
INTRODUCTION
Gastric cancer (GC) is a multifactorial disease in which both genetic and environmental factors are involved. According to the statistic, GC is the fourth cause of cancer death worldwide, with a median overall survival of ≤12 months for advanced stage [1]. It is rare in the younger population, where less than 10% of patients suffer from GC before 45 years of age [2–6]. It is highly heterogeneous disease with different molecular and genetic alterations [7].
The most common classification of gastric cancer is the Lauren classification. It differentiates two types of GC - intestinal and diffuse which have distinguishing features like morphology, genetics, clinical characteristics, progression pattern and epidemiology [8]. Diffuse-type GC is composed of poorly cohesive single cells without gland formation. Intestinal-type GC consists glandular or tubular components with various degrees of differentiation [9, 10]. While the incidence of GC has decreased worldwide, the incidence of GC with signet-ring cell is rising. In the past, GC with signet-ring cell was classified as “diffuse type” according to Lauren’s classification [8]. Now, signet-ring cell carcinoma is defined as a poorly cohesive carcinoma composed predominantly of tumor cells with prominent cytoplasmic mucin and a crescent-shaped nucleus eccentrically placed [11]. It is important to understand that not all gastric cancers classified as “undifferentiated” or “diffuse” are signet-ring cell cancers.
Gastric cancer is a multifactorial disease, whereas Helicobacter pylori (H. pylori) infection is one of the risk factors in developing the cancer, but not exclusive. The infection with H. pylori increases the risk of developing GC about six-fold [10, 12]. Therefore, the World Health Organization (WHO) classified H. pylori as a class I carcinogen in 1994 [10, 13]. H. pylori, also known as Campylobacter pylori, is a bacterium detected in 1982 by Australian scientists in human mucous membrane in digestive system [14]. The name of this pathogen, from Latin, in which helix means spiral, refers to the characteristic shape of it, which probably facilitates its penetration into the mucous membrane of the digestive system. The second part of the name of H. pylori comes from pylorus because the bacteria is detected primarily in the distal part of the stomach - the pylorus. It was found that the gastric acid does not damage H. pylori and it even seemed to be essential to its growth. Later, it was shown that H. pylori could also infect other tissues, e.g. the liver or the eye [15, 16].
In further studies, it was found that there was a relationship between the presence of H. pylori in the upper gastrointestinal tract and gastric and duodenal ulcers. However, that bacterial infection alone is not synonymous with the development of peptic ulcer disease. Only a small percentage of infected people will have gastric cancer. H. pylori infection has been shown to range from approximately 60% in the general population to approximately 84% in patients with gastric cancer [17]. There must be other critical cofactors affecting risk, which may also explain the difference in morbidity between races and sex. Possible factors are specific genetic alterations, age at the onset of infection by H. pylori, differences in gastric acidity, and environmental factors like diet including salt consumption and smoking [17]. Nearly half of humanity (more in developing countries) is infected with H. pylori (in Poland about 80% of adults and about 30% of children) [18]. Meanwhile, 5-10% of the adult population falls ill for peptic ulcer of the stomach and duodenum. This means that only about one in ten people infected with H. pylori will develop peptic ulcer disease. In addition, H. pylori is not found in some people suffering from peptic ulcer disease. Therefore, H. pylori cannot be the cause of all gastric and duodenal ulcers, and alone cannot explain the pathogenesis of gastric carcinoma [19].
The review summarizes the data about early-onset gastric cancer (EOGC) and the role of H. pylori in the developing this subtype of gastric carcinoma.
Epidemiology of H. pylori infection in Europe and Asia regions
Various H. pylori infection prevalence was noted, based on the study region and period of such statistics. The infection ratio is based mostly on the hygienic lifestyle standards, which are better in Europe. Screening investigations are mostly performed in Asia regions, in Europe the infection control is decreased, as the disease rates are rather low, and the interventions are extravagant [20]. Better controlling of the lifestyle agents may cause a significant reduction of H. pylori associated diseases, like stomach cancer.
It was postulated as the lowest supremacy of infection was found in Northern Europe, the highest were in Southern and Eastern Europe [21]. The total prevalence of H. pylori infection in adults European is about 20%- 40% [22]. The studies on epidemiology among European population showed no important difference of H. pylori infection correlated with the gender. If there was a gender difference, the infection was increased in men. However, the age is affected with the H. pylori action, occurring mostly in older age groups [23]. There are some exceptions, like the Spanish investigation, which displayed lower infection ration among oldest individuals [24]. Additionally, in Swedish study, blood donors infected with the bacterium showed decreasing level after age of 50 [25]. In Southern and Eastern Europe, the infection of H. pylori is remarkably higher in comparison to other parts of the continent, mostly in Latvia, Poland and Portugal [18, 26, 27]. The prevalence is higher than 50% in the population. Studies conducted in Northern Europe, apart from Ireland, displayed quite decreased level of H. pylori infection [28].
Among the Asia–Pacific region, the distribution of H. pylori infection is various between countries, as well as chosen regions within individual communities. This prevalence depends on socioeconomic lifestyle levels, which is lower in more developed regions. In Vietnam, the H. pylori seroprevalence rate was 74.6% [29]. In India, the reported rate was even higher, reaching around 79% [30]. Bangladesh was one of the region, highly affected by H. pylori occurrence, with 92% of prevalence among population [31]. In Australia, which is more developed region the seroprevalence rate reached 15, 4% [32]. Within East Asian regions, the overall seroprevalence rate was 39.3% in Japan, 54.5% in Taiwan, 58.07% in China and 59.6% in South Korea [33–36]. Southeast Asia countries displayed the infection rate, assigned to 31% in Singapore, 35.9% in Malaysia and 57% in Thailand [37–39].
Action of H. pylori in the stomach
H. pylori is the most common infection in the world and the etiological factor causes GCs [40]. In total, H. pylori infection almost doubles the overall risk of gastric cancer. GC develops through a cascade of well-defined and well-recognizable steps: inflammation, atrophy, intestinal metaplasia, dysplasia, and carcinogenesis, and is closely associated with the environment, diet and gene mutations [41–44]. The recognition patterns of gastric cancer include genetic alterations among cell cycle regulators, factors that regulates apoptosis, microsatellite instability, multidrug resistance proteins, factors that influence cell membrane properties, module of HER2 expression, and agents with impact on the progression of gastric cancer and peritoneal metastasis [45].
Cells of gastric mucosa produce hydrochloric acid and the digestion of food in the stomach takes place at low pH conditions, but they are not fully acid-resistant. Therefore, under normal conditions, they are covered with a thin layer of protective mucus [46]. Nonsteroidal anti-inflammatory drugs, e.g. acetylsalicylic acid, damage the gastric mucosa and lead to the development of ulcers, among others because they inhibit the synthesis of prostaglandins, which in turn activate the production of mucus in the stomach [47]. H. pylori can adapt to the local conditions prevailing in the stomach, especially in the pylorus. It avoids areas with extremely low pH, thus the light of the stomach away from its walls. It migrates instead towards the epithelium of the mucous membrane where it penetrates the mucus layer covering the cell. Among people secreting larger amount of hydrochloric acid, the bacterium is more likely to colonize the pylorus, especially in the place where the stomach passes into the duodenum. In this way, the central part of the stomach is avoided, where the pH is clearly low. When hydrochloric acid secretion is lower, H. pylori colonizes the whole stomach [48].
Movement of the bacteria is active, it occurs due to the characteristic shape. Then H. pylori adheres to epithelial cells and even penetrates them. An important factor determining the effects of H. pylori infection is the way in which the immune system reacts to the bacteria. Another known toxic product of H. pylori metabolism is ammonia. It is a weak alkali which helps bacteria to neutralize hydrochloric acid in its immediate environment and is the cause of survival. It is made of urea which is produced by urease, the enzyme which H. pylori releases [49].
The geographically dependent pathogenesis of h. pylori associated GCs
The genomes of H. pylori are incredibly diverse, various virulence factors have impact on host epithelium in different ways, including direct and indirect investigations like eliciting immune response. Genetic alterations of host, smoking, different food consumption, gastric microbiota and long-term intake of proton pump inhibitors (PPIs) impact on the progression of H. pylori associated gastric cancers [50]. The pathogenesis of H. pylori related gastric lesions is a multi-step and complex action, which the progression is based on combining of environmental, host and bacterial agents [51].
Mechanism of H. pylori associated gastric carcinogenesis has not been completely described. H. pylori infection commonly lasts for decades, causing a bunch of histological variations, encompassing apoptosis and proliferation of epithelial cells, destruction of intercellular junctions, and malignant transformation [52]. H. pylori genetic alterations, outcomes in different H. pylori products on gastric epithelium and cellular signalling processes, which have been broadly studied in recent decades [53]. It is assumed that the bacteria themselves do not cause mutations that lead to cancer, rather the infection stimulates a local inflammatory reaction, in which there is a white cell infiltration and local production of free radicals, damaging DNA and initiating the development of cancer cells [54]. Another mechanism of developing cancer cells, could result from the death of epithelial cells, damaged by H. pylori. The defects are filled by replacing these cells with new, mutated, potentially cancerous cells, which can enter the subdivision cycle [55]. According to Guven-Maiorov et al., 2017 others survey pathogens often interact with the host through proteins. H. pylori proteins interfere with multiple host pathways, as they target several host proteins and thereby alter the host signalling. It may mimic host proteins at different stages - the sequence, structure, motif or interface levels [56].
H. pylori is a heterogeneous bacterium and its virulence factors are different among geographic regions. Various geographic incidence in gastric cancer development might be explained, mainly by the occurrence of different H. pylori virulence agents, like CagA, VacA and OipA [57]. H. pylori infection also might take part in development of duodenal ulcer. This variance is possibly connected with H. pylori virulence factor DupA [58].
CagA is the most popular and broadly studied H. pylori virulence factor. There are two types of clinical H. pylori isolate: CagA-producing (cagA positive) strains and CagA-nonproducing (cagA negative) strains. In Western countries, it has been postulated that cases infected with cagA-positive strains of H. pylori are displaying an increased risk of gastric cancer or peptic ulcer in comparison to those affected by cagA-negative strains [59, 60]. The pathogenic difference in East Asia is not so obvious to explain, by taking into consideration the occurrence or absence of the cagA gene alone, as most strains of H. pylori have the cagA gene irrespective of the disease [61]. It is worth to mention that cagA is a polymorphic gene, therefore the various numbers of repeat sequences located in the 3′ region of the cagA gene of different H. pylori strains are present [62]. Each repeat region of the CagA protein encompasses Glu-Pro-Ile-Tyr-Ala (EPIYA) motifs, containing a tyrosine phosphorylation site. Additionally, each CagA is allotted a sequence type containing the names of the EPIYA segments in its sequence (that is, ABC, ABCC or ABCCC for Western-type CagA and ABD for East-Asian-type CagA) [62, 63, 64].
VacA is the second very popular H. pylori virulence factor. Additionally, to promote vacuolation, VacA can start multiple cellular activities, like cytochrome c release from mitochondria causing the apoptosis process, membrane-channel formation, as well as binding to cell-membrane receptors followed by initiation of a proinflammatory response [65, 66]. The difference in the vacuolating actions of different H. pylori strains are displayed mostly because of differences in the vacA gene structure at the signal (s) region (s1 and s2) and the middle (m) region (m1 and m2) [67]. In Western countries, mostly Latin America, the Middle East and Africa, the studies performed claim that individuals infected with m1 or s1 H. pylori strains have a chance for higher risk of developing gastric cancer or peptic ulcer in comparison to those underwent the infection with m2 or s2 strains [68, 69]. In East Asia, H. pylori strains have an s1-type s region, that’s why the pathogenesis cannot be examined by the type of s region present [61]. However, m1 strains are mostly present in parts of north East Asia, like Japan or South Korea, m2 strains are popular in parts of south East Asia, such as Taiwan and Vietnam [70, 71].
OipA was firstly described as a proinflammatory-response-inducing protein, mostly because of the role of oipA isogenic mutants in reducing the production of IL-8 from gastric epithelial cell lines [72]. In vivo performed investigations using human gastric biopsies also results in the same conclusion that a functional oipA was importantly correlated with high mucosal IL-8 levels [60]. Finally, it stayed obvious that one of the OipA role is to promote inflammation and actin dynamics through phosphorylation process of multiple signalling pathways [73, 74]. Additionally, as and adhesin, OipA is committed in the attachment of H. pylori to gastric epithelial cells in vitro [73]. It is significant to mention that to all East Asian strains are producing functional OipA protein [72].
H. pylori virulence factors and further consequences in gastric carcinogenesis
Cytotoxin associated factor (CagA) and vacuolizing cytotoxin (VacA) are specific strains of H. pylori, which are mainly virulence factors involved in increasing the risk for gastric carcinoma development [75]. CagA - protein is a 120- to 140-kDa protein that is translocated into host cells by the type IV cag secretion system, after the attachment of H. pylori and in the result changes the cell-signalling mechanisms in gastric cells. VacA protein is a cytotoxin produced by bacteria and inducing vacuolation of the epithelial cells. The gene is present in all strains but has different variations of vacuolating activity [76]. This aspect is characteristic to variations in vacA gene structures within the three regions: the signal sequence region (s-region) (s1 or s2), mid-region (m-region) (m1 or m2) and the intermediate-region (i-region) (i1, i2 or i3) [40, 77, 78]. These two virulent factors and their polymorphism have been considered in many research [79–82].
In the study Matos et al., 2013 summarized a possible association between these genotypes and the risk for developing different gastric phenotypes. Scientists checked forty-four studies, all with either a case–control (n=13) or cross-sectional (n=31) design, including a total of 17 374 patients: 107 in the dysplasia group, 4994 in the peptic ulcer disease group, 5564 in the gastritis group and 6709 in the gastric cancer group. They confirmed an increased risk for gastric cancer among patients infected with CagA+ H. pylori strains and infected with VacA s1 and m1 strains [75]. Pandey et al., 2014 reviewed the comparative study of H. pylori infection and the possible influence of carcinogenic CagA+ strains in the progression of gastric cancer. But not all strains of H.pylori are pathogenic. The virulence is primarily determined by a gene cagA and these strains have strong association with gastric cancer [78, 83, 84]. H. pylori virulence factors are listed in Table 1. They discussed the connection between gastric cancer and stimulant substances like tobacco and alcohol. According to their study the number of H. pylori infection achieved higher amount in precancerous and cancerous gastric lesions and was related to the habit of tobacco use. It was because the infection was acquired orally [83].
Table 1: Helicobacter pylori virulence factors and further consequences to GC development
Risk Factors Action |
Consequences |
Authors |
|
|---|---|---|---|
H. pylori virulence factors |
Activation and secretion of cytokines in epithelial cells such as IL-8 by cag pathogenicity island (cagPAI) |
• CagPAI- and inflammation-driven cancerogenesis |
Stein et al., 2017 |
Influence of CagA on the tumor suppressor function of apoptosis-stimulating protein of p53 (ASPP2) |
• The interaction between CagA and ASPP2 |
Buti et al., 2011 |
|
CagA-dependent loss of polarity and activation of aberrant Erk signalling after the delivery into epithelial cells |
• Senescence and mitogenesis in epithelial cells, both nonpolarized and polarized |
Saito et al., 2010 |
|
East Asian-type CagA has a higher binding affinity for the Src homology-2 domain-containing phosphatase 2 (SHP2) |
• Greater risk of peptic ulcer development and/or gastric cancer when compared to its Western counterpart |
Hatakeyama et al., 2004; Higashi et al., 2002; Jones et al., 2009; Vilaichone et al., 2004 |
|
CagA-positive strains with EPIYA motifs; |
• Reduction variety of intracellular signalling systems after the infection of gastric epithelial cells; |
H. pylori which is virulent, can cause proinflammatory signalling which induces the activation and secretion of cytokines in epithelial cells such as IL-8. Cag pathogenicity island is responsible for this signalling. It is the codetermination of the risk for gastric cancer [85]. On the encoded cagPAI the Cag type IV secretion system (CagT4SS), can change the place of various molecules in cells, the effector protein CagA, metabolites and DNA. Although these translocated molecules are known to contribute to cellular responses to some extent, a major part of the cagPAI-induced signalling leading to IL-8 secretion remains unexplained [86]. Stein et al., 2017 reported that biosynthesis of an intermediate metabolite of LPS inner heptose core, heptose-1,7-bisphosphate (HBP), is translocated into host cells dependent on the CagT4SS. It is also the main factor which leads to the activation of cellular responses and contributes in the induction of proinflammatory signal and IL-8 secretion in human epithelial cells. This response pathway is linked with the human cellular adaptor protein TIFA. The knowledge of this will enable to understand the way of modulating the immune response in human host by H. pylori. Mutants defective in the genes required for synthesis of HBP exhibited a more than 95% reduction of IL-8 induction and impaired CagT4SS-dependent cellular signalling. It was not possible to enrich the active metabolite in bacterial supernatants, but it was present in H. pylori lysates. These results will help to understand H. pylori cagPAI-dependent signalling which is mediated by intracellular pattern recognition receptors. Stein et al., 2017 also advance the knowledge of immunomodulatory activities by H. pylori and make the possibilities of intervention in cagPAI- and inflammation-driven cancerogenesis more realistic [86]. The Cag Type IV secretion system, which leads to inflammation and cancerogenesis during chronic infection, is one of the main virulence factors of the bacterial gastric pathogen H. pylori. However, the mechanisms lead to cagPAI-dependent signal transduction and cytokine.
Buti et al., 2011 disproved that cytotoxin-associated gene A has influence on the tumor suppressor function of apoptosis-stimulating protein of p53 (ASPP2) [87]. Different pathogens which may cause persistent infections have the property to demonstrate strategies which can interfere with signalling pathways and immune responses of the host. Type I strains of H. pylori encode a type four secretion system that allows the pathogen to produce the effector protein (CagA) straight to the host cells [88, 89, 90, 91]. It is the most important factor which connects the infection of the bacteria with the gastric cancer. After translocation into the host cell, CagA increases cell motility, changes cells shape, and promotes transition-like phenotype from an epithelial to mesenchymal. CagA leads to the damage of p53 and its activity in an ASPP2-dependent manner is down-regulated. Cells which are infested with H. pylori treated with drug Doxorubicin with activated the p53 are more resistant to apoptosis than uninfected cells. They require this effect from ASPP2. This is an example of bacterial protein that disproves the p53 tumor suppressor pathway in a manner like DNA tumor viruses [87].
The findings made by Saito et al., 2010 suggest that CagA uses a polarity-signalling pathway to cause oncogenesis. CagA produced by H. pylori constitutes a very important factor in gastric carcinogenesis. It induces loss of polarity and activates aberrant Erk signalling after the delivery into epithelial cells. Scientists claimed that CagA-induced Erk activation effects on the senescence and mitogenesis in epithelial cells, both nonpolarized and polarized [92].
In the study Subsomwong et al., 2017 compared the amount of incidence in gastric cancer in Thailand regions, in the North and South [93]. They showed that the prevalence of H. pylori infection was three times higher in patients living in the North (50.4%) than in the South (17.6%) [93, 94]. However, they claimed that those in the South had more severe lesions of precancerous and cancerous stages of gastric cancer. To check the virulence genotypes of H. pylori they made further analysis. The outcome which they achieved was that patients from the South may have a higher amount of risk to induce gastric cancer because of the infection with H. pylori. In the South there are Western-type CagA strains which could develop more precancerous lesions and histopathological changes in the antrum and the corpus of the gaster even after checking the differences between age, sex, location, and birthplace. The second repeat of the C-terminal region of CagA sequences has a difference between East Asian-type CagA and Western-type CagA. East Asian-type CagA has a higher binding affinity for the Src homology-2 domain-containing phosphatase 2 (SHP2), resulting in a greater risk of peptic ulcer development and/or gastric cancer when compared to its Western counterpart [95–98]. The scientists checked the ability of gastric cells to induce IL-8. They did not find the relationship between the cagA genotypes and the location on IL-8 production. However, they agreed that further studies must be made to measure other pathological markers than IL-8.
Aftab et al., 2017 focused on Bangladesh population with high number of H. pylori infection but a low morbidity to gastric cancer. H. pylori strains can be divided in two groups cagA-positive or -negative [99]. CagA expression in cagA-positive strains relates to inflammation and the higher risk for more severe clinical outcomes against to cagA-negative strains [100]. CagA-positive strains that have EPIYA motifs, tyrosine-phosphorylated by Src and Abl family kinases, reduce a variety of intracellular signalling systems after the infection of gastric epithelial cells [101]. There is a higher risk of gastric cancer among individuals infected with strains possessing cagA with an EPIYA-D segment (an East Asian-type cagA-positive strain) than with strains possessing an EPIYA-C segment (a Western-type CagA-positive strain) [100]. The scientists analysed CagA and VacA subtypes and their association with severe histology type. They segregated Bangladeshi strains into two populations of different genotypes. They claimed that in H. pylori populations, hpAsia2 strains were associated with higher activity and inflammation in the antrum than in the hpEurope strains. The cause of lower level of gastric cancer among Bangladesh population might be the high number of less-virulent genotypes. It could be a better risk factor of gastric cancer than the ancestral origin of the H. pylori strains. Moreover, the vacA m region may be use as a better virulence marker than other regions [99].
Genetic variants and H. pylori associated gastric cancer
Genetic variants in genes from cytokines and their receptors associated with inflammation are perceived to take part in tumor initiation and promotion. Considering genetic polymorphisms in gastric cancer and H.pylori infection, the growing interest in this field has become more expanded in recent years. In this process different cytokines take part e.g. IL-1, IL-17 (it is associated with a higher risk of developing gastric cancer connected with H. pylori colonization), tumor necrosis factor (TNF) α, toll-like receptors (TLRs) [77, 102–114]. The next occurrence of SNP’s is protection against pathogens which attacks (MUC1), the repair of DNA damage caused by H. pylori (XPA, XPC, ERCC2) and cell-to-cell adhesion (CDH1) [13, 94, 115–121]. SNP’s also has influence on metabolic processes of foliate (methylenetetrahydrofolate reductase), polycyclic aromatic hydrocarbons (GSTT1, SULT1A1, NAT2, EPHX1), hormones e.g. estrogen and androgen and xenobiotics (Cyp2e1) [122–126].
At a gene level, hypoxia-inducible factor 1 (HIF-1) is the primary transcriptional activator, very sensitive to oxygen, helping cells to survive in low oxygen tension [127]. Overexpression of HIF-1α is important in the activation the bunch of genes involved in cancer biology, encompassing cell proliferation, survival and apoptosis, glucose metabolism, erythropoiesis, as well as angiogenesis [128]. It was displayed that the expression of around 20 genes is regulated by HIF-1α, including NFκB1, which is involved in regulation of inflammation and cancer [129].
It was postulated that the transmembrane protein CD133 is overexpressed in 57% of gastric cancer and positively connected with the expression of Ki-67 [130]. One of the member of the cadherin family, CDH17, was described to be a marker for gastric cancer in early stage [131]. The overexpression of CD168 was assigned to be correlated with the depth of cancer invasion and metastasis stage [132]. In another investigation, Xie et al., 2015 described that tumor stem cell surface marker CD44 (CD44v6), which relates to metastasis in GC [133].
Matrix metalloproteinases (MMPs) are enzymes involved in multiple processes, covering degradation of extracellular matrix, inflammation, tumor invasion and metastasis. The meta-analysis performed by Long et al., 2014 displayed significant increment of MMP-7 expression in GC cases, also the positive correlation with lymph node metastasis and invasion of the tumor [134].
Epithelial–mesenchymal transition (EMT) it is important in tumor progression and invasion and Snail regulates the EMT in different carcinomas. The Snail overexpression was observed to be correlated with increased invasion, cell migration and tumor progression in gastric cancer [135].
Other cytokines like tumor necrosis factor α (TNF-α), IL-6, IL-8, IL-10, Toll-like receptor 4 (TLR-4 and transforming growth factor β (TGF-β) are suspected to affect tendency to be infected by H. pylori or to change the pathways of local inflammation [86, 136–138]. However, it is possible that several low-penetrant alleles in combination may result in familial aggregation, rather than dominant cancer genes with high penetration [139].
Cellular and molecular pathogenesis of Helicobacter pylori infection in gastric carcinogenesis is shown on Figure 1. The phosphorylated cagA active in the SHP-2/MAPK pathway regulates NF-κB, RAS/cMyc and MEK/extracellular signal-regulated kinase (ERK) pathways, which is the result of the regulation of genes like HIF-1α, MUCs, inducible nitric oxide synthase (iNOS), BCL2, suppressor of cytokine signalling (SOCS), signal transducer and activator of transcription 3 (STAT3), COX-2, MMPs and SNAIL provoking proliferation, differentiation, increased migration, invasion and metastasis of cancer cells [140].
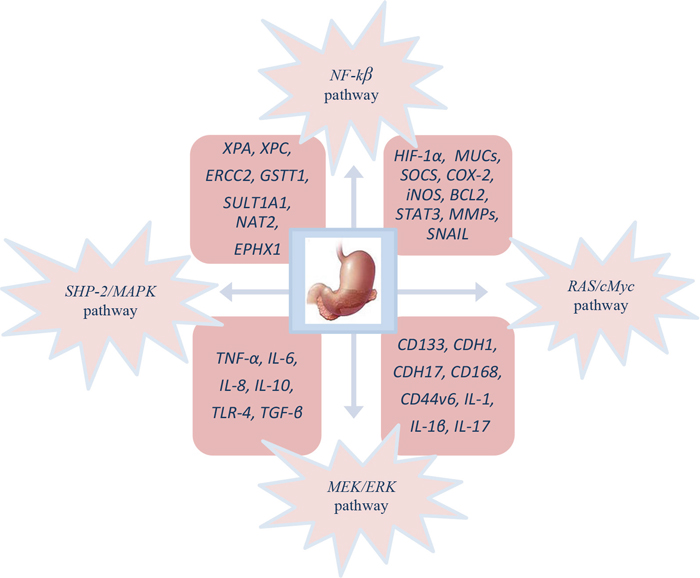
Figure 1: Molecular pathogenesis of GC patient’s infected with H. pylori.
Epidemiological studies indicated the connection between the H. pylori infections and the gastric mucosa inflammation, which change into chronic gastritis and intestinal type of gastric cancer. Interleukin-1β (IL-1β) deregulation is responsible for the higher risk of gastric carcinoma (Table 2) [40]. The latest studies claimed that the IL-1β-31 and -511T alleles links to hypochlorhydria and precancerous lesions, so they are closely associated with carcinogenesis process, but the leading mechanism is still unknown. H. pylori infection generates IL-1β and suppresses the secretion acid in the gastric mucosa while the eradication decreases the expression of IL-1β [141, 142]. El-Omar et al., 2003 first reported an association between the IL-1β-31C and IL-1RN 2/2 genotypes with a greater risk of chronic low gastric acid secretion and gastric cancer [143]. The presence of IL-1β-511T, IL-1RN*2/*2, TNF-α-308A, and IL-10 (haplotype ATA/ATA) was connected to a higher risk of noncardia gastric cancer with H. pylori infection [86, 138]. Moreover, Hong et al., 2016 found that H. pylori infection has a similar effect on the evolution of gastric cancer with IL-1β gene polymorphisms, and the highest amount of severe gastric anomalies occur in patients with high-risk genotypes (cagA(+)/vacAs1(+)/IL-1β-511T) both host and bacterial [40]. H. pylori synergistic with IL-1β gene polymorphisms seem to promote gastric cancer by their involvement in precancerous gastric lesions and hypochlorhydria. The histological type of gastric cancer may be also dependent on IL-1β [144, 145]. Intestinal type against to the diffuse or mixed-type of gastric cancer is more often in those with IL-1β-511T genotype [144, 146–148]. What is more, the levels of mucosal IL-1β are higher in H. pylori-infected gastric cancer patients with IL-1β-31TT as against those with IL-1β-31CT and IL-1β-31CC [149]. However, the infection by H. pylori is more likely among patients with IL-1β-31 CT and TT genotypes in regions of Asia and Latin America than in those with IL-1β-31 CC [121]. A higher number of intestinal metaplasia occurs in patients with H. pylori infection, especially in those with vacA m1 strain [150]. H. pylori induces the expression of IL-1β, which starts gastric carcinogenesis by affecting two types of cells inflammatory and epithelial [151]. But scientists agreed that further research is needed to expand the study area in different populations and subtypes of gastric cancer and to explain alternative underlying mechanisms.
Table 2: IL-1β genotypes and higher risk of gastric carcinoma
Genotypes |
Phenotypes |
Authors |
|---|---|---|
IL-1β-31 and -511T |
Induction of hypochlorhydria and precancerous lesions |
|
IL-1β-31C and IL-1RN 2/2 |
Greater risk of chronic low gastric acid secretion and gastric cancer |
[El-Omar et al., 2003] [143] |
IL-1β-511T, IL-1RN*2/*2, TNF-α-308A, and IL-10 (haplotypeATA/ATA) |
Higher risk of noncardia gastric cancer |
|
(cagA(+)/vacAs1(+)/IL-1β-511T) both host and bacterial |
Severe gastric anomalies |
[Hong et al., 2016] |
IL-1β-511T |
Frequent occurrence of intestinal type against to the diffuse or mixed-type of gastric |
[Yu et al., 2010; Kamangar et al., 2006; Ruzzo et al., 2005; Wang et al., 2007] |
IL-1β-31TT |
Higher levels of mucosal IL-1β in H. pylori-infected gastric cancer patients |
[Chang et al., 2005] [149] |
IL-1β-31 CT and TT |
Increased possibility of infection by H. pylori |
[Sun et al., 2015] |
H. pylori and early-onset gastric cancer
It is considered that gastric cancer arises as a combination of environmental and genes factors and affects mainly older patients. An inherited component contributes to <3% of gastric cancers and most of genetic changes associated with gastric cancer are acquired [120]. Over the past few decades, advances in technology and high-throughput analysis have improved understanding of the molecular aspects of the pathogenesis of gastric cancer but the categorization of carcinogenic events is highly complicated. The current scientific challenge is to distinguish which alterations of GC are crucial, what is the connection between these alterations and how to prevent their incidence [120, 152]. Gastric cancer at the age before 45 years (without cancer history in the family) is very rare and is called early-onset gastric carcinoma (EOGC). It is thought that these patients develop gastric cancer with a molecular genetic profile which is different from that occurring at a later age. What is more the impact of environmental factors being less important [6]. H. pylori positive gastritis is considered to occur among different gastric cancer forms in young patients, like nodular gastritis, atrophic and hyperplastic gastritis, diffuse and intestinal metaplasia, mucosal atrophy, distal GC and advanced stage [153–156] (Figure 2). This chapter describes the particular role of H. pylori infection and early-onset gastric cancer development.
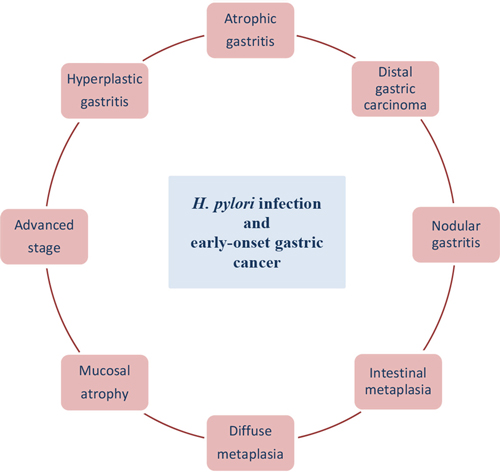
Figure 2: H. pylori infection at the age before 45 years.
Host-related factors have been more relevant than impact of environment factors in developing early-onset gastric cancers. In the study conducted by Rugge et al. 1999 [156], the possible correlation between H. pylori infected patients by cagA positive strains was shown as an important factor in developing gastric carcinoma among young patients. The group of 105 patients with gastric cancer in the age between 16-40 years, as well as control group were analysed. The H. pylori infection occurrence was assessed by histological analysis as well as using standard PCR method to confirm cagA genotype and ureC gene, which are indicators of H. pylori infection. The important correlation between intestinal, as well as diffuse GCs and infection by H. pylori was confirmed among young Italian, where the bacterium has an etiologic significance in both types of GC [156].
The H. pylori infection is higher in older patients in the asymptomatic population in Japan and is significantly increased in GC cases older than 40 years [154]. The researchers were interested in finding the association between H. pylori positive patients younger than 40 years. The group of 40 Japanese GCs in age below 40 years were compared with the equal number of controls. H. pylori infection in gastric mucosa was observed to be importantly increased among patients with cancer in comparison to control group. Moreover, the analysis was performed with 18 cases of intestinal type of young GCc and 70 cases of diffuse, showing the connection between acute, chronic inflammation and mucosal atrophy, intestinal metaplasia in the background of mucosa in both analysed types of GC, with the higher level when compared to the control group.
According to Marcos-Pinto et al., 2013 first-degree relatives of early-onset gastric cancer is supposed to be a population with a different molecular and phenotypic profile, in connection with the precancerous stages of gastric cancer. H. pylori was present in 82% of cases (vs 62% in controls; p=0.004) with vacA s1 and vacA m1 + strains with the presence of atrophy. They showed the increased prevalence of high-risk of precancerous conditions, which may be associated with high virulence H. pylori strains and pro-inflammatory host genotypes [157].
Masuda et al., 2008 [155] found that gastric cancer occurring in young age is significantly correlated with the H. pylori infection but not with the family history or the genetic background. The patients with gastric carcinoma had an increased level of H. pylori infection than those with normal mucosa. Additionally, nodular gastritis, atrophic gastritis and hyperplastic gastritis were assigned using endoscopy method as closely correlated with the H. pylori positive gastritis. Polymorphism of P4502E1 (CYP2E1) and family history were not variable among different age groups.
Helicobacter pylori infection in young population of patients with diagnosed GC was investigated by Kokkola et al., 1996 [158]. Biopsy samples were analysed among 50 cases of GCs, up to 45 years old, compared to controls, and H. pylori infection was confirmed by immunostaining. Obtained results showed that bacterium is a risk factor of early-onset gastric cancer, was found in 72% of GC cases and 43% of controls. The association between infection with CagA + and CagA - strains of H. pylori and young patients with GCs was investigated by Kikuchi et al., 1999 [153]. Among Japanese population, CagA IgG antibodies were measured in sera of 101 GC patients younger than age 43, 103 <40 years old and 100 in patients with benign diseases. In comparison to the H. pylori - /CagA - cases, both the H. pylori + /CagA - and the H. pylori + /CagA + groups revealed high odds ratios for early, advanced, distal, intestinal and diffuse type of GC.
CONCLUSIONS
Helicobacter pylori is a gastric pathogen that colonizes around 50% of the world's population. H. pylori infection provokes mostly chronic inflammation, as well as importantly increases the chance of developing duodenal and gastric ulcer disease and gastric cancer. Infection with the bacterium is one of the risk factor for GC, which is the fourth leading cause of cancer-related deaths worldwide. It is now clear that cancer risk is the combining effect of the polymorphic nature of the host genotype, bacterial population in the host, as well as environmental factors. Based on the current knowledge, encompassing genome sequences, both human and H. pylori, detectable phenotypes (CagA phosphorylation) and animal models, might be possible to describe fundamental biological basis of H. pylori-associated cancer, especially in early age, which should have direct clinical applications. Currently it is important to expand the knowledge about the pathogenesis of H. pylori provoked gastric adenocarcinomas, not only to apply more precise treatments, but also because it might deliver the paradigm for the impact of chronic inflammation on the genesis of other malignancies that developed within the gastrointestinal tract.
ACKNOWLEDGMENTS
This work was partly supported by grant from the Polish Ministry of Science and Higher Education, No. NN402423838 and DS201.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
REFERENCES
1. Zhang XY, Zhang PY. Gastric cancer: somatic genetics as a guide to therapy. J Med Genet. 2017; 54:305–12. https://doi.org/10.1136/jmedgenet-2016-104171.
2. Kong X, Wang JL, Chen HM, Fang JY. Comparison of the clinicopathological characteristics of young and elderly patients with gastric carcinoma: a meta analysis. J Surg Oncol. 2012; 106:346–52. https://doi.org/10.1002/jso.23004.
3. Kunisaki C, Akiyama H, Nomura M, Matsuda G, Otsuka Y, Ono HA, Takagawa R, Nagahori Y, Takahashi M, Kito F, Shimada H. Clinicopathological features of gastric carcinoma in younger and middle-aged patients: a comparative study. J Gastrointest Surg. 2006; 10:1023–32. https://doi.org/10.1016/j.gassur.2006.03.001.
4. Llanos O, Butte JM, Crovari F, Duarte I, Guzmán S. Survival of young patients after gastrectomy for gastric cancer. World J Surg. 2006; 30:17–20. https://doi.org/10.1007/s00268-005-7935-5.
5. Takatsu Y, Hiki N, Nunobe S, Ohashi M, Honda M, Yamaguchi T, Nakajima T, Sano T. Clinicopathological features of gastric cancer in young patients. Gastric Cancer. 2016; 19:472–78. https://doi.org/10.1007/s10120-015-0484-1.
6. Milne AN, Sitarz R, Carvalho R, Carneiro F, Offerhaus GJ. Early onset gastric cancer: on the road to unraveling gastric carcinogenesis. Curr Mol Med. 2007; 7:15–28. https://doi.org/10.2174/156652407779940503.
7. Gigek CO, Calcagno DQ, Rasmussen LT, Santos LC, Leal MF, Wisnieski F, Burbano RR, Lourenço LG, Lopes-Filho GJ, Smith MA. Genetic variants in gastric cancer: risks and clinical implications. Exp Mol Pathol. 2017; 103:101–11. https://doi.org/10.1016/j.yexmp.2017.07.004.
8. Lauren P. The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol Microbiol Scand. 1965; 64:31–49. https://doi.org/10.1111/apm.1965.64.1.31.
9. Bosman FT, Carneiro F, Hruban RH, Theise ND. WHO Classification of Tumours of the Digestive System. 4th ed. Lyon, France: IARC; 2010.
10. Vogelaar IP, van der Post RS, van Krieken JH, Spruijt L, van Zelst-Stams WA, Kets CM, Lubinski J, Jakubowska A, Teodorczyk U, Aalfs CM, van Hest LP, Pinheiro H, Oliveira C, et al. Unraveling genetic predisposition to familial or early onset gastric cancer using germline whole-exome sequencing. Eur J Hum Genet. 2017; 25:1246–52. https://doi.org/10.1038/ejhg.2017.138.
11. Lauwers G, Carneiro F, Graham D, Curado M, Franceschi S. Classification of Tumours of the Digestive System. 4th ed. Lyon: IARC Press; 2010. pp. 48–58.
12. Webb PM, Law M, Varghese C, et al, and Helicobacter and Cancer Collaborative Group. Gastric cancer and Helicobacter pylori: a combined analysis of 12 case control studies nested within prospective cohorts. Gut. 2001; 49:347–53. https://doi.org/10.1136/gut.49.3.347.
13. Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med. 2002; 347:1175–86. https://doi.org/10.1056/NEJMra020542.
14. Warren JR, Marshall B. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet. 1983; 1:1273–75.
15. Fox JG, Dewhirst FE, Tully JG, Paster BJ, Yan L, Taylor NS, Collins MJ Jr, Gorelick PL, Ward JM. Helicobacter hepaticus sp. nov., a microaerophilic bacterium isolated from livers and intestinal mucosal scrapings from mice. J Clin Microbiol. 1994; 32:1238–45.
16. Saccà SC, Vagge A, Pulliero A, Izzotti A. Helicobacter pylori infection and eye diseases: a systematic review. Medicine (Baltimore). 2014; 93:e216. https://doi.org/10.1097/MD.0000000000000216.
17. Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, Sibley RK. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med. 1991; 325:1127–31. https://doi.org/10.1056/NEJM199110173251603.
18. Łaszewicz W, Iwańczak F, Iwańczak B, Annabhani A, Bała G, Bąk-Romaniszyn L, Budzyńska A, Cader J, Celiński K, Cichy W, Czerwionka-Szaflarska M, Czkwianianc E, Czosnek R, et al, and Task Force of the Polish Society of Gastroenterology, and Task Force of the Polish Society of Gastroenterology. Seroprevalence of Helicobacter pylori infection in Polish children and adults depending on socioeconomic status and living conditions. Adv Med Sci. 2014; 59:147–50. https://doi.org/10.1016/j.advms.2014.01.003.
19. Parsonnet J. The epidemiology of C. pylori. In: Blaser MJ, editor. Campylobacter pylori in gastritis and peptic ulcer disease. New York: Igaku-Shoin; 1989. pp. 51–60.
20. Suzuki H, Mori H. Different pathophysiology of gastritis between East and West? An Asian perspective. Inflamm Intest Dis. 2016; 1:123–28. https://doi.org/10.1159/000446301.
21. Eusebi LH, Zagari RM, Bazzoli F. Epidemiology of Helicobacter pylori infection. Helicobacter. 2014; 19:1–5. https://doi.org/10.1111/hel.12165.
22. Peleteiro B, Bastos A, Ferro A, Lunet N. Prevalence of Helicobacter pylori infection worldwide: a systematic review of studies with national coverage. Dig Dis Sci. 2014; 59:1698–709. https://doi.org/10.1007/s10620-014-3063-0.
23. Pilotto A, Franceschi M. Helicobacter pylori infection in older people. World J Gastroenterol. 2014; 20:6364–73. https://doi.org/10.3748/wjg.v20.i21.6364.
24. Macenlle García R, Gayoso Diz P, Sueiro Benavides RA, Fernández Seara J. Prevalence of Helicobacter pylori infection in the general adult population of the province of Ourense. Rev Esp Enferm Dig. 2006; 98:241–48.
25. Sörberg M, Nyrén O, Granström M. Unexpected decrease with age of Helicobacter pylori seroprevalence among Swedish blood donors. J Clin Microbiol. 2003; 41:4038–42. https://doi.org/10.1128/JCM.41.9.4038-4042.2003.
26. Leja M, Cine E, Rudzite D, Vilkoite I, Huttunen T, Daugule I, Rumba-Rozenfelde I, Pimanov S, Liepniece-Karele I, Pahomova J, Purmalis K, Eglitis J, Pirags V, et al. Prevalence of Helicobacter pylori infection and atrophic gastritis in Latvia. Eur J Gastroenterol Hepatol. 2012; 24:1410–17. https://doi.org/10.1097/MEG.0b013e3283583ca5.
27. Bastos J, Peleteiro B, Barros R, Alves L, Severo M, de Fátima Pina M, Pinto H, Carvalho S, Marinho A, Guimarães JT, Azevedo A, La Vecchia C, Barros H, Lunet N. Sociodemographic determinants of prevalence and incidence of Helicobacter pylori infection in Portuguese adults. Helicobacter. 2013; 18:413–22. https://doi.org/10.1111/hel.12061.
28. Roberts SE, Morrison-Rees S, Samuel DG, Thorne K, Akbari A, Williams JG. Review article: the prevalence of Helicobacter pylori and the incidence of gastric cancer across Europe. Aliment Pharmacol Ther. 2016; 43:334–45. https://doi.org/10.1111/apt.13474.
29. Hoang TT, Bengtsson C, Phung DC, Sörberg M, Granström M. Seroprevalence of Helicobacter pylori infection in urban and rural Vietnam. Clin Diagn Lab Immunol. 2005; 12:81–85.
30. Graham DY, Adam E, Reddy GT, Agarwal JP, Agarwal R, Evans DJ Jr, Malaty HM, Evans DG. Seroepidemiology of Helicobacter pylori infection in India. Comparison of developing and developed countries. Dig Dis Sci. 1991; 36:1084–88. https://doi.org/10.1007/BF01297451.
31. Ahmad MM, Rahman M, Rumi AK, Islam S, Huq F, Chowdhury MF, Jinnah F, Morshed MG, Hassan MS, Khan AK, Hasan M. Prevalence of Helicobacter pylori in asymptomatic population—a pilot serological study in Bangladesh. J Epidemiol. 1997; 7:251–54. https://doi.org/10.2188/jea.7.251.
32. Moujaber T, MacIntyre CR, Backhouse J, Gidding H, Quinn H, Gilbert GL. The seroepidemiology of Helicobacter pylori infection in Australia. Int J Infect Dis. 2008; 12:500–04. https://doi.org/10.1016/j.ijid.2008.01.011.
33. Fujisawa T, Kumagai T, Akamatsu T, Kiyosawa K, Matsunaga Y. Changes in seroepidemiological pattern of Helicobacter pylori and hepatitis A virus over the last 20 years in Japan. Am J Gastroenterol. 1999; 94:2094–99. https://doi.org/10.1111/j.1572-0241.1999.01283.x.
34. Teh BH, Lin JT, Pan WH, Lin SH, Wang LY, Lee TK, Chen CJ. Seroprevalence and associated risk factors of Helicobacter pylori infection in Taiwan. Anticancer Res. 1994; 14:1389–92.
35. Wang KJ, Wang RT. [Meta-analysis on the epidemiology of Helicobacter pylori infection in China]. Zhonghua Liu Xing Bing Xue Za Zhi. 2003; 24:443–46.
36. Yim JY, Kim N, Choi SH, Kim YS, Cho KR, Kim SS, Seo GS, Kim HU, Baik GH, Sin CS, Cho SH, Oh BH. Seroprevalence of Helicobacter pylori in South Korea. Helicobacter. 2007; 12:333–40. https://doi.org/10.1111/j.1523-5378.2007.00504.x.
37. Fock KM. Helicobacter pylori infection—current status in Singapore. Ann Acad Med Singapore. 1997; 26:637–41.
38. Goh KL, Parasakthi N. The racial cohort phenomenon: seroepidemiology of Helicobacter pylori infection in a multiracial South-East Asian country. Eur J Gastroenterol Hepatol. 2001; 13:177–83. https://doi.org/10.1097/00042737-200102000-00014.
39. Deankanob W, Chomvarin C, Hahnvajanawong C, Intapan PM, Wongwajana S, Mairiang P, Kularbkaew C, Sangchan A. Enzyme-linked immunosorbent assay for serodiagnosis of Helicobacter pylori in dyspeptic patients and volunteer blood donors. Southeast Asian J Trop Med Public Health. 2006; 37:958–65.
40. Hong JB, Zuo W, Wang AJ, Lu NH. Helicobacter pylori Infection Synergistic with IL-1β Gene Polymorphisms Potentially Contributes to the Carcinogenesis of Gastric Cancer. Int J Med Sci. 2016; 13:298–303. https://doi.org/10.7150/ijms.14239.
41. Correa P, Haenszel W, Cuello C, Tannenbaum S, Archer M. A model for gastric cancer epidemiology. Lancet. 1975; 2:58–60. https://doi.org/10.1016/S0140-6736(75)90498-5.
42. Cokkinides VE, Bandi P, Siegel RL, Jemal A. Cancer-related risk factors and preventive measures in US Hispanics/Latinos. CA Cancer J Clin. 2012; 62:353–63. https://doi.org/10.3322/caac.21155.
43. Correa P. Helicobacter pylori and gastric carcinogenesis. Am J Surg Pathol. 1995; 19:S37–43.
44. Herrera V, Parsonnet J. Helicobacter pylori and gastric adenocarcinoma. Clin Microbiol Infect. 2009; 15:971–76. https://doi.org/10.1111/j.1469-0691.2009.03031.x.
45. Machlowska J, Maciejewski R, Sitarz R. The Pattern of Signatures in Gastric Cancer Prognosis. Int J Mol Sci. 2018; 19:19. https://doi.org/10.3390/ijms19061658.
46. Helander HF, Keeling DJ. Cell biology of gastric acid secretion. Baillieres Clin Gastroenterol. 1993; 7:1–21. https://doi.org/10.1016/0950-3528(93)90029-R.
47. Kwiecień S, Magierowska K, Śliwowski Z, Wójcik D, Magierowski M, Brzozowski T. New insight into the mechanisms of gastroduodenal injury induced by nonsteroidal anti-inflammatory drugs: practical implications. Pol Arch Med Wewn. 2015; 125:191–98. https://doi.org/10.20452/pamw.2715.
48. Schreiber S, Konradt M, Groll C, Scheid P, Hanauer G, Werling HO, Josenhans C, Suerbaum S. The spatial orientation of Helicobacter pylori in the gastric mucus. Proc Natl Acad Sci USA. 2004; 101:5024–29. https://doi.org/10.1073/pnas.0308386101.
49. Smoot DT. How does Helicobacter pylori cause mucosal damage? Direct mechanisms. Gastroenterology. 1997;113: S31-4; discussion S50. https://doi.org/10.1016/S0016-5085(97)80008-X.
50. Zhang RG, Duan GC, Fan QT, Chen SY. Role of Helicobacter pylori infection in pathogenesis of gastric carcinoma. World J Gastrointest Pathophysiol. 2016; 7:97–107. https://doi.org/10.4291/wjgp.v7.i1.97.
51. Conteduca V, Sansonno D, Lauletta G, Russi S, Ingravallo G, Dammacco F. H. pylori infection and gastric cancer: state of the art (review). Int J Oncol. 2013; 42:5–18. review https://doi.org/10.3892/ijo.2012.1701.
52. Wroblewski LE, Peek RM Jr. Helicobacter pylori in gastric carcinogenesis: mechanisms. Gastroenterol Clin North Am. 2013; 42:285–98. https://doi.org/10.1016/j.gtc.2013.01.006.
53. Naumann M, Crabtree JE. Helicobacter pylori-induced epithelial cell signalling in gastric carcinogenesis. Trends Microbiol. 2004; 12:29–36. https://doi.org/10.1016/j.tim.2003.11.005.
54. Baik SC, Youn HS, Chung MH, Lee WK, Cho MJ, Ko GH, Park CK, Kasai H, Rhee KH. Increased oxidative DNA damage in Helicobacter pylori-infected human gastric mucosa. Cancer Res. 1996; 56:1279–82.
55. Tsuji S, Kawai N, Tsujii M, Kawano S, Hori M. Review article: inflammation-related promotion of gastrointestinal carcinogenesis—a perigenetic pathway. Aliment Pharmacol Ther. 2003; 18:82–89. https://doi.org/10.1046/j.1365-2036.18.s1.22.x.
56. Guven-Maiorov E, Tsai CJ, Ma B, Nussinov R. Prediction of Host-Pathogen Interactions for Helicobacter pylori by Interface Mimicry and Implications to Gastric Cancer. J Mol Biol. 2017; 429:3925–41. https://doi.org/10.1016/j.jmb.2017.10.023.
57. Sallas ML, Melchiades JL, Zabaglia LM, Rasmussen LT. Prevalence of Helicobacter pylori vacA, cagA, dupA and oipA Genotypes in Patients with Gastric Disease. Advances in Microbiology. 2017; 7:1-9.
58. Alam J, Maiti S, Ghosh P, De R, Chowdhury A, Das S, Macaden R, Devarbhavi H, Ramamurthy T, Mukhopadhyay AK. Significant association of the dupA gene of Helicobacter pylori with duodenal ulcer development in a South-east Indian population. J Med Microbiol. 2012; 61:1295–302. https://doi.org/10.1099/jmm.0.038398-0.
59. van Doorn LJ, Figueiredo C, Sanna R, Plaisier A, Schneeberger P, de Boer W, Quint W. Clinical relevance of the cagA, vacA, and iceA status of Helicobacter pylori. Gastroenterology. 1998; 115:58–66. https://doi.org/10.1016/S0016-5085(98)70365-8.
60. Yamaoka Y, Kikuchi S, el-Zimaity HM, Gutierrez O, Osato MS, Graham DY. Importance of Helicobacter pylori oipA in clinical presentation, gastric inflammation, and mucosal interleukin 8 production. Gastroenterology. 2002; 123:414–24. https://doi.org/10.1053/gast.2002.34781.
61. Yamaoka Y, Kodama T, Gutierrez O, Kim JG, Kashima K, Graham DY. Relationship between Helicobacter pylori iceA, cagA, and vacA status and clinical outcome: studies in four different countries. J Clin Microbiol. 1999; 37:2274–79.
62. Yamaoka Y, Kodama T, Kashima K, Graham DY, Sepulveda AR. Variants of the 3′ region of the cagA gene in Helicobacter pylori isolates from patients with different H. pylori-associated diseases. J Clin Microbiol. 1998; 36:2258–63.
63. Yamaoka Y, El-Zimaity HM, Gutierrez O, Figura N, Kim JG, Kodama T, Kashima K, Graham DY. Relationship between the cagA 3′ repeat region of Helicobacter pylori, gastric histology, and susceptibility to low pH. Gastroenterology. 1999; 117:342–49. https://doi.org/10.1053/gast.1999.0029900342.
64. Yamaoka Y, Osato MS, Sepulveda AR, Gutierrez O, Figura N, Kim JG, Kodama T, Kashima K, Graham DY. Molecular epidemiology of Helicobacter pylori: separation of H. pylori from East Asian and non-Asian countries. Epidemiol Infect. 2000; 124:91–96. https://doi.org/10.1017/S0950268899003209.
65. Cover TL, Blanke SR. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat Rev Microbiol. 2005; 3:320–32. https://doi.org/10.1038/nrmicro1095.
66. Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006; 19:449–90. https://doi.org/10.1128/CMR.00054-05.
67. Atherton JC, Cao P, Peek RM Jr, Tummuru MK, Blaser MJ, Cover TL. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem. 1995; 270:17771–77. https://doi.org/10.1074/jbc.270.30.17771.
68. Sugimoto M, Zali MR, Yamaoka Y. The association of vacA genotypes and Helicobacter pylori-related gastroduodenal diseases in the Middle East. Eur J Clin Microbiol Infect Dis. 2009; 28:1227–36. https://doi.org/10.1007/s10096-009-0772-y.
69. Sugimoto M, Yamaoka Y. The association of vacA genotype and Helicobacter pylori-related disease in Latin American and African populations. Clin Microbiol Infect. 2009; 15:835–42. https://doi.org/10.1111/j.1469-0691.2009.02769.x.
70. Yamaoka Y, Orito E, Mizokami M, Gutierrez O, Saitou N, Kodama T, Osato MS, Kim JG, Ramirez FC, Mahachai V, Graham DY. Helicobacter pylori in North and South America before Columbus. FEBS Lett. 2002; 517:180–84. https://doi.org/10.1016/S0014-5793(02)02617-0.
71. Uchida T, Nguyen LT, Takayama A, Okimoto T, Kodama M, Murakami K, Matsuhisa T, Trinh TD, Ta L, Ho DQ, Hoang HH, Kishida T, Fujioka T, et al. Analysis of virulence factors of Helicobacter pylori isolated from a Vietnamese population. BMC Microbiol. 2009; 9:175. https://doi.org/10.1186/1471-2180-9-175.
72. Yamaoka Y, Kwon DH, Graham DY. A M(r) 34,000 proinflammatory outer membrane protein (oipA) of Helicobacter pylori. Proc Natl Acad Sci USA. 2000; 97:7533–38. https://doi.org/10.1073/pnas.130079797.
73. Yamaoka Y, Kudo T, Lu H, Casola A, Brasier AR, Graham DY. Role of interferon-stimulated responsive element-like element in interleukin-8 promoter in Helicobacter pylori infection. Gastroenterology. 2004; 126:1030–43. https://doi.org/10.1053/j.gastro.2003.12.048.
74. Tabassam FH, Graham DY, Yamaoka Y. Helicobacter pylori activate epidermal growth factor receptor- and phosphatidylinositol 3-OH kinase-dependent Akt and glycogen synthase kinase 3β phosphorylation. Cell Microbiol. 2009; 11:70–82. https://doi.org/10.1111/j.1462-5822.2008.01237.x.
75. Matos JI, de Sousa HA, Marcos-Pinto R, Dinis-Ribeiro M. Helicobacter pylori CagA and VacA genotypes and gastric phenotype: a meta-analysis. Eur J Gastroenterol Hepatol. 2013; 25:1431–41. https://doi.org/10.1097/MEG.0b013e328364b53e.
76. El Khadir M, Alaoui Boukhris S, Benajah DA, El Rhazi K, Ibrahimi SA, El Abkari M, Harmouch T, Nejjari C, Mahmoud M, Benlemlih M, Bennani B. VacA and CagA Status as Biomarker of Two Opposite End Outcomes of Helicobacter pylori Infection (Gastric Cancer and Duodenal Ulcer) in a Moroccan Population. PLoS One. 2017; 12:e0170616. https://doi.org/10.1371/journal.pone.0170616.
77. Hold GL, Rabkin CS, Chow WH, Smith MG, Gammon MD, Risch HA, Vaughan TL, McColl KE, Lissowska J, Zatonski W, Schoenberg JB, Blot WJ, Mowat NA, et al. A functional polymorphism of toll-like receptor 4 gene increases risk of gastric carcinoma and its precursors. Gastroenterology. 2007; 132:905–12. https://doi.org/10.1053/j.gastro.2006.12.026.
78. Huang JQ, Zheng GF, Sumanac K, Irvine EJ, Hunt RH. Meta-analysis of the relationship between cagA seropositivity and gastric cancer. Gastroenterology. 2003; 125:1636–44. https://doi.org/10.1053/j.gastro.2003.08.033.
79. Asrat D, Nilsson I, Mengistu Y, Kassa E, Ashenafi S, Ayenew K, Wadström T, Abu-Al-Soud W. Prevalence of Helicobacter pylori vacA and cagA genotypes in Ethiopian dyspeptic patients. J Clin Microbiol. 2004; 42:2682–84. https://doi.org/10.1128/JCM.42.6.2682-2684.2004.
80. Breurec S, Michel R, Seck A, Brisse S, Côme D, Dieye FB, Garin B, Huerre M, Mbengue M, Fall C, Sgouras DN, Thiberge JM, Dia D, Raymond J. Clinical relevance of cagA and vacA gene polymorphisms in Helicobacter pylori isolates from Senegalese patients. Clin Microbiol Infect. 2012; 18:153–59. https://doi.org/10.1111/j.1469-0691.2011.03524.x.
81. González CA, Figueiredo C, Lic CB, Ferreira RM, Pardo ML, Ruiz Liso JM, Alonso P, Sala N, Capella G, Sanz-Anquela JM. Helicobacter pylori cagA and vacA genotypes as predictors of progression of gastric preneoplastic lesions: a long-term follow-up in a high-risk area in Spain. Am J Gastroenterol. 2011; 106:867–74. https://doi.org/10.1038/ajg.2011.1.
82. Ortiz-Princz D, Guariglia-Oropeza V, Avila M, Correnti M, Perrone M, Gutierrez B, Torres J, Megraud F, Cavazza ME. Helicobacter pylori cagA and vacA genotypes in Cuban and Venezuelan populations. Mem Inst Oswaldo Cruz. 2010; 105:331–35. https://doi.org/10.1590/S0074-02762010000300016.
83. Pandey A, Tripathi SC, Mahata S, Vishnoi K, Shukla S, Misra SP, Misra V, Hedau S, Mehrotra R, Dwivedi M, Bharti AC. Carcinogenic Helicobacter pylori in gastric pre-cancer and cancer lesions: association with tobacco-chewing. World J Gastroenterol. 2014; 20:6860–68. https://doi.org/10.3748/wjg.v20.i22.6860.
84. Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997; 388:539–47. https://doi.org/10.1038/41483.
85. Peek RM Jr, Crabtree JE. Helicobacter infection and gastric neoplasia. J Pathol. 2006; 208:233–48. https://doi.org/10.1002/path.1868.
86. Stein SC, Faber E, Bats SH, Murillo T, Speidel Y, Coombs N, Josenhans C. Helicobacter pylori modulates host cell responses by CagT4SS-dependent translocation of an intermediate metabolite of LPS inner core heptose biosynthesis. PLoS Pathog. 2017; 13:e1006514. https://doi.org/10.1371/journal.ppat.1006514.
87. Buti L, Spooner E, Van der Veen AG, Rappuoli R, Covacci A, Ploegh HL. Helicobacter pylori cytotoxin-associated gene A (CagA) subverts the apoptosis-stimulating protein of p53 (ASPP2) tumor suppressor pathway of the host. Proc Natl Acad Sci USA. 2011; 108:9238–43. https://doi.org/10.1073/pnas.1106200108.
88. Asahi M, Azuma T, Ito S, Ito Y, Suto H, Nagai Y, Tsubokawa M, Tohyama Y, Maeda S, Omata M, Suzuki T, Sasakawa C. Helicobacter pylori CagA protein can be tyrosine phosphorylated in gastric epithelial cells. J Exp Med. 2000; 191:593–602. https://doi.org/10.1084/jem.191.4.593.
89. Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, Rappuoli R, Covacci A. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci USA. 1996; 93:14648–53. https://doi.org/10.1073/pnas.93.25.14648.
90. Odenbreit S, Püls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000; 287:1497–500. https://doi.org/10.1126/science.287.5457.1497.
91. Stein M, Rappuoli R, Covacci A. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc Natl Acad Sci USA. 2000; 97:1263–68. https://doi.org/10.1073/pnas.97.3.1263.
92. Saito Y, Murata-Kamiya N, Hirayama T, Ohba Y, Hatakeyama M. Conversion of Helicobacter pylori CagA from senescence inducer to oncogenic driver through polarity-dependent regulation of p21. J Exp Med. 2010; 207:2157–74. https://doi.org/10.1084/jem.20100602.
93. Subsomwong P, Miftahussurur M, Uchida T, Vilaichone RK, Ratanachu-Ek T, Mahachai V, Yamaoka Y. Prevalence, risk factors, and virulence genes of Helicobacter pylori among dyspeptic patients in two different gastric cancer risk regions of Thailand. PLoS One. 2017; 12:e0187113. https://doi.org/10.1371/journal.pone.0187113.
94. Suwanrungruang K, Sriamporn S, Wiangnon S, Rangsrikajee D, Sookprasert A, Thipsuntornsak N, Satitvipawee P, Poomphakwaen K, Tokudome S. Lifestyle-related risk factors for stomach cancer in northeast Thailand. Asian Pac J Cancer Prev. 2008; 9:71–75.
95. Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer. 2004; 4:688–94. https://doi.org/10.1038/nrc1433.
96. Higashi H, Tsutsumi R, Fujita A, Yamazaki S, Asaka M, Azuma T, Hatakeyama M. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci USA. 2002; 99:14428–33. https://doi.org/10.1073/pnas.222375399.
97. Jones KR, Joo YM, Jang S, Yoo YJ, Lee HS, Chung IS, Olsen CH, Whitmire JM, Merrell DS, Cha JH. Polymorphism in the CagA EPIYA motif impacts development of gastric cancer. J Clin Microbiol. 2009; 47:959–68. https://doi.org/10.1128/JCM.02330-08.
98. Vilaichone RK, Mahachai V, Tumwasorn S, Wu JY, Graham DY, Yamaoka Y. Molecular epidemiology and outcome of Helicobacter pylori infection in Thailand: a cultural cross roads. Helicobacter. 2004; 9:453–59. https://doi.org/10.1111/j.1083-4389.2004.00260.x.
99. Aftab H, Miftahussurur M, Subsomwong P, Ahmed F, Khan AK, Matsumoto T, Suzuki R, Yamaoka Y. Two populations of less-virulent Helicobacter pylori genotypes in Bangladesh. PLoS One. 2017; 12:e0182947. https://doi.org/10.1371/journal.pone.0182947.
100. Yamaoka Y. Mechanisms of disease: helicobacter pylori virulence factors. Nat Rev Gastroenterol Hepatol. 2010; 7:629–41. https://doi.org/10.1038/nrgastro.2010.154.
101. Backert S, Moese S, Selbach M, Brinkmann V, Meyer TF. Phosphorylation of tyrosine 972 of the Helicobacter pylori CagA protein is essential for induction of a scattering phenotype in gastric epithelial cells. Mol Microbiol. 2001; 42:631–44. https://doi.org/10.1046/j.1365-2958.2001.02649.x.
102. Camargo MC, Mera R, Correa P, Peek RM Jr, Fontham ET, Goodman KJ, Piazuelo MB, Sicinschi L, Zabaleta J, Schneider BG. Interleukin-1beta and interleukin-1 receptor antagonist gene polymorphisms and gastric cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006; 15:1674–87. https://doi.org/10.1158/1055-9965.EPI-06-0189.
103. Kimang’a AN. IL-1B-511 Allele T and IL-1RN-L/L Play a Pathological Role in Helicobacter Pylori (H. Pylori) Disease Outcome in the African Population. Ethiop J Health Sci. 2012; 22:163–69.
104. Loh M, Koh KX, Yeo BH, Song CM, Chia KS, Zhu F, Yeoh KG, Hill J, Iacopetta B, Soong R. Meta-analysis of genetic polymorphisms and gastric cancer risk: variability in associations according to race. Eur J Cancer. 2009; 45:2562–68. https://doi.org/10.1016/j.ejca.2009.03.017.
105. Vincenzi B, Patti G, Galluzzo S, Pantano F, Venditti O, Santini D, Ruzzo A, Schiavon G, Caraglia M, Marra M, Graziano F, Tonini G. Interleukin 1beta-511T gene (IL1beta) polymorphism is correlated with gastric cancer in the Caucasian population: results from a meta-analysis. Oncol Rep. 2008; 20:1213–20.
106. Xue H, Lin B, Ni P, Xu H, Huang G. Interleukin-1B and interleukin-1 RN polymorphisms and gastric carcinoma risk: a meta-analysis. J Gastroenterol Hepatol. 2010; 25:1604–17. https://doi.org/10.1111/j.1440-1746.2010.06428.x.
107. Zhao JD, Geng PL, Li ZQ, Cui S, Zhao JH, Wang LJ, Li JZ, Ji FX, Li GY, Shen GS, Lin MZ, Shen CF, Cao CZ. Associations between interleukin-1 polymorphisms and gastric cancers among three ethnicities. World J Gastroenterol. 2012; 18:7093–99. https://doi.org/10.3748/wjg.v18.i47.7093.
108. Qinghai Z, Yanying W, Yunfang C, Xukui Z, Xiaoqiao Z. Effect of interleukin-17A and interleukin-17F gene polymorphisms on the risk of gastric cancer in a Chinese population. Gene. 2014; 537:328–32. https://doi.org/10.1016/j.gene.2013.11.007.
109. Rafiei A, Hosseini V, Janbabai G, Ghorbani A, Ajami A, Farzmandfar T, Azizi MD, Gilbreath JJ, Merrell DS. Polymorphism in the interleukin-17A promoter contributes to gastric cancer. World J Gastroenterol. 2013; 19:5693–99. https://doi.org/10.3748/wjg.v19.i34.5693.
110. Shibata T, Tahara T, Hirata I, Arisawa T. Genetic polymorphism of interleukin-17A and -17F genes in gastric carcinogenesis. Hum Immunol. 2009; 70:547–51. https://doi.org/10.1016/j.humimm.2009.04.030.
111. Zhang X, Zheng L, Sun Y, Zhang X. Analysis of the association of interleukin-17 gene polymorphisms with gastric cancer risk and interaction with Helicobacter pylori infection in a Chinese population. Tumour Biol. 2014; 35:1575–80. https://doi.org/10.1007/s13277-013-1217-x.
112. Gorouhi F, Islami F, Bahrami H, Kamangar F. Tumour-necrosis factor-A polymorphisms and gastric cancer risk: a meta-analysis. Br J Cancer. 2008; 98:1443–51. https://doi.org/10.1038/sj.bjc.6604277.
113. Zhang J, Dou C, Song Y, Ji C, Gu S, Xie Y, Mao Y. Polymorphisms of tumor necrosis factor-alpha are associated with increased susceptibility to gastric cancer: a meta-analysis. J Hum Genet. 2008; 53:479–89. https://doi.org/10.1007/s10038-008-0273-3.
114. Fukata M, Abreu MT. Role of Toll-like receptors in gastrointestinal malignancies. Oncogene. 2008; 27:234–43. https://doi.org/10.1038/sj.onc.1210908.
115. Xu Q, Yuan Y, Sun LP, Gong YH, Xu Y, Yu XW, Dong NN, Lin GD, Smith PN, Li RW. Risk of gastric cancer is associated with the MUC1 568 A/G polymorphism. Int J Oncol. 2009; 35:1313–20.
116. Zheng L, Zhu C, Gu J, Xi P, Du J, Jin G. Functional polymorphism rs4072037 in MUC1 gene contributes to the susceptibility to gastric cancer: evidence from pooled 6,580 cases and 10,324 controls. Mol Biol Rep. 2013; 40:5791–96. https://doi.org/10.1007/s11033-013-2682-4.
117. Capellá G, Pera G, Sala N, Agudo A, Rico F, Del Giudicce G, Plebani M, Palli D, Boeing H, Bueno-de-Mesquita HB, Carneiro F, Berrino F, Vineis P, et al. DNA repair polymorphisms and the risk of stomach adenocarcinoma and severe chronic gastritis in the EPIC-EURGAST study. Int J Epidemiol. 2008; 37:1316–25. https://doi.org/10.1093/ije/dyn145.
118. Dong Z, Guo W, Zhou R, Wan L, Li Y, Wang N, Kuang G, Wang S. Polymorphisms of the DNA repair gene XPA and XPC and its correlation with gastric cardiac adenocarcinoma in a high incidence population in North China. J Clin Gastroenterol. 2008; 42:910–15. https://doi.org/10.1097/MCG.0b013e3180f6262c.
119. Li WQ, Zhang L, Ma JL, Zhang Y, Li JY, Pan KF, You WC. Association between genetic polymorphisms of DNA base excision repair genes and evolution of precancerous gastric lesions in a Chinese population. Carcinogenesis. 2009; 30:500–05. https://doi.org/10.1093/carcin/bgp018.
120. McLean MH, El-Omar EM. Genetics of gastric cancer. Nat Rev Gastroenterol Hepatol. 2014; 11:664–74. https://doi.org/10.1038/nrgastro.2014.143.
121. Sun X, Xu Y, Zhang F, Jing T, Han J, Zhang J. Association between the IL1B -31C > T polymorphism and Helicobacter pylori infection in Asian and Latin American population: A meta-analysis. Microb Pathog. 2015; 86:45-52.
122. Dong LM, Potter JD, White E, Ulrich CM, Cardon LR, Peters U. Genetic susceptibility to cancer: the role of polymorphisms in candidate genes. JAMA. 2008; 299:2423–36. https://doi.org/10.1001/jama.299.20.2423.
123. Zintzaras E. Association of methylenetetrahydrofolate reductase (MTHFR) polymorphisms with genetic susceptibility to gastric cancer: a meta-analysis. J Hum Genet. 2006; 51:618–24. https://doi.org/10.1007/s10038-006-0405-6.
124. Agudo A, Sala N, Pera G, Capellá G, Berenguer A, García N, Palli D, Boeing H, Del Giudice G, Saieva C, Carneiro F, Berrino F, Sacerdote C, et al. Polymorphisms in metabolic genes related to tobacco smoke and the risk of gastric cancer in the European prospective investigation into cancer and nutrition. Cancer Epidemiol Biomarkers Prev. 2006; 15:2427–34. https://doi.org/10.1158/1055-9965.EPI-06-0072.
125. Boccia S, De Lauretis A, Gianfagna F, van Duijn CM, Ricciardi G. CYP2E1PstI/RsaI polymorphism and interaction with tobacco, alcohol and GSTs in gastric cancer susceptibility: A meta-analysis of the literature. Carcinogenesis. 2007; 28:101–06. https://doi.org/10.1093/carcin/bgl124.
126. Freedman ND, Ahn J, Hou L, Lissowska J, Zatonski W, Yeager M, Chanock SJ, Chow WH, Abnet CC. Polymorphisms in estrogen- and androgen-metabolizing genes and the risk of gastric cancer. Carcinogenesis. 2009; 30:71–77. https://doi.org/10.1093/carcin/bgn258.
127. Solaini G, Baracca A, Lenaz G, Sgarbi G. Hypoxia and mitochondrial oxidative metabolism. Biochim Biophys Acta. 2010; 1797:1171–77. https://doi.org/10.1016/j.bbabio.2010.02.011.
128. Fan L, Li J, Yu Z, Dang X, Wang K. The hypoxia-inducible factor pathway, prolyl hydroxylase domain protein inhibitors, and their roles in bone repair and regeneration. BioMed Res Int. 2014; 2014:239356. https://doi.org/10.1155/2014/239356.
129. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008; 453:807–11. https://doi.org/10.1038/nature06905.
130. Zhao P, Li Y, Lu Y. Aberrant expression of CD133 protein correlates with Ki-67 expression and is a prognostic marker in gastric adenocarcinoma. BMC Cancer. 2010; 10:218. https://doi.org/10.1186/1471-2407-10-218.
131. Lee HJ, Nam KT, Park HS, Kim MA, Lafleur BJ, Aburatani H, Yang HK, Kim WH, Goldenring JR. Gastroenterology. 2010; 139:213-25.e3. https://doi.org/10.1053/j.gastro.2010.04.008.
132. Ishigami S, Ueno S, Nishizono Y, Matsumoto M, Kurahara H, Arigami T, Uchikado Y, Setoyama T, Arima H, Yoshiaki K, Kijima Y, Kitazono M, Natsugoe S. Prognostic impact of CD168 expression in gastric cancer. BMC Cancer. 2011; 11:106. https://doi.org/10.1186/1471-2407-11-106.
133. Xie JW, Chen PC, Zheng CH, Li P, Wang JB, Lin JX, Lu J, Chen QY, Cao LL, Lin M, Lin Y, Huang CM. Evaluation of the prognostic value and functional roles of CD44v6 in gastric cancer. J Cancer Res Clin Oncol. 2015; 141:1809–17. https://doi.org/10.1007/s00432-015-1964-8.
134. Long ZW, Wang JL, Wang YN. Matrix metalloproteinase-7 mRNA and protein expression in gastric carcinoma: a meta-analysis. Tumour Biol. 2014; 35:11415–26. https://doi.org/10.1007/s13277-014-2441-8.
135. Shin NR, Jeong EH, Choi CI, Moon HJ, Kwon CH, Chu IS, Kim GH, Jeon TY, Kim DH, Lee JH, Park DY. Overexpression of Snail is associated with lymph node metastasis and poor prognosis in patients with gastric cancer. BMC Cancer. 2012; 12:521. https://doi.org/10.1186/1471-2407-12-521.
136. Backert S, Naumann M. What a disorder: proinflammatory signaling pathways induced by Helicobacter pylori. Trends Microbiol. 2010; 18:479–86. https://doi.org/10.1016/j.tim.2010.08.003.
137. Koch M, Mollenkopf HJ, Meyer TF. Macrophages recognize the Helicobacter pylori type IV secretion system in the absence of toll-like receptor signalling. Cell Microbiol. 2016; 18:137–47. https://doi.org/10.1111/cmi.12492.
138. Sokolova O, Maubach G, Naumann M. MEKK3 and TAK1 synergize to activate IKK complex in Helicobacter pylori infection. Biochim Biophys Acta. 2014; 1843:715–24. https://doi.org/10.1016/j.bbamcr.2014.01.006.
139. Linz B, Balloux F, Moodley Y, Manica A, Liu H, Roumagnac P, Falush D, Stamer C, Prugnolle F, van der Merwe SW, Yamaoka Y, Graham DY, Perez-Trallero E, et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007; 445:915–18. https://doi.org/10.1038/nature05562.
140. Rivas-Ortiz CI, Lopez-Vidal Y, Arredondo-Hernandez LJ, Castillo-Rojas G. Genetic Alterations in Gastric Cancer Associated with Helicobacter pylori Infection. Front Med (Lausanne). 2017; 4:47. https://doi.org/10.3389/fmed.2017.00047.
141. Takashima M, Furuta T, Hanai H, Sugimura H, Kaneko E. Effects of Helicobacter pylori infection on gastric acid secretion and serum gastrin levels in Mongolian gerbils. Gut. 2001; 48:765–73. https://doi.org/10.1136/gut.48.6.765.
142. Wang M, Furuta T, Takashima M, Futami H, Shirai N, Hanai H, Kaneko E. Relation between interleukin-1beta messenger RNA in gastric fundic mucosa and gastric juice pH in patients infected with Helicobacter pylori. J Gastroenterol. 1999; 34:10–17.
143. El-Omar EM, Rabkin CS, Gammon MD, Vaughan TL, Risch HA, Schoenberg JB, Stanford JL, Mayne ST, Goedert J, Blot WJ, Fraumeni JF Jr, Chow WH. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology. 2003; 124:1193–201. https://doi.org/10.1016/S0016-5085(03)00157-4.
144. Lee KA, Ki CS, Kim HJ, Sohn KM, Kim JW, Kang WK, Rhee JC, Song SY, Sohn TS. Novel interleukin 1beta polymorphism increased the risk of gastric cancer in a Korean population. J Gastroenterol. 2004; 39:429–33. https://doi.org/10.1007/s00535-003-1315-4.
145. Yu J, Zeng Z, Wang S, Tian L, Wu J, Xue L, Lee CW, Zhang M, Goggins WB, Chen M, Hu P, Sung JJ. IL-1B-511 polymorphism is associated with increased risk of certain subtypes of gastric cancer in Chinese: a case-control study. Am J Gastroenterol. 2010; 105:557–64. https://doi.org/10.1038/ajg.2009.644.
146. Kamangar F, Cheng C, Abnet CC, Rabkin CS. Interleukin-1B polymorphisms and gastric cancer risk—a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006; 15:1920–28. https://doi.org/10.1158/1055-9965.EPI-06-0267.
147. Ruzzo A, Graziano F, Pizzagalli F, Santini D, Battistelli V, Panunzi S, Canestrari E, Catalano V, Humar B, Ficarelli R, Bearzi I, Cascinu S, Naldi N, et al. Interleukin 1B gene (IL-1B) and interleukin 1 receptor antagonist gene (IL-1RN) polymorphisms in Helicobacter pylori-negative gastric cancer of intestinal and diffuse histotype. Ann Oncol. 2005; 16:887–92. https://doi.org/10.1093/annonc/mdi184.
148. Wang P, Xia HH, Zhang JY, Dai LP, Xu XQ, Wang KJ. Association of interleukin-1 gene polymorphisms with gastric cancer: a meta-analysis. Int J Cancer. 2007; 120:552–62. https://doi.org/10.1002/ijc.22353.
149. Chang YW, Jang JY, Kim NH, Lee JW, Lee HJ, Jung WW, Dong SH, Kim HJ, Kim BH, Lee JI, Chang R. Interleukin-1B (IL-1B) polymorphisms and gastric mucosal levels of IL-1beta cytokine in Korean patients with gastric cancer. Int J Cancer. 2005; 114:465–71. https://doi.org/10.1002/ijc.20724.
150. Leung WK, Chan MC, To KF, Man EP, Ng EK, Chu ES, Lau JY, Lin SR, Sung JJ. H. pylori genotypes and cytokine gene polymorphisms influence the development of gastric intestinal metaplasia in a Chinese population. Am J Gastroenterol. 2006; 101:714–20. https://doi.org/10.1111/j.1572-0241.2006.00560.x.
151. Shigematsu Y, Niwa T, Rehnberg E, Toyoda T, Yoshida S, Mori A, Wakabayashi M, Iwakura Y, Ichinose M, Kim YJ, Ushijima T. Interleukin-1β induced by Helicobacter pylori infection enhances mouse gastric carcinogenesis. Cancer Lett. 2013; 340:141–47. https://doi.org/10.1016/j.canlet.2013.07.034.
152. Skierucha M, Milne AN, Offerhaus GJ, Polkowski WP, Maciejewski R, Sitarz R. Molecular alterations in gastric cancer with special reference to the early-onset subtype. World J Gastroenterol. 2016; 22:2460–74. https://doi.org/10.3748/wjg.v22.i8.2460.
153. Kikuchi S, Crabtree JE, Forman D, Kurosawa M, and Research Group on Prevention of Gastric Carcinoma Among Young Adults. Association between infections with CagA-positive or -negative strains of Helicobacter pylori and risk for gastric cancer in young adults. Am J Gastroenterol. 1999; 94:3455–59. https://doi.org/10.1111/j.1572-0241.1999.01607.x.
154. Koshida Y, Koizumi W, Sasabe M, Katoh Y, Okayasu I. Association of Helicobacter pylori-dependent gastritis with gastric carcinomas in young Japanese patients: histopathological comparison of diffuse and intestinal type cancer cases. Histopathology. 2000; 37:124–30. https://doi.org/10.1046/j.1365-2559.2000.00948.x.
155. Masuda G, Tokunaga A, Shirakawa T, Togashi A, Kiyama T, Kato S, Matsukura N, Bou H, Watanabe M, Tajiri T. Helicobacter pylori infection, but not genetic polymorphism of CYP2E1, is highly prevalent in gastric cancer patients younger than 40 years. Gastric Cancer. 2007; 10:98–103. https://doi.org/10.1007/s10120-007-0414-y.
156. Rugge M, Busatto G, Cassaro M, Shiao YH, Russo V, Leandro G, Avellini C, Fabiano A, Sidoni A, Covacci A. Patients younger than 40 years with gastric carcinoma: helicobacter pylori genotype and associated gastritis phenotype. Cancer. 1999; 85:2506–11. https://doi.org/10.1002/(SICI)1097-0142(19990615)85:12%3c2506::AID-CNCR3%3e3.0.CO;2-I.
157. Marcos-Pinto R, Dinis-Ribeiro M, Carneiro F, Wen X, Lopes C, Figueiredo C, Machado JC, Ferreira RM, Reis CA, Canedo P, Durães C, Ferreira J, Pedroto I, Areias J. First-degree relatives of early-onset gastric cancer patients show a high risk for gastric cancer: phenotype and genotype profile. Virchows Arch. 2013; 463:391–99. https://doi.org/10.1007/s00428-013-1458-5.
158. Kokkola A, Valle J, Haapiainen R, Sipponen P, Kivilaakso E, Puolakkainen P. Helicobacter pylori infection in young patients with gastric carcinoma. Scand J Gastroenterol. 1996; 31:643–47. https://doi.org/10.3109/00365529609009143.