
INTRODUCTION
The immune system is capable of targeted tumor cell killing through the process of immunosurveillance. Although tumors often develop ways to escape immunosurveillance, the growing interest and understanding of molecular interactions that occur between the tumor and the immune system have resulted in treatment strategies aimed at harnessing the immune system to target cancers. Recent advances in tumor immunology have produced immune checkpoint inhibitors (ICIs), cancer treatments designed to relieve the checkpoint restraints on adaptive immunity [1]. ICIs have revolutionized treatments for many types of cancer [1–3]. Despite these successes, not all patients respond to ICI therapy, for reasons that are varied and incompletely understood. It is thought that ICIs may be less effective in tumors that are poorly immunogenic, as defined by low levels of tumor infiltrating lymphocytes (TILs), minimal cross-presentation of tumor neoantigens, and high levels of immune suppressive leukocytes such as regulatory T-cells (TRegs), tumor associated macrophages (TAMs) and myeloid derived suppressor cells (MDSCs) [4–7]. Innovative strategies to increase immunogenicity in tumors are being explored through a variety of approaches. One emerging strategy is based on activation of innate immunity in the tumor microenvironment (TME) [8, 9]. Innate immunity is a powerful arm of the immune system responsible for rapid anti-microbial immunity, often inducing programmed cell death of an infected cell. Innate immunity functions beyond the infected cell as well, by modulating the expression of cytokines and chemokines that recruit T-lymphocytes to the affected tissue, enhance antigen presentation, and increase cross-priming to antigen-specific T-cells [8, 10]. This idea is being explored extensively in regards to the pattern recognition receptor (PRR) known as Stimulator of Interferon Genes (STING) [11, 12]. Synthetic STING ligands potently induce anti-tumor immunity in several cancers, including breast cancer, chronic lymphocytic leukemia, colon cancer, and squamous cell carcinoma [13–17]. However, there is increasing evidence that STING signaling might be defective in some cancers, due to mutations, promoter methylation, and decreased expression of STING pathway effectors [18, 19], thus limiting their potential efficacy in the tumor cell compartment of the TME. However, other cells of the TME, particularly cells of the immune compartment, may retain STING signaling even when the STING pathway is defective within the tumor cells, per se, allowing STING ligands to induce innate immunity within the TME under these circumstances [20].
Viral nucleic acid sensors, such as the RNA helicase known as retinoic acid-inducible gene I (RIG-I, encoded by the gene DDX58) [21], are expressed in most cells of the human body, including tumor cells [22]. When infected by an RNA virus, double-stranded RNA replication intermediates derived from the virus bind to RIG-I [23–26] and activate a RIG-I inflammasome leading to pyroptosis, a highly immunogenic mechanism of programmed cell death [27–29]. A hallmark of pyroptosis is the formation of pores in the plasma membrane [30], leading to hypotonic cell swelling and leakage of intracellular contents, including danger associated molecular patterns (DAMPs), into the microenvironment. RIG-I signaling simultaneously induces expression of pro-inflammatory cytokines [8, 10]. Together, DAMPs and pro-inflammatory cytokines stimulate a local acute inflammatory immune response aimed at removal of virus and virally-infected cells [31]. Interestingly, viral nucleotide motifs can be mimicked using synthetic, non-infectious oligonucleotides. These RIG-I agonists are capable of triggering RIG-I signaling, pyroptosis, and acute inflammation [26, 32–35]. In the cancer setting, RIG-I activation could thus provide a three-pronged attack: 1.) direct activation of tumor cell death; 2.) cytokine-mediated activation of innate immune effectors (e.g., macrophages, natural killer cells), and 3.) increased recruitment and cross priming of adaptive immune effectors (e.g., CD8+ T-lymphocytes) through a cytokine-enriched microenvironment and enhanced activity of professional antigen presenting cells [APCs, e.g., dendritic cells (DCs) or macrophages] (Figure 1). Synthetic RIG-I agonists are being explored as a therapeutic approach in a diverse range of cancers [27, 33, 34, 36]. Here, we review studies of RIG-I signaling in the tumor microenvironment, and preclinical studies investigating RIG-I agonists for cancer treatment.
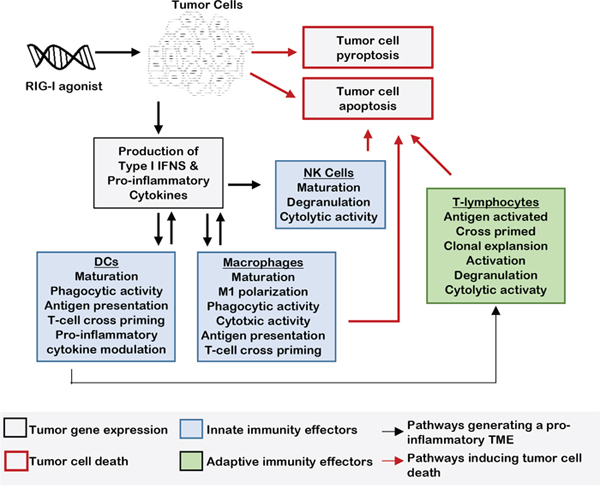
Figure 1: RLR activation signals innate immunity in the TME. When tumor cells are treated with an RIG-I mimetic, inflammatory cytokine and type I IFN expression is rapidly upregulated, inducing innate immune responses in the tumor microenvironment. The cytolytic activity of leukocytes, such NK cells and macrophages, is increased in response to this IFN-enriched microenvironment. Maturation and activation of macrophages and DCs result in enhanced antigen presentation to T-lymphocytes in tumor draining lymph nodes. T-regulatory cell differentiation is decreased by the pro-inflammatory microenvironment produced by RIG-I activation.
Activation of RIG-I induces pro-inflammatory signaling in a cell-intrinsic manner
RIG-I was first identified as a cytosolic DExD/H box RNA helicase activated in response to certain RNA viruses [21]. RIG-I is activated upon recognition of its ligand, double-stranded RNA sequences modified with a 5’-triphosphate (5’-3pRNA) or 5’-diphosphate (5’-2pRNA) motif [24, 26, 27, 37]. RIG-I activation may occur in response to other RNA motifs, including blunt dsRNAs [38], monomeric RNA within defective human immunodeficiency virus (HIV)-1 particles [39], cytoplasmic long non-coding RNAs [40], small nuclear RNAs [41–44], or endogenous retroviral transcripts. In addition to the DexD/H box RNA helicase domain, RIG-I is characterized by an amino-terminal Caspase Activation and Recruitment Domain (CARD) domain, and a Carboxy-Terminal Domain (CTD) [45–47]. Once activated by its ligand, RIG-I undergoes an ATP-dependent conformational change, exposing its CARD domain for polyubiquitylation [48] by ubiquitin ligases such as TRIM25, Riplet and others [49–52]. Once polyubiquitylated, a mitochondrial signalosome, comprised of the proteins WHIP, PPP6C and TRIM14, recruits RIG-I to the mitochondrial surface where the CARD domain of RIG-I interacts with the CARD domain of Mitochondrial Anti-Viral Signaling (MAVS), a requisite RIG-I co-factor [49, 53–55].
Once engaged, MAVS signaling activates three kinases that serve as regulators of inflammation, Inhibitor of κB-Kinase (IKK)-γ, TANK-Binding Kinase (TBK)-1 and IKK-ε [56–58]. These kinases phosphorylate Interferon (IFN) Regulatory Factor (IRF)-1, IRF-3, IRF-7, and Nuclear Factor (NF)-κB [59–61], transcription factors that drive expression of a pro-inflammatory transcriptional program that includes type I IFNs and pro-inflammatory cytokines [45, 62]. Importantly, IFN-α, IFN-β, and other pro-inflammatory cytokines produced in response to RIG-I activation drive a feed-forward signaling loop that maintains high expression levels of RIG-I, IFNs and additional pro-inflammatory IFN-stimulated genes (ISGs), by maintaining phosphorylation and activation of the transcription factors IRF-3, IRF-7, and NF-κB, and by phosphorylation of the transcription factor Signal Transducer and Activator of Transcription (STAT)-1, which occurs in response to IFN-α/β receptor (IFNAR)-mediated activation of JAK-STAT signaling (Figure 2) [62]. This feed-forward signaling model amplifies inflammatory cytokine production in the infected and neighboring cells, while recruiting leukocytes to the infected area, including pro-inflammatory lymphocytes. Since a ‘T-cell inflamed’ microenvironment is often associated with an improved prognosis for several cancers, and correlates with increased tumor sensitivity to ICIs, the pro-inflammatory phenotype induced by RIG-I activation may be an attractive treatment approach to increase tumor immunogenicity and clinical success of ICIs.
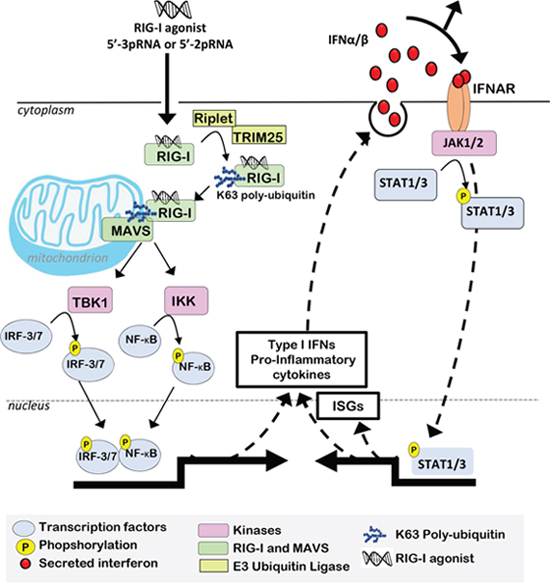
Figure 2: RIG-I activation induces Type I IFNs, which support pro-inflammatory transcriptional reprogramming. RIG-I binding to 5’-3pRNA or 5’-2pRNA induces a conformational change, allowing RIG-I CARD domains to be polyubiquitylated by E3 ligases (e.g., Riplet or TRIM25). Polyubiquitylated RIG-I is recruited to mitochondria outer membranes, where it interacts with MAVS, which then activates IKK-ε, IKK-γ, and TBK1, kinases responsible for phosphorylation/activation of transcription factors (ATF-1, c-Jun, CBP, IRF-3, NF-κB). These transcription factors induce an expression profile that includes Type I I IFNs and additional pro-inflammatory cytokines. Type I IFNs bind to IFNAR, activating the intracellular tyrosine kinase JAK1/2, which in turn phosphorylates pro-inflammatory STAT transcription factors, thus driving expression of additional ISGs and amplifying the IFN-inducible positive feedback loop to support and maintain a pro-inflammatory microenvironment.
Two RIG-I-like receptors (RLRs) with structural similarity to RIG-I have been identified. One of these RLRs, Melanoma Differentiation Associated (MDA)-5, harbors an amino-terminal CARD domain, a DexD/H box motif, and a CTD domain [63, 64]. Like RIG-I, MDA-5 induces type I IFNs and other pro-inflammatory cytokines in response to viral nucleotides, albeit viral nucleotide motifs that are distinct from those that activate RIG-I. MDA-5 is activated by blunt-ended, long double-stranded RNA [e.g. polyinosinic-polycytidylic acid, or poly(I:C)], a ligand that also activated some Toll-Like Receptors (TLRs). In contrast to RIG-I and MDA-5, the other RLR known as Laboratory of Genetics and Physiology (LGP)-2 lacks the CARD domain shared by RIG-I and MDA-5, but is otherwise similar to the other RLRs [65]. Without the CARD domain, LGP-2 is unable to interact directly with MAVS to initiate a pro-inflammatory response. There are reports suggesting that LGP-2 activation interferes with RIG-I signaling, but that MDA-5 signaling may be enhanced by LGP2 [48, 66–69]. The implications of LGP2 expression and signaling in the context of cancer therapy, and how LGP2 might affect therapeutic responses to RIG-I agonists, are currently unclear.
RIG-I signaling potently activates programmed cell death
In the context of viral infection, RIG-I signaling is capable of inducing programmed cell death (PCD) as a mechanism to eliminate virally-infected cells. Cellular mechanisms by which RIG-I induces PCD include activation of the intrinsic apoptosis pathway, the extrinsic apoptosis pathway, and a type of programmed necrosis termed ‘pyroptosis.’ The molecular factors governing the mode of RIG-I mediated cell death may depend to some extent on cell type. For example, RLR activation in keratinocytes, melanoma cells, glioblastoma cells, and many leukemia cells cause mitochondrial outer membrane permeabilization (MOMP), cytochrome-C release from mitochondria, and activation of caspase-9 and Apaf-1, the irreversible molecular switch that governs the intrinsic apoptotic pathway [27]. However, RIG-I signaling in pancreatic and prostate cancer cells robustly induces expression of several factors that activate the extrinsic apoptotic pathway, including Fas, Fas Ligand, Tumor Necrosis Factor (TNF), TNF-related apoptosis-inducing ligand (TRAIL), and the TRAIL receptors Death Receptor (DR)-4 and DR-5, causing caspase-8 activation and extrinsic apoptosis. The mechanism by which RIG-I signaling upregulates TRAIL, FAS and other extrinsic apoptosis-activating factors are not entirely clear, although it is likely that IFN signaling is involved, given that Fas, TRAIL, and caspase-8 are known ISGs [70, 71].
Another mode of programmed cell death induced upon RIG-I activation is termed “pyroptosis,” an immunogenic form of cell death occurring in response to activation of the inflammasome, a multi-protein holoenzyme comprised of capsase-1 oligomers, adaptor proteins known as ASC (Apoptosis-associated Speck with a Caspase-recruitment domain), and a molecular sensor of pathogens, such as RIG-I (Figure 3). RIG-I can interact, via its CARD domain, with the CARD domains of inflammasome components [72], resulting in auto-cleavage and activation of caspase-1 [29, 73], which then allows proteolysis of the pro-inflammatory cytokines interleukin (IL)-1β and IL-18 [73], which amplify inflammatory signaling in the local environment while activating natural killer (NK) cells and recruiting leukocytes to the affected tissue. Caspase-1 activation also results in cleavage of Gasdermin-D, removing the auto-inhibitory domain from Gasdermin-D to allow oligomerization at the plasma membrane and pore formation. Plasma membrane permeabilization by Gasdermin-D pores allows water to enter and swell the cell, a hallmark of necrosis. Once membrane integrity is lost, intracellular contents, including DAMPs, permeate the extracellular environment, inducing danger responses in neighboring cells, which amplifies the inflammatory response.
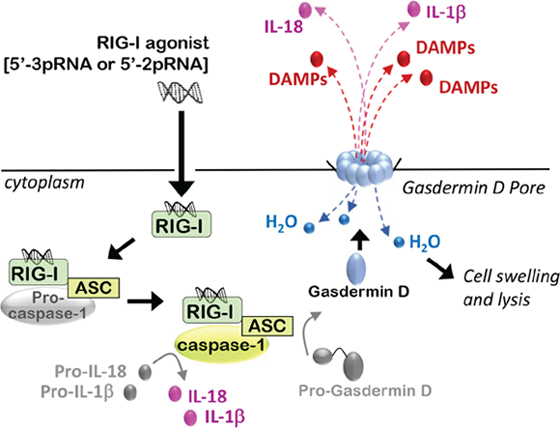
Figure 3: RIG-I activation induces immunogenic modes of programmed cell death. Activated RIG-I recruits the inflammasome adaptor protein ASC, which facilitates binding and oligomerization of Caspase-1, leading to caspase-1 auto-cleavage and activation. Caspase-1 cleaves protein precursors of IL-1β and IL-18 to generate their mature, pro-inflammatory isoforms, which are then secreted. Caspase-1 activity also drives cleavage of the auto-inhibitory domain from Gasdermin-D, liberation the amino-terminal pore-forming domain of Gasdermin-D to translocate to the plasma membrane and oligomerize, forming pores that initiate hypotonic cellular swelling and lysis, followed by release of DAMPs into the extracellular space, thus inducing an inflammatory response from surrounding cells.
RIG-I signaling in tumor cells affects the complex tumor microenvironment
The capacity for RIG-I signaling to induce cell death, while inducing pro-inflammatory responses, makes therapeutic use of RIG-I mimetics a highly attractive option in cancers. A growing number of studies show that the molecular responses to RIG-I or RLR signaling are retained in tumor cells and in non-tumor cells of the tumor microenvironment, and support innate immune responses against tumor cells [34]. For example, RIG-I activation in ovarian cancer cells enables NK-mediated tumor cell killing in culture [36]. Further, RIG-I signaling within the tumor cell increases phagocytosis of the affected tumor cell by professional APCs, including macrophages and DCs, thus providing tumor antigens for presentation to lymphocytes [32]. At the same time, the IFN-enriched microenvironment generated by tumor cell RIG-I signaling increases expression of major histocompatibility complex (MHC)-II antigen presentation molecules in macrophages and DCs, which may further increase tumor antigen cross-presentation. In support of this idea, it is reported that DCs presented pancreatic cancer-derived antigens more robustly to T-cells if RIG-I signaling was activated in pancreatic cancer cells prior to their co-culture with DCs [32, 36]. Similar results were observed upon RIG-I activation ovarian cancer cells prior to co-culture with macrophages [74].
RIG-I mimetics are gaining traction as a possible cancer treatment in pre-clinical studies
Through direct activation of intrinsic immunity in cancer cells, and accompanying indirect activation of leukocytes in the TME, synthetic RIG-I mimetics are under investigation for cancer treatment in pre-clinical studies in hepatocellular carcinoma [75], leukemias [76], melanomas [27], prostate cancers [77] and others. RIG-I agonists that are stable and functional in vivo are under current development. For example, a minimal 5’-triphophosphorylated stem-loop RNA (SLR) sequence for intra-venous delivery to mice was recently reported [25]. The stem-loop structure enhances structural stability of the complex, a key determinant of RIG-I ligand potency. Delivery of SLR sequences to mice in vivo activated RIG-I signaling, IFN induction, and expression of genes required for potent anti-viral immunity, although this RIG-I mimetic has not yet been studied in tumors grown in vivo. A pre-clinical compound specific for RIG-I is RGT100 (Merck/Rigontec), currently in phase I clinical trials for treatment of advanced solid tumors and lymphomas (NCT03065023), although peer-reviewed preclinical reports for RGT100 were not identified, to our knowledge. Another compound which activates RIG-I by unknown mechanisms is SB-9200 [78], which is currently under investigation as an anti-viral agent, but has not yet been tested in the pre-clinical setting of cancer treatment.
In addition to RIG-I specific mimetics, synthetic RLR mimetics are being investigated in pre-clinical and early clinical studies. The compound Hiltonol [polyinosinic-polycytidylic acid stabilized with poly-l-lysine and carboxymethylcellulose (poly-ICLC)] [79, 80] was tested in combination with chemotherapy for patients with Stage IV anaplastic astrocytoma, resulting in increased overall survival (OS) to >8 years, versus the expected survival of two years on conventional chemotherapy alone [81]. Another trial tested poly-ICLC in combination with radiation and temozolomide in newly diagnosed adult glioblastoma patients. In these studies, intramuscular poly-ICLC increased OS to 18.3 months from 14.6 months [82–84]. Further, poly-ICLC is being tested as a tumor vaccine adjuvant in several cancer types, with a growing number of successes in Phase I and II clinical trials for gliomas [85], breast cancer [86], pancreatic cancer [87], ovarian cancer [88, 89], multiple myeloma [90], and others, highlighting the potential advances that Poly-ICLC may achieve across a spectrum of cancers. Although poly-ICLC potently activates MDA-5, it also activates Toll-like Receptor (TLR)-3, making the specific contributions of RLR signaling to the therapeutic effects of poly-ICLC, and to patient outcome, difficult to dissect.
The future of RIG-I agonists in cancer
Exciting innovations within the field of RIG-I agonists are emerging. For example, a powerful, bimodal application of RNAi-based silencing of intra-tumoral gene targets using a 5’-triphosphate modified dsRNA sequence would allow for RIG-I activation and simultaneous gene targeting. This approach was demonstrated in melanomas, using 5’-3p-siRNA sequences specific to the anti-apoptotic gene BCL2. Delivery of this construct to cells potently stimulated IFN production and NK activation, while enhancing tumor cell killing through Bcl-2 ablation [34]. This concept was validated using a 5’-3p-siRNA targeting transforming growth factor (TGF)-β in pancreatic cancer cells, resulting in tumor cell apoptosis, IFN induction, and enhanced CD8+ T cell responses [36]. A similar approach was used in models of non-small cell lung cancer, using 5’-3p-siRNA sequences against vascular endothelial growth factor (VEGF), resulting in reduced tumor angiogenesis while enhancing anti-tumor immunity [91]. Defining the most appropriate gene silencing target may be a difficult task, but the use of siRNA paves the pathway for targeting certain oncogenes (e.g., MYC) that are currently ‘undruggable.’
Despite the potential success of RIG-I and RLR agonists, the immune system is powerful and incompletely understood, warranting cautious optimism and thorough examination of the caveats associated with innate immune activation, including possible on-target induction of autoimmunity, or induction of a cytokine ‘storm’ which could pose a threat to patient safety [92–94]. It is important to note that, since RIG-I is expressed in most cells of the human body, the consequences of RIG-I activation might be widespread, driving symptoms like fatigue, depression and cognitive impairment. In ICI-based therapies, these side-effects are generally managed by corticosteroid immunosuppression.
Delivery of small nucleotide sequences to tumor cells and leukocytes within the TME is another major obstacle to the widespread utility of RIG-I or RLR-based therapeutics in the cancer setting. Studies aimed at generating stable, specific and potent RIG-I ligands that retain functionality in vivo have been reported only recently. For example, a study employing a minimal 5’-triphophosphorylated stem-loop RNA (SLR) sequence delivered by intra-venous delivery to mice activated in RIG-I signaling, IFN induction, and expression of genes required for potent anti-viral immunity in vivo. A recently described ‘conditional’ RIG-I ligand, in which the 5’-triphosphorylated terminus of the RNA duplex remained shielded until release by predetermined molecular cues in vivo, could enhance delivery of RIG-I agonist to tumors, and minimize RIG-I activation outside of the TME [95]. However, the efficacy of RIG-I ligands, including SLRs and conditional RIG-I ligands, not yet been tested in animal models of cancer [25].
CONCLUSION
Therapeutic RIG-I and RLR agonists are emerging as a novel approach to engage the immune system in the fight against cancer. Importantly, RIG-I signaling directly promotes tumor cell killing through three distinct modes of action: intrinsic apoptosis, extrinsic apoptosis, and pyroptosis. Further, simultaneous activation of the innate and adaptive arms of the immune system may generate durable therapeutic responses. The multi-faceted mechanisms by which RLR agonists eliminate cancer cells represent the well-rounded arsenal of weapons required to fight aggressive and metastatic cancers effectively.
Abbreviations
APC: antigen presenting cell; ASC: Apoptosis-associated Speck with a Caspase-recruitment domain; CARD: caspase activation and recruitment domain; CTD: carboxy-terminal domain; DAMP: danger associated molecular patterns; DC: dendritic cell; DR: death receptor; HIV: human immunodeficiency virus; ICI: immune checkpoint inhibitors; IFN: interferon; IFNAR: interferon α/β receptor; IKK: inhibitor of κB-kinase; IL: interleukin; IRF: interferon response factor; ISG: interferon stimulated genes; MAVS: mitochondrial anti-viral signaling; MDSC: myeloid derived suppressor cell; MDA-5 melanoma differentiation antigen-5; MHC: major histocompatibility complex; MOMP: mitochondrial outer membrane permeabilization; NF: nuclear factor; NK: natural killer; OS: overall survival; PCD: programmed cell death; Poly(I:C) polyinosinic-polycytidylic acid; Poly-ICLC polyinosinic-polycytidylic acid stabilized with poly-l-lysine and carboxymethylcellulose; PRR: pattern recognition receptor; RIG-I retinoic acid-inducible gene I; RLR: RIG-I-like receptor; STAT: Signal transducer and transcription factor; STING: Stimulator of Interferon Genes; TAM: tumor associated macrophage; TBK: tank binding kinase; TGF: Transforming growth factor; TLR: toll-like receptor; TME: tumor microenvironment; TNF: tumor necrosis factor; TRAIL: TNF-related apoptosis-inducing ligand; TReg: regulatory T-cells; VEGF: vascular endothelial growth factor; 5’-3pRNA: 5’-triphosphate RNA; 5’-2pRNA 5’-diphosphate.
Author contributions
D.E. and R.S.C. prepared the manuscript. D.E. and R.S.C have reviewed and agree to this information.
ACKNOWLEDGMENTS
We would like to acknowledge the shared resources at Vanderbilt University, Vanderbilt University Medical Center, and the Vanderbilt-Ingram Cancer Center that contributed to the studies reported herein.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest with the materials described herein.
FUNDING
This work was supported by Specialized Program of Research Excellence (SPORE) grant NIH P50 CA098131 (VICC; to R. S. Cook), Cancer Center Support grant NIH P30 CA68485 (VICC; to R.S. Cook), CTSA UL1TR000445 (to R.S. Cook) from the National Center for Advancing Translational Sciences, W81XWH-161-0063 (to R.S. Cook) from the Congressionally Directed Medical Research Program.
REFERENCES
1. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012; 12:252-264.
2. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010; 363:711-723.
3. Ellis PM, Vella ET, Ung YC. Immune Checkpoint Inhibitors for Patients With Advanced Non-Small-Cell Lung Cancer: A Systematic Review. Clin Lung Cancer. 2017; 18:444-459 e441.
4. Luen S, Virassamy B, Savas P, Salgado R, Loi S. The genomic landscape of breast cancer and its interaction with host immunity. Breast. 2016; 29:241-250.
5. Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol. 2000; 74:181-273.
6. Garrido F, Algarra I. MHC antigens and tumor escape from immune surveillance. Adv Cancer Res. 2001; 83:117-158.
7. Gatalica Z, Snyder C, Maney T, Ghazalpour A, Holterman DA, Xiao N, Overberg P, Rose I, Basu GD, Vranic S, Lynch HT, Von Hoff DD, Hamid O. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol Biomarkers Prev. 2014; 23:2965-2970.
8. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010; 140:805-820.
9. Shalapour S, Karin M. Immunity, inflammation, and cancer: an eternal fight between good and evil. J Clin Invest. 2015; 125:3347-3355.
10. Okamoto M, Tsukamoto H, Kouwaki T, Seya T, Oshiumi H. Recognition of Viral RNA by Pattern Recognition Receptors in the Induction of Innate Immunity and Excessive Inflammation During Respiratory Viral Infections. Viral Immunol. 2017; 30:408-420.
11. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008; 455:674-678.
12. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009; 461:788-792.
13. Chandra D, Quispe-Tintaya W, Jahangir A, Asafu-Adjei D, Ramos I, Sintim HO, Zhou J, Hayakawa Y, Karaolis DK, Gravekamp C. STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer. Cancer Immunol Res. 2014; 2:901-910.
14. Li T, Cheng H, Yuan H, Xu Q, Shu C, Zhang Y, Xu P, Tan J, Rui Y, Li P, Tan X. Antitumor Activity of cGAMP via Stimulation of cGAS-cGAMP-STING-IRF3 Mediated Innate Immune Response. Sci Rep. 2016; 6:19049.
15. Tang CH, Zundell JA, Ranatunga S, Lin C, Nefedova Y, Del Valle JR, Hu CC. Agonist-Mediated Activation of STING Induces Apoptosis in Malignant B Cells. Cancer Res. 2016; 76:2137-2152.
16. Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, Gaide O, Michielin O, Hwu P, Petrova TV, Martinon F, Modlin RL, Speiser DE, Gilliet M. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci U S A. 2015; 112:15408-15413.
17. Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ, Lauer P, Liu W, Sivick KE, Zeng Q, Soares KC, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015; 7:283ra252.
18. Konno H, Yamauchi S, Berglund A, Putney RM, Mule JJ, Barber GN. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene. 2018; 37:2037-2051.
19. Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep. 2016; 14:282-297.
20. Pepin G, Gantier MP. cGAS-STING Activation in the Tumor Microenvironment and Its Role in Cancer Immunity. Adv Exp Med Biol. 2017; 1024:175-194.
21. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004; 5:730-737.
22. Loo YM, Gale M Jr. Immune signaling by RIG-I-like receptors. Immunity. 2011; 34:680-692.
23. Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, Schuberth C, Van der Veen AG, Fujimura T, Rehwinkel J, Iskarpatyoti JA, Barchet W, Ludwig J, et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5'-diphosphates. Nature. 2014; 514:372-375.
24. Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G. 5'-Triphosphate RNA is the ligand for RIG-I. Science. 2006; 314:994-997.
25. Linehan MM, Dickey TH, Molinari ES, Fitzgerald ME, Potapova O, Iwasaki A, Pyle AM. A minimal RNA ligand for potent RIG-I activation in living mice. Sci Adv. 2018; 4:e1701854.
26. Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, Barchet W, Coch C, Janke M, Mihailovic A, Wardle G, Juranek S, Kato H, Kawai T, et al. Recognition of 5' triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity. 2009; 31:25-34.
27. Besch R, Poeck H, Hohenauer T, Senft D, Hacker G, Berking C, Hornung V, Endres S, Ruzicka T, Rothenfusser S, Hartmann G. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J Clin Invest. 2009; 119:2399-2411.
28. Liu LW, Nishikawa T, Kaneda Y. An RNA Molecule Derived From Sendai Virus DI Particles Induces Antitumor Immunity and Cancer Cell-selective Apoptosis. Mol Ther. 2016; 24:135-145.
29. Rintahaka J, Wiik D, Kovanen PE, Alenius H, Matikainen S. Cytosolic antiviral RNA recognition pathway activates caspases 1 and 3. J Immunol. 2008; 180:1749-1757.
30. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018; 25:486-541.
31. Ramos HJ, Gale M Jr. RIG-I like receptors and their signaling crosstalk in the regulation of antiviral immunity. Curr Opin Virol. 2011; 1:167-176.
32. Duewell P, Steger A, Lohr H, Bourhis H, Hoelz H, Kirchleitner SV, Stieg MR, Grassmann S, Kobold S, Siveke JT, Endres S, Schnurr M. RIG-I-like helicases induce immunogenic cell death of pancreatic cancer cells and sensitize tumors toward killing by CD8(+) T cells. Cell Death Differ. 2014; 21:1825-1837.
33. Glas M, Coch C, Trageser D, Dassler J, Simon M, Koch P, Mertens J, Quandel T, Gorris R, Reinartz R, Wieland A, Von Lehe M, Pusch A, et al. Targeting the cytosolic innate immune receptors RIG-I and MDA5 effectively counteracts cancer cell heterogeneity in glioblastoma. Stem Cells. 2013; 31:1064-1074.
34. Poeck H, Besch R, Maihoefer C, Renn M, Tormo D, Morskaya SS, Kirschnek S, Gaffal E, Landsberg J, Hellmuth J, Schmidt A, Anz D, Bscheider M, et al. 5'-Triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nat Med. 2008; 14:1256-1263.
35. van den Boorn JG, Hartmann G. Turning tumors into vaccines: co-opting the innate immune system. Immunity. 2013; 39:27-37.
36. Ellermeier J, Wei J, Duewell P, Hoves S, Stieg MR, Adunka T, Noerenberg D, Anders HJ, Mayr D, Poeck H, Hartmann G, Endres S, Schnurr M. Therapeutic efficacy of bifunctional siRNA combining TGF-beta1 silencing with RIG-I activation in pancreatic cancer. Cancer Res. 2013; 73:1709-1720.
37. Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5'-phosphates. Science. 2006; 314:997-1001.
38. Marques JT, Devosse T, Wang D, Zamanian-Daryoush M, Serbinowski P, Hartmann R, Fujita T, Behlke MA, Williams BR. A structural basis for discriminating between self and nonself double-stranded RNAs in mammalian cells. Nat Biotechnol. 2006; 24:559-565.
39. Solis M, Nakhaei P, Jalalirad M, Lacoste J, Douville R, Arguello M, Zhao T, Laughrea M, Wainberg MA, Hiscott J. RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J Virol. 2011; 85:1224-1236.
40. Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, Azzam DJ, Twyman-Saint Victor C, Wiemann BZ, Ishwaran H, Ter Brugge PJ, Jonkers J, Slingerland J, Minn AJ. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell. 2014; 159:499-513.
41. Ranoa DR, Parekh AD, Pitroda SP, Huang X, Darga T, Wong AC, Huang L, Andrade J, Staley JP, Satoh T, Akira S, Weichselbaum RR, Khodarev NN. Cancer therapies activate RIG-I-like receptor pathway through endogenous non-coding RNAs. Oncotarget. 2016; 7:26496-26515. https://doi.org/10.18632/oncotarget.8420.
42. Zeng Y, Wang PH, Zhang M, Du JR. Aging-related renal injury and inflammation are associated with downregulation of Klotho and induction of RIG-I/NF-kappaB signaling pathway in senescence-accelerated mice. Aging Clin Exp Res. 2016; 28:69-76.
43. Zhao L, Zhu J, Zhou H, Zhao Z, Zou Z, Liu X, Lin X, Zhang X, Deng X, Wang R, Chen H, Jin M. Identification of cellular microRNA-136 as a dual regulator of RIG-I-mediated innate immunity that antagonizes H5N1 IAV replication in A549 cells. Sci Rep. 2015; 5:14991.
44. Karlsen TA, Brinchmann JE. Liposome delivery of microRNA-145 to mesenchymal stem cells leads to immunological off-target effects mediated by RIG-I. Mol Ther. 2013; 21:1169-1181.
45. Barral PM, Sarkar D, Su ZZ, Barber GN, DeSalle R, Racaniello VR, Fisher PB. Functions of the cytoplasmic RNA sensors RIG-I and MDA-5: key regulators of innate immunity. Pharmacol Ther. 2009; 124:219-234.
46. Bork P, Koonin EV. An expanding family of helicases within the 'DEAD/H' superfamily. Nucleic Acids Res. 1993; 21:751-752.
47. Jankowsky E, Jankowsky A. The DExH/D protein family database. Nucleic Acids Res. 2000; 28:333-334.
48. Saito T, Hirai R, Loo YM, Owen D, Johnson CL, Sinha SC, Akira S, Fujita T, Gale M Jr. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci U S A. 2007; 104:582-587.
49. Gack MU, Kirchhofer A, Shin YC, Inn KS, Liang C, Cui S, Myong S, Ha T, Hopfner KP, Jung JU. Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proc Natl Acad Sci U S A. 2008; 105:16743-16748.
50. Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007; 446:916-920.
51. Gao D, Yang YK, Wang RP, Zhou X, Diao FC, Li MD, Zhai ZH, Jiang ZF, Chen DY. REUL is a novel E3 ubiquitin ligase and stimulator of retinoic-acid-inducible gene-I. PLoS One. 2009; 4:e5760.
52. Oshiumi H, Matsumoto M, Hatakeyama S, Seya T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J Biol Chem. 2009; 284:807-817.
53. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005; 122:669-682.
54. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005; 6:981-988.
55. Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005; 437:1167-1172.
56. Matsuda A, Suzuki Y, Honda G, Muramatsu S, Matsuzaki O, Nagano Y, Doi T, Shimotohno K, Harada T, Nishida E, Hayashi H, Sugano S. Large-scale identification and characterization of human genes that activate NF-kappaB and MAPK signaling pathways. Oncogene. 2003; 22:3307-3318.
57. Huang J, Liu T, Xu LG, Chen D, Zhai Z, Shu HB. SIKE is an IKK epsilon/TBK1-associated suppressor of TLR3- and virus-triggered IRF-3 activation pathways. EMBO J. 2005; 24:4018-4028.
58. Chariot A, Leonardi A, Muller J, Bonif M, Brown K, Siebenlist U. Association of the adaptor TANK with the I kappa B kinase (IKK) regulator NEMO connects IKK complexes with IKK epsilon and TBK1 kinases. J Biol Chem. 2002; 277:37029-37036.
59. O'Neill LA, Bowie AG. Sensing and signaling in antiviral innate immunity. Curr Biol. 2010; 20:R328-333.
60. Panne D. The enhanceosome. Curr Opin Struct Biol. 2008; 18:236-242.
61. Schroder M, Baran M, Bowie AG. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J. 2008; 27:2147-2157.
62. Baum A, Garcia-Sastre A. Induction of type I interferon by RNA viruses: cellular receptors and their substrates. Amino Acids. 2010; 38:1283-1299.
63. Kovacsovics M, Martinon F, Micheau O, Bodmer JL, Hofmann K, Tschopp J. Overexpression of Helicard, a CARD-containing helicase cleaved during apoptosis, accelerates DNA degradation. Curr Biol. 2002; 12:838-843.
64. Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci U S A. 2002; 99:637-642.
65. Cui Y, Li M, Walton KD, Sun K, Hanover JA, Furth PA, Hennighausen L. The Stat3/5 locus encodes novel endoplasmic reticulum and helicase-like proteins that are preferentially expressed in normal and neoplastic mammary tissue. Genomics. 2001; 78:129-134.
66. Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M Jr, Akira S, Yonehara S, Kato A, Fujita T. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005; 175:2851-2858.
67. Komuro A, Horvath CM. RNA- and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J Virol. 2006; 80:12332-12342.
68. Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S, Fitzgerald KA. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol. 2005; 175:5260-5268.
69. Venkataraman T, Valdes M, Elsby R, Kakuta S, Caceres G, Saijo S, Iwakura Y, Barber GN. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J Immunol. 2007; 178:6444-6455.
70. de Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, Silverman RH, Williams BR. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol. 2001; 69:912-920.
71. Leaman DW, Chawla-Sarkar M, Jacobs B, Vyas K, Sun Y, Ozdemir A, Yi T, Williams BR, Borden EC. Novel growth and death related interferon-stimulated genes (ISGs) in melanoma: greater potency of IFN-beta compared with IFN-alpha2. J Interferon Cytokine Res. 2003; 23:745-756.
72. Yu HB, Finlay BB. The caspase-1 inflammasome: a pilot of innate immune responses. Cell Host Microbe. 2008; 4:198-208.
73. Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, Hannesschlager N, Schlee M, Rothenfusser S, Barchet W, Kato H, Akira S, Inoue S, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol. 2010; 11:63-69.
74. Kubler K, Gehrke N, Riemann S, Bohnert V, Zillinger T, Hartmann E, Polcher M, Rudlowski C, Kuhn W, Hartmann G, Barchet W. Targeted activation of RNA helicase retinoic acid-inducible gene-I induces proimmunogenic apoptosis of human ovarian cancer cells. Cancer Res. 2010; 70:5293-5304.
75. Hou J, Zhou Y, Zheng Y, Fan J, Zhou W, Ng IO, Sun H, Qin L, Qiu S, Lee JM, Lo CM, Man K, Yang Y, et al. Hepatic RIG-I predicts survival and interferon-alpha therapeutic response in hepatocellular carcinoma. Cancer Cell. 2014; 25:49-63.
76. Li D, Gale RP, Liu Y, Lei B, Wang Y, Diao D, Zhang M. 5'-Triphosphate siRNA targeting MDR1 reverses multi-drug resistance and activates RIG-I-induced immune-stimulatory and apoptotic effects against human myeloid leukaemia cells. Leuk Res. 2017; 58:23-30.
77. Kawaguchi Y, Miyamoto Y, Inoue T, Kaneda Y. Efficient eradication of hormone-resistant human prostate cancers by inactivated Sendai virus particle. Int J Cancer. 2009; 124:2478-2487.
78. Jones M, Cunningham ME, Wing P, DeSilva S, Challa R, Sheri A, Padmanabhan S, Iyer RP, Korba BE, Afdhal N, Foster GR. SB 9200, a novel agonist of innate immunity, shows potent antiviral activity against resistant HCV variants. J Med Virol. 2017; 89:1620-1628.
79. Zhu X, Nishimura F, Sasaki K, Fujita M, Dusak JE, Eguchi J, Fellows-Mayle W, Storkus WJ, Walker PR, Salazar AM, Okada H. Toll like receptor-3 ligand poly-ICLC promotes the efficacy of peripheral vaccinations with tumor antigen-derived peptide epitopes in murine CNS tumor models. J Transl Med. 2007; 5:10.
80. Salem ML, El-Naggar SA, Kadima A, Gillanders WE, Cole DJ. The adjuvant effects of the toll-like receptor 3 ligand polyinosinic-cytidylic acid poly (I:C) on antigen-specific CD8+ T cell responses are partially dependent on NK cells with the induction of a beneficial cytokine milieu. Vaccine. 2006; 24:5119-5132.
81. Salazar AM, Levy HB, Ondra S, Kende M, Scherokman B, Brown D, Mena H, Martin N, Schwab K, Donovan D, Dougherty D, Pulliam M, Ippolito M, et al. Long-term treatment of malignant gliomas with intramuscularly administered polyinosinic-polycytidylic acid stabilized with polylysine and carboxymethylcellulose: an open pilot study. Neurosurgery. 1996; 38:1096-1103; discussion 1103-1094.
82. Rosenfeld MR, Chamberlain MC, Grossman SA, Peereboom DM, Lesser GJ, Batchelor TT, Desideri S, Salazar AM, Ye X. A multi-institution phase II study of poly-ICLC and radiotherapy with concurrent and adjuvant temozolomide in adults with newly diagnosed glioblastoma. Neuro Oncol. 2010; 12:1071-1077.
83. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009; 10:459-466.
84. Grossman SA, Ye X, Piantadosi S, Desideri S, Nabors LB, Rosenfeld M, Fisher J. Survival of patients with newly diagnosed glioblastoma treated with radiation and temozolomide in research studies in the United States. Clin Cancer Res. 2010; 16:2443-2449.
85. Okada H, Butterfield LH, Hamilton RL, Hoji A, Sakaki M, Ahn BJ, Kohanbash G, Drappatz J, Engh J, Amankulor N, Lively MO, Chan MD, Salazar AM, et al. Induction of robust type-I CD8+ T-cell responses in WHO grade 2 low-grade glioma patients receiving peptide-based vaccines in combination with poly-ICLC. Clin Cancer Res. 2015; 21:286-294.
86. Dillon PM, Petroni GR, Smolkin ME, Brenin DR, Chianese-Bullock KA, Smith KT, Olson WC, Fanous IS, Nail CJ, Brenin CM, Hall EH, Slingluff CL Jr. A pilot study of the immunogenicity of a 9-peptide breast cancer vaccine plus poly-ICLC in early stage breast cancer. J Immunother Cancer. 2017; 5:92.
87. Mehrotra S, Britten CD, Chin S, Garrett-Mayer E, Cloud CA, Li M, Scurti G, Salem ML, Nelson MH, Thomas MB, Paulos CM, Salazar AM, Nishimura MI, et al. Vaccination with poly(IC:LC) and peptide-pulsed autologous dendritic cells in patients with pancreatic cancer. J Hematol Oncol. 2017; 10:82.
88. Tsuji T, Sabbatini P, Jungbluth AA, Ritter E, Pan L, Ritter G, Ferran L, Spriggs D, Salazar AM, Gnjatic S. Effect of Montanide and poly-ICLC adjuvant on human self/tumor antigen-specific CD4+ T cells in phase I overlapping long peptide vaccine trial. Cancer Immunol Res. 2013; 1:340-350.
89. Sabbatini P, Tsuji T, Ferran L, Ritter E, Sedrak C, Tuballes K, Jungbluth AA, Ritter G, Aghajanian C, Bell-McGuinn K, Hensley ML, Konner J, Tew W, et al. Phase I trial of overlapping long peptides from a tumor self-antigen and poly-ICLC shows rapid induction of integrated immune response in ovarian cancer patients. Clin Cancer Res. 2012; 18:6497-6508.
90. Rapoport AP, Aqui NA, Stadtmauer EA, Vogl DT, Xu YY, Kalos M, Cai L, Fang HB, Weiss BM, Badros A, Yanovich S, Akpek G, Tsao P, et al. Combination immunotherapy after ASCT for multiple myeloma using MAGE-A3/Poly-ICLC immunizations followed by adoptive transfer of vaccine-primed and costimulated autologous T cells. Clin Cancer Res. 2014; 20:1355-1365.
91. Yuan D, Xia M, Meng G, Xu C, Song Y, Wei J. Anti-angiogenic efficacy of 5'-triphosphate siRNA combining VEGF silencing and RIG-I activation in NSCLCs. Oncotarget. 2015; 6:29664-29674. https://doi.org/10.18632/oncotarget.4869.
92. Buers I, Nitschke Y, Rutsch F. Novel interferonopathies associated with mutations in RIG-I like receptors. Cytokine Growth Factor Rev. 2016; 29:101-107.
93. Lee-Kirsch MA. The Type I Interferonopathies. Annu Rev Med. 2017; 68:297-315.
94. Trinchieri G. Type I interferon: friend or foe? J Exp Med. 2010; 207:2053-2063.
95. Palmer CR, Jacobson ME, Fedorova O, Pyle AM, Wilson JT. Environmentally Triggerable Retinoic Acid-Inducible Gene I Agonists Using Synthetic Polymer Overhangs. Bioconjug Chem. 2018; 29:742-747.