
INTRODUCTION
Hepatocellular carcinoma (HCC) is a major malignant cancer worldwide and the third leading cause of cancer death [1]. The mortality rate in most countries is almost equal to the incidence of HCC, indicating the lack of effective treatment. Currently, surgical resection is the most common treatment for HCC. However, the clinical symptoms of HCC are not clear in the early stage, and the disease is often diagnosed at an advanced stage. Patients with inoperable HCC are commonly treated with cisplatin chemotherapy [2], which is effective, but toxic to normal tissue. Therefore, development of a drug with low toxicity that is relatively selective for HCC and with a synergistic effect with established chemotherapeutic drugs may prolong survival of patients with HCC, particularly those in an advanced stage.
Short-chain fatty acids (SCFAs) are a subgroup of fatty acids including six or less carbons. The most important SCFAs are acetate (C2), propionate (C3), and butyrate (C4). In humans, SCFAs are produced exclusively in the gastrointestinal (GI) tract via fermentation of dietary fibers by colonic bacteria. The SCFAs then enter the bloodstream, where butyrate is the main energy source for colonic epithelial cells, propionate is mainly absorbed by the liver, and acetate reaches peripheral tissues [3]. SCFAs are key regulators of immune and metabolic cells, and prevent progression of diseases such as airway disease, colitis, and metabolic disorders [4–6]. SCFAs also affect the cell cycle by inhibiting proliferation and inducing differentiation and apoptosis in human cancer cells [7–9].
SCFAs are thought to exert their effects in two ways: by activating G-protein coupled receptors (GPCRs) and by inhibiting histone deacetylases (HDACs) [10–14]. Recently, two GPCRs, GPR41 and GPR43, have been identified as receptors for SCFAs. In the past decade, SCFAs have been identified as extracellular signaling molecules that mediate regulation of several cellular processes. GPR41 couples to Gi/o signaling pathways and is expressed in gut, adipose tissue, and the peripheral nervous system, while GPR43 couples to Gi/o or Gq signaling pathways [15] and is expressed in adipose tissue, immune cells, and the intestine [16, 17]. Recent data indicate that activation of GPR41 and GPR43 affects tumor growth [8, 9]. Expression of GPR43 is low in most colorectal adenocarcinoma tissues, but high in normal colon tissues [8]. In addition, activation of GPR41 and GPR43 by propionate changes the invasive phenotype of breast cancer cells to a non-invasive phenotype [9].
SCFAs also inhibit cell proliferation by inhibiting HDAC activity and inducing apoptosis [18]. Epigenetic modulation of gene transcription through acetylation and deacetylation of histones plays an important role in the pathogenesis of HCC [19]. Expression of HDAC1-5 and 8 is upregulated in human HCC and enhanced expression of HDACs plays a critical role in malignant growth and immune escape [19, 20]. In contrast to genetic alterations, such epigenetic changes are reversible, and therefore are good therapeutic targets [21]. However, whether HDAC inhibition by SCFAs depends on GPR41 or GPR43 remains unclear. Hence, we hypothesize that SCFAs may contribute to regulation of cancer development by inhibiting HDAC activity through GPR41/43-dependent mechanisms.
The chemotherapeutic agent cisplatin binds to DNA to form intrastrand and interstrand crosslinks between purine bases. A tight chromatin structure prevents cisplatin from approaching DNA, and relaxation of chromatin by HDAC inhibitors increases the accessibility of cisplatin to DNA [22]. However, few studies have examined whether SCFAs enhance the effect of cisplatin chemotherapy by inhibiting HDAC activity by agonism of GPR41 or GPR43. We postulated that a combination of cisplatin with SCFAs would increase the anticancer efficacy of cisplatin. Here, we test this hypothesis by examining augmentation of cisplatin activity by a SCFA, sodium propionate (NaP), in HepG2 cells, and by determining the mechanism underlying the synergistic effect of the combined treatment.
RESULTS
Expression of GPR41 and GPR43 in human HCC tissues and cell lines
GPR41 and GPR43 were expressed at measurable levels in three human HCC tissues with different pathological grades (Figure 1A: grade 1, well differentiated; Figure 1B: grade 2, moderately differentiated; Figure 1C: grade 3, poorly differentiated). Immunoblot analyses showed that GPR41 and GPR43 were present as bands of approximately 39 kDa and 43 kDa, respectively, in all HCC cell lines, including HepG2, HuH-7, JHH-4 and HLE (Figure 1D). Both GPRs seem to be expressed widely in HCC.
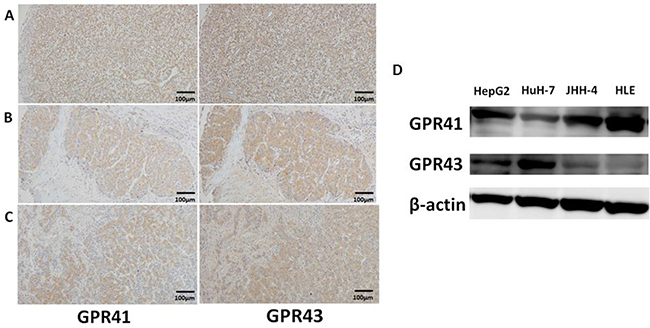
Figure 1: Protein expression of GPR41 and GPR43 in human HCC. (A–C) HCC were immunostained for GPR41 and GPR43. Original magnification ×100. (D) GPR41 and GPR43 were analyzed by immunoblotting in whole-cell lysates from confluent HepG2 cells, HuH-7 cells, JHH-4 cells and HLE cells.
Effects of NaP combined with cisplatin on proliferation and apoptosis of HCC cell lines
A MTS assay was used to determine the effects of cisplatin (25 μM), NaP (1, 10 mM), and combinations of cisplatin (25 μM) and NaP (0.1, 1, 10 mM) on proliferation of HCC cell lines cells for 24 h. NaP alone did not inhibit proliferation, whereas cisplatin alone significantly inhibited proliferation at 25 μM in HepG2 cells, HuH-7 cells, and JHH-4 cells. The inhibitory effect of NaP + cisplatin on proliferation of HCC cell lines was significantly higher than that of cisplatin alone (p < 0.01) with NaP at 0.1 to 10 mM in HepG2 cells and HuH-7 cells (Figure 2A, 2B) and with NaP at 1 to 10 mM in JHH-4 cells and HLE cells (Figure 2C, 2D). In FACS analysis to examine whether NaP (1 mM) enhanced the sensitivity of HCC cell lines to cisplatin, the apoptotic rate at 48 h was significantly higher with NaP + cisplatin than with cisplatin alone in all HCC cell lines (Figure 3A–3D).
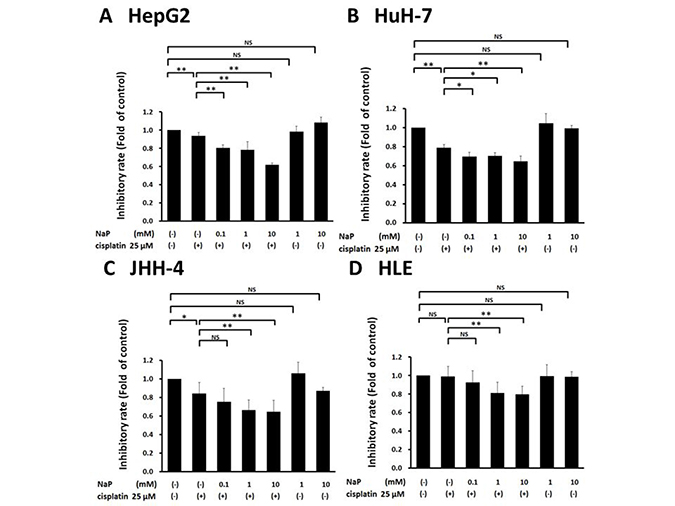
Figure 2: Effects of NaP combined with cisplatin on proliferation rate of HCC cell lines. (A–D) A MTS assay was used to determine the effects of NaP (1, 10 mM) alone, cisplatin (25 μM) alone, or cisplatin (25 μM) + NaP (0.1, 1, 10 mM) on proliferation of HCC cell lines for 24 h. Data are shown as mean ± SD of % apoptosis from three independent experiments. *P < 0.05, **P < 0.01 by one-way ANOVA with a Scheffe post hoc test.
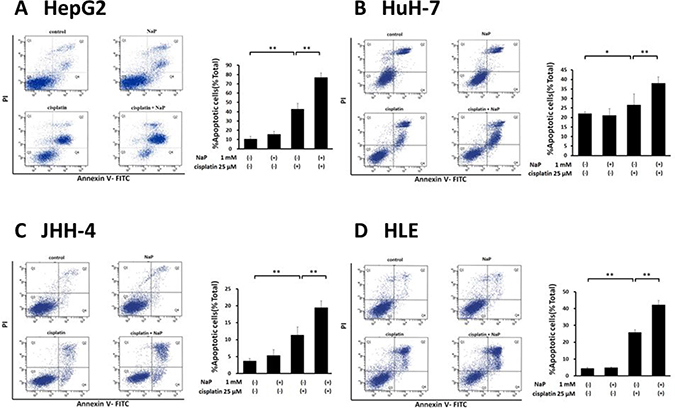
Figure 3: Effects of NaP combined with cisplatin on apoptotic rate of HCC cell lines. (A–D) HCC cells were treated with cisplatin (25 μM), NaP (1 mM), or both agents for 48 h. Cells were then stained with annexin V and PI, followed by cytometry analysis. Data are shown as mean ± SD of % apoptosis from three independent experiments. *P < 0.05, **P < 0.01 by one-way ANOVA with a Scheffe post hoc test.
NaP enhances cisplatin-induced apoptosis via GPR41 in HepG2 cells
Apoptosis is regulated by highly coordinated processes that involves activation of caspases, which are cysteine proteases [23]. Caspase-3 activation is responsible for DNA fragmentation and myonuclear apoptosis [23, 24]. Therefore, we measured the cleaved, active form of caspase-3 in HepG2 cells by Western blot analysis (Figure 4). NaP + cisplatin significantly increased expression at 0.1 mM NaP (Figure 4A). Next, we examined whether NaP + cisplatin enhanced expression of cleaved caspase-3 via GPR41 or GPR43. Enhancement by NaP was completely blocked by treatment with pertussis toxin (PTX), a Gi/o-type G protein inhibitor [25] (Figure 4A), and was blocked by Gallein, a Gβγ blocker (Figure 4A). We further investigated whether a selective agonist of GPR41 or GPR43 enhanced cisplatin-stimulated expression of cleaved caspase-3. CPC, a GPR41-selective agonist, significantly enhanced cleavage of caspase-3 at 100 μM and the enhancement effect of CPC + cisplatin was blocked by treatment with PTX (Figure 4B). In contrast, CFMB, a GPR43-selective agonist, significantly reduced cleavage at 10 μM (Figure 4C). These data indicate that the enhancement effect of CPC + cisplatin was dependent on a Gi/o signal pathway. GPR41 gene silencing in HepG2 cells using two siRNAs (siRNA-1 and siRNA-2) was performed to clarify whether NaP-mediated enhancement of cisplatin-induced apoptosis was dependent on GPR41. Significant decreases in GPR41 mRNA and protein were found in HepG2 cells treated with siRNAs against GPR41 (Figure 4D, 4E). GPR41 silencing in HepG2 cells significantly blocked NaP-induced enhancement of cisplatin-induced cleaved caspase-3 expression (Figure 4D, 4E). Taken together, these results demonstrate that NaP enhances activation of caspase-3 by cisplatin via a GPR41-mediated pathway.
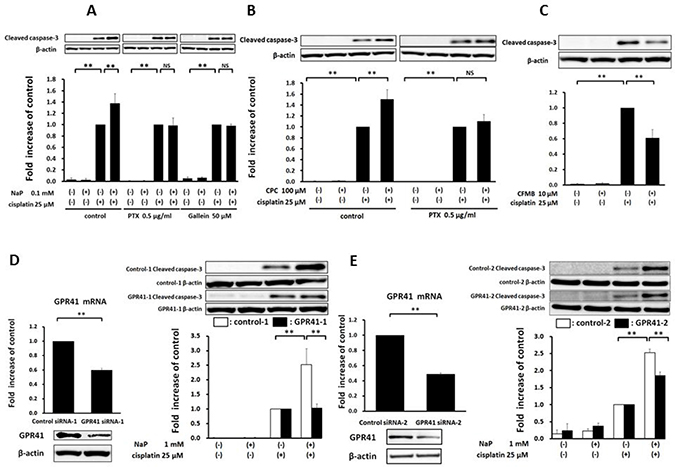
Figure 4: Enhancement of cisplatin-induced apoptosis by NaP in a GPR41-dependent manner in HepG2 cells. (A) HepG2 cells were treated with cisplatin (25 μM) with or without NaP (0.1 mM) for 24 h. HepG2 cells were treated with vehicle or cisplatin (25 μM) with or without NaP (0.1 mM), and with or without PTX (0.5 mg/ml), or Gallein (50 μM) for 24 h. (B) HepG2 cells were treated with vehicle or cisplatin (25 μM) with or without a GPR41 agonist (100 μM CPC) and in the presence of PTX (0.5 mg/ml) for 24 h. (C) HepG2 cells were treated with vehicle or cisplatin (25 μM) with or without a GPR43 agonist (10 μM CFMB) for 24 h. (D, E) Representative mRNA expression (upper panel) and immunoblot of GPR41 protein (lower panel) in siRNA-1-mediated (D) or siRNA-2-mediated (E) GPR41 knockdown cells. HepG2 cells were treated with vehicle or cisplatin (25 μM) with or without NaP (1 mM) in cells treated with control or GPR41 siRNA-1 or siRNA-2. The cleaved caspase-3 level relative to β-actin in cells treated with cisplatin alone was set to 1.0. Data are expressed as the mean ± SD of three separate experiments. **P < 0.01, NS: not significant by one-way ANOVA with a Scheffe post hoc test (A–E) and Mann–Whitney test (D, E).
NaP enhances cisplatin-induced apoptosis via a TNF-α-induced apoptotic pathway
To investigate the molecular mechanism of NaP enhancement of cisplatin-induced apoptosis in HepG2 cells, immunoblotting was performed for caspase family members that might function during apoptosis (Figure 5A). Cisplatin treatment induced cleavage of caspase-8 and -9, and the protein level of activated caspase-8 with NaP + cisplatin was significantly higher than that with cisplatin alone. NaP at 0.1 and 1 mM significantly induced expression of mRNA for TNF-α, a pro-apoptotic cytokine, in a dose-dependent manner in HepG2 cells treated with cisplatin (Figure 5B). Enhancement of cisplatin-induced TNF-α expression by NaP was blocked by treatment with PTX (Figure 5C). GPR41 silencing in HepG2 cells significantly reduced NaP-induced enhancement of cisplatin-induced expression of mRNA for TNF-α (Figure 5D). Expression of mRNA for TNF-α with NaP + cisplatin was significantly higher than that with cisplatin alone from 3 h to 48 h (Figure 5E). NaP at 1 mM significantly increased secretion of TNF-α, and this was significantly blocked by PTX (Figure 5F). Recombinant TNF-α significantly enhanced cisplatin-induced production of cleaved caspase-3 in a dose-dependent manner (Figure 5G) and the apoptotic rate at 24 h was significantly higher with TNF-α + cisplatin than with cisplatin alone in FACS analysis (Figure 5H). Enhancement of cleaved caspase-3 by NaP + cisplatin was significantly blocked by treatment with a TNF-α antagonist (Figure 5I). Taken together, these results show that NaP enhances cisplatin-induced apoptosis through a TNF-α-induced extrinsic apoptotic pathway via GPR41.
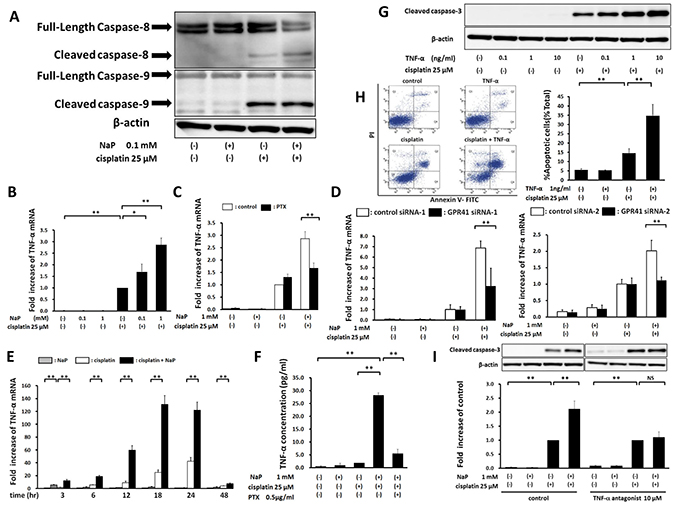
Figure 5: Enhancement of cisplatin-induced apoptosis by NaP through the TNF-α-induced extrinsic apoptotic pathway. (A) HepG2 cells were treated with vehicle or cisplatin (25 μM) with or without NaP (0.1 mM) for 24 h. Cell lysates were analyzed by western blot for the apoptosis-related proteins, caspase-8 and caspase-9. (B) HepG2 cells were treated with vehicle or cisplatin (25 μM) in the presence of the indicated concentration of NaP for 24 h. TNF-α mRNA levels were quantified by TaqMan real-time PCR, with the level in cells treated with cisplatin alone set to 1.0. (C) HepG2 cells were treated with vehicle or cisplatin (25 μM) with or without NaP (1 mM) and with or without PTX (0.5 mg/ml) for 24 h. TNF-α mRNA levels were quantified by TaqMan real-time PCR, with the level in cells treated with cisplatin alone set to 1.0. (D) HepG2 cells were treated with vehicle or cisplatin (25 μM) with or without NaP (1 mM) for 24 h in cells treated with control or GPR41 siRNA-1/2. TNF-α mRNA levels were quantified by TaqMan real-time PCR, with the level in cells treated with cisplatin alone set to 1.0. (E) HepG2 cells were treated with vehicle or cisplatin (25 μM) with or without NaP (1 mM) for 3, 6, 12, 18, 24, and 48 h. TNF-α mRNA levels were quantified by TaqMan real-time PCR, with the level in cells treated with vehicle set to 1.0. (F) HepG2 cells were treated with vehicle or cisplatin (25 μM) with or without NaP (1 mM) or PTX (0.5 mg/ml) for 24 h. TNF-α in supernatants was quantified by ELISA. (G) HepG2 cells were treated with vehicle or human recombinant TNF-α (0.1, 1, 10 ng/ml) or cisplatin (25 μM) in the presence of the indicated concentration of human recombinant TNF-α for 24 h. (H) HepG2 cells were treated with cisplatin (25 μM), TNF-α (1 ng/ml), or both agents for 24 h. Cells were then stained with annexin V and PI, followed by cytometry analysis. (I) HepG2 cells were treated with vehicle or cisplatin (25 μM) with or without NaP (1 mM) and with or without a TNF-α antagonist (10 μM) for 24 h. Cell lysates were analyzed by western blot for cleaved caspase-3, with the cleaved caspase-3 level relative to β-actin in cells treated with cisplatin alone set to 1.0. Data are expressed as the mean ± SD of three separate experiments. *P < 0.05, **P < 0.01, NS: not significant by one-way ANOVA with a Scheffe post hoc test (B–F, H, I).
NaP enhances cisplatin-induced acetylation of histone H3 via GPR41 in HepG2 cells
Next, we investigated whether NaP enhanced cisplatin-induced apoptosis through a HDAC inhibitory pathway via GPR41. NaP significantly enhanced cisplatin-induced acetylation of histone H3 at 0.1 mM (Figure 6A), and this effect was completely blocked by PTX (Figure 6A). CPC also enhanced acetylation of histone H3 (Figure 6B). TSA, a pan-HDAC inhibitor, significantly enhanced cisplatin-induced expression of cleaved caspase-3 at 1 μM (Figure 6C), and significantly enhanced cisplatin-induced TNF-α expression (Figure 6D). To investigate whether treatment with cisplatin or NaP + cisplatin decreased expression of HDACs, which lead to acetylation of histones, Western blot analysis was used to examine expression of HDACs in HepG2 cells. Cisplatin alone decreased the levels of HDAC3, 4, 5, 6, and 8, and NaP + cisplatin further decreased HDAC3 and 6 (Figure 6E, 6F). This indicates that enhancement of acetylation of histone H3 by NaP occurs partially through a HDAC3, 6-dependent pathway. Taken together, these results show that NaP enhances acetylation of histone H3 through reducing the expression of several HDACs in combination treatment with cisplatin in a GPR41-dependent manner.
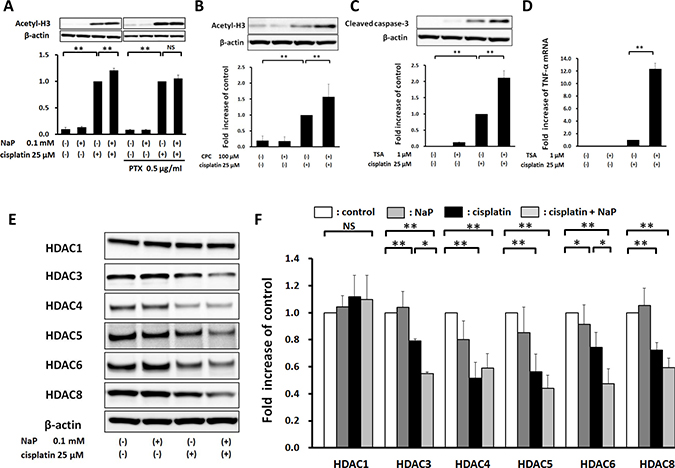
Figure 6: Elevation of cisplatin-induced acetylation of histone H3 by NaP in a GPR41-dependent manner in HepG2 cells. (A) HepG2 cells were treated with vehicle or cisplatin (25 μM) with or without NaP (0.1 mM) and with or without PTX (0.5 mg/ml) for 24 h. Cell lysates were analyzed by western blot for acetylation of histone H3. (B) HepG2 cells were treated with vehicle or cisplatin (25 μM) with or without a GPR41 agonist (100 μM CPC) for 24 h. Cell lysates were analyzed by western blot for acetylation of histone H3. (C, D) HepG2 cells were treated with vehicle or cisplatin (25 μM) with or without TSA (1 μM) for 24 h. Cell lysates were analyzed by western blot for an apoptosis-related protein, cleaved caspase-3. TNF-α mRNA levels were quantified by TaqMan real-time PCR, with the level in cells treated with cisplatin alone set to 1.0. (E, F) HepG2 cells were treated with vehicle or cisplatin (25 μM) with or without NaP (0.1 mM) for 24 h. Cell lysates were analyzed by western blot for HDAC1, 3, 4, 5, 6 and 8. The levels of acetyl-H3 and cleaved caspase-3 (all relative to β-actin) were set to 1.0 in cells treated with cisplatin alone. The levels of HDACs (all relative to β-actin) were set to 1.0 in cells treated with vehicle. Data are expressed as the mean ± SD of three separate experiments. *P < 0.05, **P < 0.01, NS not significant by one-way ANOVA with a Scheffe post hoc test (A–D, F).
Efficacy of NaP combined with cisplatin in hepatocellular carcinoma HepG2 xenografts
A HepG2 xenograft model was used for evaluation of therapeutic efficacy in vivo. NaP (250 mg/kg) + cisplatin or CPC (50 mg/kg) + cisplatin significantly suppressed growth of HepG2 xenografts (p < 0.01) (Figure 7A). NaP + cisplatin significantly enhanced acetylation of histone H3 (Figure 7B) and expression of TNF-α mRNA and in the xenografts at day 4 (Figure 7C).
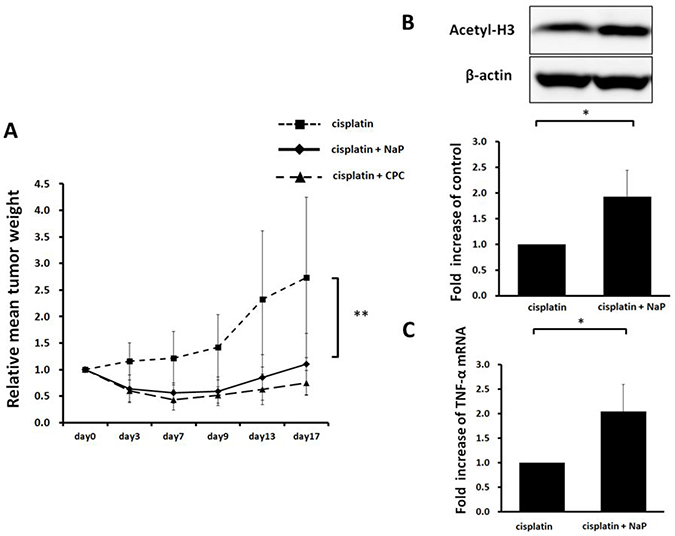
Figure 7: Therapeutic efficacy of NaP + cisplatin in HCC HepG2 xenografts. Six-week-old female SHO nude mice were subcutaneously injected with 1.0 x 106 cells. When the tumor size reached 256-500 mm3, mice were treated by intraperitoneal injection of 8 mg/kg cisplatin or 8 mg/kg cisplatin + 250 mg/kg NaP or 8 mg/kg cisplatin + 50 mg/kg CPC dissolved in saline. (A) Xenografts were measured as described in the Materials and Methods. Results are expressed as the mean ± SD (n = 5). (B) Acetylation of histone H3 in HepG2 tumors was evaluated by Western blotting at 4 days after treatment with 8 mg/kg cisplatin or 8 mg/kg cisplatin + 250 mg/kg NaP (n = 5). (C) Expression of mRNA for TNF-α in HepG2 tumors was evaluated by Western blotting at 4 days after treatment with 8 mg/kg cisplatin or 8 mg/kg cisplatin + 250 mg/kg NaP (n = 5). *P < 0.05, **P < 0.01, NS: not significant by Student t-test.
DISCUSSION
In this study, we showed that GPR41 and GPR43 are moderately expressed in human HCC, and we found that a SCFA, propionate, enhanced the chemosensitivity of HepG2 cells to cisplatin by inducing TNF-α expression via GPR41 activation involving Gi/o and particularly Gβ/γ. To the best of our knowledge, this is the first report to describe enhancement of the effect of cisplatin chemotherapy by propionate in HCC cell.
SCFAs are produced by microbial fermentation of indigestible fiber by anaerobic gut flora and are important energy sources for the host. GPR41 and GPR43 are recently identified GPCRs for SCFAs [15], and there is growing evidence that SCFAs have therapeutic potential for protection against various inflammatory disease via GPR41 or GPR43 [26–28]. We also recently reported that SCFAs, especially propionate, attenuate TNF-α-stimulated monocyte chemotactic protein-1 (MCP-1) expression by inhibiting phosphorylation of p38 and JNK in human renal cortical epithelial cells [29], and we showed that this inhibitory effect occurred in a GPR41/GPR43-dependent manner [29]. Furthermore, recent data suggest that GPR41 and GPR43 play a role in tumor suppression [7, 8]. Propionate, the most potent agonist of GPR41 and GPR43, reduces proliferation and promotes apoptosis in colon cancer [8] and leukemia [7], but it is unclear if propionate enhances the effect of chemotherapy on cancer cells. In this study, we showed that a combination of cisplatin and propionate (NaP) significantly inhibited growth and increased apoptosis in HepG2 cells.
GPR41 and GPR43 are SCFA receptors that couple with Gi/o and with Gi/o or Gq, respectively [15]. In the intestine, GPR43 promotes glucagon-like peptide-1 (GLP-1) secretion in L cells through a Gq-coupled pathway [30], and a recent study showed that acetate activates p38 phosphorylation in human monocytes, probably via Gq, but not Gi/o, signaling [31]. In the current study, NaP and CPC, a selective agonist of GPR41, significantly enhanced the cisplatin-induced activation of caspases. In contrast, CFMB, a selective agonist of GPR43, reduced the activation, and this reduction was significantly blocked in a Gq inhibitor (YM254890)-sensitive manner (data not shown). Based on these findings, enhancement of apoptosis by NaP is dependent on GPR41 activation involving Gi/o. On the other hand, selective stimulation of GPR43 may reduce cisplatin-induced apoptosis involving Gq.
A key finding in the current study is that NaP enhanced cisplatin-induced apoptosis by inhibiting HDAC, an epigenetic modifier, via GPCR activation. A recent study showed that SCFAs inhibited production of pro-inflammatory cytokines, oxidative cellular stress, and cell infiltration by inhibiting HDAC, independent of GPR41 and GPR43, in mice with ischemic nephropathy [32], and in neutrophils, SCFAs inhibit HDAC independently of GPR41 and GPR43 [33]. On the other hand, in colon tissue, HDAC inhibition by SCFAs is partially dependent on GPR43 [34]. The current study showed that NaP inhibited HDACs via GPR41 in HepG2 cells. Thus, effects of GPR41 and GPR43 on HDAC inhibition may be cell-specific.
Another major finding of the study was the enhancement of cisplatin-induced production of TNF-α by NaP via HDAC inhibition in HepG2 cells. Cisplatin is actively transported into cells, and this is followed by activation of several stress signaling cascades such as the p53 signaling pathway, mitogen-activated protein kinase (MAPK) signaling pathway, and both intrinsic and extrinsic apoptosis pathways [35, 36] The extrinsic pathway for apoptosis is induced through activation of death receptors including CD95 receptor (Fas) and tumor necrosis factor receptor 1 (TNFR1), leading to activation of caspase-8 [37]. In the current study, we found that a combination of NaP and cisplatin enhanced cleavage of caspase-3 and -8 significantly compared with cisplatin alone. In addition, expression of TNF-α mRNA and protein were enhanced significantly by NaP and cisplatin compared with cisplatin alone, and this enhancement was blocked by treatment with PTX. Recombinant TNF-α and cisplatin enhanced cleavage of caspase-3 in a dose-dependent manner, and further enhancement of NaP with cisplatin was completely blocked by a TNF-α antagonist.
Modulation of autocrine TNF-α can sensitize cancer cells to cisplatin [38]. We found that cisplatin alone decreased the levels of HDAC3, 4, 5, 6, and 8 and that addition of NaP to cisplatin further decreased HDAC3 and 6. A previous study showed that suppression of HDAC3 or 5 induced apoptosis in HCC cell lines [39, 40]. Similarly, knockdown of HDAC4 enhanced radiation-induced cell death, that of HDAC6 reduced the migration and invasion activities of all HCC cell lines [41], and that of HDAC8 repressed tumor cell growth and induced apoptosis [42]. A recent study showed that NaP at 0.1 mM reduced expression of mRNA of HDAC6 in mouse colonic regulatory T cells [34]. In the current study, we found that NaP at 0.1 mM did not reduce HDAC6 mRNA expression significantly (data not shown). Therefore, the effects of NaP on HDAC expression may be cell and/or species-dependent. We also found that a combination of cisplatin and TSA, a potent specific pan-HDAC inhibitor [43], including HDAC1, 2, 3, 4, 6 and 10, increased TNF-α expression significantly. These findings suggest that a combination of cisplatin and NaP enhances apoptosis by increasing expression of autocrine TNF-α via reduction of HDACs in HepG2 cells. However, the molecular mechanism downstream of GPR41 to HDACs remains unclear. Finally, in an in vivo study in xenograft tumors, we found combination treatment of cisplatin and NaP significantly suppressed growth of HepG2 cells and enhanced acetylation of histone H3, compared with cisplatin alone.
In conclusion, the synergism of propionate and cisplatin significantly enhances inhibition of proliferation and induction of apoptosis of HepG2 cells by increasing expression of autocrine TNF-α via reduction of HDACs in a GPR41-dependent manner (summarized in Figure 8). Our data suggest that combined treatment of propionate with cisplatin may exert better therapeutic effects on HCC compared with conventional treatment. Finally, a selective agonist of GPR41 might serve as a novel adjuvant therapeutic agent for treatment of HCC.
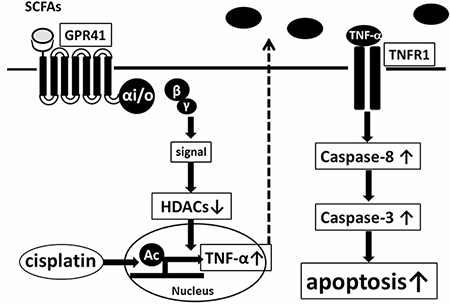
Figure 8: Model of enhancement of cisplatin-induced apoptosis in HepG2 cells mediated by GPR41, a SCFA receptor. Propionate, a gut-microbiota product, enhances proliferation of cisplatin-induced cell apoptosis of HepG2 cells by increasing expression of autocrine TNF-α via reduction of HDACs in a GPR41-dependent manner.
MATERIALS AND METHODS
Materials
(S)-2-(4-chlorophenyl)-3,3-dimethyl-N-(5-phenylthiazol-2-yl) butanamide (CFMB) and TNF-α antagonist R-7050 [44] (Calbiochem, Darmstadt, Germany); sodium propionate (NaP) (Sigma-Aldrich, St. Louis, MO, USA); pertussis toxin (PTX) (Wako, Osaka, Japan); gallein, cyclopropanecarboxylic acid (CPC) and trichostatin A (TSA) (Tokyo Chemical Industry, Tokyo, Japan); human recombinant TNF-α (PeproTech, Rocky Hill, NJ, USA); polyclonal rabbit antibodies against human β-actin (Abcam), acetyl-histone H3 (Lys9/Lys14), cleaved caspase-3 (Asp175), cleaved caspase-9 (all Cell Signaling Technology, Boston, MA USA), GPR41 (Abcam), and GPR43 (Bioss, Boston, MA, USA); monoclonal rabbit antibodies against HDAC1, HDAC4, HDAC5 (Cell Signaling Technology) and HDAC8 (Abcam); monoclonal mouse antibodies against HDAC3 and caspase-8 (Cell Signaling Technology); and horseradish peroxidase-conjugated anti-mouse, anti-goat and anti-rabbit immunoglobulins (Dako, Glostrup, Denmark) were used in the study.
Cell cultures
HepG2 cells, HuH-7 cells, JHH-4 cells and HLE cells, which are all human hepatoma cell lines, were obtained from the Japanese Collection of Research Bioresources Cell Bank. HepG2, HuH-7 and HLE cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal calf serum supplemented with 10% FBS and 1% penicillin/streptomycin, and JHH-4 cells were grown in Eagle’s minimal essential medium with 10% fetal calf serum supplemented with 10% FBS and 1% penicillin/streptomycin at 37° C in a humidified atmosphere containing 5% CO2 and 18% O2. When cells reached a logarithmic phase of growth, they were digested with trypsin and seeded in different culture plates based on experimental needs.
Cell proliferation assays
Cell proliferation was evaluated by MTS assay (Promega, Madison, WI, USA). HCC cells were seeded in 96-well plates at 5 × 103 cells per well, and treated with cisplatin (25 μM) alone, NaP (1, 10 mM) alone, or a combination of cisplatin (25 μM) and NaP (0.1, 1, 10 mM) at 37° C under 5% CO2 for 24 h. At the end of the 24-h period, 20 μl MTS reagent supplied in the CellTiter 96 Aqueous Assay was added to each well, and the plates were incubated for a further 4 h at 37° C under 5% CO2. The absorbance of the product was measured at 490 nm using a microplate reader, with a reference wavelength of 650 nm.
TaqMan real-time PCR assay
TaqMan real-time RT-PCR was performed as previously reported [45]. Specific primers and TaqMan MGB probes (Applied Biosystems) were used to detect human TNF-α (assay ID: Hs00174128_m1). A TaqMan human β-actin MGB control reagent kit (Applied Biosystems) was used to detect human β-actin. Expression of TNF-α mRNA was normalized to that of β-actin mRNA.
Knockdown of GPR41 in HepG2 cells
Gene silencing of GPR41 in HepG2 cells was performed as described previously [45]. Small interfering RNA (siRNA) against GPR41 (siGENOME Human FFAR3: GPR41 siRNA-1; and ON-TARGET plus Human FFAR3: GPR41 siRNA-2), and control siRNA (siGENOME Non-siRNA: control siRNA-1; and ON-TARGET plus Non-targeting: control siRNA-2) were purchased from GE Healthcare Dharmacon (Lafayette, CO, USA). HepG2 cells (70% confluence) were transfected with control siRNA or siRNA against GPR41 at a final concentration of 50 nmol/L, using a transfection reagent (DharmaFECT; Dharmacon). After a 72-h incubation, HepG2 cells were refreshed with DMEM and then treated for a further 24 h with cisplatin with or without NaP.
Determination of human TNF-α concentrations
The TNF-α level in each cell culture supernatant was measured by immunoassay using an ELISA kit (Quantikine Human TNF-α; R&D Systems, Minneapolis, MN, USA).
Immunoblot analysis
HepG2 cells were lysed in RIPA buffer with phosphatase inhibitors (Sigma-Aldrich). Lysates (5 μg protein) were analyzed by immunoblot analysis, first using antibodies targeting GPR41 (1:1000), GPR43 (1:1000), β-actin (1:1000), cleaved caspase-3 (1:1000), caspase-8 (1:1000), caspase-9 (1:1000), acetyl-H3 (1:1000), HDAC1 (1:1000), HDAC3 (1:1000), HDAC4 (1:1000), HDCA5 (1:1000), HDAC6 (1:1000), and HDAC8 (1:5000) for 24 h, and then with appropriate horseradish peroxidase-conjugated secondary antibodies (1:1000) at room temperature for 1 h. Immunoreactive bands were visualized as previously reported [45].
Immunohistochemistry
HCC samples were purchased from Biomax. Immunoperoxidase staining was performed with rabbit polyclonal antibodies against GPR41 (1:200) and GPR43 (1:200) as primary antibodies to determine the localization of GPR41 and GPR43. Positive staining was detected using an Envision kit/HRP (DAB) (Dako).
Cell apoptosis quantification
An annexin V kit (MBL, Nagoya, Japan) was used to evaluate apoptosis in HCC cells. Cells were resuspended in 85 μl of binding buffer. The suspension was incubated with 10 μl of Annexin V-FITC and 5 μl of propidium iodide (PI) for 15 min at room temperature in the dark. Then, 400 μl of binding buffer was added to each tube and the percentage of apoptotic cells was quantified by flow cytometry using a BD FACSCanto™ II Flow Cytometer.
In vivo xenograft study
Animals were housed in appropriate pathogen-free conditions. All experimental procedures conformed to the Regulations for Animal Research at the University of Fukui, and were reviewed by the Animal Research Committee of the University of Fukui. Mice were housed under standard conditions (12 h light/12 h dark). Six-week-old male SHO nude mice (Charles River, Japan) were subcutaneously injected with 1.0 × 106 HepG2 cells. When the tumor size reached 256–500 mm3, mice were randomly allocated to two groups (n = 5 per group) and treated by intraperitoneal injection of 8 mg/kg cisplatin or 8 mg/kg cisplatin plus 250 mg/kg NaP, both dissolved in saline. The tumor size was measured every 3 days with calipers. The tumor weight was estimated as (length × width2)/2. To standardize the variability in tumor weights among the treated groups, the relative tumor weights (RTWs) at different times were obtained using the formula TWi/TWo, where TWi is the mean tumor weight of a group on day n and TWo is the mean tumor weight on day 0. The relative mean body weight was calculated using the formula BW17/BWo, where BW17 is the mean body weight of a group on day 17 and BWo is the mean body weight on day 0. In an independent experiment, HepG2 tumor homogenates were prepared by collecting tumors 4 day after IP treatment with 8 mg/kg cisplatin or 8 mg/kg cisplatin plus 250 mg/kg NaP or 8 mg/kg cisplatin plus 50 mg/kg CPC (n = 5 per group). Tumors were frozen with liquid nitrogen and stored at –80° C prior to analysis.
Statistical analysis
Data are expressed as the mean ± the standard deviation (SD). The significance of differences between two groups was evaluated by Man–Whitney test or Student t-test, and that among more than two groups was assessed using analysis of variance (ANOVA) with a Scheffe post hoc test. Results were considered to be significant at P < 0.05.
Abbreviations
CFMB: (S)-2-(4-chlorophenyl)-3,3-dimethyl-N-(5-phenylthiazol-2-yl) butanamide; CPC: cyclopropanecarboxylic acid; DMEM: Dulbecco’s modified Eagle’s medium; GLP-1: glucagon-like peptide-1; GPCR: G-protein coupled receptor; HCC: Hepatocellular carcinoma; HDAC: histone deacetylase; MAPK: mitogen-activated protein kinase; MCP-1: monocyte chemotactic protein-1; NaP: sodium propionate; PTX: pertussis toxin; RTWs: relative tumor weights; SCFA: Short-chain fatty acid; siRNA: small interfering RNA; TNFR1: tumor necrosis factor receptor 1; TSA: trichostatin A.
Author contributions
MK performed the cell culture experiments, western blotting and flow cytometry. DM designed the study, analyzed the data, performed the immunohistochemistry, and wrote the paper. JU, TY, and HK performed the siRNA and TaqMan PCR experiments. KK performed xenograft experiments. TT and MI designed the study, analyzed the data and wrote the paper. All authors reviewed and approved the final manuscript.
ACKNOWLEDGMENTS
This work was supported in part by JSPS KAKENHI Grant Number 18K06946 (Grant-in-Aid for Scientific Research (C)), Grant Number 18K15971, Grant Number 16K19012 (Grant-in-Aid for Young Scientists (B)) granted by Japan Society for the Promotion of Science, Yamaguchi Endocrine Research Foundation, the Grant of National Center for Child Health and Development and the Smoking Research Foundation of Japan.
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest.
REFERENCES
1. Bruix J, Reig M, Sherman M. Evidence-based diagnosis, staging, and treatment of patients with hepatocellular carcinoma. Gastroenterol. 2016; 150:836–53.
2. Buecher B, Bleiberg H. Non-systemic chemotherapy in the treatment of colorectal cancer - Portal vein, hepatic arterial and intraperitoneal approaches. Aliment Pharmacol Ther. 2001; 15:1527–41.
3. Guarner F, Malagelada JR. Gut flora in health and disease. Lancet. 2003; 360:512–9.
4. Donohoe DR, Garge N, Zhang X, Sun W, O’Connell TM, Bunger MK, Bultman SJ. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011; 13:517–26.
5. Harig JM, Soergel KH, Komorowski RA, Wood CM. Treatment of diversion colitis with short-chain-fatty acid irrigation. N Engl J Med. 1989; 320:23–8.
6. Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, Tobe T, Clarke JM, Topping DL, Suzuki T, Taylor TD, Itoh K, Kikuchi J, et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature. 2011; 469:543–7.
7. Bindels LB, Porporato P, Dewulf EM, Verrax J, Neyrinck AM, Martin JC, Scott KP, Buc Calderon P, Feron O, Muccioli GG, Sonveaux P, Cani PD, Delzenne NM. Gut microbiota-derived propionate reduces cancer cell proliferation in the liver. Br J Cancer. 2012; 107:1337–44.
8. Tang Y, Chen Y, Jiang H, Robbins GT, Nie D. G-protein-coupled receptor for short-chain fatty acids suppresses colon cancer. Int J Cancer. 2011; 128:847–56.
9. Thirunavukkarasan M, Wang C, Rao A, Hind T, Teo YR, Siddiquee AAM, Goghari MAI, Kumar AP, Herr DR. Short-chain fatty acid receptors inhibit invasive phenotypes in breast cancer cells. PLoS One. 2017; 12:e0186334.
10. Candido EPM, Reeves R, Davie JR. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell. 1978; 14:105–13.
11. Sealy L, Chalkley R. The effect of sodium butyrate on histone modification. Cell. 1978; 14:115–21.
12. Vidali G, Boffa LC, Bradbury EM, Allfrey VG. Butyrate suppression of histone deacetylation leads to accumulation of multiacetylated forms of histones H3 and H4 and increased DNase I sensitivity of the associated DNA sequences. Proc Natl Acad Sci USA. 1978; 75:2239–43.
13. Boffa IC, Vidali G, Mann RS, Allfrey VG. Suppression of histone deacetylation in vivo and in vitro by sodium butyrate. J Biol Chem. 1978; 253:3364–6.
14. Hinnebusch BF, Meng S, Wu JT, Archer SY, Hodin RA. The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. J Nutr. 2002; 132:1012–7.
15. Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, Muir AI, Wigglesworth MJ, Kinghorn I, Fraser NJ, Pike NB, Strum JC, Steplewski KM, et al. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem. 2003; 278:11312–9.
16. Hong YH, Nishimura Y, Hishikawa D, Tsuzuki H, Miyahara H, Gotoh C, Choi KC, Feng DD, Chen C, Lee HG, Katoh K, Roh SG, Sasaki S. Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology. 2005; 146:5092–9.
17. Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, MacKay F, Artis D, Xavier RJ, Teixeira MM, MacKay CR. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009; 461:1282–6.
18. Koprinarova M, Markovska P, Iliev I, Anachkova B, Russev G. Sodium butyrate enhances the cytotoxic effect of cisplatin by abrogating the cisplatin imposed cell cycle arrest. BMC Mol Biol. 2010; 11:49.
19. Buurman R, Grlevik E, Schffer V, Eilers M, Sandbothe M, Kreipe H, Wilkens L, Schlegelberger B, Khnel F, Skawran B. Histone deacetylases activate hepatocyte growth factor signaling by repressing microRNA-449 in hepatocellular carcinoma cells. Gastroenterology. 2012; 143:811–820.e15.
20. Shin S, Kim M, Lee SJ, Park KS, Lee CH. Trichostatin a sensitizes hepatocellular carcinoma cells to enhanced NK cell-mediated killing by regulating immune-related genes. Cancer Genomics Proteomics. 2017; 14:349–62.
21. Rivera S, Leteur C, Mégnin F, Law F, Martins I, Kloos I, Depil S, Modjtahedi N, Perfettini JL, Hennequin C, Deutsch E. Time dependent modulation of tumor radiosensitivity by a pan HDAC inhibitor: abexinostat. Oncotarget. 2017; 8:56210–27. https://doi.org/10.18632/oncotarget.14813.
22. Kim MS, Blake M, Baek JH, Kohlhagen G, Pommier Y, Carrier F. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res. 2003; 63:7291–300.
23. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007; 35:495–516.
24. Powers SK, Hudson MB, Nelson WB, Talbert EE, Min K, Szeto HH, Kavazis AN, Smuder AJ. Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit Care Med. 2011; 39:1749–59.
25. Mangmool S, Kurose H. Gi/o protein-dependent and -independent actions of pertussis toxin (ptx). Toxins (Basel). 2011; 3:884–99.
26. Park JS, Lee EJ, Lee JC, Kim WK, Kim HS. Anti-inflammatory effects of short chain fatty acids in IFN-gamma-stimulated RAW 264.7 murine macrophage cells: involvement of NF-kappaB and ERK signaling pathways. Int Immunopharmacol. 2007; 7:70–7.
27. Vinolo MAR, Rodrigues HG, Hatanaka E, Sato FT, Sampaio SC, Curi R. Suppressive effect of short-chain fatty acids on production of proinflammatory mediators by neutrophils. J Nutr Biochem. 2011; 22:849–55.
28. Tedelind S, Westberg F, Kjerrulf M, Vidal A. Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: A study with relevance to inflammatory bowel disease. World J Gastroenterol. 2007; 13:2826–32.
29. Kobayashi M, Mikami D, Kimura H, Kamiyama K, Morikawa Y, Yokoi S, Kasuno K, Takahashi N, Taniguchi T, Iwano M. Short-chain fatty acids, GPR41 and GPR43 ligands, inhibit TNF-α-induced MCP-1 expression by modulating p38 and JNK signaling pathways in human renal cortical epithelial cells. Biochem Biophys Res Commun. 2017; 486:499–505.
30. Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010; 328:228–31.
31. Ang Z, Er JZ, Tan NS, Lu J, Liou YC, Grosse J, Ding JL. Human and mouse monocytes display distinct signalling and cytokine profiles upon stimulation with FFAR2/FFAR3 short-chain fatty acid receptor agonists. Sci Rep. 2016; 6:34145.
32. Andrade-Oliveira V, Amano MT, Correa-Costa M, Castoldi A, Felizardo RJF, de Almeida DC, Bassi EJ, Moraes-Vieira PM, Hiyane MI, Rodas ACD, Peron JPS, Aguiar CF, Reis MA, et al. Gut bacteria products prevent aki induced by ischemia-reperfusion. J Am Soc Nephrol. 2015; 26:1877–88.
33. Aoyama M, Kotani J, Usami M. Butyrate and propionate induced activated or non-activated neutrophil apoptosis via HDAC inhibitor activity but without activating GPR-41/GPR-43 pathways. Nutrition. 2010; 26:653–61.
34. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic treg cell homeostasis. Science. 2013; 341:569–73.
35. Benedetti G, Fredriksson L, Herpers B, Meerman J, Van De Water B, De Graauw M. TNF-α-mediated NF-κB survival signaling impairment by cisplatin enhances JNK activation allowing synergistic apoptosis of renal proximal tubular cells. Biochem Pharmacol. 2013; 85:274–86.
36. Pabla N, Dong Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008; 73:994–1007.
37. van Horssen R, Ten Hagen TLM, Eggermont AMM. TNF-alpha in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist. 2006; 11:397–408.
38. Sun H, Zhong YJ, Zheng XL, Wang Q, Yang L, Shi F, Yan JQ, He F, Liao LC, Lin Y, Zhang L, Wang X. Critical role of CD40-mediated autocrine tumor necrosis factor-alpha in potentiation of cisplatin-induced cytotoxicity in cancer cells. Cancer Sci. 2012; 103:197–202.
39. Liu J, Li G, Wang X, Wang L, Zhao R, Wang J, Kong Y, Ding J, Li J, Zhang L. Droxinostat, a histone deacetylase inhibitor, induces apoptosis in hepatocellular carcinoma cell lines via activation of the mitochondrial pathway and downregulation of FLIP. Transl Oncol. 2016; 9:70–8.
40. Fan J, Lou B, Chen W, Zhang J, Lin S, Lv FF, Chen Y. Down-regulation of HDAC5 inhibits growth of human hepatocellular carcinoma by induction of apoptosis and cell cycle arrest. Tumour Biol. 2014; 35:11523–32.
41. Kanno K, Kanno S, Nitta H, Uesugi N, Sugai T, Masuda T, Wakabayashi G, Maesawa C. Overexpression of histone deacetylase 6 contributes to accelerated migration and invasion activity of hepatocellular carcinoma cells. Oncol Rep. 2012; 28:867–73.
42. Wu J, Du C, Lv Z, Ding C, Cheng J, Xie H, Zhou L, Zheng S. The up-regulation of histone deacetylase 8 promotes proliferation and inhibits apoptosis in hepatocellular carcinoma. Dig Dis Sci. 2013; 58:3545–53.
43. Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999; 401:188–93.
44. Gururaja TL, Yung S, Ding R, Huang J, Zhou X, McLaughlin J, Daniel-Issakani S, Singh R, Cooper RDG, Payan DG, Masuda ES, Kinoshita T. A class of small molecules that inhibit TNFα-induced survival and death pathways via prevention of interactions between TNFαRI, TRADD, and RIP1. Chem Biol. 2007; 14:1105–18.
45. Mikami D, Kimura H, Kamiyama K, Torii K, Kasuno K, Takahashi N, Yoshida H, Iwano M. Telmisartan activates endogenous peroxisome proliferator-activated receptor-δ and may have anti-fibrotic effects in human mesangial cells. Hypertens Res. 2014; 37:422–31.