast Cancers: Combined Exploratory Biomarker Analysis From BOLERO-1 and BOLERO-3. J Clin Oncol. 2016; 34:2115–24.
8. Baselga J, Im SA, Iwata H, Cortés J, De Laurentiis M, Jiang Z, Arteaga CL, Jonat W, Clemons M, Ito Y, Awada A, Chia S, Jagiełło-Gruszfeld A, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017; 18:904–916.
9. Dickler MN, Saura C, Richards DA, Krop IE, Cervantes A, Bedard PL, Patel MR, Pusztai L, Oliveira M, Ware JA, Jin H, Wilson TR, Stout T, et al. A phase II study of the PI3K inhibitor taselisib (GDC-0032) combined with fulvestrant (F) in patients (pts) with HER2-negative (HER2-), hormone receptor-positive (HR+) advanced breast cancer (BC). J Clin Oncol. 2016; 34:520.
10. Baselga J, Lewis Phillips GD, Verma S, Ro J, Huober J, Guardino AE, Samant MK, Olsen S, de Haas SL, Pegram MD. Relationship between Tumor Biomarkers and Efficacy in EMILIA, a Phase III Study of Trastuzumab Emtansine in HER2-Positive Metastatic Breast Cancer. Clin Cancer Res. 2016; 22:3755–63.
11. Fribbens C, O’Leary B, Kilburn L, Hrebien S, Garcia-Murillas I, Beaney M, Cristofanilli M, Andre F, Loi S, Loibl S, Jiang J, Bartlett CH, Koehler M, et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor-Positive Advanced Breast Cancer. J Clin Oncol. 2016; 34:2961–68.
12. Wang T, Xu Y, Sheng S, Yuan H, Ouyang T, Li J, Wang T, Fan Z, Fan T, Lin B, Xie Y. HER2 somatic mutations are associated with poor survival in HER2-negative breast cancers. Cancer Sci. 2017; 108:671–677.
13. Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, Monsey J, Goel N, Aronson AB, Li S, Ma CX, Ding L, Mardis ER, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013; 3:224–37.
14. Ma CX, Bose R, Gao F, Freedman RA, Telli ML, Kimmick G, Winer E, Naughton M, Goetz MP, Russell C, Tripathy D, Cobleigh M, Forero A, et al. Neratinib Efficacy and Circulating Tumor DNA Detection of HER2 Mutations in HER2 Nonamplified Metastatic Breast Cancer. Clin Cancer Res. 2017; 23:5687–95. https://doi.org/10.1158/1078-0432.CCR-17-0900.
15. Hyman DM, Piha-Paul SA, Won H, Rodon J, Saura C, Shapiro GI, Juric D, Quinn DI, Moreno V, Doger B, Mayer IA, Boni V, Calvo E, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature. 2018; 554:189–94. https://doi.org/10.1038/nature25475.
16. Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, Harris CC, McLellan MD, Fulton RS, Fulton LL, Abbott RM, Hoog J, Dooling DJ, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010; 464:999–1005.
17. Navin NE. The first five years of single-cell cancer genomics and beyond. Genome Res. 2015; 25:1499–507.
18. Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, Allred DC, Bartlett JM, Bilous M, Fitzgibbons P, Hanna W, Jenkins RB, Mangu PB, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013; 1:3997–4013.
19. Karakas B, Bachman KE, Park BH. Mutation of the PIK3CA oncogene in human cancers. Br J Cancer. 2006; 94:455–59.
20. Ligresti G, Militello L, Steelman LS, Cavallaro A, Basile F, Nicoletti F, Stivala F, McCubrey JA, Libra M. PIK3CA mutations in human solid tumors: role in sensitivity to various therapeutic approaches. Cell Cycle. 2009; 8:1352–58.
21. Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011; 472:90–94.
22. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012; 366:883–92.
23. Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012; 72:4875–82.
24. Badve S, Nakshatri H. Breast-cancer stem cells-beyond semantics. Lancet Oncol. 2012; 13:e43–8.
25. Tsimberidou AM, Wen S, Hong DS, Wheler JJ, Falchook GS, Fu S, Piha-Paul S, Naing A, Janku F, Aldape K, Ye Y, Kurzrock R, Berry D. Personalized medicine for patients with advanced cancer in the phase I program at MD Anderson: validation and landmark analyses. Clin Cancer Res. 2014; 20 :4827–36.
26. Dai X, Huang N. Numerical simulation of drifting snow sublimation in the saltation layer. Sci Rep. 2014; 4:6611.
27. Petrelli F, Tomasello G, Barni S, Lonati V, Passalacqua R, Ghidini M. Clinical and pathological characterization of HER2 mutations in human breast cancer: a systematic review of the literature. Breast Cancer Res Treat. 2017; 166:339–349.
28. Zardavas D, Te Marvelde L, Milne RL, Fumagalli D, Fountzilas G, Kotoula V, Razis E, Papaxoinis G, Joensuu H, Moynahan ME, Hennessy BT, Bieche I, Saal LH, et al. Tumor PIK3CA Genotype and Prognosis in Early-Stage Breast Cancer: A Pooled Analysis of Individual Patient Data. J Clin Oncol. 2018; 36:981–90. https://doi.org/10.1200/JCO.2017.74.8301.
29. Loi S, Michiels S, Lambrechts D, Fumagalli D, Claes B, Kellokumpu-Lehtinen PL, Bono P, Kataja V, Piccart MJ, Joensuu H, Sotiriou C. Somatic mutation profiling and associations with prognosis and trastuzumab benefit in early breast cancer. J Natl Cancer Inst. 2013; 105:960–67. https://doi.org/10.1093/jnci/djt121.
30. Yang SX, Polley E, Lipkowitz S. New insights on PI3K/AKT pathway alterations and clinical outcomes in breast cancer. Cancer Treat Rev. 2016; 45:87–96.
31. Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, Beck JT, Ito Y, Yardley D, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012; 366:520–29. https://doi.org/10.1056/NEJMoa1109653.
32. Di Leo A, Johnston S, Lee KS, Ciruelos E, Lønning PE, Janni W, O’Regan R, Mouret-Reynier MA, Kalev D, Egle D, Csőszi T, Bordonaro R, Decker T, et al. Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2018; 19:87–100. https://doi.org/10.1016/S1470-2045(17)30688-5.
33. Chae YK, Davis AA, Jain S, Santa-Maria C, Flaum L, Beaubier N, Platanias LC, Gradishar W, Giles FJ, Cristofanilli M. Concordance of Genomic Alterations by Next-Generation Sequencing in Tumor Tissue versus Circulating Tumor DNA in Breast Cancer. Mol Cancer Ther. 2017; 16:1412–1420.
34. Kokkat TJ, Patel MS, McGarvey D, LiVolsi VA, Baloch ZW. Archived formalin-fixed paraffin-embedded (FFPE) blocks: A valuable underexploited resource for extraction of DNA, RNA, and protein. Biopreserv Biobank. 2013; 11:101–6. https://doi.org/10.1089/bio.2012.0052.
35. Lin NU, Winer EP. New targets for therapy in breast cancer: small molecule tyrosine kinase inhibitors. Breast Cancer Res. 2004; 6:204–10.
36. Sabine VS, Crozier C, Brookes CL, Drake C, Piper T, van de Velde CJ, Hasenburg A, Kieback DG, Markopoulos C, Dirix L, Seynaeve C, Rea DW, Bartlett JM. Mutational analysis of PI3K/AKT signaling pathway in tamoxifen exemestane adjuvant multinational pathology study. J Clin Oncol. 2014; 32:2951–58.
37. Kalinsky K, Jacks LM, Heguy A, Patil S, Drobnjak M, Bhanot UK, Hedvat CV, Traina TA, Solit D, Gerald W, Moynahan ME. PIK3CA mutation associates with improved outcome in breast cancer. Clin Cancer Res. 2009; 15:5049–59.
38. Zhang HY, Liang F, Jia ZL, Song ST, Jiang ZF. PTEN mutation, methylation and expression in breast cancer patients. Oncol Lett. 2013; 6:161–168.
39. Angus L, Beije N, Jager A, Martens JW, Sleijfer S. ESR1 mutations: Moving towards guiding treatment decision-making in metastatic breast cancer patients. Cancer Treat Rev. 2017; 52:33–40.
40. Niu J, Andres G, Kramer K, Kundranda MN, Alvarez RH, Klimant E, Parikh AR, Tan B, Staren ED, Markman M. Incidence and clinical significance of ESR1 mutations in heavily pretreated metastatic breast cancer patients. Onco Targets Ther. 2015; 8:3323–8.
41. Bernardis I, Chiesi L, Tenedini E, Artuso L, Percesepe A, Artusi V, Simone ML, Manfredini R, Camparini M, Rinaldi C, Ciardella A, Graziano C, Balducci N, et al. Unravelling the Complexity of Inherited Retinal Dystrophies Molecular Testing: Added Value of Targeted Next-Generation Sequencing. Biomed Res Int. 2016; 2016:6341870.