
INTRODUCTION
Neuroblastoma is the most common pediatric extracranial solid tumour originating from the sympatoadrenal lineage residing in neural crest [1–3]. It accounts for 7–15% of childhood cancers [2, 4]. Neuroblastoma is prone to spontaneous regression or differentiation into a benign ganglioneuroma, when diagnosed in infancy. However, in older patients neuroblastoma can be metastatic with rapid progression and fatal outcome. Pharmacological agents, such as retinoic acid, NO, phenylacetate, can also induce neuroblastoma cell differentiation or apoptosis in culture as well as tumor regression in clinics [5–8]. Currently, even the most intensive multimodal therapy results in only modest improvement in the cure rate of aggressive neuroblastoma [9].
The mechanisms responsible for the aggressive neuroblastoma behavior are poorly understood. Statistically, significant association between low patient survival rate and MYCN oncogene amplification, diploid DNA content, allelic loss, undifferentiated cell histology and changes in Trk receptors’ profiles have been reported [9]. A family of neurotrophic growth factors and their receptors, playing an important role in neural development, has been implicated in pathogenesis and progression of neuroblastoma [10–14]. This family consists of four members: nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), Neurotrophin-3 (NT3) and Neurotrophin-4/5 (NT4/5). Neurotrophin effects are mediated by binding to two classes of neurotrophin receptors (NTRs) – p75NTR and Trks (tyrosine kinase receptors), the latter comprising TrkA, TrkB and TrkC [15].
Several studies indicate that tumor cells utilize proteolytic enzymes for the degradation of extracellular matrix and invasion. Elevated levels of urokinase (uPA) and urokinase receptor (uPAR) have been reported in basalioma, melanoma, glioblastoma, different types of carcinomas, prostate, lung, ovarian, breast and gastrointestinal cancers [16–34]. uPA and uPAR were overexpressed in highly invasive metastatic forms of human neuroblastoma [3]. Increased expression of uPA or its receptor has been associated with elevated metastatic potential and poor prognosis in these patients. Binding of uPA to uPAR accelerates activation and increases the enzymatic activity of uPA. Experimental approaches using small interfering RNA to inhibit uPAR expression or blocking antibody to impair uPAR function can significantly interfere with glioma/glioblastoma invasion in vivo and in vitro and can also downregulate intracellular signalling leading to reduced tumor vascularization, suppress cell survival and proliferation [16, 19, 28, 29, 35]. These and other data indicate that the uPAR intervention aimed at reduction of its expression in cancer cells may represent potentially promising new approach to anticancer therapy. Although siRNA approach is effective in uPAR suppression, it has some drawbacks, since reduction in gene expression is not stable and siRNA effect drops down rapidly in actively proliferating cells.
A significant advance in genome engineering was made upon development of CRISPR/Cas9 system for nuclease-based genome editing and transcriptional regulation [36, 37]. The RNA-guided CRISPR/Cas9 (clustered regularly interspaced short palindrome repeats) technology provides an effective means for introduction of targeted loss-of function mutations into the genes of interest. These mutations, and hence, biological effects are heritable, highly specific and ensure complete gene shut-off in contrast to partial reduction of gene expression by other methods [38]. The CRISPR/Cas9 nickase (Cas9n introduces single strand breaks to DNA) genome editing system combines two plasmids each harbouring Cas9n gene and chimeric guide RNA (sgRNA). These sgRNAs are complementary to DNA sequences next to obligate PAM (protospacer adjuscent motif) trinucleotides. CRISPR-Cas9n makes two single-strand breaks with minimal off-target effects within the target DNA, followed by activation of non-homologous end joining (NHEJ) repair system. NHEJ inserts or removes a few nucleotides to Cas9n cleavage sites leading to a farameshift mutations and premature termination of translation [36, 39–43]. This approach can be used effectively for high precision loss-of-function genetic studies in cell lines and primary cultures, in animal disease models, for whole-genome mutation screening in cancer cell and genome editing in vivo [37, 39, 42, 44–46].
Recent advances using CRISPR/Cas9 system have opened new perspectives from basic research to clinical application. Inactivation of EPH1 with CRISPR/Cas9 technology suppressed ovarian cancer cell proliferation, invasion and migration in vitro [46]. In breast cancer cells, CRISPR/Cas9 system has been applied to disrupt HER2 oncogene expression. Ablation of HER2 resulted in inhibition of MAPK/Erk and PI3K/Akt signalling cascade, reduced cell proliferation and decreased tumorigenicity [45]. CRISPR/Cas9 technology has been used for genetic correction of a dominant mutation in Crygc gene that causes cataract in mice [37]. The first human trial using CRISPR/Cas9 gene editing to treat metastatic non-small-cell-lung cancer has been launched in China in 2016 [47]. In the current study we employed CRISPR/Cas9n system to target Plaur gene in Neuro 2A neuroblastoma cells. We created plasmids for uPAR gene inactivation, selected genetically modified clones and tested the efficiency of uPAR targeting using CRISPR/Cas9n. We showed that CRISPR/Cas9n targeting of Plaur gene resulted in inhibition of neuroblastoma proliferation, significant reduction in the number of Ki-67 positive cells, caspase 3 activation and PARP-1 cleavage. uPAR downregulation correlated with the decrease in TrkC mRNA level and Akt phosphorylation.
RESULTS
Targeting of Plaur by CRISPR/Cas9n and selection of modified clones
In the current study we designed pX458nickase-sg1 and pX458nickase-sg2 constructs to selectively target Plaur and disrupt uPAR function in Neuro 2A cells. These constructs also drove expression of EGFP, which was used as a selection marker to sort out cells transfected with components of CRISPR/Cas9n genome editing tool. CRISPR/Cas9n application was predicted to result in a frameshift mutation close to the start codon of Plaur and to cause premature termination of uPAR translation. Specific DNA regions recognized by sg1 and sg2 were separated by 13 nucleotides, which were sufficient to induce double-strand breaks in the Plaur and to activate the NHEJ repair (Figure 1). The analysis of on-target sites and most probable off-target sites of Plaur sgRNAs are presented in Supplementary Figure 1 and Supplementary Figure 2, respectively.
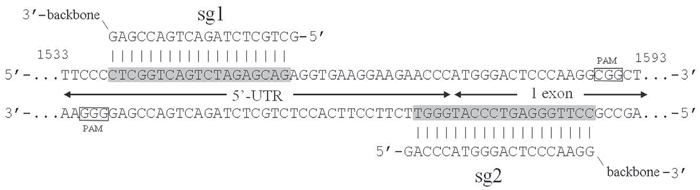
Figure 1: gRNAs and targeted region of Plaur. Designed gRNAs are complimentary to the first exon (sg2) and 5’-UTR (sg1) of the Plaur. Boundary of 5’UTR and exon1 of Plaur is shown by arrows. Binding sites of sgRNAs in genomic DNA are shadowed in grey. The start codon is highlighted, PAM sequences are framed.
To prove that the designed constructs effectively induced mutations in Plaur, we used them for Neuro 2A cell co-transfection. EGFP-positive cell with diminished uPAR cell surface expression were selected using FACS sorting. Neuro 2A is a line of immortalized tumor cells, characterized by an unstable karyotype of 94-98 chromosomes in the stemline, and 59-193 chromosomes in individual subclones (ATCC® CCL-131™, Manassas, Virginia). The copy number of loci containing Plaur gene was expected to vary from one to several. Therefore, we carried out three sequential co-transfections with pX458nickase-sg1 and pX458nickase-sg2 to maximize targeting of multiple Plaur copies. uPAR expression was assessed using immunofluorescent staining with anti-uPAR antibody of EGFP-expressing cells after each round of co-transfection. Sorting gates and results of anti-uPAR staining are presented in Figures 2 and 3. Wt, s1, s2 and s3 correspond to Neuro 2A cell subpopulations of wild type (Figure 3A), cells after the first (Figure 3B), the second (Figure 3C) and the third co-transfection (Figure 3D). The proportion of uPAR-positive cells in control culture was 88%, while after the first, the second and the third round of transfection it decreased to 64.6%, 59.1%, and 46.0%, respectively (Figure 3). The efficacy of uPAR suppression was low and not sufficient to study phenotypic and functional changes. To sort out this problem we generated uPAR-deficient single-cell clones.
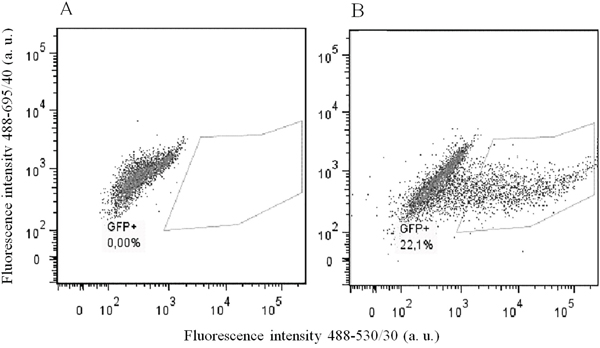
Figure 2: Flow cytometry analysis and sorting of EGFP-positive Neuro 2A cells. (A) wild type Neuro 2A cells. (B) Neuro 2A cells co-transfected with sg1 and sg2 plasmids. Sorting gate for EGFP-positive cells is shown. Fluorescence emission in 530/30 channel is shown on the x-axis, and in 695/40 channel on the y-axis; excitation - 488 nm laser.
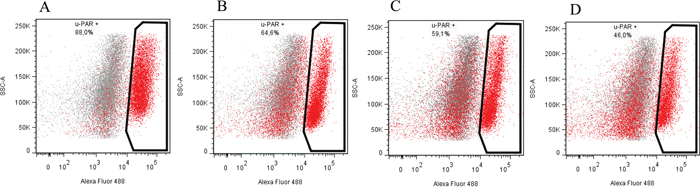
Figure 3: Subsequent co-transfection with sgRNA and Cas9n constructs reduces uPAR expression on the surface of Neuro 2A cells. (A) wild type Neuro 2A cells. (B, C, D) s1, s2 and s3 cell populations, correspondingly. Fluorescence emission in 530/30 channel is shown on the x-axis and side-scatter is shown on the y-axis; excitation – 488nm laser.
To obtain uPAR-deficient clones, single-cell dilution of EGFP-positive s3 cells into 96-well plates at 1 cell per well was performed. After single cell clone expansion, 30 clones were selected for further analysis. uPAR protein expression level was detected by Western blotting. While some clones showed only moderate decrease in uPAR expression (data not shown), others demonstartated convincing decrease or complete loss of uPAR (clones 1, 3 6, 8, 12, 15, 30) (Figure 4). uPAR mRNA expression in three chosen uPAR-deficient clones (3, 6 and 30) ranged from a significant decrease to a complete loss as confirmed by RT-PCR analysis (Supplementary Figure 3).

Figure 4: Analysis of uPAR expression in selected Neuro 2A clones by Western blot followed by detection with anti-uPAR antibodies. Control Neuro 2A cells; 1, 3, 6, 8, 12, 15, 30 – Neuro 2A clones after co-transfection and single cell clone expansion. Selected clones showed reduced uPAR expression from moderate decrease to complete uPAR loss. β3-tubulin was used as loading control. Typical results from three independent experiments are presented.
Sanger's sequencing analysis of Plaur sgRNAs on-target sites using ChromasLite and TIDE programs (https://tide.deskgen.com/) revealed the uPAR knockout in clone #6 and 75% suppression of uPAR in clone #30 (Supplementary Figure 1). Precisely, the three cycles of gene modification using CRISPR/Cas9 nickase plasmid resulted in the 62-nucleotide deletion in Plaur gene in clone #6. The detection of a single peak in clone #6 in chromatograms (Supplementary Figure 1) corresponds to the only allele variant. In clone #30, three allele variants have been identified: two alleles bearing identical inserts of 4 nucleotides; one allele with 6 nucleotide deletion potentially restoring the reading frame; and one allele with 19 nucleotide deletion.
To predict the most probable Plaur sgRNAs off-target sites COSMID software (https://crispr.bme.gatech.edu/) was implemented. No mutations in the loci of the most likely off-target activity of the CRISPR/Cas9n system targeted by the chosen sgRNAs could be detected (Supplementary Figure 2).
Thus, the strategy of pX458nickase-sg1 and pX458nickase-sg2 transfection, EGFP-based cell sorting and clonal selection was effective and allowed us to obtain cells with varying uPAR expression levels.
Downregulation of uPAR inhibits proliferation of Neuro 2A cells
Literature data indicate that uPAR expression is elevated in high-risk neuroblastomas and is associated with enhanced invasive/metastatic potential and overall negative patient’s prognosis [3]. To evaluate the pathophysiological significance of uPAR knockout on Neuro 2A cells, we compared the proliferation rate of uPAR-deficient clones (#6 and #30) to control Neuro 2A cells (wt) (Figure 5A, Figure 6, Figure 7). Decline in proliferation rate is obvious from the data showing live cell counts at different time points (equal number of cells were plated in control and experimental cultures) (Figure 5A). After 72 hours up to 5.3 times difference in live cell numbers was evident between control Neuro2A and clone #30. Similar results on the decreased cell counts were obtained for s2 and s3 subpopulations and clone #22 after 96 hours in culture. We observed an approximate 23% reduction in the number of cells for s2 and s3 subpopulations, compared to s1 and control cells; 33% decrease for clone #22 compared to the control (data not shown).
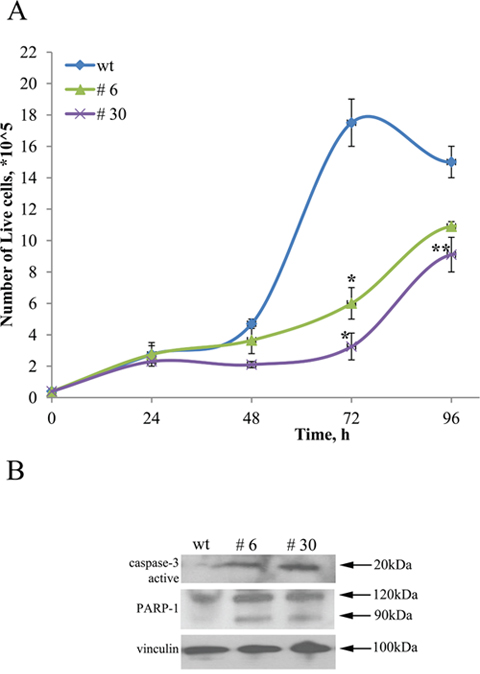
Figure 5: uPAR knockout restrains proliferation of Neuro 2A cells. (A) ControlNeuro 2A cells; #3, #30 – uPAR-deficient clones. Reproducible results of three independent experiments are presented. The data are presented as Mean ± SEM (n=3 wells per group). * - p<0.001 versus control, **- p<0.05 versus control (Newman-Keuls test). (B) Western blot demonstrating caspase-3 activation, PARP-1 cleavage and accumulation of 89 kDa fragment in uPAR-deficient clones compared to wt cells. Vinculin was used as loading control. Typical result from three independent experiments is presented.
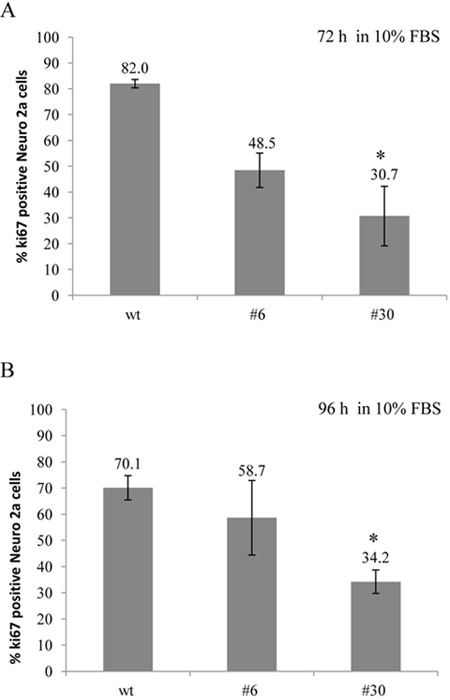
Figure 6: uPAR knockout results in a decreased amount of Ki-67-positive Neuro 2A cells. Ki-67 expression was assessed in uPAR-deficient clones (#6, #30) and in wt cells in standard culture media (10% FBS) after 72 and 96 hours. Cells were fixed, stained with antibody against Ki-67 and subjected to FACS analisys. (A) The bar chart depicts the percentage of Ki-67-positive Neuro 2A cells in each cell type after 72 hours; (B) The percentage of Ki-67-positive Neuro 2A cells in each cell type after 96 hours. The data are presented as Mean ± SD (n=4 wells per group), *p < 0.05 by ANOVA test.
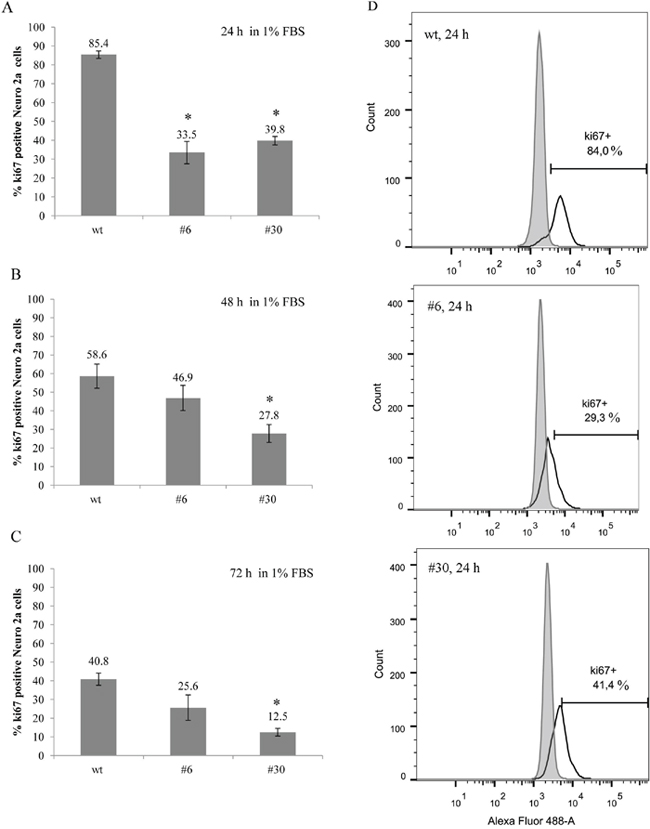
Figure 7: uPAR knockout results in a decreased amount of Ki-67-positive cells (low serum conditions). (A-C) The bar charts depict Ki-67 expression level in uPAR-deficient clones (#6, #30) and in wt cells after 24, 48 and 72 hours in low serum conditions (1% FBS). The data are presented as Mean ± SD (n=3 wells per group), * p < 0.05 ANOVA test. (D) Histograms depict the percentage of Ki-67-positive Neuro 2A cells in each cell type after 24 hours. Fluorescence emission in 530/30 channel is shown on the x-axis, and the cell count is shown on the y-axis; excitation - 488 nm laser. Typical results from three independent experiments are presented.
To further confirm these results, we used antibody against Ki-67 – a classic marker of cell proliferation that has been widely applied in cancer diagnostic and research [48]. We examined the proportion of uPAR-deficient and control cells, which entered the cell cycle after 72 and 96 hours. Ki-67 expression was determined using direct immunostaining followed by FACS analysis (Figure 6). After 72 hours the level of Ki-67 expression was lower in uPAR-deficient clones #6 and #30 (48.5±6.7% and 30.7±11.5%, correspondingly) compared to 82±1.6% in wt cells (p < 0.05, ANOVA) (Figure 6A). The decline in Ki-67 expression in clones #6 and #30 was also detected after 96 hours: 58.7±14.2% and 34.2±4.5% compared to 70.1±4.6 % Ki-67 level in wt cells (Figure 6B).
Due to the known effect of serum starvation on cancer cells in vitro resulting in their enhanced sensitization to standard chemotherapeutic agents [49], we performed the same experiment with Ki-67 cell staining and FACS analysis using low serum culture conditions (1% FBS). In low serum the difference in Ki-67 expression between uPAR-deficient clones and wt cells became even more pronounced (Figure 7). After 24 hours in culture, Ki-67 expression level in uPAR-deficient clones #6 and #30 was 33.5±5.9% and 39.8±2.3% Ki-67, respectively; while in wt cells Ki-67 was expressed in 85.4±2.0% of cells (p < 0.05, ANOVA) (Figure 7A). Further analysis revealed the gradual decline of Ki-67 expression in wt cells: from 85.4% after 24 hours to 40.8% after 72 hours in culture. Similarly, Ki-67 expression declined in uPAR-deficient clones from approximarely 30% (24 hours) to 12.5% (72 hours) (Figure 7B, 7C).
The difference in the amount of live cells in culture could be attributed not only to the decrease in cell proliferation rate but also to the induction of apoptosis. Actual execution of apoptosis depends upon activation of effector caspases, particularly caspase 3 [50]. Caspase 3 is primarily responsible for the cleavage of Poly (ADPribose) polymerase (PARP) during programmed cell death [51]. To test if uPAR knockout has any effect on the induction of apoptosis, we analysed the expression level of caspase 3 and the extent of PARP-1 cleavage in protein extracts using Western blot. We detected a significant increase of caspase 3 in uPAR-deficient clones. PARP-1 cleavage by caspase 3 (89 kDa fragment accumulation) was significantly higher in #6 and #30 clones compared to the control cells (Figure 5B).
uPAR-deficient Neuro 2A cells exhibit decreased expression of TrkC
Trk family kinases are known to be responsible for the regulation of neuroblastoma survival/apoptosis, proliferation and differentiation [11-14, 52, 53]. To identify the potential mechanisms responsible for reduced proliferation of uPAR-deficient Neuro 2A cells, we examined the mRNA expression level of Trks extracted from control cells and uPAR-deficient clones (#3, #6, #30). We also analysed the mRNA expression level of Nanog, the main pluripotency factor in Neuro 2A cells [54]. It appeared that uPAR knockout was accompanied by a 2.5-fold decrease in TrkC mRNA (Figure 8, Supplementary Figure 4), while no statistically significant difference in mRNA expression of TrkA, TrkB and p75NTR could be detected in uPAR-deficient cells compared to the control (Figure 8C, 8D, 8E). In addition, Nanog mRNA remained unchanged (data not shown). Given that the full-length and the truncated form of TrkC may be differently expressed in favourable and aggressive neuroblastomas [12, 52], we used primers designed to differentiate between the splice variants of the full-length TrkC (TrkC-FL) and truncated TrkC form (TrkC-Trunc) that lack tyrosine kinase domain (Figure 8F, 8G). No statistically significant difference could be detected in mRNA expression of TrkC-Trunc, while there was a 1.25-fold reduction in the functional TrkC-FL mRNA in uPAR-deficient clones.
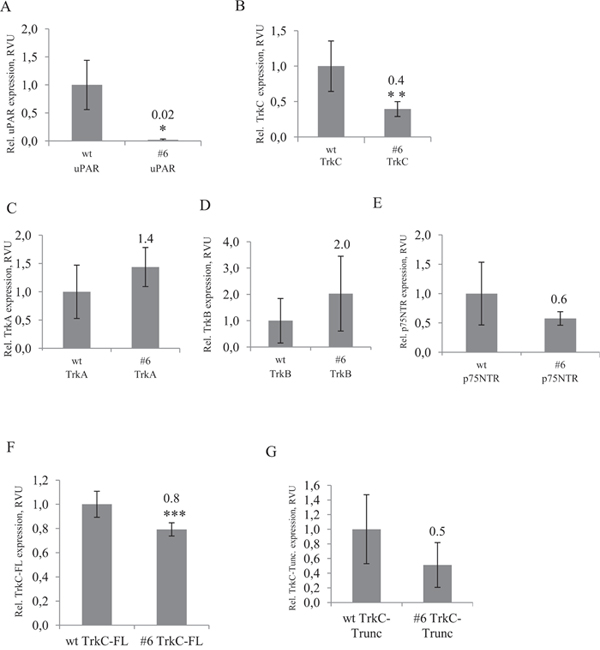
Figure 8: RT-PCR analysis of Trks mRNA expression shows significunt reduction of full length TrkC mRNA in uPAR-deficient cells. (A) uPAR expression in control Neuro2A and uPAR-deficient clone #6, (B) TrkC expression in control cells and #6 clone, (C) TrkA expression in control (wt) and #6 clone, (D) TrkB expression in control (wt) and #6 clone, (E) p75NTR expression in control (wt) and #6 clone, (F) TrkC-FL (full-length TrkC) expression in control (wt) and #6 clone, (G) TrkC-Trunc expression (truncated form of TrkC) in control (wt) and #6 clone. The data are presented as Mean ± SD (n=3 wells per group), * p < 0.006; ** p < 0.02; *** p < 0.05 (Student's t-test). The mRNA level was normalized to the expression of the housekeeping gene β-actin.
According to the classic neurotrophic view, TrkC interaction with its NT-3 ligand may result in activation of the PI3K/Akt, MAPK/Erk1/2 and p38MAPK signalling pathways in neuroblastoma cells [11, 52]. To identify critical signalling pathways involved in decreased neuroblastoma cell proliferation, we measured phosphorylation of Akt, Erk1/2 and p38MAPK using Western blot analysis. The representative results are shown in Figure 7A. There was significant decrease in p-Akt in uPAR deficient clones compared to the control: Ser473 1.8- and Thr308 3.3-fold decline in phosphorylation (Figure 9A, 9D, 9E). Erk1/2 phosphrylation level didn’t change significantly (Figure 9A and 9B), while there was approximately a 1.5-fold increase in p38MAPK phosphorylation in uPAR-deficient clones compared to the control (Figure 9A and 9C).
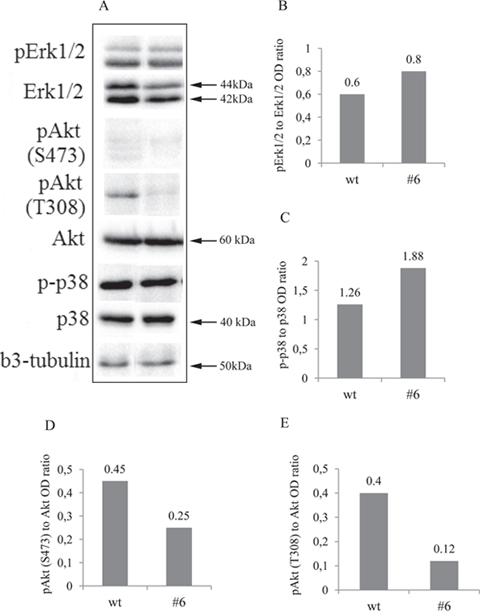
Figure 9: Western blot and densitometry analysis of Erk1/2, Akt, p38MAPK and their phosphorylated forms showed decreased activation of Akt and increased of p38MAPK. (A) Western blots showed a marked reduction of Akt phosphorylation on both, Thr308 and Ser473 residues and a slight increase in phospho-p38MAPK in uPAR-deficient clone #6 compared to the contol. Erk1/2 was not significuntly altered. β3-tubulin was used as loading control. Typical results from three independent experiments are presented. (B-E) densitometry analysis of the blots further confirming the obtained results.
DISCUSSION
Several reports on uPAR mRNA and uPAR protein expression, on kinetic analysis of uPA-uPAR binding, and on the implementation of RNAi and uPAR antisense-expressing plasmids to inhibit uPAR expression, point to the important role of uPAR in the progression of malignant tumors of the nervous system such as astrocytoma, neuroblastoma, glioma and glioblastoma [3, 16, 55, 56]. In the present study we used CRISPR/Cas9 technology to suppress uPAR in neuroblastoma cells in order to gain a better understanding of the mechanisms responsible for neuroblastoma proliferation. The designed gRNAs were proved to selectively target Plaur in Neuro 2A cells. Analysis of the sequence data revealed uPAR complete knockout in clone #6 and 75% suppression in clone #30 (Supplementary Figure 1). We also analyzed the possible CRISPR/Cas9 off-target sites. No mutations in the loci of the most likely off-target activity of the CRISPR/Cas9n system targeted by the chosen sgRNAs could be detected (Supplementary Figure 2).
Targeting of the Plaur results in significant reduction in proliferative activity of Neuro 2A cells with the maximum effect in uPAR-deficient clones (Figures 5, 6, and 7). Proliferation rate was evaluated using live cell counts and Ki-67 immunostaining followed by FACS. Ki-67 antigen is specifically expressed in proliferating cells in G1, S, G2 and M phase of the cell cycle, whereas it is absent from non-cycling cells in G0 [48]. Both methods demonstrated the decline in the amount of live cycling cells. We also addressed a question if uPAR knockout can induce apoptotic cell death. First, we showed accumulation of active caspase 3 in uPAR-deficient clones compared to the control. Caspase mediated apoptosis is accomplished through the cleavage of several key proteins required for cell survival and functioning. PARP-1 is known to be involved in preventing DNA degradation and resisting to apoptosis. Cleavage of PARP-1 by caspases is known to be a hallmark of apoptosis [51, 67]. Accumulation of 89 kDa fragment of PARP-1 was significantly elevated in #6 and #30 clones compared to the control cells (Figure 5B). These results indicate that uPAR knockout has an impact on neuroblastoma cell survival and proliferation, which is in accordance with the previously published results on reduced tumor growth and invasion upon uPAR downregulation in glioma, glioblastoma and other cancers [16, 22, 27, 35, 57]. However, adenoviral vectors and RNAi-based strategies used in these studies have serious disadvantages, such as transient expression and inability to integrate into the genome [58]. The CRISPR/Cas9 technology used in the current study lacks these shortcomings [44, 59] and could be a promising tool for genetic engineering and cancer studies in vitro and in vivo.
Trk receptors and their ligands are known to be involved in neuroblastoma proliferation, survival/apoptosis and differentiation [2]. High TrkA and TrkC expression levels are associated with favourable clinical and biological features of neuroblastoma [52, 60]. Tumors with high level of TrkA are often subjected to spontaneous regression. TrkA-expressing tumor cells in the presence of NGF undergo neuronal differentiation, while NGF deprivation leads to apoptosis [2, 14, 53]. Co-expression of p75NTR and TrkA in neuroblastoma cells is associated with good prognosis. Overexpression of p75NTR alone leads to apoptosis in line with p75NTR structural homology to the TNF-R/Fas family of death receptors [2, 61]. TrkB and BDNF co-expression is detected in more aggressive neuroblastomas especially with MYCN amplification [2, 14]. As opposed to TrkA and TrkB, TrkC is considered to be a dependence receptor: in the presence of NT3 a positive differentiation or survival signal is transduced, while in the absence of the ligand TrkC induces apoptosis [11]. In the earlier studies, using Northern blot analysis low levels of TrkA and TrkC in Neuro 2A cells were detected [62]; the expression of p75NTR in these cells was verified using immunostaining [63]. Later it has been reported that Neuro 2A cells express p75NTR, TrkB and TrkC, but no TrkA [14]. Our data indicate that mRNA of all Trk receptors (TrkA, TrkB, TrkC, p75NTR) can be detected in Neuro 2A cells. All neurotrophins (NGF, BDNF, NT3 NT4/5) are present in Neuro 2A cells and can stimulate cell survival and proliferation [14]. The reduced proliferation of uPAR-deficient Neuro 2A cells in our experimental settings correlates with a decrease in mRNA TrkC content (Figure 8B), probably resulting from a lack of survival signalling via TrkC.
Truncated forms of Trks have an important biological role in neuroblastoma behaviour. Expression of truncated TrkB, that is generated by alternative splicing and serves as a decoy for BDNF, correlates with a more favourable outcome in neuroblastoma patients [64]. In contrast to tumor-suppressing role of full-length TrkA, a constitutively-active TrkAIII splice form in primary neuroblastomas and in SH-SY5Y cell line, antagonizes NGF/TrkA signalling and promotes tumor progression [13]. A subset of unfavourable neuroblastomas expresses a truncated form of TrkC, lacking tyrosine kinase domain [52, 60]. The main function of truncated TrkC is the inhibition of full-length TrkC signalling, achieved via a ligand-sequestering or a dominant-negative mechanism. Given that the full-length TrkC is increased while truncated TrkC is decreased in human neuroblastomas upon exposure to retinoic acid that limits proliferation and induces differentiation [12], we compared mRNA expression of full-length TrkC and truncated TrkC in control and in uPAR-deficient clones (Figure 8). We used primers designed to differentiate between the splice variants of the full-length TrkC and truncated TrkC, described earlier [65]. Statistically significant difference between control and uPAR-deficient cells was detected only for full-length TrkC, but not for truncated TrkC (Figure 8F and 8G), thus further supporting the assumption of the uPAR-mediated TrkC signaling that tranduces pro-survival and proliferating stimuli in Neuro 2A cells.
The cytoplasmic part of Trk receptors recruits signaling complexes, which activate PI3K/Akt, MAPK/Erk1/2 and p38MAPK pathways [11, 14, 52, 66]. Akt pathway is considered to be a clinically relevant and promising target for neuroblastoma treatment [4, 67, 68]. Akt is a serine/threonine kinase that is often hyperactivated in aggressive neuroblastomas, primary and metastatic tumors and cell lines [4, 67–69]. Phosphorylation of Akt at Thr308 (catalytical domain) is necessary and sufficient for Akt activation, whereas phosphorylation at Ser473 (C-terminal regulatory domain) is not sufficient but is required for optimal Akt activation [70]. Akt Thr308 up-regulates mammalian target of rapamycin (mTORC1) and p70S6K, which enhance protein synthesis. Phosphorylation of Akt at Ser473 by mTORC2 promotes anti-apoptotic and cell survival pathways [4, 68, 69]. In vitro activation of PI3K/Akt pathway is associated with enhanced survival and proliferation of SH-SY5Y, SK-N-BE(2) and Neuro 2A neuroblastoma cell lines [69, 71], while in vivo PI3K/Akt inhibition reduces tumor growth and MYCN protein expression [72]. In the current study, the decrease in cell proliferation in uPAR deficient clones was accompanied by a significant (3.3-fold) reduction of Akt phosphorylation at Thr308 residue and a less pronounced (1.8-fold) decrease in Akt phosphorylation at Ser473 (Figure 9A and 9D); the total Akt content remained unchanged. The obtained data suggest that the uPAR-dependent reduction in Neuro 2A cell proliferation can be responsible for the decrease in Akt phosphorylation.
Activating mutations in Ras-MAPK signalling pathways have been predicted in relapsing neuroblastomas resistant to chemotherapy [73]. Proliferative activity of neuroblastoma NB69 cell line can be inhibited by application of p38MAPK pharmacological inhibitors [74]. A combination of p38MAPK inhibitors with etoposide, a standard chemotherapy agent, strongly increases the sensitivity of neuroblastoma to chemotherapy and allows lowering the drug dose and overall toxicity [75]. Apoptotic pathway, activated in neuroblastoma SH-SY5Y cell line upon exposure to H2O2 can be blocked by cell pretreatment with p38MAPK pharmacological inhibitors, indicating p38MAPK involvement in regulation of cell death/survival response [76]. In our experiments there was approximately a 1.5-fold increase in p38MAPK phosphorylation (Figure 9) suggesting its role in p38MAPK in proliferation/survival pathways.
In summary, in the current study we employed the CRISPR/Cas9n technology to target the Plaur gene in Neuro 2A neuroblastoma cells. We showed that CRISPR/Cas9n targeting of Plaur inhibited cell proliferation. The extent of uPAR downregulation correlated with decrease in full-length TrkC mRNA expression level and downstream intracellular signalling involving Akt and p38MAPK.
MATERIALS AND METHODS
Guide RNA design and molecular cloning of Plaur -specific CRISPR/CAS9n vectors
pX458nickase (D10A) plasmid was generated using pX458 vector (Addgene, # 48138) containing S. pyogenes Cas9 gene and EGFP gene. The point mutation of Asp10 → Ala in the RuvCI endonuclease domain of the Cas9 gene leads to an improved version of Cas9 – Cas9n nickase with enhanced genome editing specificity [43]. For targeting the sequences in the first exon and 5’-UTR of Plaur, a pair of gRNAs was chosen using the CRISPR/Cas9n-MIT web tool (http://crispr.mit.edu/) (Figure 1). The oligonucleotide sequences are as follows: sg1 (gRNA1 targeted to 5’-UTR of Plaur) 5’CACCGCTGCTCTAGACTGACCGAG 3’; sg1c (complementary to sg1) 5’ AAACCTCGGTCAGTCTAGAGCAGC 3’; sg2 (gRNA2 targeted to the first exon of Plaur) 5’CACCGACCCATGGGACTCCCAAGG3’; sg2c (complementary to sg2) 5’ AAACCCTTGGGAGTCCCATGGGTC 3’. The oligonucleotides sg1, sg1c, sg2, sg2c were phosphorylated, denatured and annealed according to the protocol [37]. The obtained 24 bp DNA duplexes were ligated using T4 DNA ligase (Thermo Scientific #EL0014) into linearized by BstV2I (SibEnzime # E298) pX458nickase vector. Colony PCR method was applied to determine the presence of the insert DNA in the transformed E.coli DH5α [77]. Vector specific primer and primers specific to the minus-strand of the DNA duplex (sg1c or sg2c) (https://www.neb.com/applications/cloning-and-synthetic-biology/dna-analysis/colony-pcr) were used. An amplicon of 700 bp length confirmed the correct orientation. Sequence verification was carried out using seqPrimer 5’ GTTTCGCCACCTCTGACTTGA 3’ (Evrogen).
Transfection of Neuro 2A cells, flow cytometry and cell sorting
Neuro 2A cells (ATCC® CCL-131™) were maintained in DMEM (PanEko) supplemented with 4.5g/L glucose (Hyclone), 10% FBS (Hyclone), 1% non-essential amino acids (Hyclone) and 100μg/ml penicillin/streptomycin (Gibco®) 5% CO2 at 37°C. Cells were transfected using Lipofectamine 2000 according to the manufacturer's protocol (Life Technologies). For effective uPAR suppression, we performed three consecutive co-transfection using pX458nickase-sg1 and pX458nickase-sg2 plasmids, followed by cell sorting.
For cell sorting, cells were detached by 0.25% Trypsin/EDTA solution (Gibco®), centrifuged at 250g for 5 min and re-suspended with DPBS (Dulbecco's Phosphate-Buffered Saline, PanEko) supplemented with 0.1% BSA. BD FACS Aria III cell sorter equipped with blue laser (488 nm) and BD FacsDiva software was used to select EGFP-positive cells. EGFP-enriched cell populations after the first, second and third sorting were marked s1, s2 and s3, respectively, and cultured for 2 days.
Cell surface expression of uPAR in s1, s2 and s3 cell populations was analyzed using flow cytometry. Cells were washed with DPBS, detached using Versene solution (PanEko), centrifuged at 250g for 5 min and re-suspended in 100 μl DPBS, containing 0.1% BSA. Cells were incubated sequentially with anti-uPAR antibody (SC-10815, Santa Cruz) and with secondary antibody (AlexaFluor 488, Jackson ImmuneResearch) at 4°C for 30 minutes. Non-specific IgG were used as a negative control. Cells were washed and re-suspended in 500μl DPBS/0.1% BSA.
Electrophoresis and Western blot analysis
Cells were lysed in RIPA buffer (50 мM HEPES, 100 мM NaCl, 1% sodium deoxycholate, 1% Triton X-100, 0,1% SDS, pH 7.2) containing 4% β-mercaptoethanol and Protease Inhibitor Cocktail (Thermo Scientific). Total protein content was quantified using Bradford assay. After separation by 10% SDS-PAGE electrophoresis, the proteins were transferred to PVDF membrane (GE Healthcare) in transfer buffer (1.92 M Tris/glycine buffer, 10% SDS and 20% methanol). Nonspecific binding was blocked in 5% non-fat dried milk in PBS buffer (Phosphate-Buffered Saline, Sigma), containing 0,1% Tween-20) overnight at 4°C. Proteins were probed with anti-uPAR (Santa Cruz), anti-Akt, anti-p (ser347)-Akt (Cell Signaling #587F11) and anti-p (Thr308)-Akt (Cell Signaling #9275), anti-p38MAPK (Cell Signalling #9212) and anti-phospho-p38MAPK (Abcam #ab32557), β3-tubulin (Santa Cruz #sc-51670), vinculin (Sigmaaldrich V9131), PARP-1 (Santa Cruz sc-7150), caspase 3 (Abcam #ab32351) primary antibodies (1:1000) at room temperature for 2 hours, followed by incubation with secondary antibodies (IMTEK), (1:3000). The proteins were detected using an enhanced chemiluminescence reagent kit (SuperSignal WestPico, Thermo Scientific) and a Chemi-Doc imaging system (Bio-Rad). The proteins were detected using an enhanced chemiluminescence reagent kit (SuperSignal WestPico, Thermo Scientific) and a ChemiDoc™ MP Imaging system (Bio-Rad). Densitometric analysis of blots was performed using ImageJ.
Quantitative real-time polymerase chain reaction analysis (RT-PCR)
Total RNA from Neuro 2A cells was extracted using an RNeasy® Mini Kit (Qiagen, Germany). To generate cDNA, 1μg of total RNA and MMLV RT kit (Evrogen, Moscow, Russia) was used. PCR was carried out using qPCRmix-HS SYBR (Evrogen, Russia) on a DT-96 real-time PCR device (DNA-technology, Russia). The thermal cycling program for template denaturation, primer annealing and primer extension was 94°C for 15 sec, 62°C for 15 sec and 72°C for 20 sec for 40 cycles, respectively. A relative transcript level of uPAR was calculated using the 2−ΔΔCt method. The following primers for murine uPAR, and β−actin [78], TrkA, TrkB, TrkC, TrkC-FL (full-length) and TrkC-Truncated [65], p75NTR, were obtained from Evrogen (Russia): uPAR-forward 5′-CGCCACAAACCTCTGCAAC-3′, uPAR-reverse 5′-CTCTGTAGGATAGCGGCATTG-3′, β-actin-forward 5′-AGTGTGACGTTGACATCCGTA-3′, β-actin-reverse 5′-GCCAGAGCAGTAATCTCCTTCT-3′, TrkA forward 5’-GCCTAACCATCGTGAAGAGTG-3’, TrkA reverse 5’-CCAACGCATTGGAGGACAGAT-3’, TrkB forward 5’-TGGGACGTTGGGAATTTGGT-3’, TrkB reverse 5’-AGTTGGCGGAAAAAGCACAG-3’, TrkC forward 5’-AGCCACGTCAACCTGACTG-3’, TrkC reverse 5’-CCTCGCTCGTCACGTTCAC-3’, TrkC-FL forward 5’-TGATCCTCGTGGATGGACAG-3’, TrkC-FL reverse 5’-CTTCACTAGTAGATTGGCTCC-3’, TrkC-Truncated forward 5’-CCACTTCCTGAAGGAGCCCT-3’, TrkC-Truncated reverse 5’-CCCACTCTGGACCTCAGGTT-3’, p75NTR forward 5’-ACCCTGCCTGGACAGTGTTA-3’, p75NTR reverse 5’-AGAACACGAGTCCTGAGCCC-3’.
Neuro 2A cell proliferation analysis in vitro
For cell counting analysis, Neuro 2A cells were seeded into 2x12-well at 4x104 cells/well. The cell number was calculated every 24 hours for each cell type and time point using an automatic Countess® Automated Cell Counter (Thermo Fisher Scientific). The proliferation curves were plotted based on the obtained data. Data are presented as the dependence of total cell number on the time interval. The experiment was performed in 2 parallels and repeated three times. Data are presented as the mean ± standard error of the mean, p < 0.05.
Neuro 2A cells were fixed using Fixation&Permeabilization Reagents (eBioscience™) and immunostained with monoclonal antibody (Abcam ab15580) against Ki-67. The percentage of Ki-67-positive Neuro 2A cells in wt culture and uPAR-deficient clones (#6, #30) was analysed using FACS BD FACSCanto™ II Flow Cytometry System. The amount of Ki-67-positive cells, cultured in standard culture media and in low serum conditions (1% FBS), were evaluated after 24, 72 and 96 hours. Data are presented as the mean ± standard deviation, p < 0.05.
Statistical analysis
Statistical analysis was carried out using statistics program STATISTICA. For pairwise comparisons in case of a normal value distribution, the Student's t-test was implemented; in other cases the Mann-Whitney rank-sum test was used. For multiple comparisons, data were tested for significance using ANOVA (Newman-Keuls test). Differences were considered statistically significant when P < 0.05 was achieved.
Abbreviations
BSA: Bovine serum albumin; CRISPR: clustered regularly interspaced short palindromic repeats; EGFP: enhanced green fluorescent protein; gRNA: guidance RNA; siRNA: small interfering RNA; uPA: urokinase plasminogen activator; uPAR: uPA receptor; Plaur: uPAR gene; 5’-UTR: the untranslated 5' terminal region of the mRNA.
Authors’ contributions
Rysenkova K. performed the experiments and analysed the data; Semina E. conceived and planned the experiments, contributed to data analysis and interpretation; Rubtsov Y. and Karagyaur M. carried out the guide RNA design and molecular cloning of CRISPR/CAS9n vectors; Shmakova A., Dyikanov D., Vasiluev P. contributed to the data acquisition; Rubina K. wrote the manuscript and contributed to data analysis and interpretation; Tkachuk V. made a substantial contribution to the conception of the manuscript. All of the authors approved the final version of the manuscript.
CONFLICTS OF INTEREST
The authors declare no competing interests.
FUNDING
Source of funding: Grant №14-24-00086 of the Russian Science Foundation and Grant №14-50-00029 of the Russian Foundation of Basic Research. The study was conducted using biomaterials collected and preserved in the frame of the project “Scientific basis for national bank-depositary of living systems” (RSF agreement #14-50-00029) using the equipment purchased as a part of Moscow State University Program of Development.
REFERENCES
1. Gatta G, Botta L, Rossi S, Aareleid T, Bielska-Lasota M, Clavel J, Dimitrova N, Jakab Z, Kaatsch P, Lacour B, Mallone S, Marcos-Gragera R, Minicozzi P, et al. Childhood cancer survival in Europe 1999-2007: results of EUROCARE-5-a population-based study. Lancet Oncol. 2014; 15: 35-47. https://doi.org/10.1016/S1470-2045(13)70548-5.
2. Brodeur GM, Bagatell R. Mechanisms of neuroblastoma regression. Nat Rev Clin Oncol. 2014; 11: 704-13. https://doi.org/10.1038/nrclinonc.2014.168.
3. Li P, Gao Y, Ji Z, Zhang X, Xu Q, Li G, Guo Z, Zheng B, Guo X. Role of urokinase plasminogen activator and its receptor in metastasis and invasion of neuroblastoma. J Pediatr Surg. 2004; 39: 1512-9. https://doi.org/10.1016/j.jpedsurg.2004.06.011.
4. Opel D, Poremba C, Simon T, Debatin KM, Fulda S. Activation of Akt predicts poor outcome in neuroblastoma. Cancer Res. 2007; 67: 735-45. https://doi.org/10.1158/0008-5472.CAN-06-2201.
5. Sidell N. Retinoic acid-induced growth inhibition and morphologic differentiation of human neuroblastoma cells in vitro. J Natl Cancer Inst. 1982; 68: 589-96. http://www.ncbi.nlm.nih.gov/pubmed/7040765.
6. Han S, Wada RK, Sidell N. Differentiation of human neuroblastoma by phenylacetate is mediated by peroxisome proliferator-activated receptor gamma. Cancer Res. 2001; 61: 3998-4002. http://cancerres.aacrjournals.org/content/61/10/3998.long.
7. Ciani E, Severi S, Contestabile A, Bartesaghi R. Nitric oxide negatively regulates proliferation and promotes neuronal differentiation through N-Myc downregulation. J Cell Sci. 2004; 117: 4727-37. https://doi.org/10.1242/jcs.01348.
8. Qiao J, Paul P, Lee S, Qiao L, Josifi E, Tiao JR, Chung DH. PI3K/AKT and ERK regulate retinoic acid-induced neuroblastoma cellular differentiation. Biochem Biophys Res Commun. 2012; 424: 421-6. https://doi.org/10.1016/j.bbrc.2012.06.125.
9. Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003; 3: 203-16. https://doi.org/10.1038/nrc1014.
10. Pahlman S, Hoehner JC, Nanberg E, Hedborg F, Fagerstrom S, Gestblom C, Johansson I, Larsson U, Lavenius E, Ortoft E, Söderholm H. Differentiation and survival influences of growth factors in human neuroblastoma. Eur J Cancer. 1995; 31A: 453-8. https://doi.org/10.1016/0959-8049(95)00033-F.
11. Bouzas-Rodriguez J, Cabrera JR, Delloye-Bourgeois C, Ichim G, Delcros JG, Raquin MA, Rousseau R, Combaret V, Benard J, Tauszig-Delamasure S, Mehlen P. Neurotrophin-3 production promotes human neuroblastoma cell survival by inhibiting TrkC-induced apoptosis. J Clin Invest. 2010; 120: 850-8. https://doi.org/10.1172/JCI41013.
12. Guidi M, Muinos-Gimeno M, Kagerbauer B, Marti E, Estivill X, Espinosa-Parrilla Y. Overexpression of miR-128 specifically inhibits the truncated isoform of NTRK3 and upregulates BCL2 in SH-SY5Y neuroblastoma cells. BMC Mol Biol. 2010; 11: 95. https://doi.org/10.1186/1471-2199-11-95.
13. Tacconelli A, Farina AR, Cappabianca L, Desantis G, Tessitore A, Vetuschi A, Sferra R, Rucci N, Argenti B, Screpanti I, Gulino A, Mackay AR. TrkA alternative splicing: a regulated tumor-promoting switch in human neuroblastoma. Cancer Cell. 2004; 6: 347-60. https://doi.org/10.1016/j.ccr.2004.09.011.
14. Evangelopoulos ME, Weis J, Kruttgen A. Neurotrophin effects on neuroblastoma cells: correlation with trk and p75NTR expression and influence of Trk receptor bodies. J Neurooncol. 2004; 66: 101-10. https://doi.org/10.1023/B:NEON.0000013492.37426.0c.
15. Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001; 24: 677-736. https://doi.org/10.1146/annurev.neuro.24.1.677.
16. Lakka SS, Gondi CS, Dinh DH, Olivero WC, Gujrati M, Rao VH, Sioka C, Rao JS. Specific interference of urokinase-type plasminogen activator receptor and matrix metalloproteinase-9 gene expression induced by double-stranded RNA results in decreased invasion, tumor growth, and angiogenesis in gliomas. J Biol Chem. 2005; 280: 21882-92. https://doi.org/10.1074/jbc.M408520200.
17. Rubina KA, Sysoeva VY, Zagorujko EI, Tsokolaeva ZI, Kurdina MI, Parfyonova YV, Tkachuk VA. Increased expression of uPA, uPAR, and PAI-1 in psoriatic skin and in basal cell carcinomas. Arch Dermatol Res. 2017; 309: 433-42. https://doi.org/10.1007/s00403-017-1738-z.
18. Swiercz R, Wolfe JD, Zaher A, Jankun J. Expression of the plasminogen activation system in kidney cancer correlates with its aggressive phenotype. Clin Cancer Res. 1998; 4: 869-77. http://www.ncbi.nlm.nih.gov/pubmed/9563880.
19. Mohan PM, Chintala SK, Mohanam S, Gladson CL, Kim ES, Gokaslan ZL, Lakka SS, Roth JA, Fang B, Sawaya R, Kyritsis AP, Rao JS. Adenovirus-mediated delivery of antisense gene to urokinase-type plasminogen activator receptor suppresses glioma invasion and tumor growth. Cancer Res. 1999; 59: 3369-73.
20. Margheri F, Luciani C, Taddei ML, Giannoni E, Laurenzana A, Biagioni A, Chilla A, Chiarugi P, Fibbi G, Del Rosso M. The receptor for urokinase-plasminogen activator (uPAR) controls plasticity of cancer cell movement in mesenchymal and amoeboid migration style. Oncotarget. 2014; 5: 1538-53. https://doi.org/10.18632/oncotarget.1754.
21. Laurenzana A, Biagioni A, D'Alessio S, Bianchini F, Chilla A, Margheri F, Luciani C, Mazzanti B, Pimpinelli N, Torre E, Danese S, Calorini L, Del Rosso M, et al. Melanoma cell therapy: Endothelial progenitor cells as shuttle of the MMP12 uPAR-degrading enzyme. Oncotarget. 2014; 5: 3711-27. https://doi.org/10.18632/oncotarget.1987.
22. Montuori N, Pesapane A, Rossi FW, Giudice V, De Paulis A, Selleri C, Ragno P. Urokinase type plasminogen activator receptor (uPAR) as a new therapeutic target in cancer. Transl Med UniSa. 2016; 15: 15-21. http://www.ncbi.nlm.nih.gov/pubmed/27896223.
23. Duffy MJ, McGowan PM, Harbeck N, Thomssen C, Schmitt M. uPA and PAI-1 as biomarkers in breast cancer: validated for clinical use in level-of-evidence-1 studies. Breast Cancer Res. 2014; 16: 428. https://doi.org/10.1186/s13058-014-0428-4.
24. Ciavarella S, Laurenzana A, De Summa S, Pilato B, Chilla A, Lacalamita R, Minoia C, Margheri F, Iacobazzi A, Rana A, Merchionne F, Fibbi G, Del Rosso M, et al. u-PAR expression in cancer associated fibroblast: new acquisitions in multiple myeloma progression. BMC Cancer. 2017; 17: 215. https://doi.org/10.1186/s12885-017-3183-y.
25. Heissig B, Dhahri D, Eiamboonsert S, Salama Y, Shimazu H, Munakata S, Hattori K. Role of mesenchymal stem cell-derived fibrinolytic factor in tissue regeneration and cancer progression. Cell Mol Life Sci. 2015; 72: 4759-70. https://doi.org/10.1007/s00018-015-2035-7.
26. Festuccia C, Dolo V, Guerra F, Violini S, Muzi P, Pavan A, Bologna M. Plasminogen activator system modulates invasive capacity and proliferation in prostatic tumor cells. Clin Exp Metastasis. 1998; 16: 513-28. https://doi.org/10.1023/A:1006590217724.
27. Boonstra MC, Verspaget HW, Ganesh S, Kubben FJ, Vahrmeijer AL, van de Velde CJ, Kuppen PJ, Quax PH, Sier CF. Clinical applications of the urokinase receptor (uPAR) for cancer patients. Curr Pharm Des. 2011; 17: 1890-910. https://doi.org/10.2174/138161211796718233.
28. Raghu H, Gondi CS, Dinh DH, Gujrati M, Rao JS. Specific knockdown of uPA/uPAR attenuates invasion in glioblastoma cells and xenografts by inhibition of cleavage and trafficking of Notch -1 receptor. Mol Cancer. 2011; 10: 130. https://doi.org/10.1186/1476-4598-10-130.
29. Raghu H, Nalla AK, Gondi CS, Gujrati M, Dinh DH, Rao JS. uPA and uPAR shRNA inhibit angiogenesis via enhanced secretion of SVEGFR1 independent of GM-CSF but dependent on TIMP-1 in endothelial and glioblastoma cells. Mol Oncol. 2012; 6: 33-47. https://doi.org/10.1016/j.molonc.2011.11.008.
30. Miyake H, Hara I, Yamanaka K, Gohji K, Arakawa S, Kamidono S. Elevation of serum levels of urokinase-type plasminogen activator and its receptor is associated with disease progression and prognosis in patients with prostate cancer. Prostate. 1999; 39: 123-39. https://doi.org/10.1002/(SICI)1097-0045(19990501)39:2<123::AID-PROS7>3.0.CO;2-2.
31. Tecimer C, Doering DL, Goldsmith LJ, Meyer JS, Abdulhay G, Wittliff JL. Clinical relevance of urokinase-type plasminogen activator, its receptor and inhibitor type 1 in ovarian cancer. Int J Gynecol Cancer. 2000; 10: 372-81. https://doi.org/10.1046/j.1525-1438.2000.010005372.x.
32. Borstnar S, Vrhovec I, Svetic B, Cufer T. Prognostic value of the urokinase-type plasminogen activator, and its inhibitors and receptor in breast cancer patients. Clin Breast Cancer. 2002; 3: 138-46. https://doi.org/10.3816/CBC.2002.n.018.
33. Seetoo DQ, Crowe PJ, Russell PJ, Yang JL. Quantitative expression of protein markers of plasminogen activation system in prognosis of colorectal cancer. J Surg Oncol. 2003; 82: 184-93. https://doi.org/10.1002/jso.10210.
34. Okusa Y, Ichikura T, Mochizuki H, Shinomiya N. Urokinase type plasminogen activator and its receptor regulate the invasive potential of gastric cancer cell lines. Int J Oncol. 2000; 17: 1001-15. https://doi.org/10.3892/ijo.17.5.1001.
35. Gondi CS, Lakka SS, Dinh DH, Olivero WC, Gujrati M, Rao JS. Downregulation of uPA, uPAR and MMP-9 using small, interfering, hairpin RNA (siRNA) inhibits glioma cell invasion, angiogenesis and tumor growth. Neuron Glia Biol. 2004; 1: 165-76. https://doi.org/10.1017/s1740925x04000237.
36. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013; 339: 823-36. https://doi.org/10.1126/science.1232033.
37. Wu Y, Liang D, Wang Y, Bai M, Tang W, Bao S, Yan Z, Li D, Li J. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell. 2013; 13: 659-62. https://doi.org/10.1016/j.stem.2013.10.016.
38. Nemudryi AA, Valetdinova KR, Medvedev SP, Zakian SM. TALEN and CRISPR/Cas Genome Editing Systems: Tools of Discovery. Acta Naturae. 2014; 6: 19-40. http://www.ncbi.nlm.nih.gov/pubmed/25349712.
39. Canver MC, Bauer DE, Dass A, Yien YY, Chung J, Masuda T, Maeda T, Paw BH, Orkin SH. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem. 2014; 289: 21312-24. https://doi.org/10.1074/jbc.M114.564625.
40. Sakuma T, Nishikawa A, Kume S, Chayama K, Yamamoto T. Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci Rep. 2014; 4: 5400. https://doi.org/10.1038/srep05400.
41. Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014; 343: 84-7. https://doi.org/10.1126/science.1247005.
42. Kabadi AM, Ousterout DG, Hilton IB, Gersbach CA. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res. 2014; 42: e147. https://doi.org/10.1093/nar/gku749.
43. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013; 8: 2281-308. https://doi.org/10.1038/nprot.2013.143.
44. Chen S, Sun H, Miao K, Deng CX. CRISPR-Cas9: from Genome Editing to Cancer Research. Int J Biol Sci. 2016; 12: 1427-36. https://doi.org/10.7150/ijbs.17421.
45. Wang M, Zuris JA, Meng F, Rees H, Sun S, Deng P, Han Y, Gao X, Pouli D, Wu Q, Georgakoudi I, Liu DR, Xu Q. Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc Natl Acad Sci U S A. 2016; 113: 2868-73. https://doi.org/10.1073/pnas.1520244113
46. Cui Y, Wu BO, Flamini V, Evans BAJ, Zhou D, Jiang WG. Knockdown of EPHA1 Using CRISPR/CAS9 Suppresses Aggressive Properties of Ovarian Cancer Cells. Anticancer Res. 2017; 37: 4415-24. https://doi.org/10.21873/anticanres.11836.
47. Cyranoski D. Chinese scientists to pioneer first human CRISPR trial. Nature. 2016; 535: 476-7. https://doi.org/10.1038/nature.2016.20302.
48. Bologna-Molina R, Mosqueda-Taylor A, Molina-Frechero N, Mori-Estevez AD, Sanchez-Acuna G. Comparison of the value of PCNA and Ki-67 as markers of cell proliferation in ameloblastic tumors. Med Oral Patol Oral Cir Bucal. 2013; 18: 174-9. https://doi.org/10.4317/medoral.18573.
49. Shi Y, Felley-Bosco E, Marti TM, Orlowski K, Pruschy M, Stahel RA. Starvation-induced activation of ATM/Chk2/p53 signaling sensitizes cancer cells to cisplatin. BMC Cancer. 2012; 12: 571. https://doi.org/10.1186/1471-2407-12-571.
50. ten Berge RL, Meijer CJ, Dukers DF, Kummer JA, Bladergroen BA, Vos W, Hack CE, Ossenkoppele GJ, Oudejans JJ. Expression levels of apoptosis-related proteins predict clinical outcome in anaplastic large cell lymphoma. Blood. 2002; 99: 4540-6. https://doi.org/10.1182/blood.V99.12.4540.
51. Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, Munday NA, Raju SM, Smulson ME, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995; 376: 37-43. https://doi.org/10.1038/376037a0.
52. Ryden M, Ibanez CF. Binding of neurotrophin-3 to p75LNGFR, TrkA, and TrkB mediated by a single functional epitope distinct from that recognized by trkC. J Biol Chem. 1996; 271: 5623-7. https://doi.org/10.1074/jbc.271.10.5623.
53. Hoehner JC, Olsen L, Sandstedt B, Kaplan DR, Pahlman S. Association of neurotrophin receptor expression and differentiation in human neuroblastoma. Am J Pathol. 1995; 147: 102-13 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1869894/.
54. Monajemzadeh M, Soleimani V, Vasei M, Koochakzadeh L, Karbakhsh M. Expression and prognostic significance of Oct4 and Nanog in neuroblastoma. APMIS. 2014; 122: 734-41. https://doi.org/10.1111/apm.12207.
55. Go Y, Chintala SK, Mohanam S, Gokaslan Z, Venkaiah B, Bjerkvig R, Oka K, Nicolson GL, Sawaya R, Rao JS. Inhibition of in vivo tumorigenicity and invasiveness of a human glioblastoma cell line transfected with antisense uPAR vectors. Clin Exp Metastasis. 1997; 15: 440-56. https://doi.org/10.1023/A:1018410523635.
56. Mohanam S, Chintala SK, Go Y, Bhattacharya A, Venkaiah B, Boyd D, Gokaslan ZL, Sawaya R, Rao JS. In vitro inhibition of human glioblastoma cell line invasiveness by antisense uPA receptor. Oncogene. 1997; 14: 1351-9. https://doi.org/10.1038/sj.onc.1200963.
57. Lakka SS, Rajagopal R, Rajan MK, Mohan PM, Adachi Y, Dinh DH, Olivero WC, Gujrati M, Ali-Osman F, Roth JA, Yung WK, Kyritsis AP, Rao JS. Adenovirus-mediated antisense urokinase-type plasminogen activator receptor gene transfer reduces tumor cell invasion and metastasis in non-small cell lung cancer cell lines. Clin Cancer Res. 2001; 7: 1087-93. http://www.ncbi.nlm.nih.gov/pubmed/11309361
58. Boutros M, Ahringer J. The art and design of genetic screens: RNA interference. Nat Rev Genet. 2008; 9: 554-66. https://doi.org/10.1038/nrg2364.
59. Mehrabian M, Brethour D, MacIsaac S, Kim JK, Gunawardana CG, Wang H, Schmitt-Ulms G. CRISPR-Cas9-based knockout of the prion protein and its effect on the proteome. PLoS One. 2014; 9: e114594. https://doi.org/10.1371/journal.pone.0114594.
60. Svensson T, Ryden M, Schilling FH, Dominici C, Sehgal R, Ibanez CF, Kogner P. Coexpression of mRNA for the full-length neurotrophin receptor trk-C and trk-A in favourable neuroblastoma. Eur J Cancer. 1997; 33: 2058-63. https://doi.org/10.1016/S0959-8049(97)00290-6.
61. Kuner P, Hertel C. NGF induces apoptosis in a human neuroblastoma cell line expressing the neurotrophin receptor p75NTR. J Neurosci Res. 1998; 54: 465-74. https://doi.org/10.1002/(SICI)1097-4547(19981115)54:4<465::AID-JNR4>3.0.CO;2-T.
62. Pastor R, Bernal J, Rodriguez-Pena A. Unliganded c-erbA/thyroid hormone receptor induces trkB expression in neuroblastoma cells. Oncogene. 1994; 9: 1081-89. http://www.ncbi.nlm.nih.gov/pubmed/8134111.
63. Haapasalo A, Saarelainen T, Moshnyakov M, Arumae U, Kiema TR, Saarma M, Wong G, Castren E. Expression of the naturally occurring truncated trkB neurotrophin receptor induces outgrowth of filopodia and processes in neuroblastoma cells. Oncogene. 1999; 18: 1285-96. https://doi.org/10.1038/sj.onc.1202401.
64. Aoyama M, Asai K, Shishikura T, Kawamoto T, Miyachi T, Yokoi T, Togari H, Wada Y, Kato T, Nakagawara A. Human neuroblastomas with unfavorable biologies express high levels of brain-derived neurotrophic factor mRNA and a variety of its variants. Cancer Lett. 2001; 164: 51-60. https://doi.org/10.1016/S0304-3835(00)00715-1.
65. Brahimi F, Maira M, Barcelona PF, Galan A, Aboulkassim T, Teske K, Rogers ML, Bertram L, Wang J, Yousefi M, Rush R, Fabian M, Cashman N, et al. The Paradoxical Signals of Two TrkC Receptor Isoforms Supports a Rationale for Novel Therapeutic Strategies in ALS. PLoS One. 2016; 11: e0162307. https://doi.org/10.1371/journal.pone.0162307.
66. Sartelet H, Oligny LL, Vassal G. AKT pathway in neuroblastoma and its therapeutic implication. Expert Rev Anticancer Ther. 2008; 8: 757-69. https://doi.org/10.1586/14737140.8.5.757.
67. Smith JR, Moreno L, Heaton SP, Chesler L, Pearson AD, Garrett MD. Novel pharmacodynamic biomarkers for MYCN protein and PI3K/AKT/mTOR pathway signaling in children with neuroblastoma. Mol Oncol. 2016; 10: 538-52. https://doi.org/10.1016/j.molonc.2015.11.005.
68. Sartelet H, Rougemont AL, Fabre M, Castaing M, Duval M, Fetni R, Michiels S, Beaunoyer M, Vassal G. Activation of the phosphatidylinositol 3'-kinase/AKT pathway in neuroblastoma and its regulation by thioredoxin 1. Hum Pathol. 2011; 42: 1727-39. https://doi.org/10.1016/j.humpath.2011.01.019.
69. Navratilova J, Karasova M, Kohutkova Lanova M, Jirakova L, Budkova Z, Pachernik J, Smarda J, Benes P. Selective elimination of neuroblastoma cells by synergistic effect of Akt kinase inhibitor and tetrathiomolybdate. J Cell Mol Med. 2017; 21: 1859-69. https://doi.org/10.1111/jcmm.13106.
70. Welch H, Eguinoa A, Stephens LR, Hawkins PT. Protein kinase B and rac are activated in parallel within a phosphatidylinositide 3OH-kinase-controlled signaling pathway. J Biol Chem. 1998; 273: 48-56. https://doi.org/10.1074/jbc.273.18.11248.
71. Gomez-Villafuertes R, Garcia-Huerta P, Diaz-Hernandez JI, Miras-Portugal MT. PI3K/Akt signaling pathway triggers P2X7 receptor expression as a pro-survival factor of neuroblastoma cells under limiting growth conditions. Sci Rep. 2015; 5: 18417. https://doi.org/10.1038/srep18417.
72. Chesler L, Schlieve C, Goldenberg DD, Kenney A, Kim G, McMillan A, Matthay KK, Rowitch D, Weiss WA. Inhibition of phosphatidylinositol 3-kinase destabilizes Mycn protein and blocks malignant progression in neuroblastoma. Cancer Res. 2006; 66: 8139-46. https://doi.org/10.1158/0008-5472.CAN-05-2769.
73. Eleveld TF, Oldridge DA, Bernard V, Koster J, Daage LC, Diskin SJ, Schild L, Bentahar NB, Bellini A, Chicard M, Lapouble E, Combaret V, Legoix-Ne P, et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet. 2015; 47: 864-71. https://doi.org/10.1038/ng.3333.
74. Martinez MA, Ubeda A, Moreno J, Trillo MA. Power Frequency Magnetic Fields Affect the p38 MAPK-Mediated Regulation of NB69 Cell Proliferation Implication of Free Radicals. Int J Mol Sci. 2016; 17: 510. https://doi.org/10.3390/ijms17040510.
75. Marengo B, De Ciucis CG, Ricciarelli R, Furfaro AL, Colla R, Canepa E, Traverso N, Marinari UM, Pronzato MA, Domenicotti C. p38MAPK inhibition: a new combined approach to reduce neuroblastoma resistance under etoposide treatment. Cell Death Dis. 2013; 4: e589. https://doi.org/10.1038/cddis.2013.118.
76. Filomeni G, Piccirillo S, Rotilio G, Ciriolo MR. p38(MAPK) and ERK1/2 dictate cell death/survival response to different pro-oxidant stimuli via p53 and Nrf2 in neuroblastoma cells SH-SY5Y. Biochem Pharmacol. 2012; 83: 1349-57. https://doi.org/10.1016/j.bcp.2012.02.003.
77. Packeiser H, Lim C, Balagurunathan B, Wu J, Zhao H. An extremely simple and effective colony PCR procedure for bacteria, yeasts, and microalgae. Appl Biochem Biotechnol. 2013; 169: 695-700. https://doi.org/10.1007/s12010-012-0043-8.
78. Semina E, Rubina K, Sysoeva V, Rysenkova K, Klimovich P, Plekhanova O, Tkachuk V. Urokinase and urokinase receptor participate in regulation of neuronal migration, axon growth and branching. Eur J Cell Biol. 2016; 95: 295-310. https://doi.org/10.1016/j.ejcb.2016.05.003.