
INTRODUCTION
Rituximab (RTX), a monoclonal antibody (mAb) directed against the B cell molecule CD20, was the first mAb to receive approval to be used in non-Hodgkin’s lymphomas (NHL), and later in chronic lymphocytic leukemia (CLL) [1]. Efficacy of RTX varies from patient to patient and a higher rate of relapse has been observed in CLL patients compared to NHL [2]. Thus, the combination of RTX with the purine analogue, fludarabine, and the DNA alkylating agent, cyclophosphamide, represents the gold standard as first line treatment, although alternative agents such as bendamustine or chlorambucil can be proposed in combination with RTX [3, 4]. The novel combinations with RTX have become more and more effective, but relapses still remain common events, which may be attributed, in part, to CLL cell resistance to RTX.
RTX mediates different mechanisms of B cell death such as: antibody-dependent cellular cytotoxicity (ADCC); complement-dependent cytotoxicity (CDC); Ab dependent cellular phagocytosis (ADCP) through the clearance of C3b(i) opsonized cells by macrophages; and direct cell death [5-11]. ADCC activity is linked to the efficiency of Natural Killer (NK) cells to kill tumor cells, and it is notably dependent on the Fc gamma receptor (FcγR)3 polymorphism V158F [12-14]. Although ADCC is suspected to be the main mechanism of action with regards to RTX therapeutic efficacy and clinical outcomes [15-17], several authors suggested that activation of the complement pathway is also involved in the clinical response to RTX [18-21]. To further support the importance of the complement pathway, it was established that RTX therapeutic activity was strongly enhanced in CLL patients resistant to RTX after concurrent administration of fresh frozen plasma [22]. Last but not least, complement deficiencies are reported in CLL patients and suspected of limiting RTX efficacy in vivo [23].
Mechanisms controlling the RTX capacity to induce complement activation are multiple, incompletely understood and the results reported are conflicting. On one hand, the level of the target molecule CD20 appeared to be important, as well as the presence of complement regulatory molecules, which seem critical, based on the observations that blocking CD55 and/or CD59 functions are effective in increasing RTX-induced CDC [24, 25]. On the other hand, complement C1q molecule recruitment into lipid rafts mediated by RTX suggests that expression of ganglioside M1 (GM1), a marker of lipid rafts, can determine the susceptibility to RTX treatment as reported in NHL [26]. Another mechanism of resistance to RTX could be the sialylation of the cell surface. Sialic acid acts frequently via a α2-3 or α2-6 glycosidic linkage to galactose and N-acetylgalactosamine. Enzymes that support these sialylations belong to the sialyltransferases (ST) family, and they are unlikely to be expressed in normal B cells [27]. The presence of sialic acid is implicated in the recruitment of complement inhibitors [28], and, in turn, controls the mAb-capacity to induce complement activation [29].
In order to support our hypothesis that resistance to RTX-induced complement activation may have clinical implications, we decided to study in vitro B cell resistance mechanisms to CDC in 69 CLL patients including 34 who would benefit from RTX-chemotherapy. The predictive value of the in vitro CDC-resistance response was independent from the clinical, biological and cytogenetic characteristics of the patients. In contrast, when the CDC results obtained in vitro were compared to the clinical response, an association between CLL resistance to CDC and RTX-chemotherapy response was observed. We further highlighted the important role of CLL α2-6 ST activity and membrane sialylation in determining resistance and susceptibility to CDC.
RESULTS
CDC of CLL cells induced by RTX is delayed and inhibited by Eculizumab
The RTX capacity to induce in vitro CDC has been addressed by several authors but with significant differences with regards to the source and number of B cells, the time and concentration of RTX, the amount of sera, and the method for CDC detection (Table 1). Accordingly, the assay to test complement mediated killing of CLL cells sensitized by the anti-CD20 mAb RTX (10μg/ml) at 37°C was optimized in a preliminary step and, as shown in Figure 1A, CDC induced by RTX was effective after 1h when using the human B cell line Ramos (n=3) but such an effect was delayed to 24h when using CLL cells (n=8) and B cells from healthy controls (n=3). We further established that the RTX capacity to lyse CLL cells disappeared when using Eculizumab as a terminal complement inhibitor, when complement present in HSAB (human serum AB) was heat-inactivated or when HSAB was omitted (Figure 1A/B). Next, in order to differentiate CDC-normal from CDC-resistant CLL cells, the 24h time point was selected and a cut-off value of 6% was established by considering the mean of decrease in CLL survival (21.2±7.6%) minus 2 standard deviations of normal B cells from 21 healthy controls (Figure 1C). Therefore, 45 out of 69 (65.2%) CLL patients were considered resistant to complement in vitro and this is independent from the RTX capacity to induce direct apoptosis and ADCC (Table 2).
Table 1: Overview of the different assays reporting complement dependent cytotoxicity assay with rituximab (RTX) in patients with chronic lymphocytic leukemia (CLL), non Hodgkin’s lymphoma (NHL), and human B cell lines
Cell type (nb) |
Number of cells/ mL |
RTX in μg/mL |
Sera (%) |
Total incubation time |
Detection method |
Lysis (%) |
Comment |
References |
|---|---|---|---|---|---|---|---|---|
PBMC from CLL (n=33) |
5×104 |
10 μg/ml |
25% |
16h |
Alamar |
<10% in 58% |
PBMC not purified B cells (CD20: 65-98%) |
[65] |
NHL and myeloma cell lines (n=3) |
106 |
10 μg/ml |
25% |
12h |
Trypan blue and PI |
100% |
[75] |
|
CLL (n=5) |
2.5×106 |
20 μg/ml |
50% |
2.5h |
PI |
Near 0% |
[23] |
|
CLL (n=4) and Daudi cell line (n=3) |
5×106 |
10 μg/ml |
50% |
0.25h |
PI |
CLL: 10% |
[76] |
|
CLL (n=11) |
5×105 |
100 μg/ml |
6.25% |
2h |
LDH |
Near 0% |
[48] |
|
EHEB (CLL, n=3) and Raji (n=3) B cell lines |
5×104 |
10 μg/ml |
16.7% |
6h |
WST-1 |
EHEB: 0% |
[77] |
|
CLL (n=16) Daudi (n=5) SU-DHL4 (n=5) |
Not reported |
10 μg/ml |
20% |
1h to 6h |
C51 (CLL) & IP (cell lines) |
CLL: 15% |
[78] |
|
PBMC from CLL (n=6) and NHL (n=6) |
106 |
10 μg/ml |
50% |
30 min |
IP |
PBMC-CLL: 10% |
PBMC not purified B cells |
[79] |
PBMC-CLL (n=11) Raji (n=4) cell line |
104 |
10 μg/ml |
10% |
90 min |
C51 |
PBMC-CLL: 10 ± 8% |
PBMC not purified B cells |
[80] |
CLL (n=7) |
Not reported |
10 μg/ml |
50% |
30 min |
TOPRO-3 |
Near 0%, except one (78%) |
1/7 responder |
[81] |
CLL (n=10) and Daudi (n=2) |
107-108 |
100μg/ml |
50% |
15 min to 1 hour |
TOPRO-3 and IP |
CLL: 10% |
CLL cells isolated using Ficoll-Paque |
[82] |
Abbreviations: PI: propidium iodide; nb: number of experiments; LDH: lactate dehydrogenase release assay; C51: Chromium-51 release assay; CLL: purified peripheral blood B cells from CLL patients; PBMC: polymorphonuclear B cells from CLL patients.
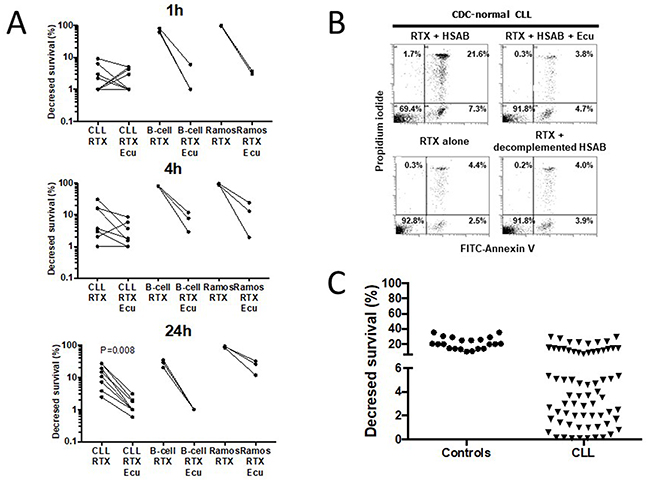
Figure 1: Rituximab (RTX)-induces in vitro complement-dependent cytotoxicity (CDC) of B lymphocytes in normal controls and in a subset of CLL. (A) In order to improve the determination of CDC induction by RTX (10μg/ml) in CLL cells, CDC was initially established after 1h (top), 4h (middle), and 24h (bottom) incubation at 37°C in CLL cells (n=9), healthy control B cells (n=4), and the human B cell line Ramos (3 experiments) using normal human serum AB (HSAB, 20%) as a source of complement, and Eculizumab (Ecu, 10μg/ml) as a terminal complement inhibitor. (B) RTX capacity to induce CDC in a representative RTX-normal CLL patient was abrogated when adding Ecu, when complement present in HSAB was heat inactivated or when HSAB was omitted. (C) B cells from 21 normal controls and 69 CLL patients were incubated 24h with RTX (10μg/ml) and HSAB at 20%. After staining with FITC-annexin V plus propidium iodide, cells were analyzed by FACS and the CDC decreased survival calculated (see material and methods).
Table 2: Complement dependent cytoxicity (CDC), direct apoptosis, antibody dependent cell cytotoxicity (ADCC) and associated factors characteristic for CDC-normal and CDC-resistant chronic lymphocytic leukemia (CLL) patients
Control B cells |
CDC-normal CLL |
CDC-resistant CLL |
Normal vs Resistant |
|
|---|---|---|---|---|
Decreased survival (%, CDC) |
21.2±1.7 (21)* |
16.1±1.4 (24) |
2.0±0.2 (45) |
p<10-3 |
Decreased survival (%, apoptosis) |
4.4±2. 8 (8) |
5.9±1.6 (9) |
4.3±1.3 (8) |
NS |
Decreased survival (%, ADCC) |
13.6±1.6 (6) |
16.1±5.6 (6) |
NS |
|
CD20 (ABC units) |
317,000±56,800 (6) |
95,700±12,000 (9) |
75,000±5,800 (39) |
NS |
GM1 (MFI) |
2.5±0.25 (6) |
2.6±0.2 (20) |
2.3±0.3 (26) |
NS |
Sphingomyelin (MFI) |
33.4±3.2 (6) |
27.5±7.8 (8) |
31.1±5.0 (16) |
NS |
CD55 (MFI) |
11.6±0.9 (6) |
8.7±1.6 (6) |
10.7±2.1 (6) |
NS |
CD59 (MFI) |
5.5±0.7 (6) |
5.6±0.8 (6) |
6.8±1.0 (6) |
NS |
Factor H binding (%) |
20.4±4.8% (6) |
13.3±3.0% (6) |
18.3±4.2% (10) |
NS |
Sambucus nigra (MFI) |
52±3 (6) |
97±32 (6) |
243±35 (8) |
p=0.01 |
Maakia amurensis (MFI) |
44.8±8.5 (6) |
38.6±17.8 (7) |
43.4±5.1 (28) |
NS |
α2-6 ST activity (OD, 9h) |
0.721±0.011 (4) |
0.730±0.009 (5) |
0.981±0.017 (5) |
p<10-4 |
Abbreviations: ND: not determined; ABC: antibody-binding capacities; MFI: mean fluorescence intensity; OD: optical density at 492nm, NS: not significant.*The number of samples tested is indicated in brackets.
Time to relapse (TTR) is reduced in the in vitro CDC-resistant CLL patient subgroup
When the whole population was taken into consideration (Table 3), no difference was observed between the CDC-resistant and -normal groups with regards to demographic, clinical and biological characteristics of the patients (age, sex, Binet stage, progression free survival [PFS], treatment free survival [TFS], lymphocytosis, lymphocyte doubling time [LDT], cytogenetic characteristics, and CD38 expression).
Table 3: Characteristics of the patients according to rituximab capacity to induce complement-dependent cytotoxicity (CDC)
CDC-normal (n=24 including 11 treated) |
CDC-resistant (n=45 including 23 treated) |
Statistics |
|
|---|---|---|---|
Demographic factors |
|||
Age, mean±SEM |
73±2 |
69±2 |
NS |
Sex Male:Female |
10:14 |
27:18 |
NS |
Clinical data |
|||
Binet A/B/C |
7/13/4 |
13/23/9 |
NS |
PFS, mean (months)* |
53 |
72 |
NS |
TFS, mean (months)* |
96 |
108 |
NS |
Tumoral cells |
|||
Lymphocytosis |
80±12 |
69±8 |
NS |
LDT, mean (months)* |
10 |
23 |
NS |
CD38 >30%, n (%) |
6/22 (27.3%) |
10/43 (23.3%) |
NS |
Cytogenetics, n (%) |
NS |
||
Low risk |
9/21 (42.9%) |
18/39 (46.2%) |
|
Intermediate risk |
5/21 (23.8%) |
6/39 (15.4%) |
|
High risk |
7/21 (33.3%) |
15/39 (38.5%) |
|
Therapeutic response |
|||
Complete response, n (%) |
11/11 (100%) |
15/23 (65.2%) |
p=0.03 |
Partial response, n (%) |
0/11 |
8/23 (34.8%) |
|
TTR, mean (months)* |
>80 |
45 |
p=0.03 |
OS, mean (months)* |
>80 |
87 |
NS |
Cytogenetics, n (%) |
|||
Low risk |
5/11 (45%) |
5/20 (25%) |
NS |
Intermediate risk |
2/11 (18%) |
4/20 (20%) |
|
High risk |
4/11 (37%) |
11/20 (55%) |
|
Complement fractions at 1st infusion |
|||
C3c low level |
2/10 |
4/19 |
NS |
C4 low level |
2/10 |
0/19 |
NS |
IGHV UM/M |
0/5 |
8/6 |
p=0.04 |
FcγR3A V158F, n (%) |
|||
FF |
5/10 (50%) |
11/20 (55%) |
NS |
VF |
4/10 (40%) |
8/20 (40%) |
|
VV |
1/10 (10%) |
1/20 (5%) |
|
Treatment |
|||
RFC:RB |
8:3 |
20:3 |
NS |
Abbreviations: PFS: Progression free survival; TFS: Treatment free survival; LDT: Lymphocyte doubling time; n: number of patients; NS: not significant; RFC: rituximab fludarabine and cyclophosphamide; RB: rituximab bendamustine; TTR: time to relapse; OS: overall survival. *Kaplan–Meyer survival analysis.
Further on, in patients tested before the initiation of RTX-chemotherapy for CDC (n=34), response to therapy was evaluated revealing in the CDC-resistant group a reduction when considering complete response (CR) (65.2% in the CDC-resistant patient group versus 100% in the CDC-normal patients group, p=0.03), and TTR (median TTR: 45 months in the CDC-resistant patient group versus >80 months in the CDC-normal patient group; Hazard Ratio 7.2, 95% CI 2.6-19.8, p=0.03) (Figure 2 and Table 3). The Cox regression model was used to determine the optimal cutoff for the CDC assay that was 6.7% and close to 6% as previously defined. In contrast, overall survival (OS) difference between the two groups was not significant due to the development of a secondary myelodysplastic syndrome leading to death in two patients from the CDC-normal patient group. Interestingly, our results further support an over-representation of CLL with unmutated IGHV in the in vitro CDC-resistant group (p=0.04), which is in agreement with Chai-Adisaksopha C and Brown JR report [30].
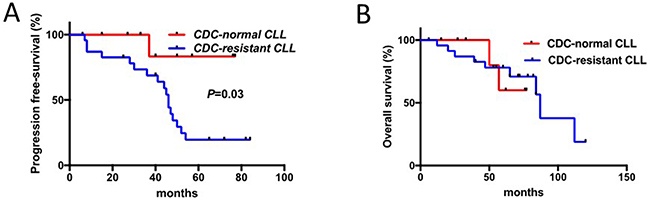
Figure 2: Response to therapy in CLL patients is associated with sensitivity to RTX-induced complement-dependent cytotoxicity (CDC). (A) TTR (time to relapse) of patients with B cells sensitive to RTX-induced CDC was compared to those resistant. (B) Overall survival (OS) of these two groups of patients was established and compared.
Rituximab incapacity to induce CDC is not related to complement inhibitor levels, CD20 expression, FcγR3A V158F polymorphism or lipid membrane disorganization
Next we compared CD20 (target antigen), GM1 and sphingomyelin (lipid raft markers) in vitro expression between the two patient groups as the ability of CD20 to translocate into lipid rafts is suspected to be important for complement activation by anti-CD20 mAb [31], and that lipid raft integrity may be compromised in CLL cells [25, 26, 32] (Figure 3A and Table 2). No differences were observed when comparing in both groups (i) the numbers of CD20 molecules between groups; (ii) the mean fluorescence intensity (MFI) of the FITC-conjugated cholera toxin subunit B recognizing GM1; and (iii) the MFI of lysenin-bound-sphingomyelin. As we have previously observed that sphingomyelin overexpression in CLL cells induced by rifampicin affects the type II anti-CD20 mAb (B1) capacity to kill CLL cells [32], the experiment was repeated with RTX instead of B1 (tositumomab), revealing that sphingomyelin overexpression had no effect on the RTX capacity to induce CDC (data not shown).
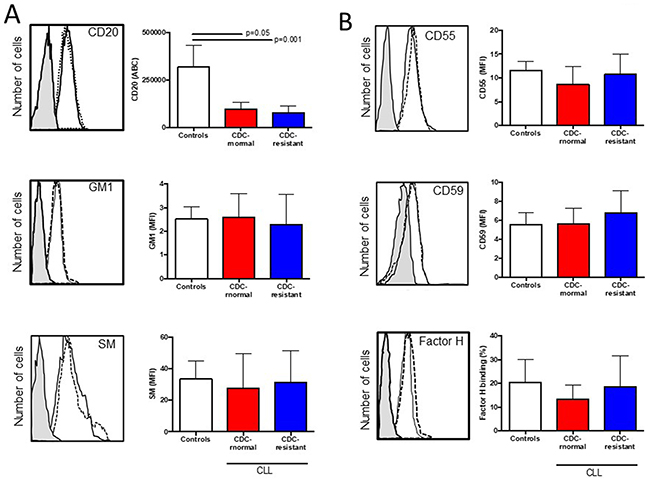
Figure 3: Expression of CD20, ganglioside M1, sphingomyelin (SM), and complement inhibitors (CD55, CD59 and factor H) in healthy control B cells and CLL cells according to their complement-dependent cytoxicity (CDC) status to rituximab (RTX). 5×105 B lymphocytes were incubated with (A) anti-CD20 antibody, cholera toxin B identifying GM1 and lysenin identifying sphingomyelin (SM). (B) Same experiments were conducted with saturated concentrations of anti-CD55, anti-CD59 and anti-factor H antibodies. For each staining, a representative example is shown on the left side. The shaded histogram corresponds to isotype control, the solid line to a representative B cell staining from the CDC-normal patient group, and the dotted line to a representative B cell staining from the CDC-resistant patient group. The number of samples tested is indicated in Table 2 and differences are indicated when p<0.05.
Regarding complement regulators, we have further explored CLL cell surface expression of the complement inhibitors CD55 and CD59 [33], and CLL capacity to recruit the fluid-phase factor H, another RTX-mediated CDC inhibitor [34] (Figure 3B and Table 2). The MFI of CD55, CD59 and the percentage for factor H binding were assessed revealing similar values between the in vitro CDC-normal patient group and the CDC-resistant patient group. In order to complete such analysis, the complement deficiency at RTX-chemotherapy initiation was based on observing C3c and C4, as well as the cytogenetic status and FcγR3A V158F polymorphism known to be critical for proper CDC and ADCC, respectively (Table 3). No difference was reported and all these factors were not considered further.
CLL lymphocytes CDC-resistant to rituximab express higher levels of terminal α2-6-linked sialic acids and their removal by neuraminidase increases activity of rituximab-induced CDC
Because sialic acid residues from the cell surface have been shown to influence Ab mediated CDC in human carcinoma cells [29], the presence of α2-3 and α2-6-linked sialic acid was tested on CLL cells and controls using specific lectins. For this purpose, two lectins from Sambucus nigra (SNA) and from Maakia amurensis (MAA) were selected, which bind to sialic acid attached to terminal galactose in α2-6 and in α2-3, respectively (Figure 4A). The CLL cell membranes of the CDC-resistant patient group expressed more α2-6 sialylated proteins (MFI of 243±35) than those of the CDC-normal patient group (MFI of 97±32, p=0.01), and controls (MFI of 52±3). No differential expression of the lectin MAA was seen in the three different groups, suggesting similar levels of α2-3 sialylated proteins.
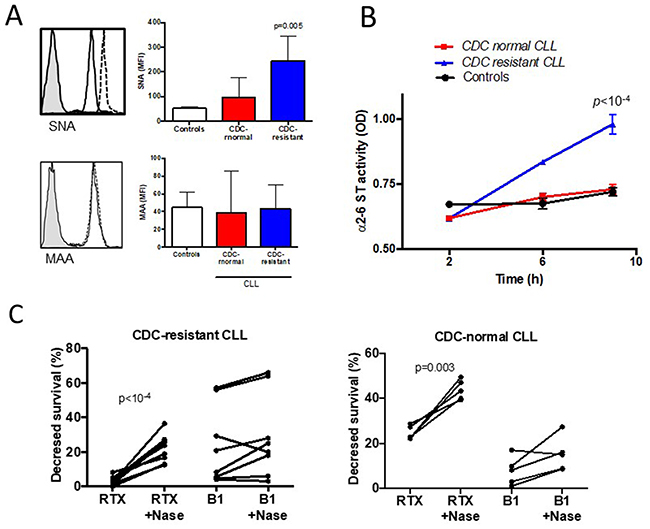
Figure 4: Hypersialylation characterizes CLL cells resistant to rituximab (RTX) induced complement-dependent cytotoxicity (CDC). (A) FACS analysis of α2-6 linked sialic acid stained with the lectin Sambucus nigra (SNA, upper panel) and α2-3 linked sialic acid stained with Maakia amureusis (MAA, lower panel). For each staining, a representative example is shown on the left side. The shaded histogram corresponds to isotype control, the solid line to CLL cells sensitive to RTX CDC, and the dotted line to CDC-resistant cells. (B) B cell lysates from five CDC-resistant patients, five CDC-normal patients and to the four controls were incubated on ELISA plates coated with an acceptor of sialic acid for 2, 6 or 9 hours. After washes, the biotinylated lectin SNA specific for sialic acid linked to α2-6 sialic acid was added. The results were visualized after incubation of streptavidin-conjugated horseradish peroxydase and 1,2 ortho-phenylenediamine. (C) B lymphocytes from ten patients within the CDC-resistant group and five patients within the CDC-normal group were treated with or without 0.05U of neuraminidase from Clostridium perfringens, washed and incubated with or without 10μg/ml of RTX or 10μg/ml of B1 (tositumomab) for 24 hours at 37°C. CDC was quantified by FACS after staining with FITC-annexinV and propidium iodide. Differences are indicated when p<0.05.
To test the hypothesis that α2-6 hypersialylation in the CDC-resistant patient group resulted from increased activity of α2-6 sialyl transferase (ST), we used a previously developed custom-made ELISA in order to measure the α2-6 ST activity in CLL cells [35]. As shown in Figure 4B, the α2-6 ST activity was found to be significantly upregulated in the CDC-resistant CLL cells treated (0.981±0.017) compared to the CDC-normal CLL cells (0.730±0.009, p<10-4) and to the B cell controls (0.721±0.011, p<10-4). In the MAA binding assay, the α2-3 ST activity was similar in the three groups (data not shown).
Finally, and to strengthen the effect of sialic acid on RTX-mediated CDC, we further selected 10 CLL patients from the CDC-resistant group and 5 patients from the CDC-normal group and cells were treated with neuraminidase to remove sialic acids from cell surface. In the CDC-resistant group, following neuraminidase treatment, RTX capacity to induce CDC was restored (p<10-4) (Figure 4C). Interestingly, the neuraminidase capacity to increase CDC activity was not restricted to the CDC-resistant subgroup as the same observation was made in the CDC-normal patient group (p=0.003). As a control and to test RTX specificity, when using B1 (tositumomab) instead of RTX, B1 capacity to directly kill CLL cells was similar following or not neuraminidase treatment (Figure 4C). Furthermore, since one may argue that desialylation can lead to membrane modifications and possibly to better accessibility of RTX to CD20 [36], we looked at the binding of RTX on the cell surface after desialylation. MFI of FITC-conjugated RTX binding to CD20 was similar with or without treatment with neuraminidase (data not shown). Altogether, these results suggest that treating CLL cells with neuraminidase can restore in vitro CDC induces by RTX, and differences between the two groups may be related to quantitative and/or qualitative differences in SA.
DISCUSSION
Both clinical and experimental studies have offered evidence that resistance to RTX, rather than resistance to chemotherapy, controls relapse and clinical outcome in CLL. Among the different possibilities, complement activation could be the predominant mechanism for killing tumoral CLL cells in vivo and this notion is supported by several considerations. First, at initial infusion, side effects may be attributed in part to complement activation. Second, rapid exhaustion of complement and C3b(i) deposition on CLL cells was observed after RTX treatment in CLL patients and even more with Ofatumumab, another type I anti-CD20 mAb [37, 38]. Third, the addition of fresh frozen plasma enhances the effect of RTX-chemotherapy [39]. Fourth, intravital complement activation following anti-CD20 mAb administration is not limited to CDC but also to opsonisation priming mAb target cells for phagocytosis in ADCP [9, 40].
As different methods were used to determine the CDC mediated RTX, we first validated the in vitro model to measure the capacity of RTX to induce CDC. Tumor cell lines sensitive to RTX, inhibitor of complement activity and different times of RTX incubation were used. Based on the in vitro capacity of RTX to induce the complement pathway, two groups of patients were established. The first group, presenting CR (100%) and a delayed median TTR over 80 months, was sensitive to CDC in vitro, while the second group, with a reduced CR rate (65.2%) and a median TTR at 45 months, was resistant to CDC in vitro. Of note, TTR in the CDC-resistant group was similar to the TTR reported in 409 treatment-naïve CLL patients receiving fludarabine and cyclophosphamide [41]. Moreover, the higher rate of CR at the end of treatment in the CDC-normal patient group (100% versus 65.2%) further supports within the CDC-resistant patient group an incomplete capacity of RTX to activate the complement pathway. The critical contribution of complement activation in RTX-chemotherapy response is further supported by the absence of an association between the RTX response and the FcγR3A V158F polymorphism status, as previously described in CLL [42]. Next, other authors have attributed the differences in response to RTX treatment in CLL to IGHV mutational status [30] and to cytogenetic modifications and in particular to deletions at 11q/ATM or 17p/TP53 loci [43, 44]. In our study and with only one patient with 17p/TP53 deletion and three with 11q/ATM deletions treated with RTX-chemotherapy, more patients have to be included to test the unfavorable impact of these deletions on CDC mechanisms with anti-CD20 mAbs. The last parameter concerns the contribution of constitutive complement deficit in RTX-chemotherapy response to enhance TTR which is again not supported in our study by the determination of C3 and C4 components in sera of all CLL patients just before treatment initiation.
After determining that a longer culture time (24h) was required to allow CDC determination in CLL cells and having confirmed the absence of CDC response when using the anti-C5 mAb Eculizumab acting downstream C3 activation or when the HSAB was omitted or decomplemented by heat, important complement resistance mechanisms were suspected in CLL cells. After observing that CDC was restored in CLL cells after treatment with neuraminidase, it was alluring to incriminate sialylation as involved mechanism. Sialic acid is used in cancers, such as breast and ovarian carcinoma, as a protection against complement activity [29], and removal of sialic acid from the cell surface by sialidase increases sensitivity to CDC [45-47]. Applied to CLL patients, we further established that the α2-6 sialylation level influences resistance to the RTX-induced CDC patient group, and predicts the in vivo clinical RTX-chemotherapy outcome. Although a high level of sialic acid could favor the recruitment of complement inhibitors and consequently the resistance of CLL cells to RTX-induced CDC [34], we were unable to associate the increased level of α2-6 sialylation with factor H binding. It was recently demonstrated that inhibiting factor H binding, through the use of a novel mAb targeting factor H, sensitizes CLL cells resistant to RTX in a subset of patients [48]. This can be explained in part by the fact that factor H binds α2-3 but not α2-6 sialic acid [49]. Sialic acid is also known to recruit other complement regulators through binding of factor I, properdin and clusterin, which needs to be further explored. Whether these molecules are involved in CLL remains unknown. Nevertheless, sialylated proteins can directly induce resistance to complement by inducing inactivation of C3b [50], and in turn reduce the clearance of RTX opsonized cells by fixed tissue macrophages [10, 11]. An elevated level of sialic acid is one of the most effective mechanisms used by the target cells to control the ADCP process by macrophages [51]. Accordingly, our results support an abnormal quantity of sialic acid available on CLL cells to control both CDC and ADCP mediated by RTX. Further studies will be required to clarify the respective role of CDC and ADCP in the therapeutic response to RTX and further provide an explanation between the low level of CDC observed in vitro and its association with a better response. Similarly, as the phenomenon of trogocytosis mediated in particular by macrophages is described in the context of therapeutic antibodies this will need to be evaluated further with the limitation that trogocytosis controls partial loss of CD20 by RTX, but not CLL cell death and complement activation [52].
Elevated levels of the main galactoside α2-6 ST (ST6Gal-I) implicated in α2-6 sialic acid overexpression at the cell surface have been associated with metastasis and therapeutic failure in leukemia [53], and colorectal cancer [54]. Therefore, ST6Gal-I knockdown regulates tumor growth and invasion [55], and reverses protection against Fas mediated apoptosis [56]. A long list of sialylated receptors (e.g. CD24, CD43, CD45, CD52, CD62L, CD75) and receptors involved in sialic acid recognition (e.g. CD22) are described in B cells [57] and most of them are deregulated in tumoral B cells [58]. Treating CLL cells with neuraminidase was shown to reveal α2-6 sialic acid masked epitopes from the cell surface CDw75 [59], and the multidrug resistance protein 1 (MDR1) [60]. MDR1 overexpression is associated with therapeutic resistance in CLL [61], and mutations in MDR1 increase the risk of CLL [62]. As a consequence, future studies are necessary to test the impact of chemotherapy associated with RTX on α2-6 ST activity and whether blocking α2-6 ST activity and/or α2-6 sialic acid would improve RTX-chemotherapy not only by improving complement activation but also by reducing drug resistance. Another important question is related to the contribution of sialic acid overexpression on therapeutic response when using new generation anti-CD20 mAb. It’s important to note that four patients from the CDC-resistant group have received Ofatumumab at relapse and that such administration was associated with an important C4 complement exhaustion [38]. A part of the patients from our cohort (47%) was also tested in vitro with the type II anti-CD20 mAb B1 (tositumomab) that triggers cell death by altering lipid organization in a mechanism independent from CDC and CLL hypersialylation ([32] and Figure 4C). Accordingly, future research is necessary to appreciate whether determining hypersialylation might be useful to propose a new generation anti-CD20 mAb as an alternative to RTX.
Other mechanisms controlling complement activation in patients receiving RTX-chemotherapy have been explored but with conflicting results reported when considering the absence of correlation between the complement inhibitors CD46, CD55 and CD59 levels and the RTX clinical outcome in NHL and CLL [18, 63], while blocking the complement regulatory molecules CD55 and CD59 improves RTX efficacy. Regarding the expression of the complement inhibitors, our results performed at baseline failed to highlight significant differences in agreement with some reports [64], but not all [18, 65] and this could be explained in part by different methodological strategies, patient selection and the fact that we have not tested CD55 and CD59 expression in the resistant CLL cells that were not cleared from the blood at completion of therapy [18]. As a consequence, the possibility that other factors control CDC needs to be taken into consideration. For this purpose, we have considered the capacity of the tumor cells to express cell membrane complement regulators, to recruit the fluid-phase complement inhibitor factor H [66], the level of the targeted antigen CD20, and the appropriate distribution of lipid rafts necessary for the recruitment of the complement protein C1q [31]. Fixation of RTX to CD20, density of CD20 molecules, GM1 and SM levels, complement inhibitor proteins CD55, CD59 and Factor H were evaluated in CLL cells in the two groups of patients.
Although these preliminary data are promising, this study has potential limitations. The developed in vitro assay necessitates long term incubations with RTX and we could not exclude that chemotherapy protocols have an effect on sialylation and CDC regulator expression. The sample size is quite small and is heterogeneous with respect to treatments. The analysis of the relationship between CDC status and clinical response were not adjusted for treatment, cytogenetic status, and immunoglobulin mutational status. In future studies it will be important to investigate dependence to these factors because existing data suggest that they are contributing factors in CLL relapse.
In conclusion, our main findings are: (i) part of the lytic effects of RTX-chemotherapy on tumor CLL cells is related to the capacity of RTX to induce the complement pathway; (ii) CDC is influenced by the level of cell surface α2-6 SA; (iii) a stronger activity of α2-6 ST characterizes the CDC-resistant patient group; and (iv) resistant capacity to CDC is not associated with distinct individual genetic groups and clinical outcome. From the therapeutic point of view and as CLL therapy is moving towards chemo-free treatments, future experiments are necessary now to develop the most appropriate biomarker between the three assays performed during this study: the CDC assay, α2-6 ST activity, and α2-6 sialic acid expression, in order to establish whether CDC resistance evaluation would help to determine early on those patients not likely to benefit from the classical first line FCR therapy.
MATERIALS AND METHODS
Patients and normal controls
Sixty-nine untreated patients fulfilling CLL diagnostic criteria were enrolled in this retrospective study including 34 tested before the RTX chemotherapy initiation (0 to 7 months) [67]. Disease assessment included Binet stage determination, CD38 expression, lymphocyte counts, LDT, and cytogenetic analysis as previously described [68, 69]. PFS was defined as the time from disease discovery to disease progression. Disease progression was considered either as the shift from Binet stage A to Binet stage B/C or as a short LDT of less than 6 months. TFS was defined as the interval between the date of disease discovery and the date of treatment initiation. Patients were segregated into cytogenetic risk groups according to the Döhner’s classification [70]: patients with isolated del(13q) were in the low risk group, normal karyotype or trisomy 12 were in the intermediate risk group, and del(11q), del(17p) or complex karyotype were in the high risk group. RTX was introduced as first line treatment in combination with fludarabine and cyclophosphamide (RFC, n=28) or with bendamustine (RB, n=6). Response to therapy was assessed according to the 2008 IW-CLL guidelines [71] by determining the CR at the end of RTX infusions, TTR was defined as the time from RTX initiation to the time of disease progression (≥5 ×109 peripheral B cells/L), and the OS rate from the inclusion to death from any cause. Blood was also withdrawn from 21 healthy volunteers. Consent was obtained from all individuals and the protocol approved by the Ethical Board at the Brest University Medical School Hospital (https://clinicaltrials.gov/ct2/show/NCT03294980; CRB Brest, collection 2008-214), in accordance with the Declaration of Helsinki.
Specimen collections and complement analysis
Blood samples were collected in order to test the RTX capacity to induce CDC and for cellular analysis. Plasma was stored at -80°C until tested for C3c (normal range (NR): 0.81-1.57 g/L) and C4 (NR: 0.13-0.39 g/L) by turbidimetry (Spa-Plus®, The binding site group limited, Birmingham, UK).
Cell preparation
Peripheral blood mononuclear cells were separated by density-gradient centrifugation on Ficoll-Hypaque, and B lymphocytes were isolated using a B-cell isolation kit according to the manufacturer’s instructions (Stem Cell Technologies, Grenoble, France). The purity estimated by flow cytometry using FITC-conjugated anti-CD19 and PE-conjugated anti-CD5 mAbs was over 95%.
Complement-dependent cell cytotoxicity assay
Based on our preliminary step optimization (Figure 1), RTX-induced CDC was evaulated in vitro as follows. Briefly, 3×105 B lymphocytes were incubated into 24-well plates at 37°C in RPMI-1640 (Sigma-Aldrich, Saint-Quentin Fallavier, France) supplemented with 2mM L-glutamine, antibiotics, 10% fetal calf decomplemented serum (Gibco, Paisley, Scotland), and 20% of HSAB decomplemented or not 20 min at 56°C (Invitrogen, Cergy Pontoise, France). In all protocols 10μg/ml RTX-stimulation (Roche, Paris, France) (saturating Ab conditions [72]) with or without 10μg/ml Eculizumab (Soliris; Alexion Pharmaceuticals, Cheshire, CT), B lymphocytes were collected, washed and stained for 15 minutes with FITC-conjugated annexin-V (AV) and propidium iodide (PI) using the Beckman-Coulter apoptosis kit. The percentage of CDC decreased survival was calculated by using the formula: 100x [(% of AV-PI- cells without RTX in HSAB)-(% of AV-PI- cells with RTX in HSAB)]/[% of AV-PI- cells without RTX in HSAB]. In selected experiments, direct apoptosis and ADCC [25, 32] were monitored as previously described; and cells were additionally treated with 0.05U of neuraminidase (Sigma-Aldrich) for 30 minutes at 37°C, and two washes were then performed to avoid any side effects of neuraminidase on RTX or B1 (10μg/ml, generous gift of Cragg MS, Southampton, UK) activity.
Mutational status of IGHV
As previously described [68, 69], the IGHV gene mutation status was determined by sequencing after conducting a PCR multiplex amplification. Briefly, for multiplex PCR, 100ng of genomic DNA, 0.25μl of Ampli Taq Gold DNA Polymerase (Applied Biosystems, Foster City, CA, USA), 10pmol of each primer, 0.2mM dNTP Mix, 1.5mM MgCl2, 1x PCR Buffer II, were adjusted to 50μl with DNase/RNase free ultrapure distilled water. Next, PCR products were visualized on 2% agarose gel, and purified with ExoSAP-IT PCR product cleanup kit (Affymetrix, High Wycombe, UK). Finally, amplicons were sequenced with a Big Dye Terminator v3.1 cycle sequencing kit (Applied Biosystems). Results were analyzed with the database IMGT/HighV-Quest (The international ImMunoGeneTics information system, Montpellier) and a homology sequence >98% defined an UM status.
Polymorphism analysis in FcγR3A V158F
The BioSprint 15 DNA blood kit was used to extract genomic DNA from mononuclear cells according to the manufacturer’s instructions (Qiagen, Valencia, CA, USA). Polymorphisms were determined as previously described [38, 73] using allele-specific polymerase chain reactions (PCRs) with one unmodified primer for FcγR3a-158V/F (ATATTTACAGAATGGCACAGG) and one locked nucleic acid modified primer for FcγR3a-158V: GAAGACACATTTTTACTCCCAA+C versus for FcγR3a-158F CTCTGAAGACACA-TTTTTACTCCCAA+A.
Flow cytometry
In this study, 5×105 freshly isolated lymphocytes were incubated for 30 minutes at 4°C with saturating concentrations of anti-CD19 mAbs combined with phycoerythrin (PE)-cyanin 7 (PCY7), and another mAb (anti-CD5, anti-CD20, anti-CD59 or anti-CD55) conjugated with fluorescein isothiocyanate (FITC). All mAb, as well as their isotype control mAb were from Beckman Coulter (Villepinte, France), biotinylated lectin SNA, specific to the α2-6 linked SA, and biotinylated lectin MAA specific to the α2-3 linked sialic acid (Vector Burlingame, CA), lysenin recognizing SM (PeptaNova, Sandhausen, Germany) or FITC-conjugated cholera toxin subunit B recognizing GM1 (Sigma-Aldrich). Samples containing biotinylated-lectin were covered for an additional 30 minutes with FITC-conjugated streptavidin (Jackson Immunoresearch Europe Ltd, Suffolk, UK). Cells stained with lysenin were incubated for 30 minutes with a polyclonal rabbit anti-lysenin Ab, and for another 30 minutes with FITC-conjugated anti-rabbit mAb (PeptaNova, Sandhausen, Germany). To study the binding of factor H on CLL cells, 3.105 cells were incubated for one hour at 37°C with normal human serum diluted 1/4 in PBS followed by a 30-minute incubation with FITC-conjugated anti-factor H mAb (Abcam, Paris, France) at 4°C. The cells were then washed with phosphate buffer saline (PBS) and analyzed on FC500 flow cytometer (Beckman Coulter). The calculation of the MFI of all markers required a minimum of 5,000 events.
The number of CD20 molecules per cell was quantified by determining the amount of mAb binding to the cells at saturating concentrations, using the Quantum kit (Flow Cytometry Standards Corp).
Assay for sialyltransferase activity
The activity of α2,6 and α2,3 sialyltransferases was evaluated by ELISA after B cell lysis as previously described [35] and the optical density (OD) was determined by a spectrometer ELISA at 492 nm.
Statistical analysis
Continuous data are described as mean ± standard error of the mean (SEM). Following normality and equality of variance tests, nominal values were compared using the student’s t test or alternatively by using a nonparametric test (Mann–Whitney rank sum test). For categorical data, differences among groups were analyzed using the Fisher’s exact test. The profile likelihood method using a Cox regression model of TTR was used in univariate analysis to determine the optimal threshold, this analysis was computed using the Survival and SurvMisc R packages [74]. LDT, TFS, PFS and TTR analyses were performed using Kaplan–Meier curves and prognosis differences between groups were assessed with a log-rank test. P values under 0.05 were considered significant. Statistical analyses were performed using GraphPad Prism 7.0 (La Jolla, CA, USA).
Abbreviations
ABC: antibody binding capacities; ADCC: antibody-dependent cellular cytotoxicity; CD: cluster of differentiation; CDC: complement-dependent cytotoxicity; CLL: chronic lymphocytic leukemia; CR: complete response; CRB: center for biological resources; FcγR3: Fc gamma receptor 3; GM1: ganglioside M1; HSAB: normal human serum AB; MAA: Maakia amurensis; mAb: monoclonal antibody; MFI: mean fluorescence intensity; NHL: non-Hodgkin’s lymphomas; NK: natural killer; NR: normal range; NS: not significant; OD: optical density; OS: overall survival; PFS: progression free survival; PR: partial response; RB: rituximab bendamustine; RFC: rituximab fludarabine cyclophosphamide; RTX: rituximab; SA: Sialic acid; SD: standard deviations; SNA: Sambucus nigra; ST: sialyltransferases; TFS: treatment free survival; TTR: time to relapse.
Author contributions
AB, AT, JOP and YR designed the study. AB, AM, BB, CBu, SD performed the research. CBa, AT, HS and CBe took care of the patients, performed the clinical data gathering, and validated clinical data accuracy. AB, CBa, JOP and YR analyzed the data. AB and YR prepared the initial draft. The final manuscript was read and approved by all authors.
ACKNOWLEDGMENTS
The authors would like to thank Mariam Hammadi and Charlotte Laot for their technical help, Geneviève Michel and Simone Forest for secretarial assistance, and Dr Wesley H Brooks for editorial assistance (Tampa, USA). The authors thank the Brest Biological Resources Center BB-0033-00037 (“CRB Santé de Brest”) for providing high quality and annotated samples and Cragg MS (Southampton, UK) for the generous gift of B1.
CONFLICTS OF INTEREST
The authors declare no financial conflicts of interest.
FUNDING
This study was supported by research funding from the “Association Laurette Fugain” (ALF 2015/03), the “Ligue contre le Cancer”, the “Region Bretagne”, the “Association pour la Recherche en Hématologie, Oncologie Pédiatrique et Hémostase” and from the “Canceropole Grand Ouest”.
REFERENCES
1. Robak T, Lech-Maranda E, Robak P. Rituximab plus fludarabine and cyclophosphamide or other agents in chronic lymphocytic leukemia. Expert Rev Anticancer Ther. 2010; 10:1529-1543.
2. Huhn D, von Schilling C, Wilhelm M, Ho AD, Hallek M, Kuse R, Knauf W, Riedel U, Hinke A, Srock S, Serke S, Peschel C, Emmerich B, and German Chronic Lymphocytic Leukemia Study Group. Rituximab therapy of patients with B-cell chronic lymphocytic leukemia. Blood. 2001; 98:1326-1331.
3. Cramer P, Langerbeins P, Eichhorst B, Hallek M. Advances in first-line treatment of chronic lymphocytic leukemia: current recommendations on management and first-line treatment by the German CLL Study Group (GCLLSG). Eur J Haematol. 2016; 96:9-18.
4. Bagacean C, Zdrenghea M, Tempescul A, Cristea V, Renaudineau Y. Anti-CD20 monoclonal antibodies in chronic lymphocytic leukemia: from uncertainties to promises. Immunotherapy. 2016; 8:569-581.
5. Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000; 6:443-446.
6. Veeramani S, Wang SY, Dahle C, Blackwell S, Jacobus L, Knutson T, Button A, Link BK, Weiner GJ. Rituximab infusion induces NK activation in lymphoma patients with the high-affinity CD16 polymorphism. Blood. 2011; 118:3347-3349.
7. Di Gaetano N, Cittera E, Nota R, Vecchi A, Grieco V, Scanziani E, Botto M, Introna M, Golay J. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol. 2003; 171:1581-1587.
8. Zent CS, Elliott MR. Maxed out macs: physiologic cell clearance as a function of macrophage phagocytic capacity. FEBS J. 2017; 284:1021-1039.
9. Grandjean CL, Montalvao F, Celli S, Michonneau D, Breart B, Garcia Z, Perro M, Freytag O, Gerdes CA, Bousso P. Intravital imaging reveals improved Kupffer cell-mediated phagocytosis as a mode of action of glycoengineered anti-CD20 antibodies. Sci Rep. 2016; 6:34382.
10. Gul N, Babes L, Siegmund K, Korthouwer R, Bogels M, Braster R, Vidarsson G, ten Hagen TL, Kubes P, van Egmond M. Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J Clin Invest. 2014; 124:812-823.
11. Montalvao F, Garcia Z, Celli S, Breart B, Deguine J, Van Rooijen N, Bousso P. The mechanism of anti-CD20-mediated B cell depletion revealed by intravital imaging. J Clin Invest. 2013; 123:5098-5103.
12. Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002; 99:754-758.
13. Anolik JH, Campbell D, Felgar RE, Young F, Sanz I, Rosenblatt J, Looney RJ. The relationship of FcgammaRIIIa genotype to degree of B cell depletion by rituximab in the treatment of systemic lupus erythematosus. Arthritis Rheum. 2003; 48:455-459.
14. Kim DH, Jung HD, Kim JG, Lee JJ, Yang DH, Park YH, Do YR, Shin HJ, Kim MK, Hyun MS, Sohn SK. FCGR3A gene polymorphisms may correlate with response to frontline R-CHOP therapy for diffuse large B-cell lymphoma. Blood. 2006; 108:2720-2725.
15. Hatjiharissi E, Xu L, Santos DD, Hunter ZR, Ciccarelli BT, Verselis S, Modica M, Cao Y, Manning RJ, Leleu X, Dimmock EA, Kortsaris A, Mitsiades C, et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the Fc{gamma}RIIIa-158 V/V and V/F polymorphism. Blood. 2007; 110:2561-2564.
16. Treon SP, Hansen M, Branagan AR, Verselis S, Emmanouilides C, Kimby E, Frankel SR, Touroutoglou N, Turnbull B, Anderson KC, Maloney DG, Fox EA. Polymorphisms in FcgammaRIIIA (CD16) receptor expression are associated with clinical response to rituximab in Waldenstrom’s macroglobulinemia. J Clin Oncol. 2005; 23:474-481.
17. Dall’Ozzo S, Tartas S, Paintaud G, Cartron G, Colombat P, Bardos P, Watier H, Thibault G. Rituximab-dependent cytotoxicity by natural killer cells: influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res. 2004; 64:4664-4669.
18. Bannerji R, Kitada S, Flinn IW, Pearson M, Young D, Reed JC, Byrd JC. Apoptotic-regulatory and complement-protecting protein expression in chronic lymphocytic leukemia: relationship to in vivo rituximab resistance. J Clin Oncol. 2003; 21:1466-1471.
19. Terui Y, Sakurai T, Mishima Y, Mishima Y, Sugimura N, Sasaoka C, Kojima K, Yokoyama M, Mizunuma N, Takahashi S, Ito Y, Hatake K. Blockade of bulky lymphoma-associated CD55 expression by RNA interference overcomes resistance to complement-dependent cytotoxicity with rituximab. Cancer Sci. 2006; 97:72-79.
20. Harjunpaa A, Junnikkala S, Meri S. Rituximab (anti-CD20) therapy of B-cell lymphomas: direct complement killing is superior to cellular effector mechanisms. Scand J Immunol. 2000; 51:634-641.
21. Seret G, Hanrotel C, Bendaoud B, Le Meur Y, Renaudineau Y. Homozygous FCGR3A-158F mutation is associated with delayed B-cell depletion following rituximab but with preserved efficacy in a patient with refractory lupus nephritis. Clin Kidney J. 2013; 6:74-76.
22. Klepfish A, Gilles L, Ioannis K, Rachmilewitz EA, Schattner A. Enhancing the action of rituximab in chronic lymphocytic leukemia by adding fresh frozen plasma: complement/rituximab interactions & clinical results in refractory CLL. Ann N Y Acad Sci. 2009; 1173:865-873.
23. Middleton O, Cosimo E, Dobbin E, McCaig AM, Clarke C, Brant AM, Leach MT, Michie AM, Wheadon H. Complement deficiencies limit CD20 monoclonal antibody treatment efficacy in CLL. Leukemia. 2015; 29:107-114.
24. Golay J, Zaffaroni L, Vaccari T, Lazzari M, Borleri GM, Bernasconi S, Tedesco F, Rambaldi A, Introna M. Biologic response of B lymphoma cells to anti-CD20 monoclonal antibody rituximab in vitro: CD55 and CD59 regulate complement-mediated cell lysis. Blood. 2000; 95:3900-3908.
25. Mankai A, Bordron A, Renaudineau Y, Martins-Carvalho C, Takahashi S, Ghedira I, Berthou C, Youinou P. Purine-rich box-1-mediated reduced expression of CD20 alters rituximab-induced lysis of chronic lymphocytic leukemia B cells. Cancer Res. 2008; 68:7512-7519.
26. Meyer zum Buschenfelde C, Feuerstacke Y, Gotze KS, Scholze K, Peschel C. GM1 expression of non-Hodgkin’s lymphoma determines susceptibility to rituximab treatment. Cancer Res. 2008; 68:5414-5422.
27. Keppler OT, Moldenhauer G, Oppenlander M, Schwartz-Albiez R, Berger EG, Funderud S, Pawlita M. Human Golgi beta-galactoside alpha-2,6-sialyltransferase generates a group of sialylated B lymphocyte differentiation antigens. Eur J Immunol. 1992; 22:2777-2781.
28. Meri S, Pangburn MK. Discrimination between activators and nonactivators of the alternative pathway of complement: regulation via a sialic acid/polyanion binding site on factor H. Proc Natl Acad Sci U S A. 1990; 87:3982-3986.
29. Donin N, Jurianz K, Ziporen L, Schultz S, Kirschfink M, Fishelson Z. Complement resistance of human carcinoma cells depends on membrane regulatory proteins, protein kinases and sialic acid. Clin Exp Immunol. 2003; 131:254-263.
30. Chai-Adisaksopha C, Brown JR. FCR achieves long-term durable remissions in patients with IGHV-mutated CLL. Blood. 2017; 130:2278-2282.
31. Cragg MS, Morgan SM, Chan HT, Morgan BP, Filatov AV, Johnson PW, French RR, Glennie MJ. Complement-mediated lysis by anti-CD20 mAb correlates with segregation into lipid rafts. Blood. 2003; 101:1045-1052.
32. Hammadi M, Youinou P, Tempescul A, Tobon G, Berthou C, Bordron A, Pers JO. Membrane microdomain sphingolipids are required for anti-CD20-induced death of chronic lymphocytic leukemia B cells. Haematologica. 2012; 97:288-296.
33. Takei K, Yamazaki T, Sawada U, Ishizuka H, Aizawa S. Analysis of changes in CD20, CD55, and CD59 expression on established rituximab-resistant B-lymphoma cell lines. Leuk Res. 2006; 30:625-631.
34. Horl S, Banki Z, Huber G, Ejaz A, Windisch D, Muellauer B, Willenbacher E, Steurer M, Stoiber H. Reduction of complement factor H binding to CLL cells improves the induction of rituximab-mediated complement-dependent cytotoxicity. Leukemia. 2013; 27:2200-2208.
35. Basset C, Durand V, Mimassi N, Pennec YL, Youinou P, Dueymes M. Enhanced sialyltransferase activity in B lymphocytes from patients with primary Sjogren’s syndrome. Scand J Immunol. 2000; 51:307-311.
36. Dai J, Allard WJ, Davis G, Yeung KK. Effect of desialylation on binding, affinity, and specificity of 56 monoclonal antibodies against MUC1 mucin. Tumour Biol. 1998; 19:100-110.
37. Kennedy AD, Beum PV, Solga MD, DiLillo DJ, Lindorfer MA, Hess CE, Densmore JJ, Williams ME, Taylor RP. Rituximab infusion promotes rapid complement depletion and acute CD20 loss in chronic lymphocytic leukemia. J Immunol. 2004; 172:3280-3288.
38. Tempescul A, Bagacean C, Riou C, Bendaoud B, Hillion S, Debant M, Buors C, Berthou C, Renaudineau Y. Ofatumumab capacity to deplete B cells from chronic lymphocytic leukaemia is affected by C4 complement exhaustion. Eur J Haematol. 2016; 96:229-235.
39. Klepfish A, Rachmilewitz EA, Kotsianidis I, Patchenko P, Schattner A. Adding fresh frozen plasma to rituximab for the treatment of patients with refractory advanced CLL. QJM. 2008; 101:737-740.
40. Skarzynski M, Niemann CU, Lee YS, Martyr S, Maric I, Salem D, Stetler-Stevenson M, Marti GE, Calvo KR, Yuan C, Valdez J, Soto S, Farooqui MZ, et al. Interactions between Ibrutinib and Anti-CD20 Antibodies: Competing Effects on the Outcome of Combination Therapy. Clin Cancer Res. 2016; 22:86-95.
41. Hallek M, Fischer K, Fingerle-Rowson G, Fink AM, Busch R, Mayer J, Hensel M, Hopfinger G, Hess G, von Grünhagen U, Bergmann M, Catalano J, Zinzani PL, et al, and International Group of Investigators, and German Chronic Lymphocytic Leukaemia Study Group. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010; 376:1164-1174.
42. Dornan D, Spleiss O, Yeh RF, Duchateau-Nguyen G, Dufour A, Zhi J, Robak T, Moiseev SI, Dmoszynska A, Solal-Celigny P, Warzocha K, Loscertales J, Catalano J, et al. Effect of FCGR2A and FCGR3A variants on CLL outcome. Blood. 2010; 116:4212-4222.
43. Sebejova L, Borsky M, Jaskova Z, Potesil D, Navrkalova V, Malcikova J, Sramek M, Doubek M, Loja T, Pospisilova S, Mayer J, Trbusek M. Distinct in vitro sensitivity of p53-mutated and ATM-mutated chronic lymphocytic leukemia cells to ofatumumab and rituximab. Exp Hematol. 2014; 42:867-874 e861.
44. Rossi D, Terzi-di-Bergamo L, De Paoli L, Cerri M, Ghilardi G, Chiarenza A, Bulian P, Visco C, Mauro FR, Morabito F, Cortelezzi A, Zaja F, Forconi F, et al. Molecular prediction of durable remission after first-line fludarabine-cyclophosphamide-rituximab in chronic lymphocytic leukemia. Blood. 2015; 126:1921-1924.
45. Lauf PK. Immunological and physiological characteristics of the rapid immune hemolysis of neuraminidase-treated sheep red cells produced by fresh guinea pig serum. J Exp Med. 1975; 142:974-988.
46. Jacobsen F. Increase of the in vitro complement-dependent cytotoxicity against autologous invasive human bladder tumor cells by neuraminidase treatment. Acta Pathol Microbiol Immunol Scand C. 1982; 90:187-192.
47. Turianskyj FH, Gyenes L. The effect of neuraminidase on the sensitivity of tumor cells toward lysis by antibody and complement or by sensitized lymphocytes. Transplantation. 1976; 22:24-30.
48. Winkler MT, Bushey RT, Gottlin EB, Campa MJ, Guadalupe ES, Volkheimer AD, Weinberg JB, Patz EF Jr. Enhanced CDC of B cell chronic lymphocytic leukemia cells mediated by rituximab combined with a novel anti-complement factor H antibody. PLoS One. 2017; 12:e0179841.
49. Blaum BS, Hannan JP, Herbert AP, Kavanagh D, Uhrin D, Stehle T. Structural basis for sialic acid-mediated self-recognition by complement factor H. Nat Chem Biol. 2015; 11:77-82.
50. Ollert MW, David K, Bredehorst R, Vogel CW. Classical complement pathway activation on nucleated cells. Role of factor H in the control of deposited C3b. J Immunol. 1995; 155:4955-4962.
51. Maganti S, Pierce MM, Hoffmaster A, Rodgers FG. The role of sialic acid in opsonin-dependent and opsonin-independent adhesion of listeria monocytogenes to murine peritoneal macrophages. Infect Immun. 1998; 66:620-626.
52. Valgardsdottir R, Cattaneo I, Klein C, Introna M, Figliuzzi M, Golay J. Human neutrophils mediate trogocytosis rather than phagocytosis of CLL B cells opsonized with anti-CD20 antibodies. Blood. 2017; 129:2636-2644.
53. Ma H, Cheng L, Hao K, Li Y, Song X, Zhou H, Jia L. Reversal effect of ST6GAL 1 on multidrug resistance in human leukemia by regulating the PI3K/Akt pathway and the expression of P-gp and MRP1. PLoS One. 2014; 9:e85113.
54. Park JJ, Lee M. Increasing the alpha 2, 6 sialylation of glycoproteins may contribute to metastatic spread and therapeutic resistance in colorectal cancer. Gut Liver. 2013; 7:629-641.
55. Meng Q, Ren C, Wang L, Zhao Y, Wang S. Knockdown of ST6Gal-I inhibits the growth and invasion of osteosarcoma MG-63 cells. Biomed Pharmacother. 2015; 72:172-178.
56. Swindall AF, Bellis SL. Sialylation of the Fas death receptor by ST6Gal-I provides protection against Fas-mediated apoptosis in colon carcinoma cells. J Biol Chem. 2011; 286:22982-22990.
57. Varki A, Gagneux P. Multifarious roles of sialic acids in immunity. Ann N Y Acad Sci. 2012; 1253:16-36.
58. Pearce OM, Laubli H. Sialic acids in cancer biology and immunity. Glycobiology. 2016; 26:111-128.
59. Guy K, Andrew JM. Expression of the CDw75 (beta-galactoside alpha 2,6-sialyltransferase) antigen on normal blood cells and in B-cell chronic lymphocytic leukaemia. Immunology. 1991; 74:206-214.
60. Maynadie M, Matutes E, Catovsky D. Quantification of P-glycoprotein in chronic lymphocytic leukemia by flow cytometry. Leuk Res. 1997; 21:825-831.
61. Guillaume N, Gouilleux-Gruart V, Claisse JF, Troussard X, Lepelley P, Damaj G, Royer B, Garidi R, Lefrere JJ. Multi-drug resistance mediated by P-glycoprotein overexpression is not correlated with ZAP-70/CD38 expression in B-cell chronic lymphocytic leukemia. Leuk Lymphoma. 2007; 48:1556-1560.
62. Sava GP, Speedy HE, Houlston RS. Candidate gene association studies and risk of chronic lymphocytic leukemia: a systematic review and meta-analysis. Leuk Lymphoma. 2014; 55:160-167.
63. Weng WK, Levy R. Expression of complement inhibitors CD46, CD55, and CD59 on tumor cells does not predict clinical outcome after rituximab treatment in follicular non-Hodgkin lymphoma. Blood. 2001; 98:1352-1357.
64. Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003; 21:3940-3947.
65. Golay J, Lazzari M, Facchinetti V, Bernasconi S, Borleri G, Barbui T, Rambaldi A, Introna M. CD20 levels determine the in vitro susceptibility to rituximab and complement of B-cell chronic lymphocytic leukemia: further regulation by CD55 and CD59. Blood. 2001; 98:3383-3389.
66. Horl S, Banki Z, Huber G, Ejaz A, Mullauer B, Willenbacher E, Steurer M, Stoiber H. Complement factor H-derived short consensus repeat 18-20 enhanced complement-dependent cytotoxicity of ofatumumab on chronic lymphocytic leukemia cells. Haematologica. 2013; 98:1939-1947.
67. Matutes E, Owusu-Ankomah K, Morilla R, Garcia Marco J, Houlihan A, Que TH, Catovsky D. The immunological profile of B-cell disorders and proposal of a scoring system for the diagnosis of CLL. Leukemia. 1994; 8:1640-1645.
68. Bagacean C, Le Dantec C, Berthou C, Tempescul A, Saad H, Bordron A, Zdrenghea M, Cristea V, Douet-Guilbert N, Renaudineau Y. Combining cytogenetic and epigenetic approaches in chronic lymphocytic leukemia improves prognosis prediction for patients with isolated 13q deletion. Clin Epigenetics. 2017; 9:122.
69. Bagacean C, Tempescul A, Le Dantec C, Bordron A, Mohr A, Saad H, Olivier V, Zdrenghea M, Cristea V, Cartron PF, Douet-Guilbert N, Berthou C, Renaudineau Y. Alterations in DNA methylation/demethylation intermediates predict clinical outcome in chronic lymphocytic leukemia. Oncotarget. 2017; 8:65699-716. https://doi.org/10.18632/oncotarget.20081.
70. Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, Dohner K, Bentz M, Lichter P. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000; 343:1910-1916.
71. Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, Hillmen P, Keating MJ, Montserrat E, Rai KR, Kipps TJ, and International Workshop on Chronic Lymphocytic Leukemia. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008; 111:5446-56.
72. Bezombes C, Grazide S, Garret C, Fabre C, Quillet-Mary A, Muller S, Jaffrezou JP, Laurent G. Rituximab antiproliferative effect in B-lymphoma cells is associated with acid-sphingomyelinase activation in raft microdomains. Blood. 2004; 104:1166-1173.
73. Cornec D, Tempescul A, Querellou S, Hutin P, Pers JO, Jamin C, Bendaoud B, Berthou C, Renaudineau Y, Youinou P. Identification of patients with indolent B cell lymphoma sensitive to rituximab monotherapy. Ann Hematol. 2012; 91:715-721.
74. Borgan Ø. Modeling survival data: extending the cox model. Terry M. Therneau and Patricia M. Grambsch, Springer-Verlag, New York, 2000. ISBN 0-387-98784-3. Stat Med. 2001; 20:2053-2054.
75. Treon SP, Mitsiades C, Mitsiades N, Young G, Doss D, Schlossman R, Anderson KC. Tumor cell expression of CD59 is associated with resistance to CD20 serotherapy in patients with B-cell malignancies. J Immunother. 2001; 24:263-271.
76. Pawluczkowycz AW, Beurskens FJ, Beum PV, Lindorfer MA, van de Winkel JG, Parren PW, Taylor RP. Binding of submaximal C1q promotes complement-dependent cytotoxicity (CDC) of B cells opsonized with anti-CD20 mAbs ofatumumab (OFA) or rituximab (RTX): considerably higher levels of CDC are induced by OFA than by RTX. J Immunol. 2009; 183:749-758.
77. Natsume A, Shimizu-Yokoyama Y, Satoh M, Shitara K, Niwa R. Engineered anti-CD20 antibodies with enhanced complement-activating capacity mediate potent anti-lymphoma activity. Cancer Sci. 2009; 100:2411-2418.
78. Teeling JL, French RR, Cragg MS, van den Brakel J, Pluyter M, Huang H, Chan C, Parren PW, Hack CE, Dechant M, Valerius T, van de Winkel JG, Glennie MJ. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood. 2004; 104:1793-1800.
79. van Meerten T, van Rijn RS, Hol S, Hagenbeek A, Ebeling SB. Complement-induced cell death by rituximab depends on CD20 expression level and acts complementary to antibody-dependent cellular cytotoxicity. Clin Cancer Res. 2006; 12:4027-4035.
80. Mamidi S, Hone S, Teufel C, Sellner L, Zenz T, Kirschfink M. Neutralization of membrane complement regulators improves complement-dependent effector functions of therapeutic anticancer antibodies targeting leukemic cells. Oncoimmunology. 2015; 4:e979688.
81. Beum PV, Lindorfer MA, Beurskens F, Stukenberg PT, Lokhorst HM, Pawluczkowycz AW, Parren PW, van de Winkel JG, Taylor RP. Complement activation on B lymphocytes opsonized with rituximab or ofatumumab produces substantial changes in membrane structure preceding cell lysis. J Immunol. 2008; 181:822-832.
82. Beurskens FJ, Lindorfer MA, Farooqui M, Beum PV, Engelberts P, Mackus WJ, Parren PW, Wiestner A, Taylor RP. Exhaustion of cytotoxic effector systems may limit monoclonal antibody-based immunotherapy in cancer patients. J Immunol. 2012; 188:3532-3541.