
INTRODUCTION
Biallelic APC mutations initiate the development of a high percentage of colorectal cancers [1, 2]. APC encodes a multi-purpose protein whose functions include negative regulation of the canonical WNT signaling pathway [3]. The APC protein inactivates canonical WNT signaling by limiting availability of β-catenin [4], a licensing factor that modifies how members of the TCF/LEF family of transcription factors regulate gene transcription [5]. APC interacts with β-catenin in a cytoplasmic complex that facilitates β-catenin degradation [6–8], while nuclear APC facilitates both β-catenin export to the cytoplasm [9–11] and β-catenin removal from specific genomic loci [12]. Interaction of APC with chromatin-associated β-catenin negatively regulates the expression of MYC, AXIN2, DKK1 and SP5 [12, 13], four known WNT targets.
The contribution of APC loss to gene expression has been assumed to be exclusively β-catenin-mediated, although this has not been broadly addressed experimentally. This study was designed to identify a more comprehensive list of genes transcriptionally regulated by chromatin-associated APC and to determine whether or not APC mediates their transcriptional repression exclusively through displacement of β-catenin from TCF/LEF family transcription factor complexes.
Chromatin immunoprecipitation of APC and next-generation sequencing were performed from HCT-116 colon cancer cells, which express wild-type APC yet can model the APC loss observed in the majority of colorectal cancers following transient siRNA-based silencing. Gene expression data were collected from HCT-116 cells in the presence or absence of siRNA targeting APC and were compared to ChIP-seq data to identify candidate genes controlled by chromatin-associated APC. High-confidence candidate genes were likely shared targets of canonical WNT signaling and surprisingly included both genes increased in expression following APC loss and decreased in expression following APC loss.
APC-associated genomic sequences identified in our initial screening step exhibited enrichment of validated transcription factor binding sites for both TCF7L2 and AP-1, and co-occurrence of these transcription factors within many of these same genomic regions. These results indicate that AP-1 modulation should be investigated as a potential therapeutic strategy for targeting the expression of a large subset of canonical WNT target genes.
RESULTS
APC ChIP-seq identified 3,985 APC-associated genomic regions
Chromatin immunoprecipitation of APC was performed in two biological replicates from HCT-116 colon cancer cells that express wild-type APC but also a degradation-resistant point mutant of β-catenin that constitutively activates canonical WNT signaling. Previously published reports identified four genes (MYC, AXIN2, SP5 and DKK1) regulated by chromatin-associated APC [12, 13], and peak-calling thresholds were adjusted to pass the stringent false discovery rate (FDR) cutoff of 1.07% (Figure 1A) while retaining peaks at three out of four of these internal positive control loci (Figure 1B). ChIP-seq data quality was consistent between replicates 1 and 2 (Figure 1C). 3,985 genomic regions enriched by APC ChIP (p < 0.00001) were identified in both replicates relative to their respective inputs (Figure 1D).
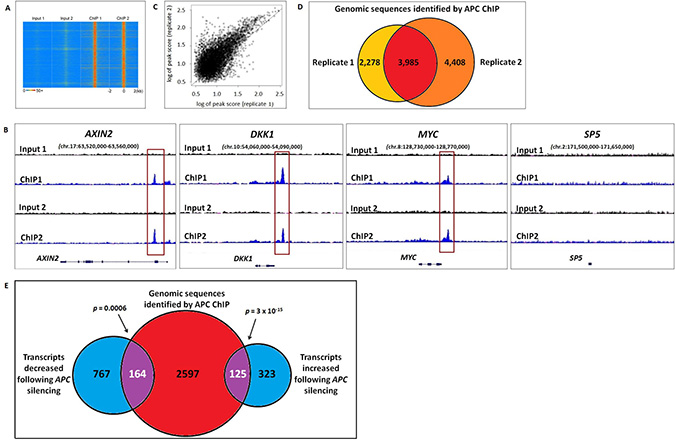
Figure 1: Genomic sequences enriched by APC ChIP overlap with transcripts altered in expression following APC loss. (A) A heat map with rows corresponding to 4-kb genomic regions centered on each peak shared between replicates 1 and 2 shows high ChIP signal intensity (red) in each ChIP-seq sample and low background (blue) in each matching input sample. (B) Sequencing data visualized using the Integrative Genomic Viewer confirm that three out of four positive control loci (AXIN2, DKK1, MYC and SP5) known to be regulated by chromatin-associated APC include genomic regions (red boxes) enriched by APC ChIP. (C) A scatter plot compares peak scores (in log scale) from ChIP-seq replicates 1 (x-axis) and 2 (y-axis), indicating high signal:background ratio and consistency in peaks scores (Pearson correlation coefficient ρ = 0.8246). (D) Genomic peaks identified in both APC ChIP-seq replicates (p < 0.00001) were defined as overlapping if their summits were separated by less than 400-bp (the median of peak width in the peak calling results). (E) 2,886 genes associated with one or more genomic sequences enriched in both APC ChIP-seq replicates were compared to 448 transcripts that increased and 931 transcripts that decreased in the same cell line following APC silencing (q < 0.05). 289 overlapping genes (purple) were identified as potential targets of direct transcriptional control by chromatin-associated APC.
RNA-seq data identify 289 APC-responsive transcripts encoded by genes located near genomic regions enriched by APC ChIP-seq
The effects of chromatin-associated APC binding on gene transcription were evaluated by RNA-seq analysis of HCT-116 cells in the presence or absence of siRNA reducing APC expression. Differential expression analysis using Cufflinks software [14] identified 1,379 transcripts altered in expression following APC silencing (q < 0.05), including two out of four positive control transcripts (AXIN2 and SP5, Figure 2B). Transcripts were compared to 3,985 genomic sequences enriched in both APC ChIP-seq replicates, which had been assigned to 2,886 different genes based on proximity to transcription start sites. Comparison of the ChIP-seq and RNA-seq analyses identified 125 genes increased and 164 genes decreased in expression following APC silencing (Figure 1E). This indicates that chromatin-associated APC may act as a transcriptional repressor or activator depending on the chromatin context, despite the fact that chromatin-associated APC antagonizes canonical WNT activation of all four target genes identified in previous studies [12, 13].
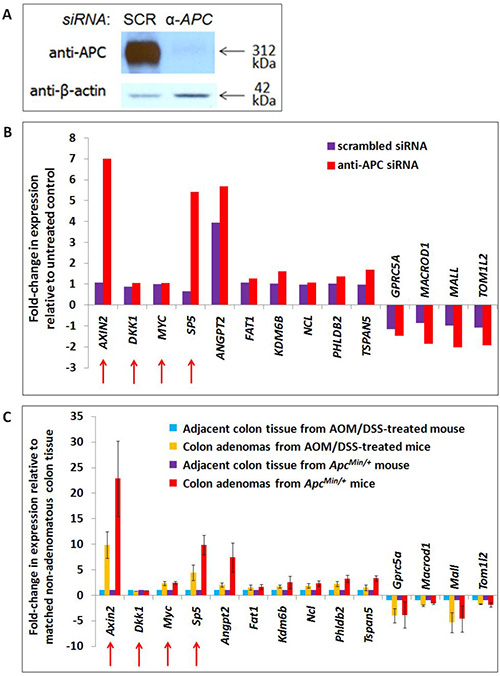
Figure 2: Transcription of APC-associated genes changes in APC-deficient cells and mouse adenomas with activated WNT signaling. A list of 2,886 genes associated with genomic regions enriched by APC ChIP were compared with 1,379 transcripts altered in expression following APC silencing by siRNA in the same cell line (Panels A, B) and 1,535 transcripts altered in expression following both Apc loss and Ctnnb1 activation in mouse colon adenomas (Panel C). Forty-nine genes satisfied both screening criteria, including ten candidates depicted in Panels B and C, and two positive controls (AXIN2 and SP5, indicated with red arrows). Two other positive controls (also indicated with red arrows) failed to satisfy these screening criteria due to an insignificant change in gene expression in either the cell line model alone (MYC) or in both models (DKK1). RNA-seq data for these genes are included in Panels B (human cell line) and C (mouse adenomas). Error bars representing standard deviation are included only for conditions for which multiple samples were sequenced.
Mouse tumor RNA-seq identifies some APC-sensitive transcripts as targets of canonical Wnt signaling
HCT-116 cells are mismatch repair-deficient and relatively unstable. To circumvent these limitations and select candidate targets shared with in vivo models of tumorigenesis, genes of interest were filtered further using colon adenoma RNA-seq data from two mouse models of colon tumorigenesis on a C57BL/6J background [15]: one expressing wild-type Apc and a degradation-resistant mutant β-catenin (as a result of treatment with azoxymethane and dextran sulfate sodium (AOM/DSS-treated) [16]) and the other with loss of function mutations of Apc (ApcMin/+) (Figure 1E). These two models of canonical Wnt-driven mouse intestinal tumorigenesis were selected for their abilities to generate tumors in the colon specifically, resulting in a better basis for comparison to the HCT-116 colon cancer cell model. The use of two adenoma types enabled identification of shared (canonical Wnt-driven) transcriptional changes with higher confidence while filtering out confounding factors (such as the inflammatory component of the AOM/DSS model) [15, 17]. While they differ in that adenomas from the ApcMin/+ model lack functional Apc whereas those from AOM/DSS-treated mice retain wild-type Apc and its chromatin-associated functions, loss of the Apc-dependent cytoplasmic mechanism promoting β-catenin degradation is a common feature of both adenoma types that makes them useful for the identification of canonical Wnt target genes. Of the 289 candidate transcripts from the previous filtering step, 49 changed in expression in the same direction as in the human RNA-seq data by at least 1.5-fold in both ApcMin/+ and AOM/DSS colon adenomas relative to non-adenoma colon tissue controls (by one-sample t-test with a significance cutoff of FDR < 0.1). These represent candidate target genes altered by canonical WNT signaling. 31 genes increased while 18 genes decreased in APC/Apc-deficient cells and adenomas. An additional filtering criterion focused on potential transcriptional regulatory elements identified by APC ChIP-seq and located within 10 kb upstream of transcription start sites or within first introns. This further reduced the number of candidate genes to 16 increased and 9 decreased following APC loss (siRNA-based, shown in Figure 2A). Ten candidate genes (Figure 2B, 2C) were selected for further study based on APC ChIP-qPCR validation of enrichment comparable to positive control peaks. APC ChIP enrichment of genomic regions in these ten loci relative to alpha-satellite repeat negative controls and comparable to AXIN2 intron 1 and MYC promoter element positive controls is clearly visible by qPCR in Supplementary Figure 3B (although the overall purpose of the figure is different). Expression values for these ten targets are depicted from the mouse (Figure 2C) and human (Figure 2B) RNA-seq datasets.
Binding sites for AP-1 and several promoter-associated transcription factors were enriched in APC peaks
We asked whether chromatin-associated APC is recruited to specific genomic sequences exclusively through a mechanism mediated by interaction with β-catenin [12] or through multiple mechanisms. Sequences enriched by APC ChIP were subjected to motif analysis using the MatInspector (from the Genomatix Software Suite) [18], MEME-ChIP [19] and Regulatory Sequence Analysis Tools (RSAT) [20] algorithms (Figure 3A). All three algorithms detected enrichment of binding sites for the AP-1 transcription factor (variations of the TPA response element, TGASTCA), while TCF7L2 binding sites (expected to predominate based on the association of APC with β-catenin and its transcription factor-binding partner TCF7L2) were significantly enriched according to MEME-ChIP and MatInspector only (Figure 3A). Enriched motifs identified by multiple algorithms (TCF7L2, AP-1, NRF1, SP1, EGR-1 and USF1/2 binding sites) were further investigated as candidates to mediate APC recruitment and transcriptional control.
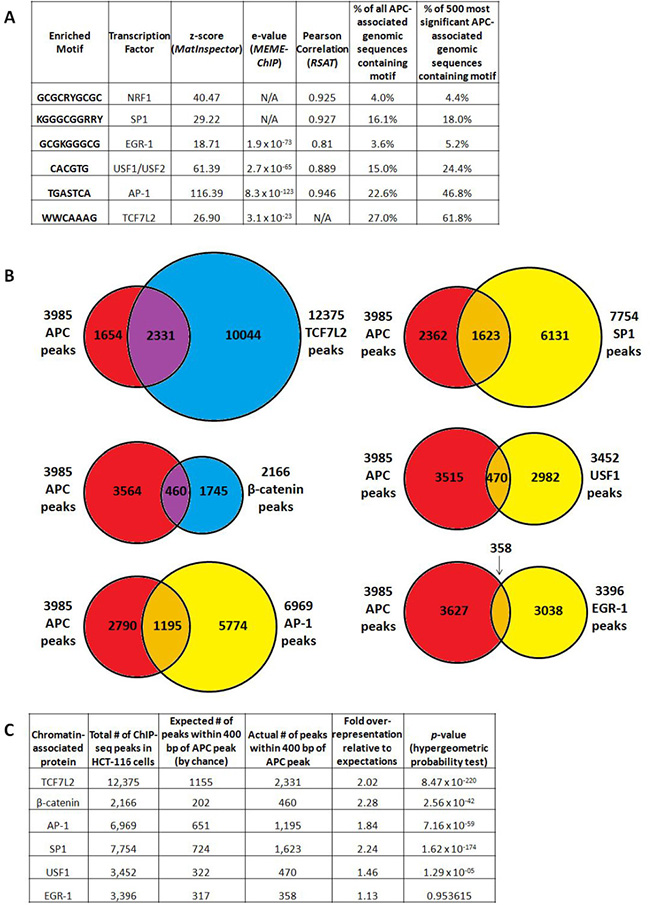
Figure 3: APC-associated genomic sequences are enriched for TCF7L2, AP-1 and SP1 transcription factor binding motifs. (A) Motif analysis of all peaks using the MatInspector (Genomatix Software Suite), MEME-ChIP and Regulatory Sequence Analysis Tools (RSAT) algorithms detected significant enrichment of predicted binding sites, particularly for the AP-1, EGR-1 and USF1/USF2 transcription factors. All transcription factor binding motifs occurred more frequently in the subset of 500 genomic sequences enriched by APC ChIP with the lowest p-values, particularly those for TCF7L2 (as expected) and AP-1. (B) 3,985 APC ChIP-seq peaks were compared with corresponding peaks from several transcription factor ChIP-seq experiments. (C) Overlap data were tabulated to calculate fold overrepresentation of each transcription factor binding site among APC-associated genomic regions, relative to expected background (p-values calculated by hypergeometric probability test).
The highest-confidence peak subset from APC ChIP-seq is highly enriched for TCF7L2 and AP-1 binding sites.
AP-1 (TGASTCA [21]) and TCF7L2 (WWCAAAG [22]) binding motifs occur in 22.6% and 27.0% of all peaks, respectively. Those for SP1 (KGGGCGGRRY [23], 16.1%), USF1/2 (CACGTG [24], 15.0%), NRF1 (GCGCRYGCGC [25], 4.0%) and EGR-1 (GCGKGGGCG [26], 3.6%) occur less frequently. Occurrences of each binding site were then counted within a subset of 500 APC-associated genomic sequences with the highest p-values for ChIP-seq enrichment (Figure 3A, columns 6 and 7). All six candidate transcription factor binding motifs show a trend of increasing enrichment as lower-confidence peaks are filtered out, with TCF7L2 (61.8%) and AP-1 (46.8%) binding sites emerging as the best represented and most likely to play roles in APC recruitment.
APC silencing modifies transcriptional activity driven by TCF7L2 motifs.
Luciferase reporter assays were then used to test the hypothesis that APC participates in transcription factor complexes organized around binding sites both for TCF7L2 and for other transcription factors, including NRF1, SP1, EGR-1, USF1/USF2 and AP-1. Three consecutive repeats of NRF1, SP1, EGR-1 USF1/2 or AP-1 consensus binding motifs were cloned into the pGL3-promoter reporter vector upstream of the firefly luciferase gene, similar to positive (TOPFLASH) and negative control (FOPFLASH) reporter vectors containing six repeats of either wild-type or mutant TCF7L2 (formerly known as TCF4) binding sites [3]. APC knockdown enhances the ability of the TOPFLASH (TCF7L2) positive control to drive luciferase expression by two-fold, while other transcription factor binding motifs show little or no sensitivity to the presence or absence of APC (Supplementary Figure 1). These results support a model in which TCF7L2 is required for gene activation, without excluding the possibility that other transcription factor binding motifs mediate transcriptional sensitivity to APC in the genomic context of a complete regulatory element.
APC ChIP data show significant overlap with published ChIP data for β-catenin, TCF7L2, AP-1 and SP1
Public ChIP-seq datasets generated from HCT-116 cells were accessed for β-catenin and TCF7L2, as well as for the transcription factors JUND and FOSL1 (components of heterodimeric AP-1 complexes), USF1, SP1 and EGR-1. Data were available from the laboratory of Dr. Richard Myers at the HudsonAlpha Institute for Biotechnology through the NCBI Gene Expression Omnibus [27], and from published work on β-catenin from the laboratories of Dr. Shannon McWeeney and Dr. Gregory Yochum [22]. Chromosomal locations of 3,985 APC-associated genomic regions were compared to 12,375 TCF7L2 peaks, 2,166 β-catenin peaks, 6,969 “AP-1” peaks (shared between JUND and FOSL1), 7,754 SP1 peaks, 3,452 USF1 peaks and 3,396 EGR-1 peaks (Figure 3B) to detect overlap. Fold over-representation was calculated for the occurrence of transcription factor binding site overlap with APC-associated genomic peaks relative to expected occurrences by random chance, with hypergeometric probability testing to calculate p-values (Figure 3C). APC peaks overlapped most significantly with TCF7L2 peaks, as well as with SP1, AP-1 and β-catenin peaks. USF1 binding sites exhibited more modest overlap with APC; overlap with EGR-1 binding was not statistically significant (Figure 3C).
Overlap of APC ChIP-seq with transcription factor binding is more pronounced near genes associated with increased expression following APC loss
Overlap was re-assessed in the subset of APC-associated genomic regions linked to 257 genes decreased and 292 genes increased in expression following APC loss in HCT-116 cells (Figure 4A). TCF7L2 binding sites showed the most significant overlap with the APC-associated regions, while β-catenin-associated sites occurred with more dramatic over-representation but with less significant p-values due to the smaller dataset (Figure 4B). USF1 and EGR-1 binding sites do not occur at higher-than-expected rates in these APC-associated regions. Surprisingly, both AP-1 and SP1 binding sites were overrepresented approximately 2- or 3-fold, and occurred more frequently and with greater significance in regions associated with increased as opposed to decreased expression following APC loss (Figure 4B). These observations were consistent with the role of chromatin-associated APC as a WNT antagonist, as β-catenin and TCF7L2 transcriptionally activate most targets while repressing transcription of a smaller subset [28–33].
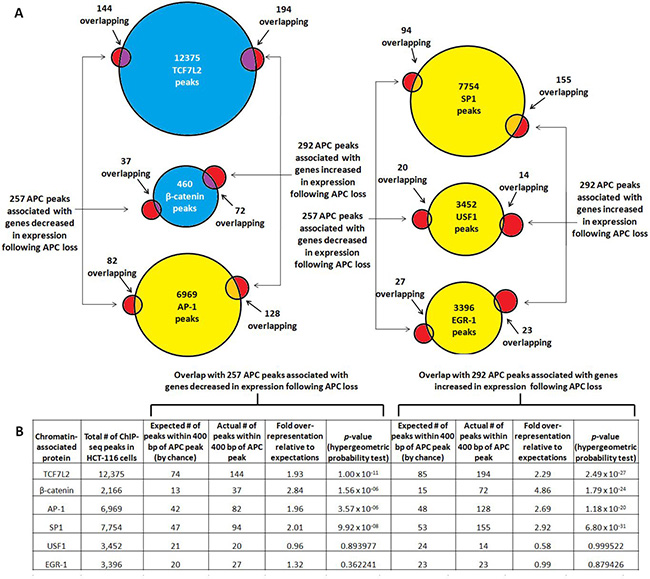
Figure 4: TCF7L2, β-catenin, AP-1 and SP1 bind genomic regions associated with increased expression following APC loss. (A) Occurrence of validated transcription factor binding sites was examined in 549 genomic sequences enriched by APC ChIP-seq and associated with genes increased (292, red circles on right) or decreased (257, red circles on left) in expression following APC silencing. (B) Overlap data were tabulated to calculate fold overrepresentation of each transcription factor binding site relative to expected background. The hypergeometric probability test indicated higher significance of overlap for all transcription factors with the subset of APC-associated genomic regions linked to increased as opposed to decreased expression following APC loss (column 10 compared to column 6).
TCF7L2 and AP-1 transcription factor binding sites frequently occur within the same or neighboring genomic regions
Co-occurrence of TCF7L2 and AP-1 binding sites was examined in order to test the hypothesis that these two transcription factor binding sites might coordinately regulate shared target genes. Frequencies were examined in a subset of 549 genomic sequences chosen because of their enrichment by APC ChIP-seq and their proximity to 280 genes altered in expression following APC silencing in HCT-116 cells (Supplementary Figure 2). Neighboring genomic regions (defined as those assigned to the same gene locus based on proximity to the nearest transcription start site) were grouped together, making the overlap between TCF7L2 and AP-1 binding sites much more striking (Supplementary Figure 2). TCF7L2 and AP-1 binding sites frequently occur within the same genomic regions, particularly in genomic peaks associated with genes transcriptionally activated following APC silencing (Supplementary Figure 2A vs. 2B). A similar co-occurrence was observed between TCF7L2 and SP1 binding sites (Supplementary Figure 2C vs. 2D). Genomic sequences associated with genes transcriptionally repressed following APC loss contained fewer binding sites for TCF7L2, AP-1 and SP1. These data indicate that TCF7L2 and AP-1 or SP1 transcription factors might bind neighboring genomic regions and converge on the same gene promoter to coordinate transcriptional regulation.
Target selection and validation
Ten genomic regions of interest were selected for further study (Figure 5) based on differential expression of their encoded transcripts following APC loss in both cell culture (Figure 2B) and mouse colon adenoma (Figure 2C) models, and successful APC ChIP-qPCR validation comparable to positive control peaks clearly visible by qPCR in Supplementary Figure 3B (although the overall purpose of the figure is different). APC-associated regions from the AXIN2 and MYC loci served as positive controls. Analysis of transcription factor ChIP-seq data reveals overlap of four candidate regions with TCF7L2 and three candidate regions with β-catenin binding sites, so that canonical WNT signaling is clearly linked to ANGPT2 intron 1, the FAT1 enhancer, PHLDB2 intron 1, TSPAN5 intron 1, MALL intron 1 and TOM1L2 intron 1. AP-1 binding sites overlap with the PHLDB2 intron 1, MALL intron 1 and TOM1L2 intron 1 regions, while SP1 binding sites overlap with the FAT1 enhancer, PHLDB2 intron 1 and MALL intron 1 regions (Figure 5). The GPRC5A enhancer contained predicted TCF7L2 binding motifs not validated by TCF7L2 ChIP (as did the MYC promoter, ANGPT2 intron 1, and MALL intron 1, Supplementary Figure 4). Five of these loci contain other APC-associated ChIP-seq peaks collectively characterized by the co-occurrence of TCF7L2 and AP-1 binding sites (Supplementary Figure 4, columns 6, 7, and 8). Regions from the KDM6B enhancer, MACROD1 promoter and NCL intron 1 were of interest due to the absence of TCF7L2 or β-catenin binding sites. Their inclusion in subsequent experiments tested the hypothesis that chromatin-associated APC may modulate the transcription of a subset of genes independently of β-catenin or canonical WNT signaling.
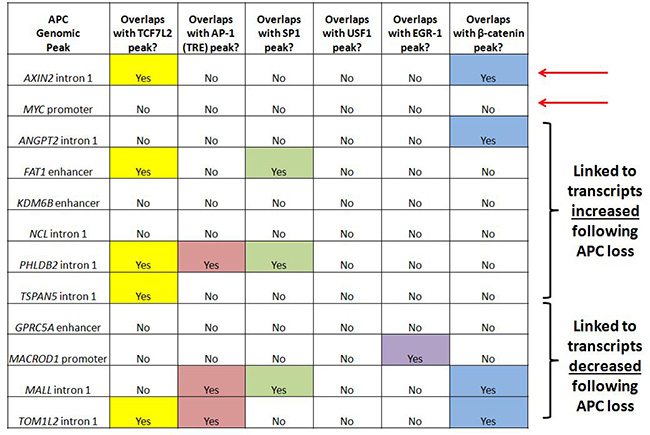
Figure 5: APC-associated candidate genomic regions contain predicted binding motifs for TCF7L2 and AP-1. Transcription factor binding sites identified by ChIP-seq are listed for ten candidate genomic sequences enriched by APC ChIP-seq and associated with transcripts altered in expression following APC loss in both in vitro and in vivo models. Since many candidate genes are associated with multiple peaks, one peak was chosen in each case based on lowest p-value and shortest distance to transcription start site. Most of these genomic sequences contain TCF7L2 and/or AP-1 transcription factor binding sites. Red arrows indicate loci located in AXIN2 intron 1 and the MYC promoter known to be transcriptionally regulated by chromatin-associated APC.
Silencing of ß-catenin expression reduces enrichment of some candidate genomic regions by APC ChIP
ChIP-qPCR experiments were designed to test whether APC requires interaction with β-catenin to associate with chromatin at candidate genomic sequences. ChIP was performed from HCT-116 cells transfected with either scrambled siRNA or siRNA silencing CTNNB1 (encoding β-catenin). ChIP antibodies targeted either β-catenin itself (Supplementary Figure 3A) or APC (Supplementary Figure 3B). Reduced enrichment by β-catenin ChIP following transfection with anti-CTNNB1 siRNA (Supplementary Figure 3A) was exhibited by both positive controls (from AXIN2 intron 1 and the MYC promoter) and six out of ten candidate sequences (from the FAT1 enhancer, PHLDB2 intron 1, TSPAN5 intron 1, the GPRC5A enhancer, MALL intron 1 and TOM1L2 intron 1). Silencing of CTNNB1 produced more subtle effects on APC ChIP enrichment of target sequences (Supplementary Figure 3B), as only one of the two positive controls (a characterized canonical WNT binding site from AXIN2 intron 1, but not a site from the MYC promoter) and two of the candidate genomic sequences (from TSPAN5 intron 1 and the GPRC5A enhancer) exhibited loss of enrichment. Collectively, these knockdown data indicate that TSPAN5, GPRC5A, FAT1, PHLDB2, MALL and TOM1L2 are shared targets of APC and β-catenin, consistent with the widely accepted model that interaction with β-catenin mediates APC recruitment to, and function in, the chromatin fraction [12, 13, 34]. The remaining three candidates (from the KDM6B enhancer, NCL intron 1 and the MACROD1 promoter) are inconclusive. Six of the seven putative targets of APC and β-catenin/canonical WNT signaling overlap with TCF7L2 or β-catenin binding sites (Figure 5), while the seventh target, the GPRC5A enhancer, has a neighboring peak that exhibits TCF7L2 binding (Supplementary Figure 4, column 7). Overall, the data in Figure 5 and Supplementary Figure 4 indicate that a substantial proportion of APC targets are likely β-catenin- and TCF7L2-dependent.
Candidate genomic regions were screened by luciferase reporter assay for APC- or β-catenin-dependent changes in their ability to drive transcription.
Previous studies have constructed a model in which APC antagonizes canonical WNT activation of target genes [12, 13]. Interestingly, we find that not all β-catenin-dependent target genes of chromatin-associated APC increase in expression following APC silencing or mutation, as GPRC5A, MALL and TOM1L2 decrease following APC loss in vitro (Figure 2B) and in vivo (Figure 2C). These data demonstrate that APC associates with chromatin at WNT-repressed target genes as well, likely via a similar mechanism of interaction with β-catenin. This was further tested using luciferase reporter assays to characterize the genomic sequences enriched by APC ChIP-seq (Figure 6). Peaks of interest (approximately 0.5–1.0 kb) were PCR-amplified, cloned into the pGL3-promoter firefly luciferase vector and transfected into HCT-116 cells to measure their ability to drive luciferase expression either in the presence or absence of siRNA to APC (Figure 6A) or CTNNB1 (Figure 6B).
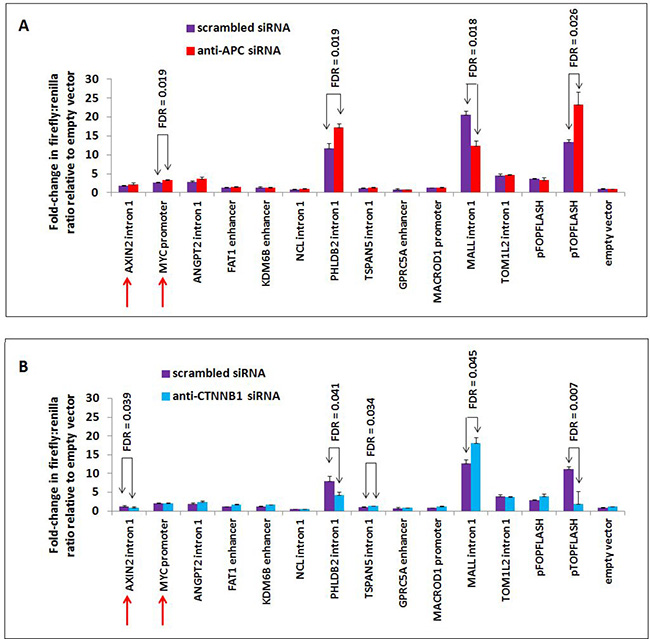
Figure 6: APC or CTNNB1 silencing alters transcriptional activity from elements in the PHLDB2 and MALL loci. Ten genomic sequences associated with APC and two positive control sequences (from AXIN2 intron 1 and MYC promoter, indicated by red arrows) were PCR-amplified, cloned into the pGL3-promoter firefly luciferase vector and transfected into HCT-116 cells previously transfected with either scrambled siRNA (purple) or siRNA targeting APC (red, panel A) or CTNNB1 (blue, panel B). The PHLDB2 intron 1 construct was more active following APC silencing (panel A) and less active following CTNNB1 silencing (panel B), similar to the pTOPFLASH positive control. The MALL intron 1 construct was less active following APC silencing and more active following CTNNB1 silencing. Errors bars are based on standard deviation, and FDR values lower than 0.05 are indicated (1-tailed Student’s t-test).
Many of the candidate luciferase constructs were not sufficient to drive transcription of the reporter above baseline levels, including the AXIN2 intron 1 and MYC promoter positive controls. Other candidates showed only limited responsiveness to siRNA reducing the expression of APC. Candidate sequences from PHLDB2 intron 1 and MALL intron 1 exhibited high transcriptional activity and strong responses to both APC and CTNNB1 silencing. APC silencing by siRNA transfection increased luciferase activity from the 541-bp PHLDB2 intron 1 construct (Figure 6A), while silencing of CTNNB1 (encoding β-catenin) decreased luciferase activity (Figure 6B). These results indicate that this genomic region contributes to the activation of PHLDB2 transcription following APC loss both observed both in vitro (Figure 2B) and in vivo (Figure 2C). The reporter construct containing a region of MALL intron 1 decreased its ability to drive transcription following APC silencing (Figure 6A) and increased its activity following CTNNB1 silencing (Figure 6B). Both observations indicate that this genomic region contributes to the transcriptional activation of MALL following APC loss (Figure 2B and 2C). It is important to note that this effect likely is mediated not only by the loss of APC function as a direct regulator of β-catenin in the partially chromatinized context of the luciferase construct, but also by the up-regulation of β-catenin protein levels that typically follows the loss of cytoplasmic APC as a negative regulator of β-catenin stability. Expression profiling data confirming that Phldb2 increases in expression while Mall decreases in expression in mouse adenomas relative to adjacent non-adenoma tissue (Figure 2C) match reports that canonical WNT signaling activates the transcription of certain genes and simultaneously represses the transcription of others [28–33]. Collectively, the results of this study indicate that chromatin-associated APC may function to reverse the effects of canonical WNT signaling on both activation and repression of targets (Figure 7).
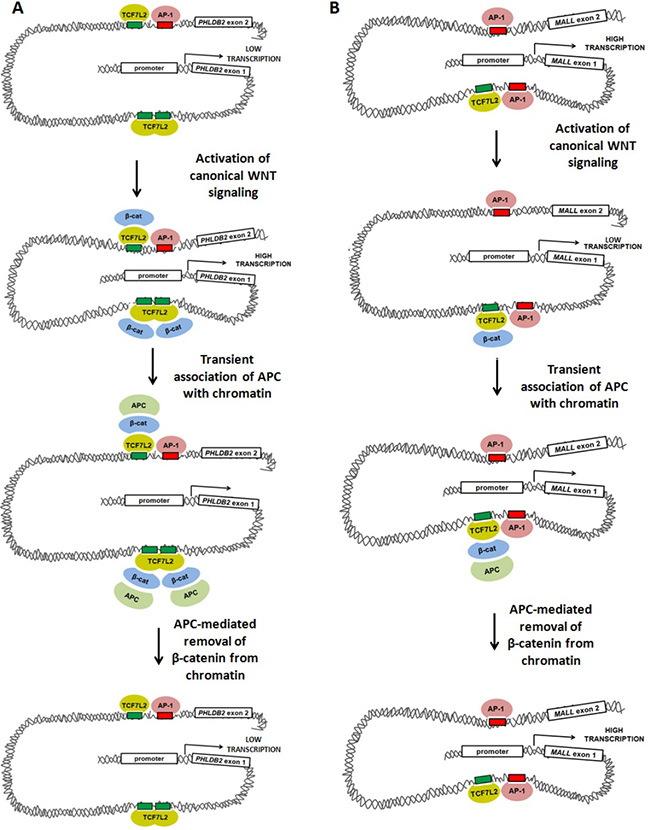
Figure 7: Models of WNT-mediated transcriptional activation and repression involving TCF7L2, AP-1, β-catenin and APC. Our data suggest at least two models in which canonical WNT-mediated transcriptional activity, APC and AP-1 alter gene expression. (A) Activation of canonical WNT signaling up-regulates transcription of PHLDB2 through a previously-characterized mechanism in which β-catenin binds to the TCF7L2 transcription factor, promoting DNA bending that brings transcriptional machinery (including AP-1) into closer association with the proximal promoter. APC disrupts this association by mediating the removal of β-catenin from the complex. (B) Activation of canonical WNT signaling may down-regulate AP-1-dependent transcription of MALL through a mechanism in which β-catenin binding to TCF7L2 disrupts interaction of transcriptional machinery with the proximal promoter. APC may relieve this disruption by removing β-catenin from the complex.
DISCUSSION
This study has identified high-confidence APC targets genes by comparing mechanistic (ChIP-seq) and functional (RNA-seq) data, similar to other studies identifying WNT-activated target genes [35, 36]. The present study adds evidence that canonical WNT signaling represses distinct target genes in human colorectal cancers and that APC antagonizes WNT repression at these loci. This pattern differs from the better-characterized canonical WNT targets but matches a smaller group of targets found in model systems such as Drosophila [28, 29], chick [30], mouse [31, 32] and human melanocytes [33].
The mechanism by which this occurs remains unclear. Published studies of chromatin-associated APC have established its ability to antagonize canonical WNT transactivation of targets, and have shown that interaction with β-catenin and recruitment of co-repressors such as CtBP-1, TLE-1 and HDAC1 are key elements of that mechanism [12, 13, 34]. APC and CtBP-1 interact with these loci transiently, and their disappearance coincides with the loss of β-catenin and the appearance of the more stable co-repressors [12]. Our findings indicate that the antagonistic relationship between chromatin-associated APC and β-catenin exists at WNT-repressed genes such as MALL as well. We hypothesize that regardless of whether canonical WNT signaling activates or represses transcription of a particular gene, APC exerts an antagonistic effect by mediating β-catenin removal (Figure 7). Further mechanistic experiments will be necessary to test this hypothesis and to identify co-activators involved in APC regulation of genes such as MALL.
Among the candidate genes identified in this study are seven characterized β-catenin-dependent targets of chromatin-associated APC. The ANGPT2, FAT1, PHLDB2 and TSPAN5 transcripts increase while GPRC5A, MALL and TOM1L2 decrease following APC loss in vitro or in vivo. An intronic regulatory element from the PHLDB2 locus is transcriptionally activated by β-catenin/TCF7L2 and repressed by APC, while an intronic regulatory element from the MALL locus is transcriptionally repressed by β-catenin/TCF7L2 and activated by APC. It is important to note that the effects of APC on their transcription are likely mediated by both cytoplasmic and nuclear functions of the APC protein that cannot be distinguished in our reporter assays. Previously published studies of the relationship between chromatin-associated APC and canonical WNT signaling indicate an antagonistic relationship that is consistent with the functions of APC in facilitating nuclear export and cytoplasmic degradation of β-catenin. The co-existence of these three seemingly redundant mechanisms emphasizes the critical importance of APC as a switch whose activation in maturing cells of the colorectal epithelium triggers rapid and tight negative regulation over the nuclear functions of β-catenin at multiple levels.
The chromatin-associated fraction of the APC tumor suppressor protein associates with a large number and variety of genomic regions, with patterns in genomic distribution and transcription factor binding site content that match ChIP-seq studies of other known components of the canonical WNT signaling pathway [22, 35]. Binding sites for the TCF7L2 transcription factor are enriched among APC-associated regions (Figures 3, 4), similar to published ChIP-seq results for β-catenin [22], a known co-regulator of TCF7L2 transcription factor complexes. These similarities are consistent with the reported co-localization of both proteins to target sites in the chromatin fraction and the interpretation that APC can be recruited to chromatin by β-catenin binding [12]. Despite similarities to related studies, it is important to note the limitations of informatics analyses that are two-dimensional and assign ChIP-seq peaks to RNA-seq hits based on the assumption that each APC-bound region controls gene expression from the nearest transcription start site, where the more realistic spatial organization of chromatin in three dimensions enables regulatory relationships that are not as long-range as they appear in two-dimensional units (bp). It is particularly important to consider these limitations in light of the heterogeneous effects of APC on “control” loci such as SP5, which lacks obvious APC-associated sites according to ChIP-seq, but nevertheless shows sensitivity to APC loss in the RNA-seq data. Three-dimensional analyses based on chromatin conformation capture (3C) methodologies are required for a more comprehensive identification of the genes targeted by APC-associated regions genome-wide.
Mounting evidence links β-catenin [22, 37, 38] and now APC to genomic regions containing predicted AP-1 binding sites. Canonical WNT signaling shares transcriptional targets with the AP-1 signaling pathway [37, 38], and previous ChIP-seq targeting β-catenin [22] similarly found AP-1 binding motif enrichment. The AP-1 component c-JUN physically interacts with TCF7L2 in HCT-116 cells, co-regulating the JUN promoter in a β-catenin-dependent manner [37]. AP-1 components co-immunprecipitate with β-catenin and co-regulate expression of the TCF7L2 target genes MYC and CCND1 [38]. Co-occurrence of predicted TCF7L2 and AP-1 binding sites in many of the same or neighboring genomic regions (Supplementary Figure 2) indicates that the two transcription factors may coordinately regulate the transcription of shared target genes in the colorectal epithelium. SP1 transcription factor binding sites are similarly over-represented among APC ChIP-seq peaks (Figures 3, 4) and co-occur with TCF7L2 binding sites (Supplementary Figure 2).
It is as yet unclear what determines whether canonical WNT signaling activates or represses a particular target gene in human cells. Some correlation exists between the presence of TCF7L2 and AP-1 transcription factor binding sites and canonical WNT activation of transcription, as both types of binding site are more prevalent within the subset of genomic sequences associated with transcripts increased in expression following APC loss (Supplementary Figure 2A vs. 2B). However, all seven loci characterized in this study include APC-associated genomic regions that contain both TCF7L2 and AP-1 binding motifs, regardless of whether they are transcriptionally increased (ANGPT2, FAT1, PHLDB2 and TSPAN5) or decreased (GPRC5A, MALL and TOM1L2) following APC silencing.
β-catenin/TCF7L2 may control transcription by modulating AP-1 binding or subsequent AP-1-dependent steps. If canonical WNT signaling exerts transcriptional effects largely by modifying AP-1 activity on shared target genes, WNT-activated targets may be those in which TCF7L2 promotes AP-1 binding or coordinates long-distance interactions between AP-1 proteins from multiple binding sites at the same promoter (Figure 7A). This would be consistent with the observation that many target genes (including PHLDB2) have multiple associated peaks, some with either TCF7L2 or AP-1 binding sites, and some with both. These regulatory elements likely interact with one another and converge on the promoter to co-regulate transcription. According to this model, WNT-repressed targets (Figure 7B) might be those in which TCF7L2 instead competes with AP-1 binding to adjacent sites or interferes with long-distance interactions between AP-1 proteins and their intended target promoters.
The MALL gene was confirmed (Figure 6) as a direct APC target repressed by canonical WNT signaling. These data imply that other targets with similar expression patterns may be similarly regulated candidates. Their value as potential markers for prognosis or as therapeutic targets may be important. MALL is down-regulated in colorectal cancers [39], with this low expression predicting tumor recurrence, metastasis and poor outcome [40]. MALL is a member of the glycolipid-enriched membrane raft family [41] and interacts with caveolin-1 [42], potentially linking it to integrin signaling and cell migration [43]. High expression of PHLDB2 in colorectal cancer is also associated with shorter metastasis-free survival. PHLDB2 is required for cell migration and invasion of the HCT-116 colon cancer cell line [44]. It is regulated by PI3K [45] and contributes to microtubule stabilization [46], and helps promote cell protrusions by linking actin and adhesion dynamics [47]. MALL, PHLDB2 and other genes responsive to APC are consistent with the emerging role of canonical WNT signaling targets in colorectal cancer invasion and progression [48–50], in addition to the better-characterized role of the pathway in colorectal tumor initiation.
Finally, this study strongly suggests that targets of the canonical WNT signaling pathway should be stratified into several categories, based on criteria such as the direction of their transcriptional response to activation of the pathway. The high rates of co-occurrence of TCF7L2 and AP-1 binding sites observed in the ChIP-seq data further indicate that the presence or absence of AP-1 binding sites may be an additional criterion by which to sort target genes. Interestingly, the AP-1 component c-Jun is required for the full phenotype of Apc loss in mice, as introduction of Jun mutations into the ApcMin/+ mouse model lead to lower polyp number, reduced polyp size and longer life span [37]. Canonical WNT signaling promotes the maintenance of progenitor cell phenotypes in the stem cell compartment of the colorectal epithelium, and may do so in part by fine-tuning the transcription of target genes shared with AP-1. Modulation of the AP-1 pathway is increasingly feasible with the emergence of the small molecule T-5224, designed to inhibit transactivation by blocking DNA binding by the leucine zipper domain of c-FOS [51, 52]. T-5224 can be one interventional strategy for WNT-driven colorectal tumors and has been characterized as an agent for the treatment of inflammatory conditions such as rheumatoid arthritis [52] and injuries of the kidneys [53] and liver [54] as well as oral cancer [55]. The natural product veratramine is an additional small molecule inhibitor of AP-1 that inhibits transactivation by interacting not with AP-1 itself but with its TGACTCA binding motifs [56]. These agents may show potential as therapeutic interventions to reduce the contribution of shared TCF7L2/AP-1 target genes to colorectal tumor progression.
MATERIALS AND METHODS
Chromatin immunoprecipitation and next-generation sequencing
APC ChIP was performed from 60–70% confluent HCT-116 cells in a 150-mm dish. Cells were maintained in McCoy’s 5A medium including glutamine and supplemented with 10% FBS. Following formaldehyde crosslinking, nuclei were isolated and resuspended in 1 mL of Lysis Buffer (25 mM Tris-HCl pH 7.5, 150 mM NaCl, 5 mM EDTA, 0.1% SDS, 1% Triton X-100, 0.5% Sodium Deoxycholate, 1 mM PMSF with mammalian protease inhibitor cocktail). Probe-based sonication was performed at 4° C over a period of 40 minutes total by 30 pulses of 10-seconds each at 35% amplitude. Average fragment size was further reduced to ~1kb using micrococcal nuclease (Affymetrix, Inc.). Input material was pre-cleared by 2-hour incubation with 5 μg rabbit IgG antibody and 40 μL of pre-equilibrated Protein G Dynabead slurry (Thermo Fisher Scientific) at 4° C with rotation. 100 μL of the resulting supernatant was saved as “pre-cleared input”, and the remainder was used for α-APC ChIP in combination with 10 μg of α-APC antibody (a polyclonal antibody recognizing the C-terminal 50 amino acids of APC, catalog #A3081A, Bethyl Laboratories, Inc.). ChIP was performed according to an established protocol [57], with the following exceptions. 40 μL of pre-equilibrated Protein G Dynabead slurry (Thermo Fisher Scientific) was used to pull down antibody-APC complexes, and each of the wash steps was performed twice. Yields from multiple parallel α-APC ChIP reactions were pooled to obtain the 10 ng required for ChIP-seq library preparation, and the PCR Purification Kit (QIAGEN) was used to clean up reactions prior to library preparation. Two separate biological replicates were performed under identical conditions several months apart, from HCT-116 cells at passage numbers between 10 and 20 since they were purchased from the American Type Culture Collection. Library preparation was performed using the NEBNext ChIP-Seq Library Prep Master Mix Set for Illumina, using adaptors AD005 and AD019 (New England Biolabs) for input and ChIP libraries, respectively. Size selection was performed by E-gel (Life Technologies, Inc.) to obtain fragments of 300-400-bp in size. Next-generation sequencing (50-bp, single-end) was performed by the OSU Genomics Shared Resource using a HiSeq 2500 instrument (Illumina, Inc.).
ChIP-seq data analysis
97–98% of reads for two ChIP-seq samples and the matching input samples passed quality control filters, and the Burrows-Wheeler Aligner generated bam alignment files using UCSC hg19. The Model-based Analysis of ChIP-Seq version 2 (MACS2) tool [58] performed peak-calling and generated peak scores, p-values and false discovery rates (FDR) for each peak. Peak score threshold was adjusted to lower the noise observed in input files while retaining internal positive control peaks in the AXIN2, DKK1 and MYC loci. The RefGene database was used to annotate peak regions, determine distances to transcription start sites and assign peaks to genes. The distribution of peak locations relative to transcription start sites is shown in Supplementary Figure 5. R software identified overlapping peaks present in both replicates with summits separated by less than 400-bp (the median of peak width in the peak calling results). ChIP-seq datasets are available through NCBI’s Gene Expression Omnibus (accession #GSE99264, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE99264). Reviewers must use the secure token uzojsoqgddktvev. MEME-ChIP [19], Regulatory Sequence Analysis Tools (RSAT) [20], and MatInspector [18] algorithms were used with standard/default settings to perform initial analyses of all ChIP-seq peak sequences from FASTA files.
Whole transcriptome profiling of HCT-116 cells
HCT-116 cells were transfected on consecutive days with pooled siRNA (Dharmacon) targeting APC (L-003869-00-0005), scrambled sequence (D-001810-10-05), or neither (mock transfection). Cells were harvested 48 hours later by standard Trizol (Life Technologies catalog # 15596-026) isolation protocol. Single-read library preparation was performed using the TruSeq RNA Library Preparation Kit (Illumina catalog # RS-122-2001). Next-generation sequencing was performed by the OSU Genomics Shared Resource using an Illumina Genome Analyzer II instrument. The Cufflinks software program [14] assigned reads to transcripts, performed quantification and calculated statistical significance. The Cuffdiff program provided transcript levels (as FPKM) and (FDR-adjusted) q-values based on fold-changes and sample sizes and identified significantly significant differences between anti-APC siRNA and scrambled siRNA-transfected conditions (q < 0.05). RNA-seq datasets are available through the NCBI Gene Expression Omnibus (accession #GSE99264).
Whole transcriptome profiling of ApcMin/+ and AOM/DSS mouse colon tumors
RNA isolation, preparation of sequencing libraries and processing of these data were described previously [15]. The presence of activating mutations in Ctnnb1 was confirmed by Sanger sequencing of cDNA from the adenomas from AOM/DSS-treated mice (Supplementary Figure 6). Transcripts of interest were identified based on 1.5-fold-changes in expression in adenomas from both ApcMin/+ and AOM/DSS-treated mice relative to their respective non-adenoma colon tissue controls (using one-sample t-test with a significance cutoff of FDR < 0.05) in the same direction as was observed in human cells following APC silencing. For multiple test correction, fold-changes in expression for individual genes were ordered by p-value and ranked, and FDR was calculated by multiplying p-values by the total number of tests and dividing by their rank (Benjamini and Hochberg method). RNA-seq datasets are available through NCBI’s Gene Expression Omnibus (accession #GSE98496, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=wpidaoiuphunvcn&acc=GSE98496).
APC ChIP-seq overlap with publicly available transcription factor ChIP-seq datasets
Public ChIP-seq datasets were obtained through accession numbers GSM1010846 (EGR-1) GSM1010756 (FOSL1), GSM1010847 (JUND), GSM803474 (POLR2A), GSM1010902 (SP1), GSM782123 (TCF7L2), and GSM1010836 (USF1). The coordinates of β-catenin ChIP-seq peaks were obtained from published work from the laboratories of Dr. Shannon McWeeney and Dr. Gregory Yochum [22]. All datasets included lists of peaks from two ChIP-seq replicates (except for β-catenin); peaks for each transcription factor were narrowed down to only those shared between both replicates. Chromosomal locations of 3,985 APC-associated genomic regions were compared to 12,375 TCF7L2 peaks, 2,166 β-catenin peaks, 6,969 “AP-1” peaks (shared between both JUND and both FOSL1 replicates), 7,754 SP1 peaks, 3,452 USF1 peaks and 3,396 EGR-1 peaks. Overlap between an APC peak and a transcription factor peak was scored only when the centers of two corresponding peaks were no more than 400-bp apart.
Luciferase reporter assays
Genomic regions were PCR amplified, restriction digested and ligated into the pGL3-promoter vector upstream of the firefly luciferase gene. Restriction enzymes pairs varied depending on the target, with Kpn I and Bgl II used for the majority. Primer sets are listed in Supplementary Figure 7. HCT-116 cells were seeded in 6-well plates and transfected on consecutive days with pooled siRNA (Dharmacon) targeting APC (L-003869-00-0005), targeting CTNNB1 (encoding β-catenin, L-003482-00-0010), or with scrambled sequence (D-001810-10-05) using Dharmafect 2 reagent (T-2002-01). 12 hours later, cells were trypsinized and re-seeded into 96-well plates at a density of 25,000 cells per well. Cells were co-transfected 12-hours post-plating with a firefly luciferase construct (9 ng per well) and a Renilla luciferase construct (1 ng per well). Luciferase assays were performed 24 hours later using the Promega Dual-Luciferase Reporter Assay System (catalog #E1910). The ratio of firefly luciferase to Renilla luciferase signal for each well was normalized to the average across all wells transfected with empty vector. Three independent experiments were performed and two-tailed Student’s t-test was used to calculate p-values to test the hypothesis that each construct was APC-sensitive or β-catenin-sensitive, relative to its scrambled siRNA control. For multiple test correction, fold-changes in expression for individual constructs were ordered by p-value and ranked, and FDR was calculated by multiplying p-values by the total number of tests and dividing by their rank (Benjamini and Hochberg method).
Ethics statement
The OSU Institutional Animal Care and Use Committee (IACUC) provided prior approval for all experiments involving mouse tissues (OLAW Assurance # A3261-01). IACUC approved the animal use protocol #2012A00000021, and animal work was conducted in accordance with its established criteria. Decisions to remove mice from the animal facility were made in conjunction with veterinary staff, and mice were observed and evaluated daily for predetermined criteria necessitating removal and euthanasia. Carbon dioxide inhalation followed by cervical dislocation was used to euthanize mice.
Author contributions
W.H. performed chromatin immunoprecipitation, RNA preparation, qPCR, in silico analyses and manuscript preparation. Z.C. and W.H. performed ChIP-seq library preparation, informatic analysis of ChIP-seq data and technical troubleshooting, and Z.C. performed manuscript revision and advised on experimental design. M.J.B. assisted with cell culture experiments and revised the manuscript. M.O.F. assisted with motif analysis and revised the manuscript. B.H. performed ChIP-seq data processing and analysis and revised the manuscript. X.L. performed data processing, informatics analysis of RNA-seq data and manuscript revision. A.G.J. performed motif analysis and revised the manuscript. J.Z. supervised B.H. for ChIP-seq data analysis and revised the manuscript. V.X.J. supervised X.L. for data processing and informatics analysis and performed manuscript revision. B.J.A. advised on the design of informatics analysis and revised the manuscript. Q.W. advised on experimental design and revised the manuscript. J.G. performed study design, data interpretation and manuscript preparation and revision.
ACKNOWLEDGMENTS
The authors thank Dr. Michelle Barton and Kendra Allton of the MD Anderson Cancer Center and Dr. Sudarshana Sharma of OSU for generously sharing ChIP protocols. We thank Dr. Jeffrey Parvin, Dr. Hui-wen Liu and Dr. Mansi Arora from OSU for expertise in troubleshooting ChIP. We thank Dr. Pearlly Yan and the Nucleic Acid Shared Resource at OSU for generating next-generation sequencing data. We thank Dr. Pierluigi Gasparini and Dr. Francesca Lovat from the lab of Dr. Carlo Croce at OSU for assistance in luciferase reporter experiment design. We thank Dr. Albert de la Chapelle, Dr. Kay Huebner, Dr. Mark Parthun and Dr. Jeffrey Parvin from OSU for helpful discussions in the development of the project.
CONFLICTS OF INTEREST
The authors declare that they have no potential conflicts of interest.
FUNDING
NIH Awards R01CA063507 (J.G.), UL1TR001070 (J.G.), F31CA174260 (W.H.), HHMI MED into GRAD (W.H.), and Pelotonia Fellowship Program (W.H.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding institutions or the National Institutes of Health.
REFERENCES
1. Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature. 1992; 359:235–237.
2. Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y. Somatic mutations of the APC gene in colorectal tumors: Mutation cluster region in the APC gene. Human Molecular Genetics. 1992; 1:229–233.
3. Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997; 275:1784–1787.
4. Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proceedings of the National Academy of Sciences of the United States of America. 1995; 92:3046–3050.
5. Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996; 382:638–642.
6. Orford K, Crockett C, Jensen JP, Weissman AM, Byers SW. Serine phosphorylation-regulated ubiquitination and degradation of beta-catenin. The Journal of Biological Chemistry. 1997; 272:24735–24738.
7. Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, Kuhl M, Wedlich D, Birchmeier W. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 1998; 280:596–599.
8. Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. Beta-catenin is a target for the ubiquitin-proteasome pathway. The EMBO Journal. 1997; 16:3797–3804.
9. Henderson BR. Nuclear-cytoplasmic shuttling of APC regulates beta-catenin subcellular localization and turnover. Nature Cell Biology. 2000; 2:653–660.
10. Neufeld KL, Zhang F, Cullen BR, White RL. APC-mediated downregulation of beta-catenin activity involves nuclear sequestration and nuclear export. EMBO Reports. 2000; 1:519–523.
11. Rosin-Arbesfeld R, Townsley F, Bienz M. The APC tumour suppressor has a nuclear export function. Nature. 2000; 406:1009–1012.
12. Sierra J, Yoshida T, Joazeiro CA, Jones KA. The APC tumor suppressor counteracts beta-catenin activation and H3K4 methylation at Wnt target genes. Genes & Development. 2006; 20:586–600.
13. Choi SH, Estarás C, Moresco JJ, Yates JR 3rd, Jones KA. α-Catenin interacts with APC to regulate β-catenin proteolysis and transcriptional repression of Wnt target genes. Genes & Development. 2013; 27:2473–2488.
14. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology. 2010; 28:511–515.
15. Hankey WC, McIlhatton MA, Ebede K, Kennedy B, Hancioglu B, Zhang J, Brock GN, Huang K, Groden JL. Mutational mechanisms that activate Wnt signaling and predict outcomes in colorectal cancer patients. Cancer Research. 2017; 78:617–630.
16. Kohno H, Suzuki R, Sugie S, Tanaka T. β-catenin mutations in a mouse model of inflammation-related colon carcinogenesis induced by 1,2-dimethylhydrazine and dextran sodium sulfate. Cancer Science. 2005; 96:69–76.
17. Kaiser S, Park YK, Franklin JL, Halberg RB, Yu M, Jessen WJ, Freudenberg J, Chen X, Haigis K, Jegga AG, Kong S, Sakthivel B, Xu H, et al. Transcriptional recapitulation and subversion of embryonic colon development by mouse colon tumor models and human colon cancer. Genome Biology. 2007; 8:R131.
18. Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. MatInspector and beyond: Promoter analysis based on transcription factor binding sites. Bioinformatics. 2005; 21:2933–2942.
19. Machanick P, Bailey TL. MEME-ChIP: Motif analysis of large DNA datasets. Bioinformatics. 2011; 27:1696–1697.
20. Medina-Rivera A, Defrance M, Sand O, Herrmann C, Castro-Mondragon JA, Delerce J, Jaeger S, Blanchet C, Vincens P, Caron C, Staines DM, Contreras-Moreira B, Artufel M, et al. RSAT 2015: Regulatory Sequence Analysis Tools. Nucleic Acids Research. 2015; 43:W50–56.
21. Eferl R, Wagner EF. AP-1: A double-edged sword in tumorigenesis. Nat Rev Cancer. 2003; 3:859–868.
22. Bottomly D, Kyler SL, McWeeney SK, Yochum GS. Identification of {beta}-catenin binding regions in colon cancer cells using ChIP-Seq. Nucleic Acids Research. 2010; 38:5735–5745.
23. Nagaoka M, Shiraishi Y, Sugiura Y. Selected base sequence outside the target binding site of zinc finger protein Sp1. Nucleic Acids Research. 2001; 29:4920–4929.
24. Luo X, Sawadogo M. Antiproliferative properties of the USF family of helix-loop-helix transcription factors. Proceedings of the National Academy of Sciences of the United States of America. 1996; 93:1308–1313.
25. Virbasius CA, Virbasius JV, Scarpulla RC. NRF-1, an activator involved in nuclear-mitochondrial interactions, utilizes a new DNA-binding domain conserved in a family of developmental regulators. Genes & Development. 1993; 7:2431–2445.
26. Christy B, Nathans D. DNA binding site of the growth factor-inducible protein Zif268. Proceedings of the National Academy of Sciences of the United States of America. 1989; 86:8737–8741.
27. Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007; 316:1497–1502.
28. Yang X, van Beest M, Clevers H, Jones T, Hursh DA, Mortin MA. Decapentaplegic is a direct target of dTcf repression in the Drosophila visceral mesoderm. Development. 2000; 127:3695–3702.
29. Piepenburg O, Vorbruggen G, Jackle H. Drosophila segment borders result from unilateral repression of hedgehog activity by wingless signaling. Molecular Cell. 2000; 6:203–209.
30. Lei Q, Jeong Y, Misra K, Li S, Zelman AK, Epstein DJ, Matise MP. Wnt signaling inhibitors regulate the transcriptional response to morphogenetic Shh-Gli signaling in the neural tube. Developmental Cell. 2006; 11:325–337.
31. Jamora C, DasGupta R, Kocieniewski P, Fuchs E. Links between signal transduction, transcription and adhesion in epithelial bud development. Nature. 2003; 422:317–322.
32. Kahler RA, Westendorf JJ. Lymphoid enhancer factor-1 and beta-catenin inhibit Runx2-dependent transcriptional activation of the osteocalcin promoter. The Journal of Biological Chemistry. 2003; 278:11937–11944.
33. Delmas V, Beermann F, Martinozzi S, Carreira S, Ackermann J, Kumasaka M, Denat L, Goodall J, Luciani F, Viros A, Demirkan N, Bastian BC, Goding CR, Larue L. Beta-catenin induces immortalization of melanocytes by suppressing p16INK4a expression and cooperates with N-Ras in melanoma development. Genes & Development. 2007; 21:2923–2935.
34. Hamada F, Bienz M. The APC tumor suppressor binds to C-terminal binding protein to divert nuclear beta-catenin from TCF. Developmental Cell. 2004; 7:677–685.
35. Hatzis P, van der Flier LG, van Driel MA, Guryev V, Nielsen F, Denissov S, Nijman IJ, Koster J, Santo EE, Welboren W, Versteeg R, Cuppen E, van de Wetering M, et al. Genome-wide pattern of TCF7L2/TCF4 chromatin occupancy in colorectal cancer cells. Molecular and Cellular Biology. 2008; 28:2732–2744.
36. Schuijers J, Mokry M, Hatzis P, Cuppen E, Clevers H. Wnt-induced transcriptional activation is exclusively mediated by TCF/LEF. The EMBO Journal. 2014; 33:146–156.
37. Nateri AS, Spencer-Dene B, Behrens A. Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature. 2005; 437:281–285.
38. Toualbi K, Guller MC, Mauriz JL, Labalette C, Buendia MA, Mauviel A, Bernuau D. Physical and functional cooperation between AP-1 and beta-catenin for the regulation of TCF-dependent genes. Oncogene. 2007; 26:3492–3502.
39. Kim K, Park U, Wang J, Lee J, Park S, Kim S, Choi D, Kim C, Park J. Gene profiling of colonic serrated adenomas by using oligonucleotide microarray. International Journal of Colorectal Disease. 2008; 23:569–580.
40. Wang X, Fan J, Yu F, Cui F, Sun X, Zhong L, Yan D, Zhou C, Deng G, Wang B, Qi X, Wang S, Qu L, et al. Decreased MALL expression negatively impacts colorectal cancer patient survival. Oncotarget. 2016; 7:22911–22927. https://doi.org/10.18632/oncotarget.8094.
41. Hatta M, Nagai H, Okino K, Onda M, Yoneyama K, Ohta Y, Nakayama H, Araki T, Emi M. Down-regulation of members of glycolipid-enriched membrane raft gene family, MAL and BENE, in cervical squamous cell cancers. The Journal of Obstetrics and Gynaecology Research. 2004; 30:53–58.
42. de Marco MC, Kremer L, Albar JP, Martinez-Menarguez JA, Ballesta J, Garcia-Lopez MA, Marazuela M, Puertollano R, Alonso MA. BENE, a novel raft-associated protein of the MAL proteolipid family, interacts with caveolin-1 in human endothelial-like ECV304 cells. The Journal of Biological Chemistry. 2001; 276:23009–23017.
43. Wary KK, Mariotti A, Zurzolo C, Giancotti FG. A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell. 1998; 94:625–634.
44. Chen G, Zhou T, Li Y, Yu Z, Sun L. p53 target miR-29c-3p suppresses colon cancer cell invasion and migration through inhibition of PHLDB2. Biochemical and Biophysical Research Communications. 2017; 487:90–95.
45. Paranavitane V, Coadwell WJ, Eguinoa A, Hawkins PT, Stephens L. LL5beta is a phosphatidylinositol (3,4,5)-trisphosphate sensor that can bind the cytoskeletal adaptor, gamma-filamin. The Journal of Biological Chemistry. 2003; 278:1328–1335.
46. Lansbergen G, Grigoriev I, Mimori-Kiyosue Y, Ohtsuka T, Higa S, Kitajima I, Demmers J, Galjart N, Houtsmuller AB, Grosveld F, Akhmanova A. CLASPs attach microtubule plus ends to the cell cortex through a complex with LL5beta. Developmental Cell. 2006; 11:21–32.
47. Astro V, Chiaretti S, Magistrati E, Fivaz M, de Curtis I. Liprin-alpha1, ERC1 and LL5 define polarized and dynamic structures that are implicated in cell migration. Journal of Cell Science. 2014; 127:3862–3876.
48. Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA, Knuechel R, Kirchner T. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98:10356–10361.
49. Hlubek F, Brabletz T, Budczies J, Pfeiffer S, Jung A, Kirchner T. Heterogeneous expression of Wnt/beta-catenin target genes within colorectal cancer. International Journal of Cancer. 2007; 121:1941–1948.
50. Todaro M, Gaggianesi M, Catalano V, Benfante A, Iovino F, Biffoni M, Apuzzo T, Sperduti I, Volpe S, Cocorullo G, Gulotta G, Dieli F, De Maria R, Stassi G. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Current Protocols in Bioinformatics. 2014; 14:342–356.
51. Tsuchida K, Chaki H, Takakura T, Kotsubo H, Tanaka T, Aikawa Y, Shiozawa S, Hirono S. Discovery of nonpeptidic small-molecule AP-1 inhibitors: lead hopping based on a three-dimensional pharmacophore model. Journal of Medicinal Chemistry. 2006; 49:80–91.
52. Aikawa Y, Morimoto K, Yamamoto T, Chaki H, Hashiramoto A, Narita H, Hirono S, Shiozawa S. Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nature Biotechnology. 2008; 26:817–823.
53. Miyazaki H, Morishita J, Ueki M, Nishina K, Shiozawa S, Maekawa N. The effects of a selective inhibitor of c-Fos/activator protein-1 on endotoxin-induced acute kidney injury in mice. BMC Nephrology. 2012; 13:153.
54. Izuta S, Ueki M, Ueno M, Nishina K, Shiozawa S, Maekawa N. T-5224, a selective inhibitor of c-Fos/activator protein-1, attenuates lipopolysaccharide-induced liver injury in mice. Biotechnology Letters. 2012; 34:2175–2182.
55. Kamide D, Yamashita T, Araki K, Tomifuji M, Tanaka Y, Tanaka S, Shiozawa S, Shiotani A. Selective activator protein-1 inhibitor T-5224 prevents lymph node metastasis in an oral cancer model. Cancer Science. 2016; 107:666–673.
56. Bai F, Liu K, Li H, Wang J, Zhu J, Hao P, Zhu L, Zhang S, Shan L, Ma W, Bode AM, Zhang W, Dong Z. Veratramine modulates AP-1-dependent gene transcription by directly binding to programmable DNA. Nucleic Acids Research. 2018; 46:546–557.
57. Chadee DN, Hendzel MJ, Tylipski CP, Allis CD, Bazett-Jones DP, Wright JA, Davie JR. Increased Ser-10 phosphorylation of histone H3 in mitogen-stimulated and oncogene-transformed mouse fibroblasts. The Journal of Biological Chemistry. 1999; 274:24914–24920.
58. Feng J, Liu T, Zhang Y. Using MACS to identify peaks from ChIP-Seq data. Current Protocols in Bioinformatics. 2011; Chapter 2:Unit 2.14.