
INTRODUCTION
Prostate cancer (PC) is the most commonly diagnosed cancer and the sixth leading cause of cancer-related death among men worldwide. Bone metastasis is the leading cause of morbidity and mortality in patients with PC and is the preferred target for therapy. Because metastatic biopsies are often difficult to obtain, using blood samples would be preferred for biological characterization of PC. Classical blood biomarkers are used for prognostic evaluation of disease progression and therapeutic response. but analysis of circulating tumor cells (CTCs), cell-free DNA, cell-free RNAs or miRNAs, exosomes and thrombocytes in liquid biopsies can provide broader molecular details of the cancer phenotype, which could be used for personalized therapy with specific targeting drugs [1].
CTCs can be used as tumor biomarkers because a high number of CTCs strongly correlate with metastatic disease and poor prognosis in metastatic PC in both the hormone naïve (HN) and castration-resistant (CR) settings [2, 3]. Furthermore, detection of specific gene expression in CTCs can provide reliable information regarding the prognosis and therapeutic resistance to treatments targeting androgen receptor signaling [3–6]. CTCs may thus reflect not only the tumor burden but also the biology of the disease.
In various cancers, similarities have been detected in the genomic compositions of CTCs and tumor tissues [7, 8]. In PC, for example, shared genomic alterations have been identified in CTCs and tumor tissue samples [9]. Moreover, androgen receptor (AR) amplification is concordant between CTCs and tumor tissue biopsies from CRPC patients [10]. It is unclear, however, whether CTCs accurately reflect the phenotype of metastatic tissue in PC. Cho et al. showed that expression of a small number of genes (present or absent) was concordant between single CTCs and micro-dissected PC cells from bone marrow biopsies in 75% of cases [11]. In colorectal cancer, gene expression in the CTCs was more similar to that in liver metastases than the primary tumors, but the concordance between the gene expression profiles of CTCs and metastases was low [12]. In breast cancer, concordance of gene expression profiles in CTCs and primary tumors was highly variable [13].
In the present study, we analyzed the gene expression profiles in paired samples of CTCs and metastatic tissue from the spines from PC patients (Table 1). Our data show that to a large extent the gene expression profile of CTCs mirrors that in the paired metastatic tissue. However, all selected genes did not perform equally well. Therefore, careful selection of genes and analysis of a biomarker panel are required to develop CTC-based liquid biopsy strategies for clinical applications in PC.
Table 1: Patient characteristics
At diagnosis |
At surgery |
Time from diagnosis to surgery (years) |
|||
|---|---|---|---|---|---|
Median age year (range) |
|||||
hormone naive (n=5) |
68 (64-83) |
68 (64-83) |
0 |
||
GnRH naive (n=1) |
82 |
87 |
5.8 |
||
GnRH initated (n=1) |
69 |
70 |
0.1 |
||
CRPC (n=15) |
70 (57-83) |
75 (59-88) |
4.5 (1.0-14.8) |
||
Median PSA ng/ml (range) |
|||||
hormone naive (n=5) |
731 (111-1200) |
731 (111-1200) |
|||
GnRH naive (n=1) |
3.12 |
93 |
|||
GnRH initated (n=1) |
1200 |
x |
|||
CRPC (n=15) |
33.5 (5-334) |
81.4 (5.8-276) |
|||
Gleason score (n) |
GS 6-7 |
GS 8-10 |
GS X |
||
hormone naive (n=5) |
0 |
1 |
4 |
||
GnRH naive (n=1) |
1 |
0 |
0 |
||
GnRH initated (n=1) |
0 |
1 |
0 |
||
CRPC (n=15)* |
7 |
6 |
2 |
||
Therapy before surgery |
None |
TAB** |
TAB+Docetaxel |
Docetaxel |
Enzalutamide |
CRPC (n=15)*** |
6 |
3 |
2 |
3 |
1 |
* Gleason score was obtained at diagnosis, not at surgery.
* TAB = total androgen blockade, GnRH analogue plus bicalutamide.
** All patients were treated with GnRH analogues.
RESULTS
Detection of gene expression in spinal metastasis tissues and CTCs from PC patients
We excluded 15 of the 46 genes in the PC-panel from further analysis. These included EPCAM used for normalizing gene expression values, the general endogenous control genes GAPDH and GUSB, as well as CD45 and CD44 which mainly reflect the white blood cell contamination in the CTC samples. MYC, TP53, and ANXA2R were excluded due to their frequent detection in CTC-negative (EPCAM- negative) samples, which was interpreted as contaminating signals from the white blood cells (data not shown). Further, ESR1, ESR2, PTCH1, MET, CYP11A1, CYP17A1, and CYP19A1 genes were excluded because they were rarely detected in the CTC samples (Table 2).
Table 2: Prostate cancer panel; detected gene expression signals in CTC and metastatic tissue and their correlation
Gene |
Detected signals in CTC |
Detected signals in metastases |
Correlation coefficient, R |
P-value |
Detected signals in CTC |
Detected signals in metastases |
|
|---|---|---|---|---|---|---|---|
AGR2 |
18 |
22 |
0.778 |
p<0.001 |
Genes not included in analysis due to low detection frequency in CTC samples |
||
AHR |
3 |
22 |
-0.500 |
0.667 |
|||
AKR1C3 |
17 |
22 |
0.704 |
p<0.01 |
CYP11A1 |
1 |
22 |
AKT2 |
11 |
22 |
-0.159 |
0.640 |
CYP17A1 |
1 |
14 |
ALDH |
13 |
22 |
0.316 |
0.293 |
CYP19A1 |
1 |
22 |
AR |
14 |
22 |
0.565 |
p<0.05 |
ESR1 |
0 |
22 |
ARV7 |
7 |
22 |
0.750 |
0.052 |
ESR2 |
0 |
13 |
AURKA |
12 |
22 |
-0.130 |
0.688 |
MET |
1 |
22 |
BCL2 |
10 |
22 |
-0.395 |
0.258 |
PTCH1 |
0 |
22 |
CDH1 |
14 |
22 |
0.575 |
p<0.05 |
Genes not included in analysis due to frequent detection in CTC negative samples |
||
CDH2 |
2 |
22 |
- |
- |
|||
DDR1 |
2 |
22 |
- |
- |
MYC |
22 |
|
EGFR |
4 |
22 |
-0.200 |
0.800 |
TP53 |
22 |
|
EMP2 |
11 |
22 |
0.359 |
0.278 |
ANXA2R |
22 |
|
FOLH1 |
16 |
22 |
0.900 |
p<0.001 |
Control genes not included in analysis |
||
HER2 |
11 |
22 |
0.712 |
p<0.05 |
GAPDH |
22 |
22 |
KLK3 |
18 |
22 |
0.451 |
0.061 |
GUSB |
22 |
22 |
KRT19 |
16 |
22 |
0.693 |
p<0.01 |
CD44 |
22 |
22 |
MDK |
18 |
22 |
0.720 |
p<0.01 |
CD45 |
2 |
2 22 |
POU5F1 |
13 |
21 |
0.478 |
0.099 |
|||
PSCA |
10 |
22 |
0.588 |
0.074 |
|||
RUNX2 |
5 |
22 |
0.872 |
0.054 |
|||
SNAI1 |
5 |
22 |
0.300 |
0.624 |
|||
SPINK1 |
11 |
22 |
0.724 |
p<0.05 |
|||
SRD5A1 |
15 |
21 |
0.445 |
0.110 |
|||
TACSTD2 |
16 |
22 |
0.379 |
0.147 |
|||
TOP2A |
14 |
21 |
0.263 |
0.363 |
|||
TUBB3 |
6 |
22 |
-0.029 |
0.957 |
|||
Twist1 |
7 |
22 |
0.179 |
0.702 |
|||
UPA |
7 |
22 |
-0.414 |
0.355 |
|||
VEGFA |
14 |
22 |
0.066 |
0.822 |
|||
Analysis are performed with Spearman rank correlation. Genes with a correlation between CTC and metastases with a p-value less than 0.1 are marked in bold text.
In the bone metastasis tissue, the 31 genes that were analyzed were detected in all samples, except for POU5F1, SRD5A1, and TOP2A (96%), CYP17A1 (67 %), and ESR2 (58 %). In the CTCs, the detection ratio of these 31 genes was much lower. The detection frequency in the CTC samples ranged from 87 % (AGR2, AKR1C3, and KLK3) to 4 % (CYP11A1, CDH11, CYP17A1, and MET) (Table 2). Prostate cancer origin of one of the CRPC patients could not be confirmed because the CTCs did not show any detectable expression of either KLK3 or FOLH1. Hence, this sample was excluded from further analyses.
Identification of genes in CTC reflecting expression in bone metastases
We assessed the potential of the included genes in the CTC-based analysis to reflect gene expression in tissue metastases by estimating the correlation in signal intensity of the expression of individual genes in the CTC and metastasis samples of all patients. Nine of the thirty-one included genes showed a statistically significant correlation between the expression in CTC and the metastasis samples (Table 2). In addition, ARV7, POU5F1, PSCA, RUNX2, and KLK3 showed moderate to a high degree of correlation (R>0.4 and p<0.100).
Gene expression profiles in the CTCs and metastatic tissue samples of individual patients
We compared the gene expression profiles between matched CTC and metastatic tissue samples of each patient to get a broader picture of the metastasis than obtaining just the expression levels of individual genes. In this analysis, we included the calculated values for low detection levels in CTCs (i.e., cases with no signal for a specific gene despite sufficient CTC content). We analyzed 14 patients whose CTC samples showed gene expression values for more than 50% of the 31 included genes. Among these, 11 patients showed good correlation (correlation coefficient ≥ 0.5) between the gene expression profiles in CTC and metastasis samples. Six patients showed bootstrap generated p-values below 0.05 while five patients showed p-values between 0.05 and 0.1 (Table 3). Three of the 14 patients showed poor correlation (0.185, 0.250 and 0.307, respectively) between gene expression in CTC and metastatic tissue samples. Two of these three patients showed metastasis in the lungs (66%), whereas only one of the eleven patients (14 %) with a correlation coefficient > 0.5 showed lung metastasis.
Table 3: Correlation between gene expression in CTCs and metastases in individual patients
Patient ID |
CTC/Mets pairs |
Correlation coefficient, R |
P-value |
|---|---|---|---|
1 |
24 |
0.532 |
<0.05 |
2 |
29 |
0.591 |
<0.05 |
5 |
28 |
0.508 |
<0.05 |
6 |
28 |
0.793 |
<0.001 |
7 |
22 |
0.676 |
<0.01 |
9 |
18 |
0.531 |
<0.05 |
10 |
28 |
0.487 |
<0.05 |
12 |
20 |
0.677 |
<0.01 |
14 |
24 |
0.25 |
0.187 |
16 |
18 |
0.185 |
0.27 |
18 |
31 |
0.307 |
0.092 |
19 |
29 |
0.593 |
<0.001 |
21 |
30 |
0.53 |
<0.001 |
22 |
16 |
0.506 |
=0.05 |
Spearman correlations and bootstrap p-values for the gene expression profiles in CTCs and metastatic tissue in individual patients.
Analysis of patient grouping based on CTC and metastatic gene expression profiles
We selected the subset of 14 genes that showed good correlation between the CTC and metastatic tissue samples (Table 2) for hierarchical clustering analysis to determine whether patient grouping using the gene expression data from CTCs is similar to their grouping when the data from metastatic tissue samples were used. However, to optimize the clustering analysis, we only included patients with expression values for more than 10 of the included genes in their CTC samples, and we only included genes with positive expression signals in more than 10 of these patients. This resulted in a matrix of 13 patients and 12 genes (ARV7 and PSCA genes were excluded). Based on expression in metastatic tissue samples, the patients were grouped into three clusters. Concurrent analysis using CTC-derived data showed that the eleven of the thirteen patients (exceptions: patient numbers 14 and 21) grouped similarly into three clusters (Figure 1).
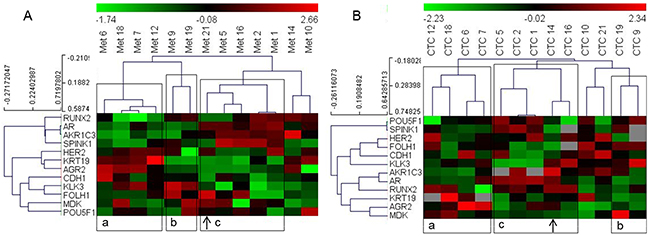
Figure 1: Heat map illustrates hierarchical clustering using significantly correlating genes in PC patients based on their expression in (A) metastatic tissue (B) and CTCs. Note: a, b and c represent groups of patient samples that cluster similarly in both metastases and CTC analyses. Arrows indicate samples switching clusters between the two analyses.
Next, we included all genes that were detected in more than 10 CTC samples and all patient samples whose CTC analyses resulted in 10 or more signals to determine the performance of the PC Panel in grouping patients based on CTC data without pre-analysis of significantly correlating genes or patients. We obtained a matrix of 15 patients and 23 genes. Hierarchical clustering of this dataset demonstrated that PC patient grouping based on the analyses of gene expression in CTCs was similar to that based on the metastatic dataset (Figure 2).
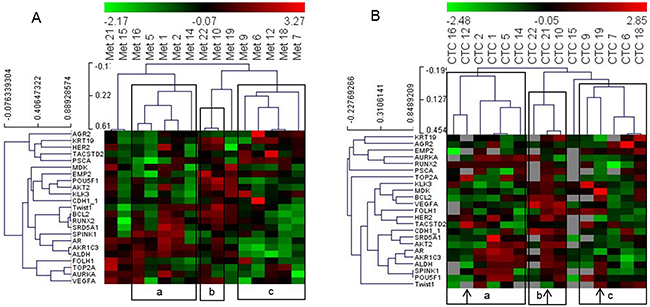
Figure 2: Heat map illustrates hierarchical clustering using significant genes without prior selection based on their expression in (A) metastatic tissue and (B) and CTCs. Note: a, b and c represent groups of patient samples that cluster similarly in analyses of both metastases and CTCs. Arrows indicate samples that switch clusters between the two analyses.
CTC analysis reflects patient treatment status
Next, we analyzed if the gene expression data from the CTCs identified phenotypes that match the clinical status of the patients. To address this, we classified the patients based on their hormonal status at sampling, i.e., if they were resistant to treatment with GnRH analogues [castration-resistant (CR), n=12] or were hormone naïve (HN; hormone naïve and GnRH naïve, n=6, Table 1). For clustering analysis, we used nine genes that were differentially expressed in the metastasis samples of these two categories. However, due to the low detection frequency of certain genes in the CTC samples, the dataset was modified to include patients with genes that displayed more than 50% detection frequency in the CTC-derived data. Thus, we obtained a matrix of 8 genes and 13 patients (5 HN and 8 CR). Clustering of these patients based on the gene expression data from metastatic tissue samples revealed two clusters, of which one included all HN and two CR patients. Clustering analysis of the CTC-derived dataset from these patients again grouped all HN samples, with two more CR patients clustered together with the HN patients (Figure 3). Notably, the two CR patients that grouped with the HN cluster in the CTC-based analysis (18 and 6) displayed high expression (green signal) of both AR and AKR1C3 genes, which was atypical for most HN samples (highlighted in Figure 3).
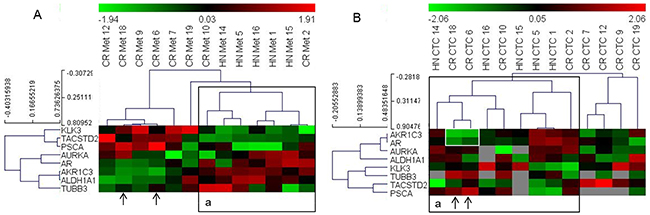
Figure 3: Heat map illustrates the hierarchical clustering using the limited gene subset discriminating HN and CR metastases, based on their expression in (A) metastases and (B) CTCs. Note: ‘A: a’ represents a cluster that includes all HN samples in the metastases analyses; ‘B: a’ represents a similar cluster in the CTCs. Arrows indicate samples that cluster differently in the CTC-based analysis; white box highlights the AR and AKR1C3 signals in two CR samples that change clusters.
DISCUSSION
CTCs have been used for a long time as biomarkers for PC prognosis and therapy response. However, the potential of obtaining phenotypical information from these circulating cancer cells has not been satisfactorily explored. In the present study, we analyzed the gene expression of CTCs to explore the potential of gaining phenotypical information regarding the metastatic disease.
The CTCs are intact cancer cells, and therefore carry biologically relevant information regarding the disease in addition to their use as biomarkers based on their enumeration. In the present study, we show a strong correlation between CTCs and bone metastasis samples from the same patient regarding the expression levels of several genes that are related to PC progression, metastasis, and therapy resistance. However, when analyzing the genes individually, it was obvious that the potential to reflect the expression level in metastases by CTC analysis differed among genes. Our analysis showed that nine out of forty-one genes displayed a strong correlation between their expression in CTCs and metastasis samples. Among these was ARV7 which expression in CTCs is related to abiraterone and enzalutamide resistance in several studies [4–6]. Previously, detection of any of the other eight genes in CTCs has not been related to any clinically relevant characteristic, and is for the first time presented to be found in CTCs in prostate cancer patients. The MDK and AGR2 genes are related to neuro-endocrine differentiation in CRPC [14, 15], which is a biological process that is associated with a non-functional AR signaling axis and correlates with poor prognosis. These tumors likely do not respond optimally to drugs that are targeted towards sustained AR signaling in CRPC. Similarly, although high expression of AR, ARV7, and AKR1C3 indicates that the tumors are dependent on activated AR-signaling their overexpression may be a part of the activated resistance mechanisms that lower sensitivity to AR targeting drugs [5, 16]. This was illustrated by our findings that show higher expression of AR and AKR1C3 the CR patients than the HN patients.
There are at least two plausible reasons for the differences in expression of several genes between CTC and metastasis samples. First, these genes may be differentially regulated in a CTC than a metastatic tumor cell. Second, the CTC and metastasis samples may contain different amounts and types of contaminating cells. In the CTCs, leukocyte contamination affects the detection levels of certain genes, whereas, in the metastatic tissue, tumor stroma is a major contaminating factor that influences detection of gene expression from tumor cells alone. This is exemplified in the present study by the poor correlation of urokinase-type plasminogen activator (UPA) and vascular endothelial growth factor A (VEGFA) genes, which are both expressed by tumor cells as well as the tumor stromal cells. Thus, care must be taken in selecting genes that enable CTC analysis to provide useful information regarding the metastatic tumor cells.
In the individual patients, the gene expression profile of CTC samples correlated with the gene expression profile of the corresponding metastatic samples in most cases. However, we also encountered patients where the correlation was poor or absent. We are unable to reliably predict the reasons for these differences between patients because the patient material in this study was too small. However, the presence of lung metastasis in two out of three patients that showed poor concordance between gene expression in the CTCs and bone metastatic tissue samples indicates that these CTC samples may also represent lung metastases, which presumably represent a different phenotype than the bone metastases. This suggests that CTCs represent the whole metastatic disease, and therefore, the gene panel should potentially represent the biological characteristics of metastases in different organs. This is supported by previous findings that the single cell CTC mutational status may be highly heterogeneous [17].
Our study also showed that patient grouping based on gene expression analysis of CTC samples was similar to grouping based on the analysis of the metastatic tissue. Moreover, patients were grouped according to the hormonal status based on the gene expression profiles of both CTC and metastasis samples. This shows that although all independent genes may not display a good correlation between the CTC and metastasis samples, a combination of genes may represent clinically relevant information regarding the tumor phenotype.
Although the analysis of CTCs from liquid biopsies has many advantages, the technology requires further optimization to overcome the limitations. We encountered two technical issues in the present study. The first relates to the contamination by leukocytes during CTC isolation, which limits the expression panel to genes that are not expressed in leukocytes. This represents a major obstacle that needs improvement in CTC isolation methodologies to develop reliable treatment prediction analysis for immune-related therapies such as programmed death-ligand 1 (PD-L1) antibody-based immunotherapy. The second technical limitation was the reliance on EPCAM and HER2 antibodies for the isolation of CTCs. This limits the detection of CTC populations with low expression of these antigens [18] or those masking these antigens by macromolecules [19]. Hence, CTCs that were not isolated by our methodology may represent other sub-populations of CTC, and their genetic profile may also be critical for evaluating the status of PC metastasis. This problem may be overcome by novel isolation methods that are not based on epithelial antigens. Therefore, it is plausible that we may overcome the insufficient sensitivity to detect expression of certain genes in the CTC samples by utilizing other isolation methods.
De Bono et al. showed that the amount of CTCs reflects the progression and treatment response of PC [20]. Since one of the technical issues encountered with CTC profiling is sensitivity as a result of the limited amount of material, the usefulness of the method is dependent on the amount of CTCs that are available for detection. Thus, patients with a non-metastatic disease or those responding to therapy are less suitable for this type of analysis. The present study includes only patients with metastatic disease, but despite their severe condition, we observed significant variations in the amount of CTCs isolated from individual patients. This affected the detection frequency of certain genes depending on their relative expression levels and limited their use in the analyses.
Another challenge we encountered in our study was about interpreting the absent gene expression signals from samples with low CTC content. It was obvious from our results that some genes were less expressed than others despite comparable CTC content. Thus, an absent signal in CTC-derived expression data cannot simply be interpreted as a low expression signal because it may be a result of a too low CTC content to allow detection of the specific gene. In the present study, we developed a strategy to identify the threshold of CTC content that would enable detection of each gene individually and avoid false low detection values. If a gene was not detected in a sample despite sufficient CTC content, it was assigned an expression value lower than the lowest detected for that gene. If the CTC content was below the threshold and the gene expression could not be detected, that absent signal was excluded from further analysis. In the future, to enable expression profiling of CTCs for clinical applications, there is a need to increase the detection sensitivity and develop robust methods to handle absent gene expression signals as a result of limited CTC content.
In conclusion, our study demonstrates the potential of CTCs to mirror the gene expression profile of PC bone metastases in individual patients. The study also points out the importance of careful selection of genes to accommodate the technical and biological aspects that limit the interpretation of expression of different genes.
MATERIALS AND METHODS
Patients
We recruited twenty-five patients undergoing surgery for spinal cord compression symptoms related to metastatic PC, between 2013 and 2016, at the Department of Orthopedics, Sahlgrenska University Hospital, Gothenburg. The study protocol was approved by the ethics committee in Gothenburg, Sweden (# 936-12 and # 455-11). We excluded two patients that had other cancer diagnoses in addition to PC and one patient that was CTC-negative. Clinical information of the included 22 patients is shown in Table 1. The hormone naïve patients (n=5) were diagnosed with metastatic PC at the time of surgery and did not receive any hormonal therapy before surgery. The patient annotated as GnRH naïve in this study received bicalutamide three months before surgery, but responded to GnRH therapy initiated after surgery. One patient annotated as GnRH initiated in this study was diagnosed with metastatic PC and was treated with GnRH antagonists for one month before surgery, after which he responded well to GnRH agonists. The CRPC patients (n=15) relapsed with metastasis in the spine after GnRH therapy alone, or after second or third line therapy (Table 1).
RNA preparation and cDNA synthesis from metastatic tissue
Metastatic tissue that was removed during surgery was immersed in RNAlater (Ambion) and frozen at -80°C. Total RNA was prepared from 40-100 mg tissue (dependent on the content of bone) using the RNeasy Plus Universal Mini kit (Qiagen) according to the manufacturer’s instructions. Briefly, the tissue was homogenized in the Qiazol Lysis Reagent in a Tissue Lyser II homogenizer at 25 Hz for5 min twice, and the resulting homogenate was treated with gDNA eliminator solution (Qiagen), extracted with chloroform. After centrifugation, the aqueous phase was mixed with 70% ethanol and directly loaded onto the RNeasy Mini spin column. After washing, the total RNA was eluted from the column in 30 μl RNase-free water, and its concentration and purity were measured in a NanoDrop (Thermo Scientific, Waltham, MA, USA). Forty nanograms of the RNA from metastatic tissue samples were converted to cDNA using the TATAA GrandScript cDNA Synthesis Kit (Cat. No. #A103a, TATAA Biocenter, Gothenburg, Sweden).
CTC isolation and cDNA synthesis
The CTCs were isolated from blood samples and detected using the AdnaTest ProstateCancerSelect/Detect kit (Qiagen Hannover GmbH, Germany) as previously described [3]. Briefly, the patient blood samples were collected immediately before surgery in AdnaCollect tubes and refrigerated at 4°C. CTC isolation was performed within 24 hours by capturing them on EPCAM- and HER2 antibody-conjugated magnetic beads. The mRNAs from lysed CTCs were isolated using oligodT-conjugated magnetic beads and transcribed into cDNA.
Design of the PC gene expression panel
As shown in Table 2, the gene expression panel, which is referred to as PC-panel, was composed of 46 genes, of which five were control genes, and 41 were PC-related genes. The genes were selected based on their role in PC progression, metastasis, steroid synthesis and signaling, stemness and neuroendocrine differentiation as reported in the literature. All the included genes were expected to be highly expressed in PC cells and negligible expression in the leukocytes (contaminating white blood cells in the CTC samples).
Gene expression profiling
We pre-amplified 2 μl of cDNA samples from metastases and CTC samples (including beads from CTC collection) using the TATAA PreAmp Primer Mix and TATAA PreAmp GrandMaster® Mix (Cat. No. #TA05, TATAA Biocenter) in a T100 BioRad thermocycler. We also pre-amplified non-template control and human gDNA (0.5 ng/μl, TATAA Biocenter) samples. The preamplified samples were centrifuged to pellet the magnetic beads, and a fraction of the supernatant was diluted 10X in a separate tube. We performed qPCR analyses of the diluted samples (45 assays) using the ValidPrime™ assay kit (TATAA Biocenter) with specific primers designed for this study as shown in Supplementary Table 1. This assay is now available as part of the GrandPerformance CTC Assay Panel at the TATAA Biocenter. The qPCR analysis was performed using the TATAA Probe GrandMaster® Mix Low ROX (TATAA Biocenter) and GE 96.96 Dynamic Array™ Sample & Assay Loading Reagent Kit (P/N 85000802-R, Fluidigm). We also included preamplified no template control (preAmp NTC) and no template control (NTC) for the qPCR, which was performed on the BioMark system (Fluidigm) using the 96.96 Dynamic Array™ IFC (Integrated Fluidic Circuit). All the samples (including the NTCs and gDNA) were analyzed in duplicates. The assays we use were designed with ISO 17025 accredited methods (TATAA Biocenter) and validated in compliance with the MIQE guidelines [21], which is considered sufficient for research and most diagnostic usage.
Preprocessing, normalization, and interpretation of gene expression data
The raw data (averaged Cq-values) was controlled and corrected for genomic DNA contamination using the GenEx software (MultiD Analyses AB) with implemented functions for the ValidPrime™ concept [22]. The averaged Cq values corrected with more than one cycle (Cq) were considered compromised due to large-scale genomic DNA contamination and removed from the analysis.
EPCAM expression was considered as a measure of epithelial cell (i.e. CTC) content. The expression of other genes was normalized to EPCAM expression to eliminate the contamination from normal bone tissue or white blood cells in metastatic tissue and CTC samples, respectively. When expression was not detected for some genes in the CTC samples, the results were not automatically interpreted as displaying low expression. Instead, we individually identified a cut-off level of CTC content (based on EPCAM expression) so that the gene expression levels could be reliably detected in CTC samples, and eliminate false low CTC expression values. If the CTC content was now high enough for the gene to be reliably detected, the low expression value was used instead of the absent signal. On the other hand, if the CTC content was lower than the cut-off detection level that was required to detect the expression of a particular gene, the absent signal was excluded from further analyses. Therefore, if a specific gene was not detected in a specific CTC sample, the Cq(EPCAM) value for that CTC sample was compared to the Cq(EPCAM) values in other CTC samples where the specific gene was detected. We interpreted the absent signal as a valid detection value if the Cq(EPCAM) value in the specific sample was at least 2Cq values lower (i.e. four times higher EPCAM expression) than the highest Cq(EPCAM) value at which the specific gene could be detected in all the CTC samples analyzed. In such a scenario, the expression of that specific gene was assigned a delta Cq value, which was one Cq value higher than the highest Cq value detected for that specific gene. If the Cq(EPCAM) value in the CTC sample was higher than the cut-off Cq(EPCAM) value, i.e., displaying a lower EPCAM expression, the absent signal was regarded as absent and excluded from further analyses.
Statistics
Spearman Rank Correlation was used to compare the gene expression levels in CTC and metastasis samples from individual patients using the IBM SPSS Statistics 22.0.0.0 software. Since expression levels of the genes included in the panel may partly be dependent on each other within individual patients, the p-values were derived using the bootstrapping method as follows: Two patients were sampled randomly and the correlation between the CTC expression values from one patient and the metastasis expression values from the second patient were calculated. This was repeated 100000 times to achieve the empirical distribution function for the correlation between independent individuals. The p-value was then derived by comparing the observed correlations within patients to this distribution. The procedure was applied to the number of available complete pairs of matched samples for each patient. Hierarchical clustering was performed using normalized and mean centered data with Spearman Rank Correlation and Average linkage in a MultiExperiment Viewer (MeV, Dana-Farber Cancer Institute, US). All statistical tests were two-sided and p-values < 0.05 were considered statistically significant.
Abbreviations
CTC, Circulating tumor cells; CR, Castration resistant; HN, Hormone naïve; PC, Prostate cancer; Cq, Cycles of qPCR reaction; qPCR, quantitative real-time polymerase chain reaction; CRPC, Castration-resistant prostate cancer; GnRH, Gonadotropin-releasing hormone; AR, androgen receptor; RNA, ribonucleic acid; DNA, deoxyribonucleic acid; cDNA, Cyclic-DNA.
Author contributions
Andreas Josefsson contributed to project planning, ethical approvals, logistics, the inclusion of patients, analysis, preparation of tables and manuscript writing.
Karin Welen contributed to study initiation, project planning, ethical approvals, logistics of sample handling, isolation of circulating tumor cells and mRNA from metastasis samples, statistical analysis, preparation of tables and figures, and manuscript writing.
Jan-Erik Damber contributed to study initiation, writing ethical approvals, project planning, interpretation of results and manuscript writing.
Helena Brisby was responsible for the inclusion of patients and collaboration at the orthopedic center, interpretation of some results and finalizing the manuscript.
Karin Larsson was responsible for handling the patient samples, logistics, isolation of circulating tumor cells and extraction of the mRNA from the metastases, and finalizing the manuscript.
Marianne Månsson performed the majority of the statistical analysis and interpreted the results.
Jens Björkman was involved in optimizing the experimental set-up, interpretation of results, writing the methods section, and finalizing the manuscript.
Eva Rohlova performed the technical work at the TATAA Biocenter, including optimization of assays and validation of the pre-amplification steps. She also participated in writing the methods section, interpretation of results and finalization of the manuscript.
Daniel Åhs was involved in patient inclusion, logistics, the collection of patient data and data evaluation, and finalization of the manuscript.
ACKNOWLEDGMENTS
The authors acknowledge Anita Fae for help with CTC isolation, Mikael Kubista and Robert Sjöback (TATAA Biocenter) for help with setting up the CTC analyses, and the staff at the orthopedic surgery unit of the Sahlgrenska University Hospital for their valued assistance with blood and tissue sampling. The authors also acknowledge the funding support for this project from the Swedish Cancer Society, the Swedish Society of Medicine, the Movember Foundation, BioCARE, the Göteborg Medical Society, the Swedish PC Federation, the County Council for West Sweden (ALF project), the Swedish Society for Strategic Research, and the Research Foundations of P. Falk, M. Ågren, U. and K-E. Winberg.
CONFLICTS OF INTEREST
J. Björkman and E. Rohlova are employed by the TATAA Biocenter AB; J. Björkman is a stock owner in TATAA Biocenter AB. All the other authors declare that there are no potential conflicts of interest.
REFERENCES
1. Bardelli A, Pantel K. Liquid biopsies, what we do not know (yet). Cancer Cell. 2017; 31:172–79. https://doi.org/10.1016/j.ccell.2017.01.002.
2. Goodman OB Jr, Symanowski JT, Loudyi A, Fink LM, Ward DC, Vogelzang NJ. Circulating tumor cells as a predictive biomarker in patients with hormone-sensitive prostate cancer. Clin Genitourin Cancer. 2011; 9:31–38.
3. Josefsson A, Linder A, Flondell Site D, Canesin G, Stiehm A, Anand A, Bjartell A, Damber JE, Welén K. Circulating tumor cells as a marker for progression-free survival in metastatic castration-naïve prostate cancer. Prostate. 2017; 77:849–58. https://doi.org/10.1002/pros.23325.
4. Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, Nakazawa M, Nadal R, Paller CJ, Denmeade SR, Carducci MA, Eisenberger MA, Luo J. Androgen Receptor Splice Variant 7 and Efficacy of taxane chemotherapy in patients with metastatic castration-resistant prostate cancer. JAMA Oncol. 2015; 1:582–91. https://doi.org/10.1001/jamaoncol.2015.1341.
5. Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, Lotan TL, Zheng Q, De Marzo AM, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014; 371:1028–38. https://doi.org/10.1056/NEJMoa1315815.
6. Scher HI, Lu D, Schreiber NA, Louw J, Graf RP, Vargas HA, Johnson A, Jendrisak A, Bambury R, Danila D, McLaughlin B, Wahl J, Greene SB, et al. Association of AR-V7 on circulating tumor cells as a treatment-specific biomarker with outcomes and survival in castration-resistant prostate cancer. JAMA Oncol. 2016; 2:1441–49. https://doi.org/10.1001/jamaoncol.2016.1828.
7. Heitzer E, Auer M, Gasch C, Pichler M, Ulz P, Hoffmann EM, Lax S, Waldispuehl-Geigl J, Mauermann O, Lackner C, Höfler G, Eisner F, Sill H, et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013; 73:2965–75. https://doi.org/10.1158/0008-5472.CAN-12-4140.
8. Maheswaran S, Sequist LV, Nagrath S, Ulkus L, Brannigan B, Collura CV, Inserra E, Diederichs S, Iafrate AJ, Bell DW, Digumarthy S, Muzikansky A, Irimia D, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med. 2008; 359:366–77. https://doi.org/10.1056/NEJMoa0800668.
9. Jiang R, Lu YT, Ho H, Li B, Chen JF, Lin M, Li F, Wu K, Wu H, Lichterman J, Wan H, Lu CL, OuYang W, et al. A comparison of isolated circulating tumor cells and tissue biopsies using whole-genome sequencing in prostate cancer. Oncotarget. 2015; 6:44781–93. https://doi.org/10.18632/oncotarget.6330.
10. Podolak J, Eilers K, Newby T, Slottke R, Tucker E, Olson SB, Lue HW, Youngren J, Aggarwal R, Small EJ, Graff JN, Alumkal JJ, Beer TM, Thomas GV. Androgen receptor amplification is concordant between circulating tumor cells and biopsies from men undergoing treatment for metastatic castration resistant prostate cancer. Oncotarget. 2017; 8:71447–55. https://doi.org/10.18632/oncotarget.16169.
11. Cho WJ, Oliveira DS, Najy AJ, Mainetti LE, Aoun HD, Cher ML, Heath E, Kim HR, Bonfil RD. Gene expression analysis of bone metastasis and circulating tumor cells from metastatic castrate-resistant prostate cancer patients. J Transl Med. 2016; 14:72. https://doi.org/10.1186/s12967-016-0829-5.
12. Onstenk W, Sieuwerts AM, Mostert B, Lalmahomed Z, Bolt-de Vries JB, van Galen A, Smid M, Kraan J, Van M, de Weerd V, Ramírez-Moreno R, Biermann K, Verhoef C, et al. Molecular characteristics of circulating tumor cells resemble the liver metastasis more closely than the primary tumor in metastatic colorectal cancer. Oncotarget. 2016; 7:59058–69. https://doi.org/10.18632/oncotarget.10175.
13. Onstenk W, Sieuwerts AM, Weekhout M, Mostert B, Reijm EA, van Deurzen CH, Bolt-de Vries JB, Peeters DJ, Hamberg P, Seynaeve C, Jager A, de Jongh FE, Smid M, et al. Gene expression profiles of circulating tumor cells versus primary tumors in metastatic breast cancer. Cancer Lett. 2015; 362:36–44. https://doi.org/10.1016/j.canlet.2015.03.020.
14. Kani K, Malihi PD, Jiang Y, Wang H, Wang Y, Ruderman DL, Agus DB, Mallick P, Gross ME. Anterior gradient 2 (AGR2): blood-based biomarker elevated in metastatic prostate cancer associated with the neuroendocrine phenotype. Prostate. 2013; 73:306–15. https://doi.org/10.1002/pros.22569.
15. Nordin A, Wang W, Welén K, Damber JE. Midkine is associated with neuroendocrine differentiation in castration-resistant prostate cancer. Prostate. 2013; 73:657–67. https://doi.org/10.1002/pros.22607.
16. Jernberg E, Thysell E, Bovinder Ylitalo E, Rudolfsson S, Crnalic S, Widmark A, Bergh A, Wikström P. Characterization of prostate cancer bone metastases according to expression levels of steroidogenic enzymes and androgen receptor splice variants. PLoS One. 2013; 8:e77407. https://doi.org/10.1371/journal.pone.0077407.
17. De Luca F, Rotunno G, Salvianti F, Galardi F, Pestrin M, Gabellini S, Simi L, Mancini I, Vannucchi AM, Pazzagli M, Di Leo A, Pinzani P. Mutational analysis of single circulating tumor cells by next generation sequencing in metastatic breast cancer. Oncotarget. 2016; 7:26107–19. https://doi.org/10.18632/oncotarget.8431.
18. Punnoose EA, Atwal SK, Spoerke JM, Savage H, Pandita A, Yeh RF, Pirzkall A, Fine BM, Amler LC, Chen DS, Lackner MR. Molecular biomarker analyses using circulating tumor cells. PLoS One. 2010; 5:e12517. https://doi.org/10.1371/journal.pone.0012517.
19. Hekimian K, Stein EL, Pachmann U, Pachmann K. Demasking of epithelial cell adhesion molecule (EpCAM) on circulating epithelial tumor cells by Tween®20 treatment in breast cancer patients. Clin Chem Lab Med. 2012; 50:701–08. https://doi.org/10.1515/cclm.2011.812.
20. de Bono JS, Scher HI, Montgomery RB, Parker C, Miller MC, Tissing H, Doyle GV, Terstappen LW, Pienta KJ, Raghavan D. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clinical Cancer Research. 2008; 14:6302–6309. https://doi.org/10.1158/1078-0432.CCR-08-0872.
21. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009; 55:611–22. https://doi.org/10.1373/clinchem.2008.112797.
22. Laurell H, Iacovoni JS, Abot A, Svec D, Maoret JJ, Arnal JF, Kubista M. Correction of RT-qPCR data for genomic DNA-derived signals with ValidPrime. Nucleic Acids Res. 2012; 40:e51. https://doi.org/10.1093/nar/gkr1259.