
INTRODUCTION
Male sex is one of the strongest risk factors for the development of colorectal cancer development. The incidence of both colorectal adenoma and cancer development is higher in males than females [1, 2]. This suggests that the process of colorectal carcinogenesis is influenced by sex hormones. The male predominance of CRC development could be driven either by a protective effect of female hormones or a tumor promoting effect of male hormones. We have previously identified two animal models in which males developed more colorectal adenomas than females. In both models, we found that sex disparity in colonic adenomagenesis is driven by tumor promotion by male hormones, not protection by female hormones [3]. This suggests that sex disparity in CRC incidence is driven by male hormones. The observation that male hormones may be driving colorectal carcinogenesis is at apparent contradiction with results from the randomized studies of the Women’s Health Initiative. In these very large randomized controlled studies, women with an intact uterus received either placebo or a combination of equine estrogen (E2) and medroxyprogesterone acetate (MPA), whereas women who underwent hysterectomy received placebo or E2 alone. The studies showed that E2/MPA combination therapy substantially reduced the risk of CRC development [4, 5] whereas treatment with E2 alone had no effect [6]. Together these studies suggest that MPA or the MPA/E2 combination therapy protect against the development of CRC. Animal models of CRC development have failed to consistently show a protective effect of female hormones on CRC. In our own experiments we have not observed an effect of either treatment with MPA or deletion of the progesterone receptor in rodent models of adenoma development. However, experiments performed by us and others had an important caveat since they relied on ovariectomy for elucidating the effect of female hormones. Although this technique reliably removes the source of female hormones, it fails to reproduce important aspects of the biology of the menopause. Human ovaries in postmenopausal women do not produce female sex hormones, however, they are still hormonally active and produce substantial amounts of androgens [7, 8]. The abundance of androgens together with decreased levels of sex hormone binding globulin in the circulation during menopause [9] provides an increased biological availability of androgens. Therefore we set out to investigate the chemopreventive effects of MPA in an adenoma development mouse model combined with a model for menopause.
RESULTS
We used the 4-vinylcyclohexene diepoxide (VCD) to obtain a model for menopause in mice [10, 11], hereinafter referred to as postmenopausal state. In this model, daily injections with VCD leads to ovarian failure as the result of follicle depletion. Ovarian exhaustion is induced after approximately 50 days. Age matched fertile female mice that had received daily vehicle (corn oil) injections were used as controls. We subsequently induced colonic adenoma formation using six injections with the carcinogen azoxymethane (AOM) [12]. In all mice, 90-day slow release pellets were implanted subcutaneously containing either 7,5 mg medroxyprogesterone (MPA) or placebo (Figure 1A, 1B). This dose was tenfold higher than the IC50 dose that resulted in ovulation inhibition in rats (1mg/ kg/day), allosterically converted between rats and mice (4mg/kg/day) [13, 14]. Mice were sacrificed at 47 weeks of age (Figure 1A). In VCD treated mice, ovarian histology confirmed atrophy and depletion of ovarian follicles (Figure 1C), as is similarly observed in postmenopausal women.
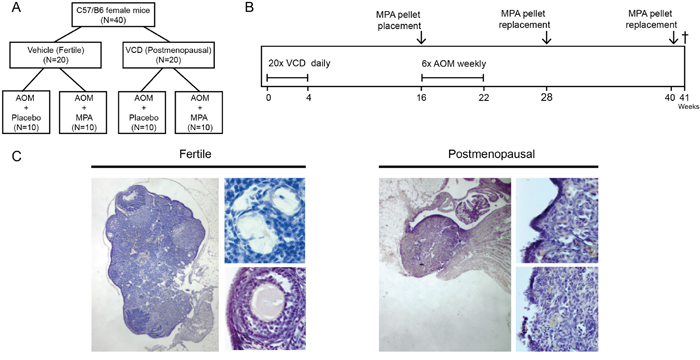
Figure 1: Experimental setup and VCD induced follicle depleted ovaria. (A) Mice were allocated to one of the four treatment groups (n=10 per group) (B) At six weeks of age mice were injected intraperitoneally in 21 subsequent days with either 4-vinylcyclohexene diepoxide (VCD, 160 mg/kg in corn oil) to induce postmenopausal state or vehicle only according as described previously (Hoyer et al) [9]. Three months after VCD treatment mice were 6 times injected with AOM (10mg/kg in 0.9%NaCl) weekly. Simultaneously with the AOM, hormonal replacement therapy was started by placing medroxy-progesteron acetate (MPA, 7,5mg in 90days) or vehicle slow release pellets subcutaneously. Pellets were replaced with new pellets after 12 and 24 weeks. 30 weeks after start AOM mice were sacrificed. (C) H&E Representative ovary of a fertile mouse (left) showing presence of follicles in all stages. Top (40X magnification) primary follicle with single layer of cuboidal cells. Bottom (20X magnification) secondary follicle with multiple layers of granulosa cells. Right panel shows a representative ovary of a VCD treated postmenopausal mouse depleted of follicles.
In control animals (fertile females that had received placebo pellets), AOM injections resulted in an incidence of colonic adenoma of 80% with an average multiplicity of 1.3, corresponding to previous literature [12, 15-16]. Long term treatment of fertile females with MPA did not alter adenoma number or size in these mice (Figure 2A, 2B). These results confirmed previous experiments reporting no effect of MPA on adenoma precursor lesions (aberrant crypt foci) or adenomas in ovariectomized rats [3, 17]. In postmenopausal mice that had received placebo pellets, the incidence of adenoma was 100% and multiplicity was significantly increased compared to untreated fertile mice (2.6 vs 1.3 P < 0.05). Surprisingly, in postmenopausal mice that had received MPA, colonic adenoma numbers had normalized to the level found in fertile mice (VCD-MPA: 0.9 vs VCD-placebo: 2.6, P < 0.001) (Figure 2A). Although postmenopausal state and treatment with MPA influenced tumor incidence, none of the conditions affected average tumor size (Figure 2B). To further examine the effect of menopause and MPA treatment on adenomagenesis, we assessed epithelial proliferation by quantification of short term BrdU incorporation in non-adenomatous colonic mucosa. We did not observe altered proliferation comparing different treatment groups (Figure 3A, 3B). Colorectal adenomas most frequently develop as the result of hyperactivation of the oncogenic Wnt pathway. The hallmark of activated Wnt signaling is nuclear accumulation of β-catenin. We did not observe gross differences in the nuclear accumulation of β-catenin in colon of different treatment groups (Figure 3C). Together, these data are compatible with the observation that hormonal status did not influence adenoma size. Our data suggest that menopausal status and MPA specifically influence tumor initiation but not growth.
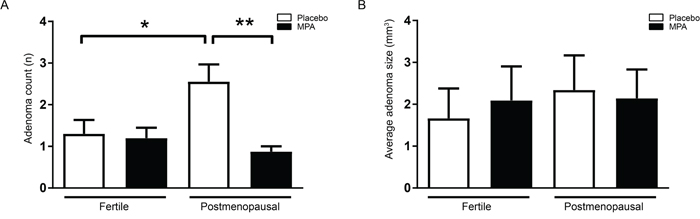
Figure 2: MPA protects from adenoma formation in VCD treated mice. (A) Total adenoma number in fertile mice colon receiving placebo pellets (n=10), fertile mice receiving MPA pellets (n=10), VCD treated postmenopausal mice receiving placebo pellets (n=9) and VCD treated postmenopausal mice receiving MPA pellets (n=8). (B) Average adenoma size in colon per mouse, measured using a ruler guide. One-way analysis of variance (ANOVA) test was used, followed by a Bonferroni post-test for multiple comparisons. Data are mean ± SEM (*= p-value (< 0,05),**= (*= p-value (< 0,01).
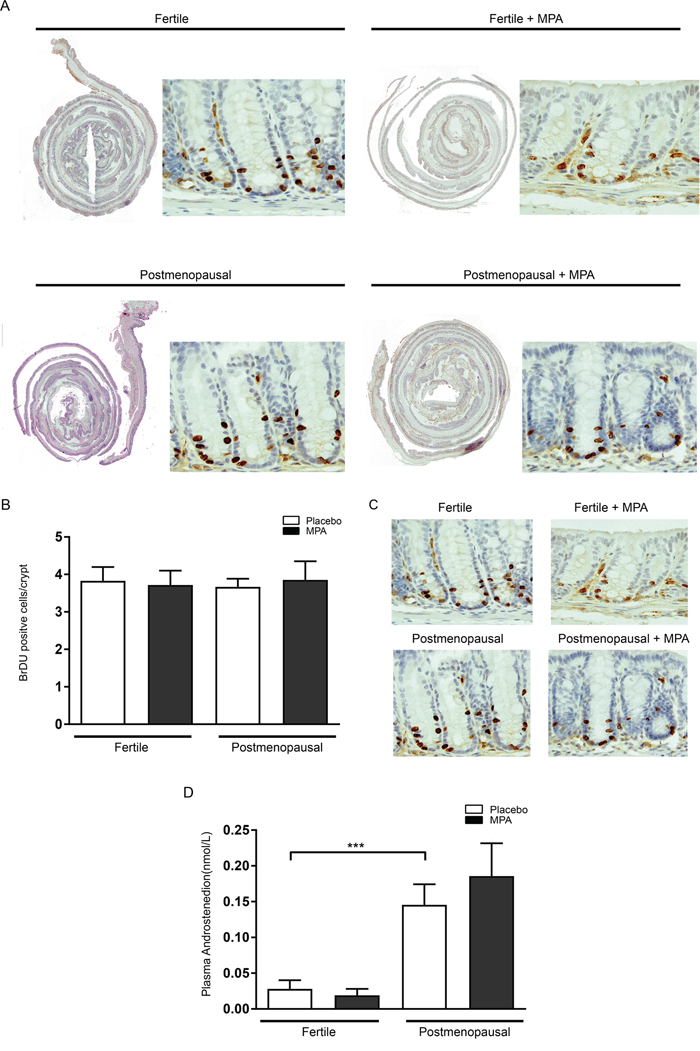
Figure 3: Epithelial proliferation is not influenced by menopause induction and MPA treatment. (A) One hour prior to sacrifice, all animals were injected with 200 μl BrdU (10 mg/ml in PBS. Colonic tissues were embedded as swiss rolls, deparaffinised in xylene and rehydrated. (B) Quantification of BrdU positive cells per crypt for all treatment groups. (C) Unaltered Wnt signalling between treatment groups. B-catenin staining on colonic tissue. (D) Menopause induction increases circulating levels of serum androstenedione. Serum levels of circulating androstenedione for all treatment groups. One-way analysis of variance (ANOVA) test was used, followed by a Bonferroni post-test for multiple comparisons. Data are mean ± SEM (*= p-value (< 0,05),**= (*= p-value (< 0,01).
Azoxymethane methylates DNA in the intestine and causes DNA double-strand breaks. To examine if the different treatments affected DNA damage, we quantified phosphorylated histone 2AX (γ-H2AX), an established marker for DNA double-strand breaks. The γ-H2AX positive stained cells were mainly located at the basal part of the colon crypts as described (Figure 4) [18]. The number of γ-H2AX positive cells was substantially increased compared to AOM untreated mice. However, the was no difference between the treatment groups, indicating that hormone status did not affect the DNA damage response.
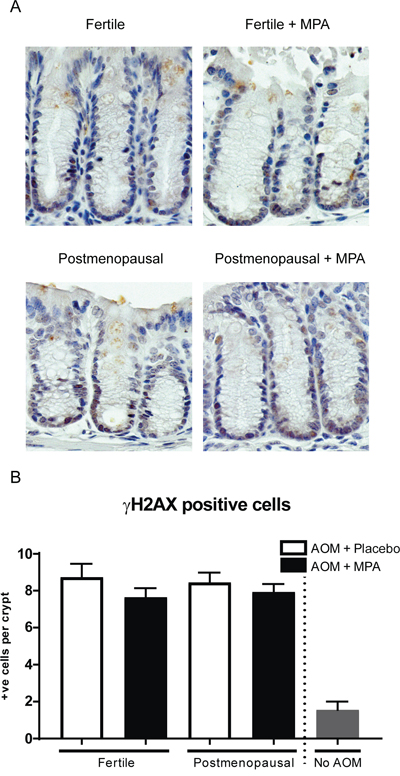
Figure 4: MPA treatment does not rescue increased DNA damage upon azomxymethane treatment shown by γ-H2AX staining. (A) γ-H2AX staining on colonic tissue 30 weeks after start AOM (B) Quantification of γ-H2AX positive cells per colonic crypt. Dotted line separates experiment mice (n=10) from mice not treated with AOM as control (n=3), per mouse approximately 20 crypts were count. One-way analysis of variance (ANOVA) test was used, followed by a Bonferroni post-test for multiple comparisons. Data are mean ± SEM (*= p-value (< 0,05),**= (*= p-value (< 0,01).
Postmenopausal ovaria exhibit increased androgen production [8], and this is reproduced in the VCD mouse model of the menopause [11]. As we have previously demonstrated that androgens are a tumor promoting factor in animal models of adenomagenesis including the AOM model in this study, we examined if menopausal status and MPA treatment influenced the level of androgen production in the ovaries in our experiment. We thus measured levels of the androgen androstenedione in our experimental groups. Indeed, VCD treated mice had significantly higher levels of serum androstenedione compared to fertile mice (0.024 vs 0.163 nmol/L, p<0.0001). MPA treatment however did not affect androgen levels (Figure 3D).
DISCUSSION
Our data show that using the VCD model of the menopause in age matched mice, provides an opportunity to understand the influence of postmenopausal status on the risk of CRC. The model may help to unravel the factors that predispose women to CRC after the menopause and the way this risk is influenced by therapeutic intervention such as hormone replacement therapy. Experiments that have examined the role of female hormones in CRC development have relied on using ovariectomies. Ovariectomies are a useful way to study the effect of acute depletion of all sex hormones in the premenopausal state. However, the intervention does not adequately recapitulate the biology of the post-menopausal state where ovaries remain an important source of androgens. This may explain why previous research has failed to demonstrate protective effects of hormone replacement therapy on adenomagenesis [3, 17]. In the model used for this study, postmenopausal mice developed increased colonic adenomas compared to their fertile counterparts. Postmenopausal ovaries increase their androgen production. As we have previously shown that androgens promote adenomagenesis and explain the biological sex difference in adenoma incidence in two rodent models in which such disparity was observed [3], the drop in female sex hormone production and increased androgen secretion may explain increased tumorigenesis.
We find that MPA reduces tumorigenesis in postmenopausal mice, but not in fertile mice. This may be related to an unique property of MPA that is unrelated to its progestagenic activity. MPA has been described as an AR-agonist harboring either androgenic or anti-androgenic effects, dependent on dose and context. MPA binds with a similar affinity to AR as the native ligand 5alfadihydrotestosteroneand can disrupt AR signaling [19–21]. However, while a role for the AR in colorectal cancer has been proposed [22], we have previously shown that the AR is not expressed in colonic epithelial cells [3]. We have pursued the hypothesis that the menopausal state and MPA treatment affects adenoma development indirectly via effects on the liver. However, in an experiment in which we examined the effect of these different interventions on the liver, we found no major effects of either VCD treatment or treatment with MPA on liver gene expression using gene expression arrays (data not shown). Alternatively, the effects may be mediated more centrally, for example through effects on the hypothalamus and/ or pituitary. This is a hypothesis that may be pursued in future experiments. Taken together, our work shows that postmenopausal status may specifically increase the risk of CRC and that MPA reduces this risk specifically in a model of the post-menopausal state. Our data suggest that MPA monotherapy may be a chemopreventive strategy to reduce the risk of CRC in postmenopausal women.
MATERIALS AND METHODS
Experiments were performed on C57B6/JOlaHsd wildtype (Harlan laboratories) at six weeks of age. The mice were housed at the animal facilities of the Amsterdam Medical Center, and experiments were performed with consent from the animal ethics committee of the University of Amsterdam (permit number ALC102969). At six weeks of age were injected intraperitoneally in 21 subsequent days with either 4-vinylcyclohexene diepoxide (VCD, 160 mg/kg in corn oil) to induce postmenopausal state or vehicle only according as described previously (Hoyer et al) [9] (n=10 per treatment group). Three months after VCD treatment mice were 6 times injected with Azoxymethane (AOM, 10mg/kg in 0.9%NaCl, Sigma-Aldrich) weekly. Simultaneously with the AOM, hormonal replacement therapy was started by placing medroxy-progesteron acetate (MPA, 7,5mg in 90days, Innovative Research of America) or vehicle slow release pellets subcutaneously. Pellets were replaced with new pellets after 12 and 24 weeks. 30 weeks after start AOM mice were sacrificed.
For immunohistology tissue was fixed in 4% ice-cold formalin and embedded in paraffin. Sections of 4 μm were deparaffinised in xylene and rehydrated. H&E staining was performed. For epithelial proliferation, BrdU assay was performed. One hour prior to sacrifice, all animals were injected with 200 μl BrdU (10 mg/ml in PBS; Sigma–Aldrich). Tissue was fixed as described above. Colonic tissues were embedded as swiss rolls, deparaffinised in xylene and rehydrated. Endogenous peroxidase was blocked using 0,3% H2o2 in methanol. Sections ere cooked in 0.01M citrate buffer pH 6.0 for 20minutes and incubated with mouse monoclonal anti-BrdU in PBS with 1% BSA and 0.1% Triton-X-100. and then stained with anti-BrdU (clone BMC9318, Roche). Antibody binding was visualized with Powervision horseradish peroxidase-labelled secondary antibodies, and diaminobenzidine for substrate development. All sections were counterstained with Mayer’s haematoxylin.
B-catenin and γ-H2AX, staining on colonic tissues was performed with mouse purifiedanti-Beta-catenin (Biosciences Pharmingen) and γ-H2AX (Abcam, ab11174) used as antibody.
Androstenedion measurement. Blood was obtained with cardiac puncture during sacrifice. Blood was spinned down and serum was collected and analysed for circulating androstenedione for all treatment groups.
Adenoma Count Tumors were macroscopically assessed after ice-cold formalin fixation, by two blinded researchers. Average adenoma size in colon per mouse, was measured using a ruler guide.
Statistics analysis
Overall, One-way analysis of variance (ANOVA) test was used, followed by a Bonferroni post-test for multiple comparisons. Data are mean ± SEM (*= p-value (< 0,05),**= (*= p-value (< 0,01).
Abbreviations
AOM: Azoxymethane; AR: Androgen receptor; CRC: Colorectal cancer; BrdU: Bromodeoxyuridine; E2: Equine Estrogen; MPA: Medroxyprogesterone acetate; PMP: Post-menopausal; VCD: 4-vinylcyclohexene diepoxide; WHI: Women’s Health Initiative.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
No funding.
REFERENCES
1. Ferlitsch M, Reinhart K, Pramhas S, Wiener C, Gal O, Bannert C, Hassler M, Kozbial K, Dunkler D, Trauner M, Weiss W. Sex-specific prevalence of adenomas, advanced adenomas, and colorectal cancer in individuals undergoing screening colonoscopy. JAMA. 2011; 306:1352–58.
2. Regula J, Rupinski M, Kraszewska E, Polkowski M, Pachlewski J, Orlowska J, Nowacki MP, Butruk E. Colonoscopy in colorectal-cancer screening for detection of advanced neoplasia. N Engl J Med. 2006; 355:1863-72.
3. Amos-Landgraf JM, Heijmans J, Wielenga MC, Dunkin E, Krentz KJ, Clipson L, Ederveen AG, Groothuis PG, Mosselman S, Muncan V, Hommes DW, Shedlovsky A, Dove WF, van den Brink GR. Sex disparity in colonic adenomagenesis involves promotion by male hormones, not protection by female hormones. Proc Natl Acad Sci U S A. 2014; 111:16514–19.
4. Chlebowski RT, Wactawski-Wende J, Ritenbaugh C, Hubbell A, Ascensao J, Rodabough RJ, Rosenberg CA, Taylor VM, Harris R, Chen C, Adams-Campbell LL, White E; Women’s Health Initiative Investigators. Estrogen plus Progestin and Colorectal Cancer in Postmenopausal Women. N Engl J Med. 2004; 350:991-1004.
5. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J, and Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA. 2002; 288:321–33.
6. Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SA, Black H, Bonds D, Brunner R, Brzyski R, Caan B, Chlebowski R, Curb D, Gass M, et al, and Women’s Health Initiative Steering Committee. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA. 2004; 291:1701–12.
7. Davison SL, Bell R, Donath S, Montalto JG, Davis SR. Androgen levels in adult females: changes with age, menopause, and oophorectomy. J Clin Endocrinol Metab. 2005; 90:3847–53.
8. Fogle RH, Stanczyk FZ, Zhang X, Paulson RJ. Ovarian androgen production in postmenopausal women. J Clin Endocrinol Metab. 2007; 92:3040–43.
9. Burger HG, Dudley EC, Cui J, Dennerstein L, Hopper JL. A prospective longitudinal study of serum testosterone, dehydroepiandrosterone sulfate, and sex hormone-binding globulin levels through the menopause transition. J Clin Endocrinol Metab. 2000; 85:2832–38.
10. Kappeler CJ, Hoyer PB. 4-vinylcyclohexene diepoxide: a model chemical for ovotoxicity. Syst Biol Reprod Med. 2012; 58:57-62.
11. Mayer LP, Devine PJ, Dyer CA, Hoyer PB. The follicle-deplete mouse ovary produces androgen. Biol Reprod. 2004; 71:130–38.
12. Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat Protoc. 2007; 2:1998–2004.
13. Zhang Z, Olland AM, Zhu Y, Cohen J, Berrodin T, Chippari S, Appavu C, Li S, Wilhem J, Chopra R, Fensome A, Zhang P, Wrobel J, et al. Molecular and pharmacological properties of a potent and selective novel nonsteroidal progesterone receptor agonist tanaproget. J Biol Chem. 2005; 280:28468–75.
14. Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008; 22:659-61.
15. Bissahoyo A, Pearsall RS, Hanlon K, Amann V, Hicks D, Godfrey VL, Threadgill DW. Azoxymethane is a genetic background-dependent colorectal tumor initiator and promoter in mice: effects of dose, route, and diet. Toxicol Sci. 2005; 88:340–45.
16. Suzuki R, Kohno H, Sugie S, Nakagama H, Tanaka T. Strain differences in the susceptibility to azoxymethane and dextran sodium sulfate-induced colon carcinogenesis in mice. Carcinogenesis. 2006; 27:162–69.
17. Heijmans J, Muncan V, Jacobs RJ, de Jonge-Muller ES, Graven L, Biemond I, Ederveen AG, Groothuis PG, Mosselman S, Hardwick JC, Hommes DW, van den Brink GR. Intestinal tumorigenesis is not affected by progesterone signaling in rodent models. PLoS One. 2011; 6:e22620.
18. Fahrer J, Frisch J, Nagel G, Kraus A, Dörsam B, Thomas AD, Reißig S, Waisman A, Kaina B. DNA repair by MGMT, but not AAG, causes a threshold in alkylation-induced colorectal carcinogenesis. Carcinogenesis. 2015; 36:1235–44.
19. Bianco-Miotto T, Trotta AP, Need EF, Lee AM, Ochnik AM, Giorgio L, Leach DA, Swinstead EE, O’Loughlin MA, Newman MR, Birrell SN, Butler LM, Harris JM, Buchanan G. Molecular and structural basis of androgen receptor responses to dihydrotestosterone, medroxyprogesterone acetate and Δ(4)-tibolone. Mol Cell Endocrinol. 2014; 382:899–908.
20. Sitruk-Ware R. Pharmacological profile of progestins. Maturitas. 2004; 47:277–83.
21. Ochnik AM, Moore NL, Jankovic-Karasoulos T, Bianco-Miotto T, Ryan NK, Thomas MR, Birrell SN, Butler LM, Tilley WD, Hickey TE. Antiandrogenic actions of medroxyprogesterone acetate on epithelial cells within normal human breast tissues cultured ex vivo. Menopause. 2014; 21:79–88.
22. D’Errico I, Moschetta A. Nuclear receptors, intestinal architecture and colon cancer: an intriguing link. Cell Mol Life Sci. 2008; 65:1523–43.