
INTRODUCTION
Canine cancers represent one of the best animal models of human cancers [1–4], because of the shared biology (e.g., intact immune system), physiology, living environment and clinical symptoms between the two species. Indeed, genomic studies from our group and others have revealed a high degree of molecular homology for histopathologically matched cancer types/subtypes between the dog and the human [5–7]. For example, the stepwise model of human colorectal tumorigenesis [8] also applies to spontaneous colorectal tumors in pet dogs [5, 9]. Furthermore, our group has successfully developed a novel dog-human comparative genomics and oncology strategy for driver-passenger discrimination, a central aim of cancer research [10], for colorectal cancer (CRC) copy number alteration [11, 12].
In humans, individuals with classic familial adenomatous polyposis (FAP) differ from the general population [13, 14]. These individuals develop significantly more (>100) adenomatous polyps in their colon, beginning at much younger age (age 16 on average). They also develop polyps in the small intestine and other places [15, 16]. The underlying pathogenic mechanism is well studied [8, 13, 14]. Most classic FAP patients inherit a dysfunctional copy of the APC gene, and then, a second mutation inactivates the other functional copy of APC. This results in translocation of β-catenin into the nucleus, activating WNT/β-catenin targets (e.g., MYC) [17] and accelerating cell proliferation. Furthermore, defective APC also interferes with cell adhesion, cytoskeleton and the establishment of epithelial apical-basolateral polarity. All these lead to extreme colon polyposis. Other variants of FAP include attenuated FAP, which is also APC mutation-associated but the patients typically develop polyps at older age, and autosomal recessive FAP, which is MUTYH mutation-associated and the patients develop fewer polyps. Hereditary nonpolyposis colorectal cancer (HNPCC), another inherited condition, is caused by mutations of DNA mismatch repair genes [13, 14, 18–20] and others [21].
Extreme intestinal polyposis in pet dogs has not yet been reported in literature, and the underlying pathogenic mechanism is unknown. We are fortunate to identify such a case. We set out to molecularly characterize this rare canine condition and compare our findings with those of human studies, as described below.
RESULTS
N14-77 represents a rare canine case of extreme intestinal polyposis
A rare canine case of extreme intestinal polyposis (Figure 1) was diagnosed at the Texas A&M University Veterinary Medical Teaching Hospital, and assigned “N14-77” as the case identifier. The detailed case information is provided in Supplementary Information and summarized below.
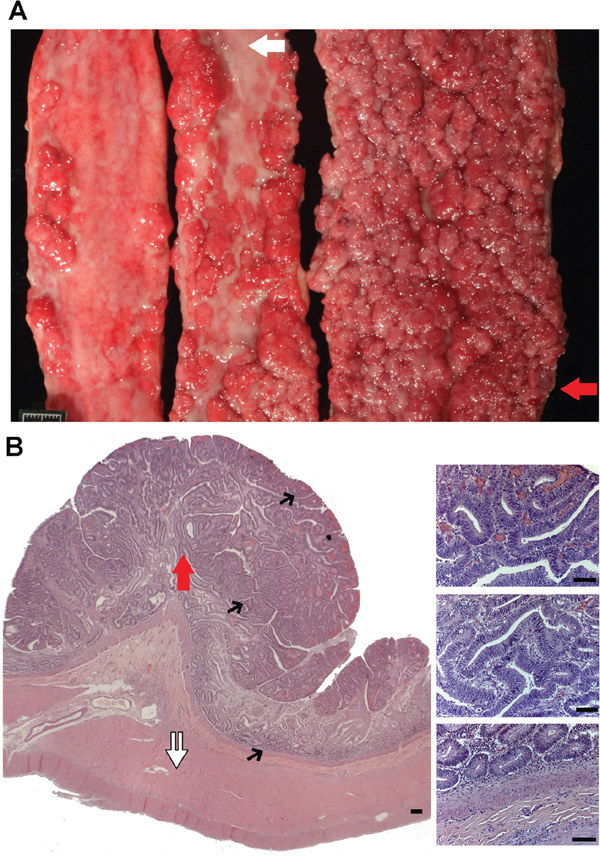
Figure 1: N14-77 represents a rare case of extreme intestinal polyposis in the dog. (A) Opened small intestinal segments from left to right are from the proximal jejunum, middle jejunum and distal jejunum-ileum junction, respectively. The red arrow indicates the area used for polyp dissection and sequencing (WGS and RNA-seq). The white arrow illustrates an unaffected inter-polyp region used for normal sample WGS. The scale bar is 1cm-long. (B) Representative H&E images of the distal jejunum-ileum junction indicate extensive cell proliferation and no invasion of proliferating enterocytes into the lamina propria or submucosa. The white double arrow exemplifies unaffected submucosa and muscularis propria tissue being dissected for normal sample RNA-seq. Images on the right are blowups of the corresponding sites pointed by black arrows on the left. Scale bar, 50μm.
At presentation, the N14-77 patient, a 9-year-old neutered male dog of Golden Retriever-mix, had a two-month history of blood-tinged, watery diarrhea and was in poor body condition. Complete blood count revealed a microcytic, hypochromic, regenerative anemia with a severe neutrophilia and hypoalbuminemia. Abdominal radiographs and ultrasounds indicated extensive intestinal changes. A rectal scraping found numerous, degenerate neutrophils containing phagocytosed bacteria and small yeast.
Euthanasia was selected. A full necropsy indicated that, while no significant abnormalities in other organ systems, about 70% of the small intestinal mucosa was affected. Specifically, intestine, extending primarily from the mid-jejunum to the ileocecal junction, was severely thickened by innumerable, 3 mm to 1.1 cm, firm nodules that progressively coalesced into large, plaque-like, 10-30 cm-long areas with a red, granular surface. The most severe region located at the distal jejunum-ileum junction (Figure 1A).
Histologic examination indicated numerous single to coalescing polyps within the mucosa of sections from the jejunum to the proximal colon, and the epithelium from crypts to mucosal surface was uniformly hyperplastic (Figure 1B). The mucosa comprising the inter-polyp regions and within the distal colon also displayed mild to moderate hyperplasia, with variable neutrophilic infiltration and mild enterocolitis. Notably, neither malignant neoplastic transformation of nor invasion of the lamina propria by enterocytes lining the intestinal villi, crypts, or colonic glands was observed (Figure 1B).
Except for the location (extending primary from the mid-jejunum to the ileocecal junction and with the distal jejunum-ileum junction being the most affected), the severity of polyposis in N14-77 resembles classic FAP patients in humans.
We performed whole genome sequencing (WGS) and RNA-seq
To characterize N14-77, we performed WGS and RNA-seq analyses with frozen polyp and normal (or rather unaffected) samples. To maximally identify molecular changes associated with extreme intestinal polyposis, we chose polyps dissected from the most affected and polyp-dense area, located at the distal jejunum-ileum junction (Figure 1A), for polyp WGS and RNA-seq. Hence, the findings represent multiple polyps but not individual ones. As controls, we performed WGS with unaffected tissue dissected from one of the inter-polyp regions of the mid-jejunum (Figure 1A), as well as RNA-seq with unaffected submucosa and muscularis propria tissue dissected away from the polyp-dense mucosa used in polyp-sequencing (Figure 1B). Thus, WGS and RNA-seq normal samples differ in their locations.
For WGS, we generated a 15X sequence coverage for the polyp sample and a 13X sequence coverage for the normal sample, with a fragment coverage at approximately 21X (Supplementary Table 1A). For RNA-seq, we acquired about 80 million paired-reads for the polyp sample and 74 million paired-reads for the normal sample (Supplementary Table 1B). For comparison, we also performed WGS and/or RNA-seq analyses with 26 intestinal normal or tumor samples from 22 dogs with spontaneous intestinal tumors, along with a healthy dog (Supplementary Table 1C). Differing from N14-77, none of these dogs have this extreme polyposis phenotype. We developed a pipeline (Supplementary Figure 1) to interrogate the data.
We corrected genomic sequence errors in the canine APC gene
Because APC mutations characterize the human FAP condition, our initial hypothesis was that APC is mutated in N14-77. We hence first investigated canine APC, but noted that it is annotated inconsistently. In the Ensembl database, APC consists of only three exons, unlike its human counterpart which has 18 exons (Figure 2A). In the Broad annotation [22], APC has more exons but only three are coding (Figure 2A). This again differs from human APC, which has 14-16 coding exons among its transcripts (Figure 2A). The human “xenoRefGene” annotation shows better resemblance (Figure 2A). This however has been achieved by mapping human APC sequences onto the dog genome, not using dog-specific data.
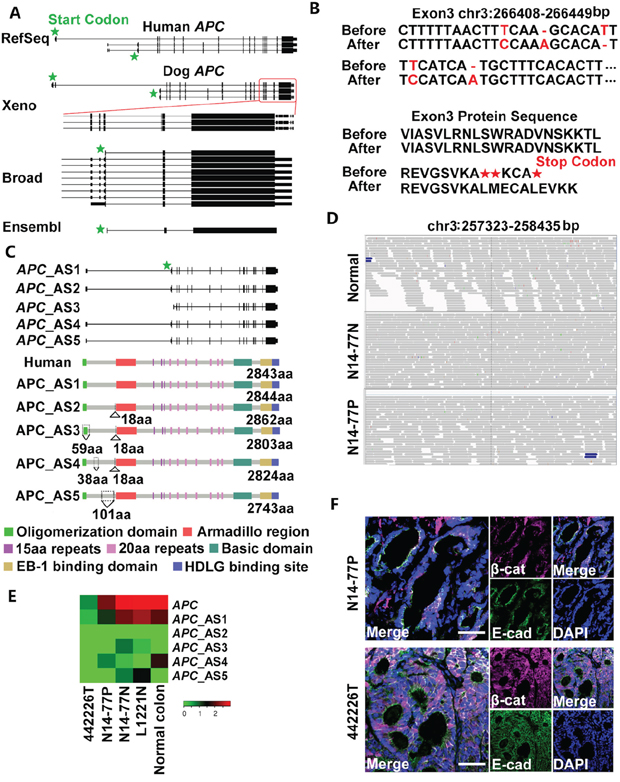
Figure 2: Neither germline nor somatic mutations of APC were found in N14-77. (A) Canine APC is annotated inconsistently. “Xeno” represents human xenoRefGene (mapping human transcript or protein sequences to the canine genome). For Broad annotation [22], only the 3'–end portion enclosed by the red square is shown. Each line designates a transcript, with coding exons, UTR exons and introns respectively represented by tall bars, short bars and the lines between the bars. (B) Five sequence errors (red) were uncovered in exon 3 of APC in the canine genome assembly canFam3.1 (top), resulting in premature stop codons (bottom). Before and After: before and after error correction. (C) Five alternatively spliced (AS) transcripts and protein isoforms were identified after the error correction in B. Also shown is the canonical isoform of human APC, with domains indicated. Amino acid (aa) insertions and deletions are indicated by Δ and ∇, respectively. (D) Representative IGV images show no convincing mutations in APC. The canine genomic region shown corresponds to the human site (codons 1061-1431) that harbors some of APC mutation hotspots. (E) The heatmap indicates the APC expression level in log2 (FPKM) (fragments per kilobase of exon per million fragments mapped). “L1221N” and “Normal colon” are normal colon epithelial tissues from two dogs, while “442226T” is a colorectal tumor with APC deletion from another dog. (F) Representative confocal images indicate no nuclear enrichment of β-catenin (β-cat) in N14-77 polyps, unlike 442226T (with APC deletion). Scale bar, 50μm.
To resolve this inconsistency, we examined our WGS and RNA-seq reads that are mapped onto the canine APC locus. These reads are from normal and tumor intestinal tissues of 23 dogs (Supplementary Table 1C), in addition to N14-77, and from other canine tissues [6, 7]. We detected 5 sequence errors in exon 3 of APC in the canine reference genome [1], including two base substitutions, two base deletions and one base insertion (Figure 2B; Supplementary Figure 2). These errors result in premature stop codons and mis-annotation of APC in Broad and Ensembl databases (Figure 2A).
After removing the errors, we remapped our RNA-seq reads (Supplementary Figure 2) and reassembled the canine APC gene and transcripts. Five transcripts were identified, with 16-17 exons in total of which 15-16 are coding (Figure 2C), better matching their human counterparts. The transcripts yield slightly different protein isoforms. One isoform is nearly identical to the canonical human APC protein (Supplementary Table 2B). The other isoforms have insertions or deletions, all occurring before the armadillo repeat domain (Figure 2C).
We found neither germline nor somatic APC mutations
To discover APC mutations in N14-77, we investigated WGS and RNA-seq reads, individually or combined, of polyp and normal samples. The combined sequence coverage reaches to 136X on average for APC coding regions (Supplementary Table 2C). We used popular software tools, GATK for germline- and MuTect for somatic mutation discovery, with the corrected APC genomic sequence (Figure 2B). Surprisingly, neither germline nor somatic mutations were detected. To confirm this, we manually examined sequence read alignment of each of the 18 APC exons (Figure 2C) with IGV, a widely-used genomics viewer. No convincing mutations were noticed. None of the changes are significantly recurrent, with most found in one or two reads (Figure 2D; Supplementary Figure 3).
Given the prominence of APC mutation in human FAP, our result is somewhat unexpected. To determine if APC alters via other mechanisms, we examined its expression and found no significant alteration (Figure 2E). This is because the N14-77 normal sample expresses a comparable level of APC_AS1, which corresponds to the canonical human APC (Supplementary Table 2A), as normal colon samples of other dogs. N14-77 polyps express a lower amount of APC_AS1, but the level is still higher than colorectal tumors with APC deletion (Figure 2E). Notably, we did not detect significant nuclear enrichment of β-catenin in N14-77 polyp cells (Figure 2F), which does not support APC inactivation.
Besides APC, we also investigated other genes with germline mutations involved in human colorectal tumor development. These include AXIN2, BMPR1A, GREM1, MUTYH, PTEN, SMAD4 and STK11, as well as others (e.g., DNA mismatch repair genes) [13, 14, 18–21]. We examined both coding regions, with average sequence coverages ranging 136-615X after combining WGS and RNA-seq reads of both polyp and normal samples (Supplementary Table 2C), as well as their promoters (22-45X sequence coverage; see Supplementary Table 2C). We only detected two germline mutations: L588P of AXIN2 and A161V of MUTYH. Both mutations have however occurred during evolution (Supplementary Table 2C) and thus are most likely natural variants - not pathogenic. We also detected three germline mutations in the AXIN2 promoter and two germline mutations in the MUTYH promoter. We however did not identify any transcription factor binding sites that are affected by these mutations. Moreover, both AXIN2 and MUTYH are expressed in N14-77 samples at a level resembling other canine intestinal normal and tumor samples (Supplementary Table 2D). These observations indicate that the identified promoter mutations are unlikely pathogenic. Other genes are also expressed in N14-77 polyps and/or normal sample at a comparable level as in other canine samples (Supplementary Table 2D), indicating that germline epigenetic silencing or activating is unlikely.
Germline mutations of other genes were identified in N14-77
STAMBPL1 G406D is the most notable germline missense mutation
After individual gene study described above, we attempted genome-wide search. GATK identified a large number of missense mutations, and we developed a pipeline (Figure 3A) to reduce false positives and to prioritize mutations. First, we selected mutations that were found in both normal and polyp samples and by both WGS and RNA-seq analyses. This step, ensuring that missense mutations of interest are indeed expressed, yields 4,329 mutations in total (Figure 3A). Second, we chose mutations that are unique to N14-77, when compared to >50 cases of sporadic canine intestinal (Supplementary Table 1C) and other cancers [6, 7], and by excluding canine SNPs from published studies [1, 23] and databases. This is because N14-77 is the only case known to have extreme intestinal polyposis. A total of 135 mutations remain after this step (Figure 3A). Third, we excluded mutations located in genes with annotation issues (e.g., retrogenes or pseudogenes). To further increase the accuracy, we selected mutations with: 1) a ≥10 WGS read coverage in both normal and polyp samples; 2) a ≥30X RNA-seq read coverage in either the normal or polyp sample; and 3) a ≥0.5 variant allele frequency in the polyp sample for either WGS or RNA-seq. These selections reduce the total mutations to 21 (Supplementary Table 3A). For heterozygous mutations, we further prioritized those being selected in polyps, i.e. with a higher mutation rate in the polyp than in the normal sample (Figure 3; Supplementary Table 3A). Lastly, we considered evolutionary conservation and excluded mutations occurring during evolution (Supplementary Table 3A). In addition, we prioritized mutations predicted to alter the protein 3D structure with modeling [24] (Supplementary Table 3A).
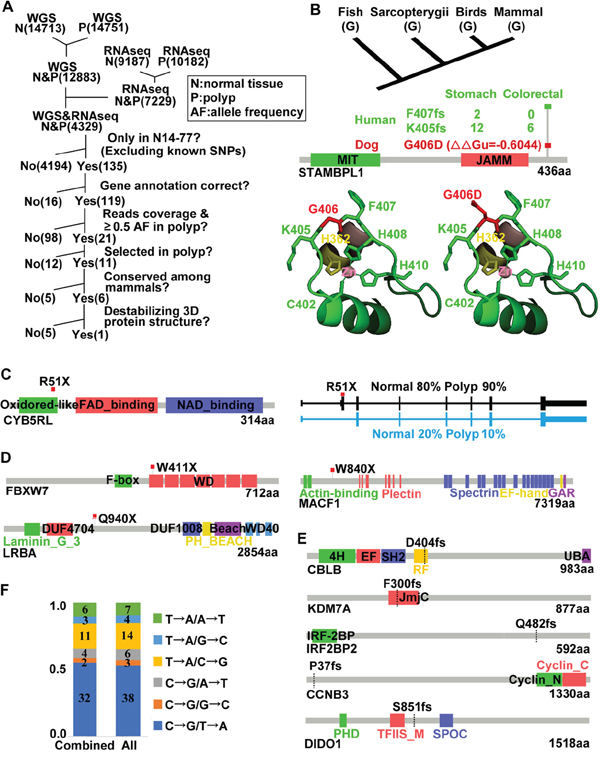
Figure 3: Notable germline and somatic mutations were identified in N14-77. (A) Outlined is our pipeline for putative pathogenic germline missense mutation discovery (see text). Numbers in parentheses indicate mutation counts. (B) G406D of STAMBPL1 is identified by the pipeline in A. The top image shows G406 conservation. Middle images (from bottom to top) indicate the protein domains, ΔΔGu (red) from modeling [24] predicting that G406D likely destabilizes the protein structure, and frameshift (fs) mutations found in human cancers (with total case numbers indicated). Bottom images are the crystal structure [25] with G406 (left) and G406D (right) in red and its flanking K405 and F407 in green, and zinc-coordinating residues C402, H362, H408 and H410 shown. (C) R51X of CYB5RL is a heterozygous germline truncation mutation. The right image indicates the two alternative splicing forms and their proportions in each sample. (D and E) Somatic truncation mutations and frameshift mutations identified. (F) C→T/G→A changes dominate. Combined: somatic missense mutations identified by combining WGS and RNA-seq reads. All: those found by WGS alone, RNA-seq alone and combined. The numbers inside the bars specify the mutation counts.
After processing through our pipeline (Figure 3A), G406D of STAMBPL1 is the only germline missense mutation that remains. This mutation is unique to N14-77 and being selected in polyps, with the mutation rate increasing from 67% in the normal sample to 82% in the polyp sample for WGS and from 50% to 70% for RNA-seq (Supplementary Table 3A). Furthermore, G406, the glycine residue at position 406, is conserved from fish to mammals for 100 species examined (Figure 3B). Based on modeling [24], the G406D change will likely destabilize the protein (Figure 3B).
STAMBPL1, also known as AMSH-LP, is a deubiquitinase (DUB) that cleaves K63-linked polyubiquitin chains. The crystal structure of human STAMBPL1 is determined [25]. As canine STAMBPL1 is highly homologous to its human counterpart, with the same length and sharing 92% similarity and 88% identity in amino acid sequence (Supplementary Figure 4A), we used the human structure to study the G406D mutation. STAMBPL1 is a zinc protease and contains two zinc centers. G406 locates in the 2nd zinc-center, neighboring the zinc-coordinating residues C402, H408 and H410 within a highly conserved peptide (C402KKK405 G406F407H408PH410) (Supplementary Figure 4B). This peptide forms a long loop, assisting the recognition and correct binding of the proximal ubiquitin of K63-linked ubiquitin chains [25]. Importantly, K405 and F407, which flank G406, are the most frequently mutated residues of STAMBPL1 in human cancers, with frameshift mutations recurrently found (Figure 3B; Supplementary Table 3B). Based on these findings, the G406D germline mutation of STAMBPL1 may be pathogenic.
CYB5RL harbors a germline truncation mutation
We followed the same procedure of Figure 3A, except skipping the steps of evolutionary conservation and protein 3D structure, for germline truncation mutation discovery. We manually confirmed the results with IGV and the UCSC and Ensembl genome browsers. With these, we detected a truncation mutation, R51X, in CYB5RL (cytochrome b5 reductase like) (Figure 3C). This mutation is selected in the polyps, although the allele frequency is rather low (25%) based on RNA-seq reads (Supplementary Table 3C). There appears to be a second, but minor, alternative splicing form that is not affected by this mutation (Figure 3C).
No germline frameshift mutations found
We applied the same strategy described above, and found no convincing germline frameshift mutations in N14-77. A small number of indels were detected, which however locate in intron regions, retrogenes, or misannotated genes based on manual examination.
Somatic mutations were identified in N14-77 polyps
Somatic truncation mutations of FBXW7, LRBA and MACF1 found
We detected 7 total somatic truncation mutations in N14-77, three of which are supported by both WGS and RNA-seq analyses (Supplementary Table 3D). The 1st mutation is W411X of FBXW7, occurring at a rate of 39% for WGS and 59% for RNA-seq (Figure 3D; Supplementary Table 3D). FBXW7 is one of the most frequently (~20%) mutated genes in human CRC [26]. The 2nd mutation is Q940X of LRBA (Figure 3D), at a rate of 90% for WGS and 38% for RNA-seq (Supplementary Table 3D). LRBA (lipopolysaccharide-responsive vesicle trafficking, beach- and anchor-containing) is linked to trafficking of immune molecules such as CTLA4. LRBA deficiency, a rare genetic disorder, is associated with autoimmunity, chronic diarrhea, and B-cell deficiency [27]. The 3rd mutation is W840X of MACF1 (microtubule-actin crosslinking factor 1) (Supplementary Table 3D), the loss of which disrupts epithelial cell polarity [28].
Somatic frameshift mutations of CBLB and other genes found
CBLB encodes an E3 ubiquitin ligase CBL-B, an immune response regulator [29]. We detected a somatic base T deletion at a rate of 50% for WGS and 29% for RNA-seq, resulting in a frameshift mutation at residue D404 (D404fs) of CBLB (Figure 3E; Supplementary Table 3D). Frameshift indels were also uncovered in DIDO1, involved in apoptosis [30], as well as within homopolymer sites (e.g., GGGGGG) of KDM7A, IRF2BP2 and CCNB3 (Figure 3E; Supplementary Table 3D).
G→A/C→T changes dominate among somatic base substitutions
We identified 72 missense mutations in total (Supplementary Table 3E). Consistent with human studies [26], G→A/C→T changes dominate over other base substitutions (Figure 3F), indicating that C/G deamination is the major somatic mutation mechanism in N14-77 polyps. Among 72 mutations, only 7 were detected by both WGS and RNA-seq analyses (Supplementary Table 3E). Furthermore, quite a few mutations could be passengers, based on evolutionary conservation and molecular modeling (Supplementary Table 3E), as well as comparison to human mutation findings (Supplementary Figure 5A). However, more studies are required to determine their driver-passenger role.
Somatic whole chromosome gains detected
Our analysis (Supplementary Figure 1B) revealed no translocations or inversions in the N14-77 polyp or normal genome. Neither did we find focal amplifications/deletions. We did, however, detect whole chromosome gain of chromosomes 4, 7-10, 13, 15, 23, and 26. These changes are clearly somatic, because they were only found in the polyp genome but not in the normal genome (Supplementary Figure 5B).
Highly- and lowly expressed genes in N14-77 polyps are enriched in specific functions
With our RNA-seq data from 28 samples of canine intestinal tumor and normal tissues (Supplementary Tables 1B and 1C), we identified genes that are highly or lowly expressed in N14-77 polyps. These are defined as genes with an expression level outside the expression mean ± one standard deviation range and being the highest or lowest among the 28 samples. A total of 528 highly expressed genes were identified, of which about 474 (90%) encode proteins (Figure 4A; Supplementary Table 4A). These genes are significantly enriched in functions of cell cycle, DNA repair, as well as transcription, mRNA processing and splicing (Figure 4A; Supplementary Table 4A). Meanwhile, 621 lowly expressed genes were discovered, of which only one appears to be noncoding and 614 encode proteins with functional annotation (Figure 4A; Supplementary Table 4A). Among them, the prominently enriched functions include protein localization, trafficking and degradation, as well as cell cycle.
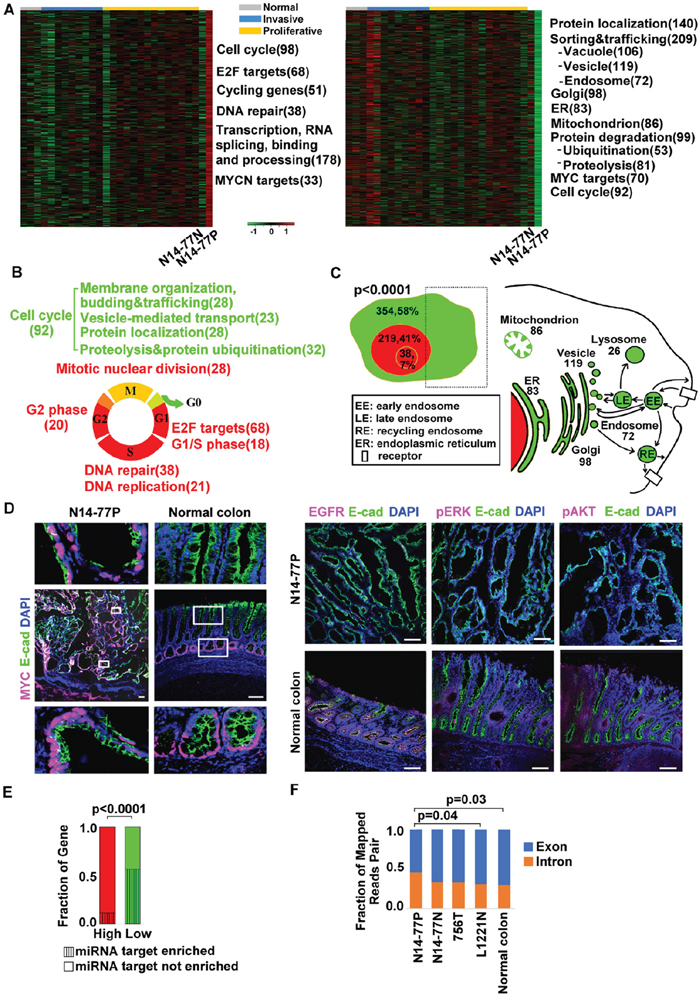
Figure 4: Highly- and lowly expressed genes in N14-77 polyps differ in function, cell cycle phase, cellular location and miRNA target site enrichment. (A) Heatmaps from left to right indicate the log2 (FPKM) values of 474 highly- and 614 lowly expressed genes in 28 canine intestinal samples grouped as shown. Significantly enriched functions are listed next to the heatmap, with the total number of genes involved shown in the parenthesis. (B) Red and green respectively designate highly and lowly expressed genes, along with their enriched functions and cell cycle phases. Yellow indicates cell cycle phases enriched in both highly and lowly expressed genes. (C) Highly expressed genes are enriched in the nucleus (red) and lowly expressed genes are enriched in the cytoplasm (green), e.g., “354, 58%” indicating 354 genes, which make up 58% of all lowly expressed genes, located in the cytoplasm. The small red circle inside the nucleus designates the nucleolus. The right image illustrates that the sub cellular locations of lowly expressed genes, e.g., 83 genes are associated with ER. (D) Representative IHC images illustrate the enrichment of nuclear protein MYC, and the depletion of membrane and cytoplasmic proteins EGFR, pERK and pAKT, in N14-77 polyps. Scale bar, 100 μm. (E) Highly and lowly expressed genes differ in enriched miRNA target sites. (F) More RNA-seq reads were mapped to intronic regions in N14-77 polyps. 756T is a canine jejunum tumor and others are described in Figure 2E.
>50 ubiquitin-related genes are lowly expressed
Ubiquitin-related genes are enriched only among lowly expressed genes of N14-77 polyps (Figure 4A). Specifically, a total of 53 such genes are lowly expressed (Supplementary Figure 6A), of which ≥29 are associated with ubiquitin ligases and ≥4 are linked with DUBs (Supplementary Table 4A). About 35 genes are associated with protein degradation, including 6 encoding F-box proteins (Supplementary Table 4A). Interestingly, 39 genes (73%) are enriched in microRNA (miRNA) target sites.
Highly and lowly expressed cell cycle genes differ in cell cycle phase and function
Although cell cycle genes are enriched in both highly and lowly expressed gene sets (Figure 4A), they differ. Highly expressed ones consist of E2F targets, and cycling genes with their expression peaking during the G1/S or G2 phase (Figure 4B; Supplementary Table 4B). DNA replication and/or repair genes, which primarily function in the S phase, and mitotic nuclear division genes are also among highly expressed (Figure 4B; Supplementary Table 4B). In contrast, 63 of the 92 lowly expressed cell cycle genes are associated with membrane organization, budding and trafficking; vesicle-mediated transport; protein localization, as well as ubiquitination and proteolysis (Figure 4B; Supplementary Table 4B). Furthermore, at least 12 lowly expressed genes encode cell cycle inhibitors, including RB1 and TSG101 (Supplementary Table 4B).
Highly and low expressed genes are enriched in different cellular locations
Approximately 41% of the highly expressed genes are annotated to be located in the nucleus (Figure 4C; Supplementary Table 4C). These include those (7%) located in the nucleolus, as well as genes functioning in DNA repair and/or replication, transcription and chromatin. This is significantly higher, when compared to both the lowly expressed genes (about 12%) and the entire gene set encoded in the genome (<25%). To the contrary, about 58% of the downregulated genes are located in the cytoplasm, associated with endosomes and other organelles (Figure 4C; Supplementary Table 4C). This is also significantly higher when compared to the highly expressed genes (~18%) and the entire gene set encoded in the genome (<40%).
Consistent with mRNA expression (Figure 4C), our immunohistochemistry (IHC) analysis reveals depletion of EGFR, a membrane protein, and of phosphorylated ERK and AKT, both cytoplasmic proteins, in N14-77 polyps, when compared to normal intestinal tissue samples (Figure 4D). This differs from MYC, a nuclear protein (Figure 4D), as described later.
Lowly expressed genes are enriched in miRNA target sites
About 56% (342 genes) of lowly expressed genes are enriched in putative miRNA target sites, compared to only 16% (77 genes) for highly expressed genes (Figure 4E). Interestingly, more noncoding RNA genes are found among highly expressed genes than among lowly expressed genes, as previously described. This is consistent with that more RNA-seq reads were mapped into intronic regions in the N14-77 polyp sample, compared to the other samples (Figure 4F; Supplementary Table 4D).
MYC network is activated in N14-77 polyps
Several analyses indicate that the MYC network is activated in N14-77 polyps. First, the MYC protein is expressed highly and more or less uniformly throughout the polyps, ranging from the bottom to the top of the intestinal mucosa (Figure 4D). This differs from normal intestinal tissues of other dogs where MYC is only expressed at the bottom layer of the mucosa (Figure 4D). Second, MYC has the highest mRNA expression level in N14-77 polyps, among 28 canine intestinal tumor and normal samples investigated (Supplementary Figure 6B).
Notably, MYC targets are enriched in both highly and lowly expressed gene sets of N14-77 polyps (Figure 4A). Specifically, 21 MYC targets, 38% of which function in DNA repair, and 33 MYCN targets, ~70% of which are E2F targets and/or associated with RNA-binding and processing, are highly expressed (Figure 5A; Supplementary Table 5A). Meanwhile, a total of 70 MYC targets are lowly expressed and, except for protein degradation, are enriched in the same functions as the entire lowly expressed gene set (Figure 5A; Supplementary Table 5A). Also like the entire gene sets, highly expressed MYC targets are enriched in the nucleus (76%) and depleted in miRNA target sites (4%), while lowly expressed ones are enriched in the cytoplasm (64%) and miRNA target sites (53%) (Figure 5B and 5C).
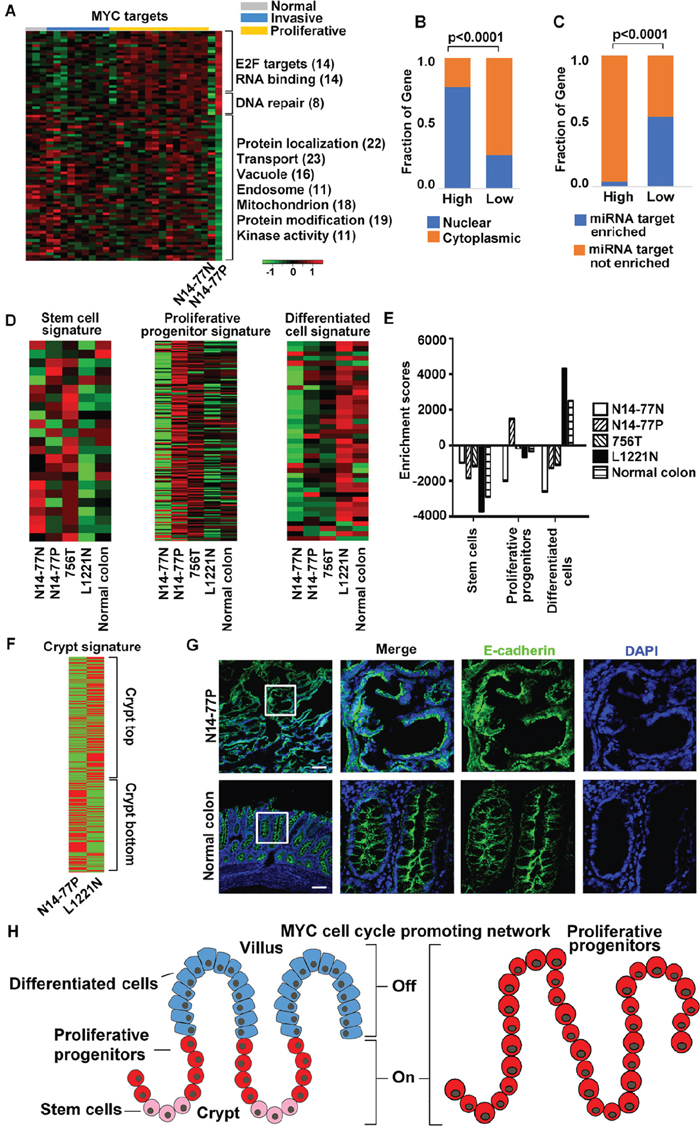
Figure 5: MYC network and crypt proliferative progenitor signature are activated in N14-77 polyps. (A) MYC targets are enriched in both highly and lowly expressed genes. The image is presented as described for Figure 4A. (B and C) Highly and lowly expressed MYC target genes differ in enriched cellular locations and miRNA target sites. (D and E) Signature of intestinal proliferative progenitors, but not of either intestinal stem cells or differentiated cells, is activated in N14-77 polyps. The heatmaps indicate the log2 (FPKM values of signature genes [31] (D), and the bar plot indicate the corresponding ssGSEA results (E). (F) Signature [32] of the crypt bottom, but not of the crypt top, is activated in N14-77 polyps. (G) Representative IHC images indicate the lack of well-established epithelial apical-basolateral cell polarity in N14-77-polyp cells. Scale bar, 100 μm. (H) Cartoons illustrate the differentiation of normal intestinal epithelium (left) and indicate that N14-77 polyp cells are in the proliferative progenitor state (right).
N14-77 polyps exhibit crypt proliferative progenitor signature
We investigated published gene signatures that mark different intestinal epithelial differentiation stages [31] via single sample gene set enrichment analysis (ssGSEA). Signature genes of crypt proliferative progenitors, but not of either intestinal stem cells or differentiated epithelial cells, are significantly upregulated in N14-77 polyps (Figures 5D and 5E; Supplementary Tables 5D and 5E). This agrees with that N14-77 polyps display upregulated signature of the crypt bottom and downregulated signature of the crypt top [32] (Figure 5F; Supplementary Table 5F). Furthermore, also consistent with the ssGSEA results, our IHC analysis reveals that N14-77 polyp cells lack well-established apical-basolateral polarity, unlike fully differentiated epithelial cells (Figure 5G; Supplementary Figure 6D). These observations indicate that N14-77 polyp cells are in the proliferative progenitor state (Figure 5H).
N14-77 intestinal microbiota is enriched in bacteroidetes
As described previously, medical examination indicates extensive bacterial infection in the N14-77 intestine. To better understand this, we utilized WGS and RNA-seq data to examine the intestinal microbiota. Briefly, we first identified WGS and RNA-seq read pairs of which neither read could be mapped onto the canine reference genome (Supplementary Tables 1A and 1B), which were then searched against three microbial databases. The 1st database is the reference genomes curated by the Human Microbiome Project [33], referred to as HMP hereafter. The 2nd database contains all bacterial genomic sequences (ABG) downloaded from the NCBI. The 3rd database is simplified from ABG, consisting of genomic sequence of the longest strain of each bacterial species with genome sequencing completed. It is hence named longest bacterial genomes (LBG). We noted that the results with LBG are somewhat skewed. We thus only focus on HMP and ABG studies, as described below.
Our analysis with WGS data reveals that N14-77 samples contain more bacteria than other intestinal tumor and normal samples which we investigated (Supplementary Table 1C). More importantly, bacteroidetes is the most enriched bacterial phylum, accounting for 67-72% for polyps and 45-48% for the normal sample, followed by proteobacteria and firmicutes (Figure 6A; Supplementary Table 6A). Other phyla each makes up < 1% (Supplementary Table 6A). Our results differ from typical microbiota of canine jejunum published [34], where bacteroidetes are less enriched than proteobacteria, firmicutes, actinobacteria and spirochaetes. Instead, with bacteroidetes predominating, N14-77 samples, which are from jejunum (Figure 1), better resemble colon in microbiota [34]. This is confirmed at the family level, where bacteroidaceae, enterobacteriaceae, clostridiaceae and tannerellaceae dominate (Figure 6B; Supplementary Table 6B). Again, families of bacteroidetes, i.e. bacteroidaceae and tannerellaceae, are significantly enriched, particularly in the polyp sample. At the species level, the top enriched include Bacteroides uniformis and Clostridium perfringens (Figure 6C; Supplementary Table 6C). While both bacteria can be found in the intestine of healthy individuals, they are thousands times more enriched in N14-77 tissues, compared to other canine intestinal samples investigated (Supplementary Table 1C).
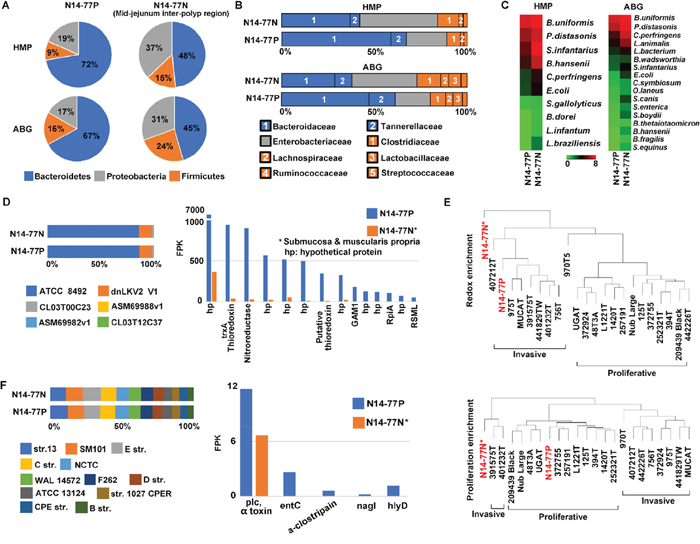
Figure 6: Bacteroidetes, B. uniformis and C. perfringens are significantly enriched in N14-77 intestinal microbiota. (A) Bacteroidetes is the dominating phylum. The pie charts indicate the composition of bacterial phyla, determined by searching WGS reads against microbial genome databases HMP and ABG. (B) Bacteriotecea is the dominant family in polyps. The colors represent bacterial phyla as shown in A. (C) B. uniformis and C. perfringens are among the top enriched species. (D) ATCC 8492 is the most enriched strain of B. uniformis (left), determined with WGS reads, and expresses abundantly thioredoxin and nitroreductase genes (right), determined with RNA-seq reads. (E) N14-77 polyps resemble invasive tumors, but not proliferative tumors, in redox gene expression. The images indicate sample clustering based on the ssGSEA enrichment scores with indicated gene sets. (F) C. perfringens strain enrichment and toxin gene expression.
B. uniformis is highly enriched and expresses thioredoxin and nitroreductase abundantly
B. uniformis is the top enriched microbial species in both normal and polyp samples of N14-77 (Figure 6C; Supplementary Table 6C). Among its 6 strains examined, ATCC 8492 is about 7-800 times more enriched than others (Figure 6D; Supplementary Table 6D). Importantly, our RNA-seq data reveal that redox genes of B. uniformis are highly expressed in N14-77 polyps. Specifically, trxA which encodes thioredoxin, a redox protein, is the 2nd most abundantly expressed gene, while a nitroreductase gene ranks the third highest expressed (Figure 6D; Supplementary Table 6D).
Although being proliferative but not invasive (Figure 1B), N14-77 polyps appear to have a tissue redox state that better resembles invasive tumors than proliferative tumors. With host redox-related gene sets (Supplementary Figure 7A and Supplementary Table 6E), the N14-77 polyp sample clusters with invasive tumors, instead of proliferative tumors (Figure 6E). With cell proliferation-related gene sets (Figure 6E; Supplementary Figure 7B) or in genome-wide expression (Supplementary Figure 7C), the opposite was observed.
C. perfringens is enriched and expresses α-toxin
C. perfringens is among the top few enriched species in N14-77 samples (Figure 6C), and is linked to conditions such as diarrhea and enteritis in dogs [35]. We hence examined C. perfringens in more depth. C. perfringens strains are classified into A, B, C, D and E types, based on major toxins produced [36]. There are 12 strains in our database: 3 type A, 2 type C, and one each for types B, D and E, plus 4 unclassified (Supplementary Table 6F). By counting WGS reads that are uniquely mapped to each strain, we note that strains 13 and SM101, both type A, and type E strain JGS1987 are slightly more enriched (Figure 6F; Supplementary Table 6F). Meanwhile, type B strain ATCC 3626 is the least enriched. Finally, we examined the expression of C. perfringens toxin genes with our RNA-seq data. As strain 13 represents the reference strain for C. perfringens and its genome is well annotated [37], we used it to identify the toxin genes and found 25 of them (except for nanH). In the polyp sample, we detected substantial expression of α-toxin, a phospholipase C, and an enterotoxin (entC), as well as trace expression of μ-toxin (nagI), α-clostripain and hemolysin (hlyD) (Figure 6F; Supplementary Table 6F).
Lastly, although medical examination suggests yeast infection, we did not find any of our WGS reads mapped to the yeast genomes in the HMP database.
DISCUSSION
N14-77 represents the first reported case of extreme intestinal polyposis in the dog. Our analysis does not support the involvement of APC mutations. Instead, we propose that this extraordinary phenotype is possibly caused by an alteration network collaborating across the genome, transcriptome and microbiome (Figure 7), as discussed below.
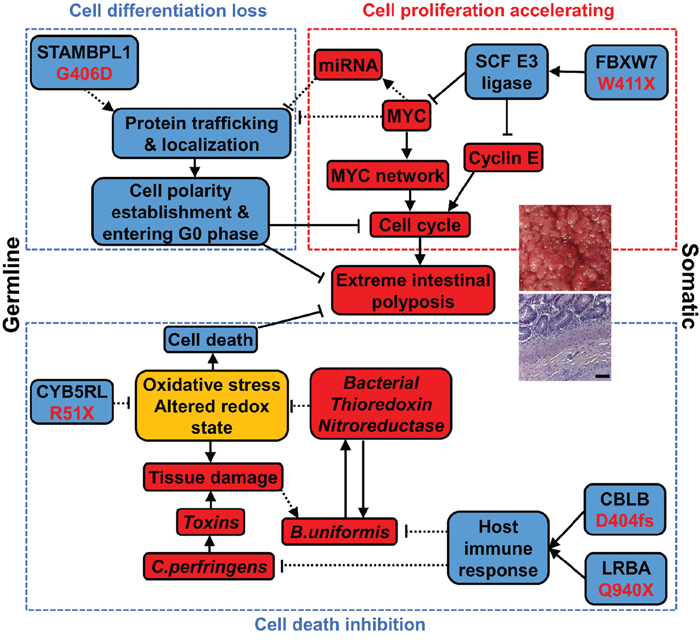
Figure 7: An alteration network collaborating across the genome, transcriptome and microbiome contributes to extreme intestinal polyposis of N14-77. Bacterial elements are shown in italics. Red, blue, and yellow designate promoting, inhibiting, and both promoting and inhibiting factors, respectively. “→”: promoting; “┤”: inhibiting. Solid lines indicate that the relationship is supported by published studies and our observations, while dashed lines indicate that the relationship requires future functional validation.
Host ubiquitin gene alterations and MYC and cell cycle-promoting network activation keep cells proliferating
FAP and many CRCs in humans follow the pathogenic pathway of APC mutation → β-catenin accumulation in the nucleus → MYC upregulation and cell-cycle activation → cell proliferation [26]. With the lack of APC mutation, our data indicates that N14-77 has likely taken a different route: FBXW7 truncation mutation → MYC protein accumulation and cell cycle activation → cell proliferation. FBXW7, a F-box protein, constitutes the substrate-recognition subunit of the SKP1-cullin-F-box (SCF) E3 ubiquitin ligase that targets MYC and cyclin E for degradation [38]. The W411X truncation mutation of FBXW7 could render this SCF complex defective and unable to ubiquitinate MYC and cyclin E for degradation. Deletion of FBXW7 in the gut has induced intestinal adenomas in mice [39].
Interestingly, 53 ubiquitin genes are downregulated in N14-77 polyps, the significance and mechanism of which clearly need further studies. Among them are genes encoding TRPC4AP and CUL4A, which constitute the MYC-targeting DDB1-CUL4 E3 ligase complex [40]. This may further lead to MYC protein accumulation.
MYC is a master transcription factor. MYC protein accumulation accelerates the transcription of numerous cell cycle promoting genes. Indeed, E2F targets, DNA repair genes, and RNA processing and slicing genes are all upregulated in N14-77 polyps. These would keep N14-77 polyp cells proliferating (Figure 7).
Ubiquitin gene alteration and MYC network activation likely inhibit epithelial polarity establishment and cell differentiation
G406D of STAMBPL1 is the most significant germline missense mutation discovered in N14-77. STAMBPL1 (AMSH-LP) is a K63-specific DUB of the JAMM/MPN+ family [41]. G406 appears critical to its DUB activity, based on strong evolutionary conservation, crystal structure [25] and human cancer mutation findings. The G406D mutation may disrupt the DUB activity by destabilizing the 2nd zinc-center, affecting substrate binding.
The function of STAMBPL1 is not well understood at present. A study indicates that it potentiates TGFβ signaling by inhibiting SMAD7 [42]. However, our ssGSEA reveals no significant difference in TGFβ signaling between N14-77 polyps and other canine intestinal tumor and normal samples (Supplementary Figure 6C). Thus, it is possible that STAMBPL1 has other functions. Its homologue STAMBP (or AMSH) is known to participate in endosomal sorting of receptors and membrane proteins [43], e.g., STAMBP knockdown enhancing EGFR degradation. Consistent with this, we observed depletion of EGFR, pAKT and pERK proteins in N14-77 polyp cells. Interestingly, like N14-77 polyps, stomach cancers [44] that harbor STAMBPL1 F407fs or K405fs mutation also display upregulation of MYC target genes and downregulation of trafficking genes (Supplementary Figure 4C). We propose that the DUB activity of STAMBPL1 is required for efficient sorting, trafficking and localization of proteins inside the cell. And this is disrupted by the G406D mutation, based on our model (Figure 7).
Intracellular sorting, trafficking and localization in N14-77 polyp cells are likely further disrupted by the downregulation of numerous genes associated with the system. MYC over-activation could be a contributing factor. First, many of these genes are known or putative MYC targets [45] and MYC can directly repress their transcription. Note that MYC co-repressors ZBTB17 (MIZ-1) and MXD3 are upregulated in N14-77 polyps. Alternatively, these genes could be downregulated via miRNAs, supported by that: 1) noncoding RNA genes are upregulated in N14-77 polyps; and 2) lowly expressed genes are enriched in miRNA target sites.
Intestinal epithelium develops through intestinal stem cells → proliferative progenitors → differentiated cells [31]. During the 2nd stage of differentiation, the cells exit the cell cycle and establish epithelial apical-basolateral polarity. The underlying molecular mechanisms are complex. However, the intracellular sorting, trafficking and localization system clearly plays a critical role. For example, it is required to target various proteins to appropriate places to build cell adherent junctions and signaling complex (e.g., PAR, crumbs, and scribble complex) for polarity establishment [12].
Our model (Figure 7) proposes the following. Because of the STAMBPL1 G406D mutation and downregulation of genes described above, the intracellular protein sorting, trafficking and location system in N14-77 polyps is defective. This deficiency inhibits epithelial polarity establishment and cell differentiation, and prevent cells from entering the G0 phase. This, in combination with cell cycle activation, keeps N14-77 polyp cells forever in the proliferative state. Consistent with our model, N14-77 polyp cells lack well-established apical-basolateral polarity, and closely resemble intestinal proliferative progenitors.
Intestinally, our downregulated genes significantly overlap with transcripts enriched in the protruding pseudopodia formed by cells in response to migrating stimulus by fibronectin [46]. Whether this is a reason behind non-invasiveness of N14-77 polyp cells requires further investigation.
Bacterial redox gene expression possibly reduces oxidative stress and cell death
R51X of CYB5RL is another noteworthy germline mutation uncovered in N14-77. CYB5RL is not well-studied, but its homologue cytochrome b5 reductase (CYB5R) is. The shorter soluble isoform of CYB5R is expressed in erythrocytes, catalyzing the reduction of methemoglobin (with Fe3+-heme) to hemoglobin (with Fe2+-heme). The longer isoform is expressed in other cell types. With a membrane-anchor domain, it constitutes the plasma membrane redox system which regulates the tissue redox state and reduces oxidative stress [47].
Our study reveals two isoforms of CYB5RL as well. The longer isoform is inactivated by the R51X mutation, which may cause oxidative stress, contributing to the altered tissue redox state of N14-77 polyps. Increased oxidative stress, along with bacterial toxins produced by C. perfringens, could result in tissue damage and lead to a faulty ileocecal valve (supported by medical examination), allowing colonic bacteria to spread to the small intestine. This may possibly explain why the N14-77 jejunum microbiota, where bacteroidetes dominate, better resembles typical microbiota of the colon rather than the jejunum. Moreover, somatic mutations of CBLB and LRBA, two key immune regulators, could alter the host immune response. We propose that, as a result of all of these, B. uniformis thrives (Figure 7). Importantly, B. uniformis bacteria express thioredoxin, especially trxA, and nitroreductase genes abundantly. The trxA gene is essential for the survival of B. fragilis under aerobic condition by reducing oxidative stress [48]. Like B. fragilis, B. uniformis is an anaerobe and normally resides in the colon, where the O2 level is lower than in the jejunum. Thus, we postulate that B. uniformis expresses thioredoxin (and nitroreductase) amply to remediate oxidative stress that is induced by the more aerobic environment of the jejunum and is exacerbated by the host CYB5RL R51X mutation. Meanwhile, host cells should also benefit. Our model proposes that by decreasing oxidative stress, these bacterial redox systems reduce host cell death and contribute to extreme polyposis (Figure 7).
In summary, we propose that three pathways lead to N14-77 extreme intestinal polyposis (Figure 7). First, MYC and cell cycle-promoting network activation, caused by a FBXW7 somatic mutation-initiated SCF E3 ubiquitin ligase defect, keeps crypt cells dividing. Second, defective intracellular trafficking and localization, originating from D406G germline mutation of STAMBPL1 and enhanced by MYC network activation, inhibit cell polarity establishment and cell differentiation, preventing cell cycle exit. Lastly, bacterial redox systems reduce the oxidative stress caused by germline mutation R51X of CYB5RL, decreasing cell death. Lastly, we emphasize that future functional studies are required to validate our model.
MATERIALS AND METHODS
Canine tissue samples
Fresh-frozen (FF) canine intestinal normal tissues and spontaneous tumors were obtained from various Veterinary Colleges (Supplementary Table 1C). Samples were collected from client-owned dogs that develop the disease spontaneously, under the guidelines of the Institutional Animal Care and Use Committee for use of residual diagnostic specimens and with owner informed consent. The breed, age, histopathologic descriptions, and other information are provided in Supplementary Table 1C.
Tissue dissection, DNA and RNA extraction, and quality control
Cryosectioning of FF tissues, H&E staining and cryomicrodissection were performed as described [5, 7] to enrich polyp/tumor cells for the polyp/tumor sample, as well as unaffected/normal cells for control/normal samples. Genomic DNA and RNA were then extracted from the dissected tissues using the AllPrep DNA/RNA Mini Kit (cat. no. 80204) from QIAGEN. Only samples with a 260/280 ratio of ~1.8 (DNA) or ~2.0 (RNA) and showing no degradation and other contaminations were subjected to further quality control with qPCR and qRTPCR analysis with a panel of genes as previously described [7, 9].
Paired-end WGS and RNA-seq
Both types of sequencing were conducted using the Illumina platform, following the protocols from the manufacturer. Paired-end 125 x 125bp WGS was performed in collaboration with the BGI-America and the High Throughput Genomics Core Facility at Huntsman Cancer Center at the University of Utah. RNA-seq was performed in collaboration with the Georgia Genomics Facility at the University of Georgia.
Sequence data analyses
The overall sequence analysis pipeline was summarized in Supplementary Figure 1 and described in details in Supplementary methods. Briefly, WGS reads were aligned to the dog reference genome canFam3.1 [1] with BWA [49] v0.7.10. RNA-seq reads were mapped to the same reference genome using either TopHat [50] 2.1.1 (for gene expression) or STAR [51] v2.4.1c (for mutation finding). Three canine gene annotation databases were used, including Ensembl and the Broad annotation [22], both RNA-seq based, and human xenoRefGene [7]. Known canine SNPs used include those reported in other canine samples by us [6, 7] and the Broad Institute [1], as well as data from the NCBI, Ensembl, and DoGSD [23] databases. Both WGS and RNA-seq reads were used for germline mutation discovery with GATK [52] v3.6 and for somatic mutation finding with MuTect [53], following pipelines recommended by the Broad Institute. WGS data were used to identify germline and somatic inversions/translocations and chimeric fusion genes as described before [5–7]. For copy number changes, correctly and uniquely mapped WGS read pairs were used to calculate mapped pair density per 1kb tiling window along a chromosome. Each density was normalized against the corresponding value of a control genome and then used for germline and somatic copy number change discovery as previously described [5–7]. Gene expression quantification with RNA-seq reads and other analyses were performed as previously described [6, 7].
Microbiome analysis
WGS and RNA-seq read pairs that could not be placed onto the canine genome were mapped with BWA v0.7.10 to three microbial genome databases – HMP, ABG and LBG. HMP is the reference genomes curated by the Human Microbiome Project [33]. HMP consists of genomic sequences of bacteria (1751 strains from 1253 species), viruses (3683 strains from 1420 species), archaea (131 strains from 97 species) and 326 lower eukaryotic species. ABG contains all bacterial genomic sequences (ABG) downloaded from the NCBI, with 2,845,483 sequences in total from 2679 species. LBG is simplified from ABG by: 1) selecting species with complete genomic sequences; and 2) for species with multiple strains having complete genomic sequences, selecting the longest strain. LBG consists of 1,576 bacterial species.
Mapped WGS read pairs were used to estimate microbial enrichment in each sample. First, the taxonomy data downloaded from the GOLD database (gold.jgi.doe.gov) were used to classify each bacterial species. Second, mapped WGS read pairs were selected as follows. For pairs with at least one read uniquely mapped, those with mapping quality Q >0 were selected. For pairs with both reads duplicatedly mapped, those that are correctly mapped (i.e., both reads mapped to the same DNA fragment, in correct orientation and spanning a reasonable genomic distance) were selected. Third, each selected read pair was assigned as follows. A read pair was assigned to a phylum and counted as one, if it was mapped to this phylum only and no matter how many times it was mapped within this phylum. If a read pair was mapped to ≥ 2 different phyla, it was discarded. Lastly, read pairs assigned to each phylum was tallied and used to estimate the phylum enrichment. The same procedure was followed to estimate the family, species and strain enrichment.
Bacterial genome annotation data were downloaded from NCBI and Ensembl. HTSeq [54] v0.6.1 was used to tally correctly and uniquely mapped RNA-seq reads pairs within each gene, which were then used to estimate the expression levels of bacterial genes.
Immunohistochemical analysis
Immunohistochemical (IHC) experiments were performed with 5-μm tissue sections as described [7]. Primary antibodies used include including those against E-cadherin (R&D Systems, AF648), β-catenin (Santa Cruz, sc-7199), MYC (Abcam, ab32072), EGFR (BioGenex, PU335-UP), phospho-Erk1/2 (Cell Signaling Technology, #4370) and phospho-Akt (Cell Signaling Technology, #4060). Alexa Fluor®488–, 647– or 594–conjugated secondary antibodies are from Jackson ImmunoResearch. Images were taken with a Zeiss LSM 710 confocal microscope.
Data access
Sequence data have been submitted to the NCBI SRA database with accession number PRJNA418842.
Abbreviations
FAP, familial adenomatous polyposis; CRC, colorectal cancer; HNPCC, hereditary nonpolyposis colorectal cancer; WGS, whole genome sequencing; IHC, immunohistochemistry; GSEA, gene set enrichment analysis; ssGSEA, single sample gene set enrichment analysis; HMP, the reference genomes curated by the Human Microbiome Project; ABG, all bacterial genomic sequences; LBG, the longest bacterial genomic sequences.
Author contributions
Conceptualization, S.Z., L.B., S.D.; Methodology, J.W., T.W., S.Z.; Software, J.W., T.W., S.Z.; Validation, T.W., H.Y.; Formal Analysis, J.W., T.W., S.Z.; Investigation, T.W., S.Z., L.B., M.A.B., J.F.E.; Resources, L.B., M.A.B., J.F.E.; Data Curation, J.W., S.Z.; Writing – Original Draft, S.Z.; Writing – Review & Editing, S.Z., L.B., M.A.B., J.F.E., T.W., J.W.; Visualization, J.W., T.W.; Supervision, S.Z.; Funding Acquisition, S.Z.
ACKNOWLEDGMENTS
We thank Ms. Yanfang Sun, Ms. Xiong Huan, Ms. Jin Qian and Ms. Ye Wang for their contribution to the study; Mr. Roger Nilsen and the Georgia Genomics Facility, Dr. Brian Dalley at the High throughout Genomics Core of the University of Utah and the BGI for sequencing; Drs. Jan Mrazek, Michael Adams and Stephen W. Ragsdale for their help and useful discussion on the microbiome study; and Drs. William Kisseberth, Carolyn J. Henry, Susan E. Lana and Nicole C. Northrup for helping collecting canine samples. Confocal imaging was performed at the UGA Biomedical Microscopy Core.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work is funded by NCI R01 CA182093, and the AKC Canine Health Foundation to S. Zhao.
REFERENCES
1. Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ 3rd, Zody MC, Mauceli E, Xie X, Breen M, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005; 438:803-819.
2. Boyko AR. The domestic dog: man's best friend in the genomic era. Genome Biol. 2011; 12:216.
3. Hayward JJ, Castelhano MG, Oliveira KC, Corey E, Balkman C, Baxter TL, Casal ML, Center SA, Fang M, Garrison SJ, Kalla SE, Korniliev P, Kotlikoff MI, et al. Complex disease and phenotype mapping in the domestic dog. Nat Commun. 2016; 7:10460.
4. Paoloni M, Khanna C. Translation of new cancer treatments from pet dogs to humans. Nat Rev Cancer. 2008; 8:147-156.
5. Tang J, Le S, Sun L, Yan X, Zhang M, Macleod J, Leroy B, Northrup N, Ellis A, Yeatman TJ, Liang Y, Zwick ME, Zhao S. Copy number abnormalities in sporadic canine colorectal cancers. Genome Res. 2010; 20:341-350.
6. Liu D, Xiong H, Ellis AE, Northrup NC, Dobbin KK, Shin DM, Zhao S. Canine spontaneous head and neck squamous cell carcinomas represent their human counterparts at the molecular level. PLoS Genet. 2015; 11:e1005277.
7. Liu D, Xiong H, Ellis AE, Northrup NC, Rodriguez CO Jr, O'Regan RM, Dalton S, Zhao S. Molecular homology and difference between spontaneous canine mammary cancer and human breast cancer. Cancer Res. 2014; 74:5045-56.
8. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996; 87:159-170.
9. Youmans L, Taylor C, Shin E, Harrell A, Ellis AE, Seguin B, Ji X, Zhao S. Frequent alteration of the tumor suppressor gene APC in sporadic canine colorectal tumors. PLoS One. 2012; 7:e50813.
10. Eifert C, Powers RS. From cancer genomes to oncogenic drivers, tumour dependencies and therapeutic targets. Nat Rev Cancer. 2012; 12:572-578.
11. Tang J, Li Y, Lyon K, Camps J, Dalton S, Ried T, Zhao S. Cancer driver–passenger distinction via sporadic human and dog cancer comparison: a proof-of-principle study with colorectal cancer. Oncogene. 2014; 33:814-822.
12. Li Y, Xu J, Xiong H, Ma Z, Wang Z, Kipreos ET, Dalton S, Zhao S. Cancer driver candidate genes AVL9, DENND5A and NUPL1 contribute to MDCK cystogenesis. Oncoscience. 2014; 1:854-865. https://doi.org/10.18632/oncoscience.107.
13. Grady WM. Genetic testing for high-risk colon cancer patients. Gastroenterology. 2003; 124:1574-1594.
14. Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011; 6:479-507.
15. Samadder NJ, Neklason DW, Boucher KM, Byrne KR, Kanth P, Samowitz W, Jones D, Tavtigian SV, Done MW, Berry T, Jasperson K, Pappas L, Smith L, et al. Effect of Sulindac and Erlotinib vs Placebo on Duodenal Neoplasia in Familial Adenomatous Polyposis: A Randomized Clinical Trial. JAMA. 2016; 315:1266-1275.
16. Johnson JC, DiSario JA, Grady WM. Surveillance and Treatment of Periampullary and Duodenal Adenomas in Familial Adenomatous Polyposis. Curr Treat Options Gastroenterol. 2004; 7:79-89.
17. Di Cecilia S, Zhang F, Sancho A, Li SD, Aguilo F, Sun YF, Rengasamy M, Zhang WJ, Del Vecchio L, Salvatore F, Walsh MJ. RBM5-AS1 Is Critical for Self-Renewal of Colon Cancer Stem-like Cells. Cancer Res. 2016; 76:5615-5627.
18. Lipkin SM, Afrasiabi K. Familial colorectal cancer syndrome X. Semin Oncol. 2007; 34:425-427.
19. Lipkin SM, Wang V, Jacoby R, Banerjee-Basu S, Baxevanis AD, Lynch HT, Elliott RM, Collins FS. MLH3: a DNA mismatch repair gene associated with mammalian microsatellite instability. Nat Genet. 2000; 24:27-35.
20. Thompson BA, Greenblatt MS, Vallee MP, Herkert JC, Tessereau C, Young EL, Adzhubey IA, Li B, Bell R, Feng B, Mooney SD, Radivojac P, Sunyaev SR, et al. Calibration of multiple in silico tools for predicting pathogenicity of mismatch repair gene missense substitutions. Hum Mutat. 2013; 34:255-265.
21. Park DJ, Tao K, Le Calvez-Kelm F, Tu ND, Robinot N, Hammet F, Odefrey F, Tsimiklis H, Teo ZL, Thingholm LB, Young EL, Voegele C, Lonie A, et al. Rare Mutations in RINT1 Predispose Carriers to Breast and Lynch Syndrome-Spectrum Cancers. Cancer Discov. 2014; 4:804-815.
22. Hoeppner MP, Lundquist A, Pirun M, Meadows JR, Zamani N, Johnson J, Sundstrom G, Cook A, FitzGerald MG, Swofford R, Mauceli E, Moghadam BT, Greka A, et al. An improved canine genome and a comprehensive catalogue of coding genes and non-coding transcripts. PLoS One. 2014; 9:e91172.
23. Bai B, Zhao WM, Tang BX, Wang YQ, Wang L, Zhang Z, Yang HC, Liu YH, Zhu JW, Irwin DM, Wang GD, Zhang YP. DoGSD: the dog and wolf genome SNP database. Nucleic Acids Res. 2015; 43:D777-783.
24. Folkman L, Stantic B, Sattar A, Zhou Y. EASE-MM: Sequence-Based Prediction of Mutation-Induced Stability Changes with Feature-Based Multiple Models. J Mol Biol. 2016; 428:1394-1405.
25. Sato Y, Yoshikawa A, Yamagata A, Mimura H, Yamashita M, Ookata K, Nureki O, Iwai K, Komada M, Fukai S. Structural basis for specific cleavage of Lys 63-linked polyubiquitin chains. Nature. 2008; 455:358-362.
26. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330-337.
27. Alkhairy OK, Abolhassani H, Rezaei N, Fang M, Andersen KK, Chavoshzadeh Z, Mohammadzadeh I, El-Rajab MA, Massaad M, Chou J, Aghamohammadi A, Geha RS, Hammarstrom L. Spectrum of Phenotypes Associated with Mutations in LRBA. J Clin Immunol. 2016; 36:33-45.
28. May-Simera HL, Gumerson JD, Gao C, Campos M, Cologna SM, Beyer T, Boldt K, Kaya KD, Patel N, Kretschmer F, Kelley MW, Petralia RS, Davey MG, Li T. Loss of MACF1 Abolishes Ciliogenesis and Disrupts Apicobasal Polarity Establishment in the Retina. Cell Rep. 2016; 17:1399-1413.
29. Liyasova MS, Ma K, Lipkowitz S. Molecular pathways: cbl proteins in tumorigenesis and antitumor immunity-opportunities for cancer treatment. Clin Cancer Res. 2015; 21:1789-1794.
30. Garcia-Domingo D, Ramirez D, Gonzalez de Buitrago G, Martinez AC. Death inducer-obliterator 1 triggers apoptosis after nuclear translocation and caspase upregulation. Mol Cell Biol. 2003; 23:3216-3225.
31. Merlos-Suarez A, Barriga FM, Jung P, Iglesias M, Cespedes MV, Rossell D, Sevillano M, Hernando-Momblona X, da Silva-Diz V, Munoz P, Clevers H, Sancho E, Mangues R, Batlle E. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell. 2011; 8:511-524.
32. Kosinski C, Li VS, Chan AS, Zhang J, Ho C, Tsui WY, Chan TL, Mifflin RC, Powell DW, Yuen ST, Leung SY, Chen X. Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. Proc Natl Acad Sci U S A. 2007; 104:15418-15423.
33. Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The Human Microbiome Project. Nature. 2007; 449:804-810.
34. Schmitz S, Suchodolski J. Understanding the canine intestinal microbiota and its modification by pro-, pre- and synbiotics - what is the evidence? Vet Med Sci. 2016; 2:71-94.
35. Silva RO, Lobato FC. Clostridium perfringens: A review of enteric diseases in dogs, cats and wild animals. Anaerobe. 2015; 33:14-17.
36. Mcdonel JL. Clostridium-Perfringens Toxins (Type-a, Type-B, Type-C, Type-D, Type-E). Pharmacol Ther. 1980; 10:617-655.
37. Shimizu T, Ohtani K, Hirakawa H, Ohshima K, Yamashita A, Shiba T, Ogasawara N, Hattori M, Kuhara S, Hayashi H. Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc Natl Acad Sci U S A. 2002; 99:996-1001.
38. Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006; 6:369-381.
39. Babaei-Jadidi R, Li N, Saadeddin A, Spencer-Dene B, Jandke A, Muhammad B, Ibrahim EE, Muraleedharan R, Abuzinadah M, Davis H, Lewis A, Watson S, Behrens A, et al. FBXW7 influences murine intestinal homeostasis and cancer, targeting Notch, Jun, and DEK for degradation. J Exp Med. 2011; 208:295-312.
40. Choi SH, Wright JB, Gerber SA, Cole MD. Myc protein is stabilized by suppression of a novel E3 ligase complex in cancer cells. Genes Dev. 2010; 24:1236-1241.
41. Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009; 10:550-563.
42. Ibarrola N, Kratchmarova I, Nakajima D, Schiemann WP, Moustakas A, Pandey A, Mann M. Cloning of a novel signaling molecule, AMSH-2, that potentiates transforming growth factor beta signal ing. BMC Cell Biol. 2004; 5:2.
43. McCullough J, Clague MJ, Urbe S. AMSH is an endosome-associated ubiquitin isopeptidase. J Cell Biol. 2004; 166:487-492.
44. Cancer Genome Atlas Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014; 513:202-209.
45. Zeller KI, Jegga AG, Aronow BJ, O'Donnell KA, Dang CV. An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol. 2003; 4:R69.
46. Mili S, Moissoglu K, Macara IG. Genome-wide screen reveals APC-associated RNAs enriched in cell protrusions. Nature. 2008; 453:115-119.
47. Hyun DH, Lee GH. Cytochrome b5 reductase, a plasma membrane redox enzyme, protects neuronal cells against metabolic and oxidative stress through maintaining redox state and bioenergetics. Age (Dordr). 2015; 37:122.
48. Reott MA, Parker AC, Rocha ER, Smith CJ. Thioredoxins in Redox Maintenance and Survival during Oxidative Stress of Bacteroides fragilis. J Bacteriol. 2009; 191:3384-3391.
49. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010; 26:589-595.
50. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105-1111.
51. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15-21.
52. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013; 43:11 10 11-33.
53. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013; 31:213-219.
54. Anders S, Pyl PT, Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166-169.