
INTRODUCTION
Cancer genomics has exploded our understanding of oncogenesis [1, 2]. Human cancer genomes harbor thousands to millions of mutations, as well as large numbers of insertions or deletions (indels) and chromosomal rearrangements [3, 4]. Human cancer genomes show signatures that provide clues as to etiological agents, e.g., benzo(a)pyrenes in tobacco smoke, ultraviolet light or endogenous APOBEC3A (A3A) or APOBEC3B (A3B) cytidine deaminases [3]. Six functional APOBEC3 cytidine deaminases (A3A-A3C and A3F-A3H) are encoded by the human genome and are made up of three related, but phylogenetically distinct zinc-finger domains referred to as Z1, Z2 and Z3 [5]. While the enzymes contain one or a pair of zinc finger domains, only the C-terminal domain is catalytically active. These enzymes convert cytidine residues to uridine in single-stranded DNA (ssDNA), a process referred to as DNA editing. Such substrate specificity distinguishes them from other cytidine deaminases involved in the metabolism of nucleotide precursors [6].
APOBEC3 proteins were first identified functionally as innate restriction factors of viruses such as human immunodeficiency virus, hepatitis B virus, herpesviruses and papillomavirus to mention a few [7–16]. It latter transpired that some A3 enzymes inhibit retrotransposition of SINEs and LINEs, endogenous retroviruses and can catabolize mitochondrial DNA leaked to the cytoplasm [17–20]. Viral restriction is linked to the fact that viral genomes are riddled with uridine residues that can result in so many mutations that genetic information is lost. The alternative is that the edited DNA is catabolized.
Human A3A and A3B are singular in that they can edit chromosomal DNA [20–24]. Interestingly, it is these two enzymes that can efficiently deaminate 5-methylcytidine residues (5MeC), which have long been described as cancer mutation hotspots [22, 25–27]. As a result of A3 editing of cytidine residues, uridine in DNA triggers damage repair response whose first step is the excision of the uracil base by uracil N-glycosylase (UNG). If both DNA strands are edited close to one another, double-strand DNA breaks (DSBs) are generated, a feature of all mammalian A3As [20, 22, 28–30]. By contrast, if the uracil base is copied before excision, it leads to a C->T substitution occurring frequently in the 5ʹTpC3ʹ or 5ʹCpC3ʹ context. Hence, this is the APOBEC3 editing signature that shows up in cancer genomes. Experimentally, A3A is more active than A3B and can induce DSBs [20–22, 29, 30]. In addition, the (in front of A3B) deletion allele is almost completely fixed in parts of Oceania [31] suggesting that A3B is dispensable for life and cancer.
The human Tribbles 3 pseudokinase (TRIB3) is part of the Rb-BRCA1-ATM network regulating genome integrity. Interestingly, TRIB3 is the only known negative interactor of human A3A [32]. TRIB3 is a nuclear protein that degrades A3A in a proteasome independent manner. Given that the A3A and A3B DNA mutator enzymes are etiological agents of human cancer, it would be useful to have a small animal model to study oncogenesis in vivo.
The A3A gene is found among most placental mammals [28], although not without a few exceptions. Sheep and dog have functional A3A enzymes but closely related pigs and cats are devoid of any homolog [28, 33]. A3A is completely lacking in Rodentia (mice, rats, guinea pigs and squirrels), which is unfortunate given the important of mice and rats as experimental models. As the rabbit is a commonly used small animal model, this prompted us to look at the rabbit A3 locus. We show here that the rabbit genome encodes a strictly orthologous A3A enzyme which is the closest relative to primate counterparts. Furthermore, the enzyme is negatively regulated by the rTRIB3, just like its human counterpart that interferes with human A3A, highlighting a connection between the two proteins that has been conserved for more than 100 million years.
RESULTS
Rabbit genome encodes an A3A gene
Through extensive BLAT, BLAST and EST screening, we identified three complete APOBEC3 like Zn-finger domains on rabbit chromosome 4. Potential exons/introns junctions were verified by PCR and sequencing of cDNA fragments (data not shown). As shown in Figure 1A, each rabbit sequence clusters with a Z1, Z2 or Z3 domain for human and mice APOBEC3 enzymes. All A3 loci to date are bounded by CBX6 and CBX7 and the rabbit locus was no exception indicating that the entire locus was probably correctly assembled (Figure 1B). The two rabbit A3 genes are organized in a head-to-head manner reminiscent of the Carnivora order. The rabbit Z2 and Z3 domains are closely juxtaposed and proximal to CBX7. As the unique mouse A3 enzyme is made of both Z2 and Z3 domains and is closely related to the rabbit Z2 and Z3 domains, we suspected that the two Z2Z3 domains could belong to the same enzyme in rabbit. We confirmed by PCR amplification of rabbit cDNA across the Z2Z3 junction that the same was true (data not shown). Accordingly, the rabbit genome encodes two APOBEC3 genes that will be referred to as A3Z1 (rA3A) and A3Z2Z3 (rA3Z2Z3). As shown in Figure 1A, the rabbit A3Z1 (rA3A) is homologous to the human A3A and A3B cancer related genes.
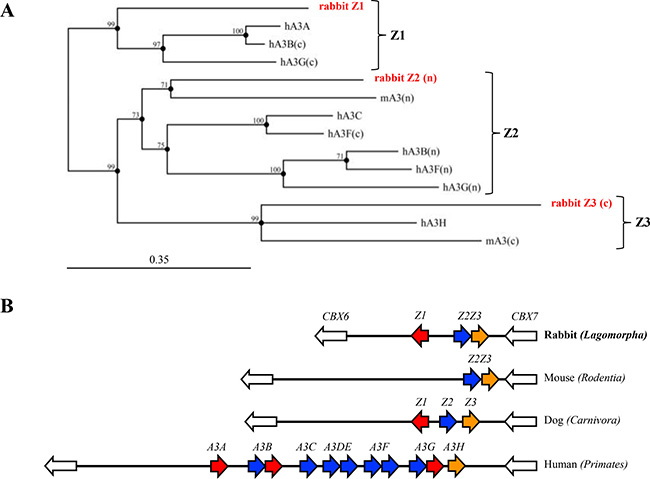
Figure 1: Rabbit APOBEC3 locus. (A) Phylogenic tree of three rabbit A3 protein domains along with mice (mA3) and human (hA3A, hA3B, hA3C, hA3F and hA3G) A3 protein sequences constructed using the Neighbor-joining method with the MCC Main Workbench 7.0.2 software. Numbers correspond to bootstrap values inferred from 1,000 replicates. Bootstrap values under the threshold of 70% are not shown. Letters n and c in brackets refer to the N-terminal or C-terminal zinc-finger domains. (B) To scale representation of the rabbit, mouse, dog and human A3 loci bounded by the CBX6 and CBX7 represented by white arrows. The Order is given in brackets.
Rabbit A3A enzyme is closely related to human A3A
The rabbit A3A amino acid sequence was aligned with those of 8 mammalian enzymes revealing subtle differences distinguished rabbit from the human APOBEC3 sequence (Supplementary Figure 1). The rabbit sequence is colinear with all others and starts with a strong Kozak AUG sequence, unlike the human gene where translation is initiated at two sites [30, 34]. Indels generally map to loops although there is a deletion of some β2 sheet residues. The rabbit sequence encodes all the key residues found in the catalytic site of A3A enzymes as well as I128 that is characteristic of all Z1 domains [5, 33]. Phylogenetic analysis of these protein sequences showed that the rA3A sequence was closest to those of the Primate A3A group being supported by a good bootstrap value (Figure 2A). Phylogenetic analyses of the rabbit Z2Z3 (rA3Z2Z3) protein sequences revealed that the Z3 sequence was an outlier to the primate group of sequences (Supplementary Figure 2A), much like the rabbit Z1 sequence (Figure 1A). By contrast, there was not enough sequence resolution to position the rabbit Z2 domain (Supplementary Figure 2B).
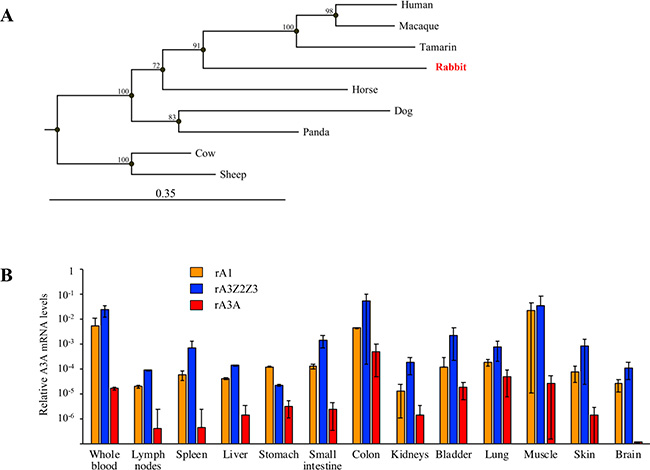
Figure 2: Rabbit A3A protein and gene expression. (A) Phylogeny of a selection of mammalian A3A protein sequences constructed using the Neighbor-joining method with the CLC Main Workbench 7.0.2 software (Qiagen). Numbers correspond to bootstrap values inferred from 1,000 replicates. (B) RT-PCR quantification of rA1 (orange), rA3Z2Z3 (blue) and rA3A (red) mRNA in blood and rabbit tissues. Results were normalized to rabbit EF1α reporter gene expression.
To study gene expression, we performed quantitative PCR on the two APOBEC3 cDNAs from a large number of rabbit tissues along with rabbit APOBEC1 (rA1) as control. Although rA1 deaminates cytidine residues primarily in dsRNA, it has been implicated in oncogenesis [35] while its murine counterpart edits both dsRNA and ssDNA [36]. Data were normalized to the housekeeping EF1α gene. Transcripts for all three genes were found in all tissues with the exception of A3A in the brain (Figure 2B). Expression of rA3Z2Z3 and rA1 were tightly related.
Rabbit A3 localization and function
To characterize these rabbit enzymes, cDNA clones where synthesized for rA3A and rA3Z2Z3 along with rabbit rA1. All three constructs were stable pcDNA3.1/V5-His-TOPO vector unlike some A3 constructs where expression proved to be toxic for bacteria [28]. Human A3A was used as positive control while an inactive catalytic mutant of rA3A was made as a negative control (rA3A*). All constructs were well expressed as assessed by Western blot (Figure 3A). Confocal microscopy analysis of transfected HeLa cells indicated a nucleo-cytoplasmic expression pattern for rA3A, rA3Z2Z3 and rA1 (Figure 3B).
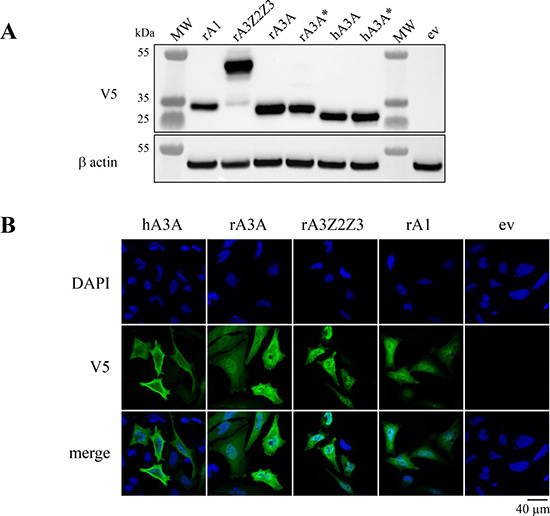
Figure 3: Rabbit A3 proteins show nucleo-cytoplasm localization. (A) Western blot detection of V5-tagged APOBEC3 proteins in HEK-293T cells. MW, molecular weight; ev, empty vector. β actin probing was used as a loading control. (B) Confocal microscopy analysis of V5-tagged A3A proteins in HeLa cells 24 hours post transfection. Nuclei are stained with DAPI.
The activity of rabbit APOBEC enzymes was assessed in vitro using a FRET based deamination assay as previously described [28]. No deaminase activity on ssDNA was observed after transfection of HEK-293T cells with rA3Z2Z3 and rA1 (Figure 4A). By contrast, rA3A and hA3A deaminate efficiently the ssDNA well above the background level provided by the inactive catalytic mutants rA3A* and hA3A*. To explore DNA editing in quail (QT6) cells which are naturally devoid of any endogenous APOBEC [37], QT6 cells were co-transfected with the A3 constructs along with a GFP reporter plasmid. Reduced GFP fluorescence due to editing of plasmid DNA was first sought by a flow cytometry. As can be seen from Figure 4B, there were more than 2-fold less GFP positive cells in rA3A co-transfected cells compared to the empty vector (ev) (p = 0.03) (Figure 4B) and consistent with transfection frequencies between 40–50%. 3D-PCR was used to recover A3 edited DNA which melts at a lower denaturation temperature (Td) [38]. No PCR products were recovered below Td = 86° C when empty vector, rA3A*, rA1, or rA3Z2Z3 plasmids were used (Figure 4C). By contrast, products were obtained at temperatures as low as 80° C and 80.2° C with hA3A and rA3A respectively, i.e. approximately 6° C below the restrictive Td, this being prima face evidence of editing. Cloning and sequencing of products obtained at 84.6° C (represented by an asterisk) confirmed that indeed cytidine deamination occurred with a clear preference for 5ʹTpC and 5ʹCpC dinucleotide contexts (Figure 4D).
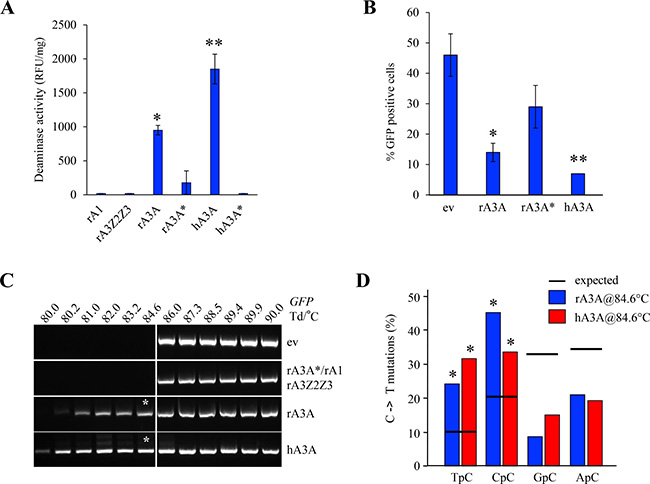
Figure 4: Rabbit A3A deaminates foreign DNA. (A) Fluorescence resonance energy transfer assay (FRET)-based in vitro deamination assay performed on FAM-TAMRA coupled oligonucleotides using transfected HEK-293T lysates. rA3A* and hA3A* catalytic mutants transfected cells used as negative controls. Results are expressed in Relative Fluorescence Unit per mg of protein (RFU/mg). A single asterisk represents a statistically significant difference (p < 0.05) and a double asterisk represents a highly statistically significant difference (p ≤ 0.005). (B) FACS analysis of HeLa cells 72 hours post transfection with an empty vector (ev) or rabbit A3A proteins. Percentage of GFP positive cells are gated on A3-V5-Tag positive cells. Single asterisks represent a statistically significant difference (p ≤ 0.05) and double asterisks represent a highly statistically significant difference (p ≤ 0.005). (C) GFP plasmid 3D-PCR gels from QT6 cells, 48 hours post co-transfection with GFP encoding plasmid and either an empty or an APOBEC3 expressing plasmid. Numbers indicates the Td for each well. Vertical white bar indicates restrictive Td. White asterisks denote PCR products that were cloned and sequenced. (D) Analysis of the 5′ nucleotide context of C->T transitions identified in cloned GFP DNA 3D-PCR products. Percentages are represented in blue for rA3A and red for hA3A. Horizontal bars represent expected values. Asterisks show a statistically significant difference (p < 0.05).
Rabbit A3A is functionally orthologous
One of the singular traits of mammalian A3A or A3B deaminases is their ability to efficiently deaminate 5MeC and small 5-substituted cytidine oxidation products [22, 25–27]. To demonstrate that rA3A can deaminate 5MeC, QT6 cells were transfected with rA3A expression plasmid and subsequently transfected by 5MeC-substituted HIV env DNA fragments [26]. As shown in Figure 5A, 3D-PCR products were recovered as low as 78° C and 79.2° C for the rA3A and hA3A co-transfections respectively, compared to 84.5° C for the empty vector or rA3A* (Figure 5A). The 83.3° C 3D-PCR products (Figure 5A, lower band with an arrow and indicated by an asterisk) were cloned, sequenced and confirmed the presence of edited 5MeC in the expected 5ʹTpC and 5ʹCpC dinucleotide context (Figure 5B). In the same rA3A transfected QT6 cells, edited cytochrome c mtDNA in the cytoplasm was also recovered again with the 5ʹTpC and 5ʹCpC context (Supplementary Figure 3A and 3B) which is in keeping with the nucleo-cytoplasmic localization of rA3A and published findings for other mammalian A3A enzymes [28].
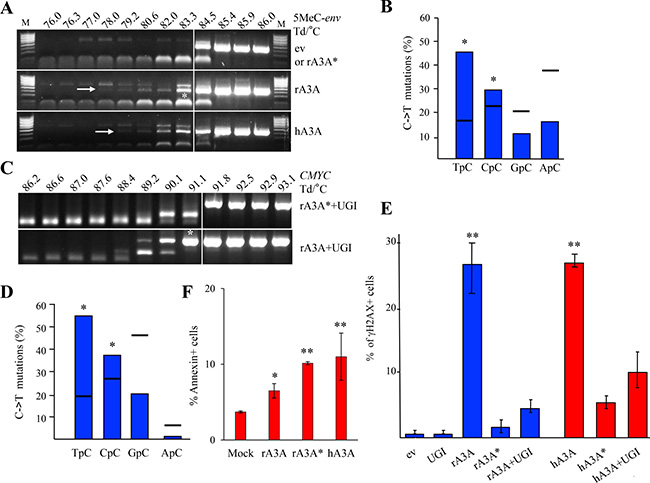
Figure 5: Rabbit A3A edits methylated DNA and generates DBSs driving cells to apoptosis. (A) QT6 cells were co-transfected with 5-methylated HIV env DNA and A3 constructs and analyzed at 48 hours by 3D-PCR. Numbers above gels indicate the Td for each well. Vertical white bar indicates restrictive Td. White asterisk denotes 3D-PCR products cloned and sequenced. (B) Analysis of the 5′ nucleotide context of C->T transitions identified in cloned methylated HIV env DNA 3D-PCR products. Horizontal bars denote expected values derived from base composition. Asterisks denote a statistically significant difference (p < 0.05). (C) Quail CMYC DNA 3D-PCR gels derived from co-transfected QT6 cells at 48 hours first with UGI and then with rA3A or rA3A* expressing plasmid. Numbers indicate the Td for each well. White asterisk denotes PCR products cloned and sequenced. (D) Analysis of the 5′ nucleotide context of C->T transitions identified in A3 edited quail CMYC sequences. Percentages correspond to results obtained after rA3A + UGI co-transfection. Horizontal bars represent expected values derived from base composition. Asterisks show a statistically significant difference (p < 0.05). (E) FACS analysis of HeLa cells 48 hours post transfection with empty vector (ev), UGI or A3A constructs alone or in combination. Percentage of γH2AX positive cells are gated on V5 positive cells except for mock and UGI transfections. Double asterisks mean a highly statistically significant difference (p ≤ 0.005). (F) FACS analysis of HeLa cells 72 hours post transfection. Annexin positive cells were gated on V5-Tag positive cells, except for the mock condition. Single asterisks represent a statistically significant difference (p ≤ 0.05) and double asterisks represent a highly statistically significant difference (p ≤ 0.005).
A3A and A3B are the sole enzymes able to target chromosomal DNA. This can be demonstrated experimentally only if the highly active UNG enzyme is inhibited [20]. As shown in Figure 5C, when rA3A was co-transfected in QT6 cells along with an expression plasmid encoding an UNG inhibitor (UGI), edited CMYC DNA was recovered by 3D-PCR at temperatures as low as 89.2° C for the rA3A+UGI compared to 91.8° C for rA3A*+UGI (Figure 5C). The 91.1° C 3D-PCR products were cloned, sequenced and confirmed the presence of C->T hypermutation in the 5ʹTpC and 5ʹCpC dinucleotide context (Figure 5D).
The ability to generate DSBs is a key feature of all mammalian A3A enzymes tested to date [29, 30]. We used a previously described flow cytometry assay using γH2AX expression as a marker of DSBs [30]. Transfection of rA3A resulted in DSBs on a par with hA3A used as reference while catalytically inactive rA3A* and hA3A* mutants failed to generate DSBs (Figure 5E). Co-transfection with equimolar amounts of UGI strongly reduced the percentage of γH2AX+ cells, confirming that DSBs indeed resulted from cytidine deamination and consequent uridine removal by the UNG enzyme during the DNA repair process (r/hA3A versus r/hA3A+UGI, Figure 5E). A3A cytidine deamination of nuDNA can lead to apoptosis (Figure 5F). The proportion of Annexin V positive cells, was ∼2-fold higher in rA3A and rA3A* than in mock transfected cells, and comparable to hA3A (Figure 5F). Once again an A3A catalytically inactive mutant was pro-apoptotic [30] presumably by binding to ssDNA in the nucleus, which could impact the cell cycle leading to cell stress and death.
Rabbit A3A forms homodimers and is negatively regulated by TRIB3
Regulation of hA3A editing is crucial considering its deleterious effect on genome integrity. Many A3 enzymes can homo- and heterodimerize including A3A [39–41]. We investigated rA3A ability to dimerize using a flow cytometry-based Försters resonance energy transfer assay (FRET-FACS). The test was based on an energy transfer from an excited donor fluorophore (a GFP-fusion protein) to a juxtaposed acceptor fluorophore (a Cherry-fusion protein), which results in specific fluorescence emission detectable by flow cytometry. Plasmids containing rA3A or rA1 fused to Cherry or GFP were constructed and transfected alone or in combination in HEK-293T cells. As shown in Figure 6A, when rA3A-Cherry and rA3A-GFP fusion plasmids were co-transfected, the percentage of fluorescent cells in the FRET channel was ~7-fold (19%) higher over background fluorescence. By contrast, cells transfected with rA3A-Cherry alone gave a background fluorescence (2.8%) whereas transfection with a positive control plasmid encoding a Cherry-GFP fusion construct generated ~5-fold more fluorescence (14%). That the gain of fluorescence resulted from a bona fide specific interaction between rA3A fusion proteins was checked by co-transfecting rA3A-Cherry with the non-homologous rA1-GFP. Only background fluorescence levels were noted due to the inability of the proteins to interact (2.6%).
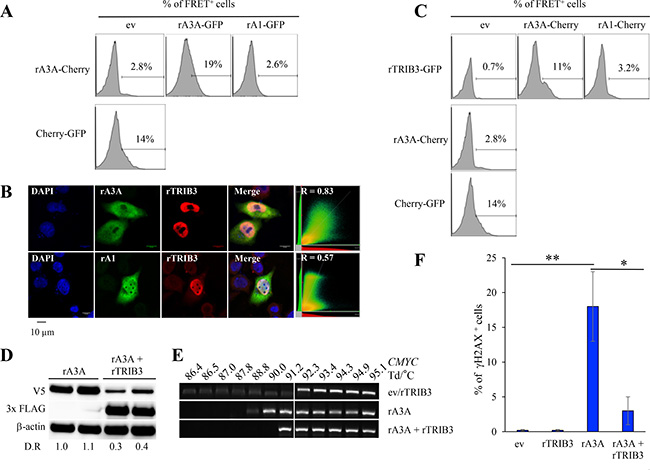
Figure 6: Rabbit A3A forms heterodimers and is negatively regulated by rabbit TRIB3. (A) Representative FRET FACS assay of HEK-293T cells 48 hours post transfection with rA3A-Cherry expression plasmid in combination with an empty vector (ev), rA3A-GFP, or rA1-GFP expression plasmid. The percentage of FRET+ cells were gated on Cherry+ cells for single transfection or Cherry+ and GFP+ cells for co-transfection. (B) Confocal microscopy analysis of V5-tagged APOBEC and 3xFLAG-tagged TRIB3 rabbit proteins in HeLa cells 24 hours post co-transfection. Nuclei are stained using DAPI. (C) Representative FRET-FACS assay of HEK293T cells 48 hours after transfection with rabbit APOBEC-Cherry or TRIB3-GFP constructs alone or in combination. A plasmid encoding a Cherry protein fused to a GFP protein was used as a positive control. Percentage of FRET+ cells are gated on Cherry+ cells for single transfection or Cherry+ and GFP+ cells for co-transfections. (D) Western blot detection of V5-tagged rA3A and 3×FLAG-tagged rTRIB3 proteins in HEK-293T transfected cells by either rA3A alone or rA3A + rTRIB3 (1/4 molar ratio). β actin was used as a loading control. Quantification of fluorescence intensity was performed using ImageJ by normalizing datas to rA3A, D.R, densitometric ratio. (E) Human CMYC DNA specific 3D-PCR gels from HEK293T-UGI cells 48 hours post transfection with either an empty vector (ev), rA3A alone and rA3A and rTRIB3 together (1/4 ratio). Numbers above the gel indicates the Td for each well. White bar indicates restrictive Td. (F) FACS analysis of HeLa cells 48 hours after transfection with an empty vector (ev), rTRIB3, rA3A and rA3A and rTRIB3 together (1/4 ratio). Percentage of γH2AX+ cells are gated on V5-Tag positive cells except for mock and rTRIB3 conditions. A single asterisk means the difference is statistically significant (p ≤ 0.05) and a double asterisk means the difference is highly statistically significant (p ≤ 0.005).
We then explored whether rA3A interacts with the rTRIB3 pseudokinase protein given that the human counterpart (hTRIB3) is the only human A3A interactor identified for the moment [32]. The rTRIB3 gene harbors the same exon/intron structure as the human gene with protein sequences displaying 78% identity. Transfection of a synthetic V5 tagged rTRIB3 expression construct in HeLa cells revealed rTRIB3 to be strictly nuclear (Figure 6B), like its human counterpart [32]. Co-transfection experiments showed that rA3A and rTRIB3 co-localized, as assessed by a Pearson’s correlation coefficient test (R = 0.83) (Figure 6B). As expected, no co-localization was observed with rA1. To confirm that colocalization corresponded to a bona fide interaction between rA3A and rTRIB3 proteins, a rabbit TRIB3-GFP construct was generated to be used in a FRET-FACS analysis as described in Figure 6A. As shown in Figure 6C, the rA3A-Cherry/rTRIB3-GFP co-transfection generated fluorescence ∼15-fold higher (11%) than background (0.7%) or rA1-Cherry/rTRIB3 co-transfection (3.2%).
Transfection of HEK-293T cells with rA3A alone or in combination with rTRIB3 at a 4/1 ratio resulted in ∼3–4 fold decrease in rA3A protein in the presence of rTRIB3 as shown by Western blot quantification (Figure 6D). HEK-293T-UGI cells, which constitutively express the UGI, were cotransfected by rA3A expression plasmids with and without rTRIB3. Transfection of rA3A alone allowed detection of edited CMYC DNA. PCR products where obtained down to 88.8° C, i.e. 3.5° C below the restrictive Td (92.3° C) (Figure 6E). However, when rA3A + rTRIB3 were co-transfected, 3D-PCR recovered DNA only down to 91.2° C, i.e. 2.4° C higher, demonstrating that rTRIB3 expression inhibited rA3A editing of CMYC DNA (Figure 6E). Finally, the percentage of γH2AX+ in HeLa cells was drastically reduced when cells where co-transfected with rA3A + rTRIB3 compared to rA3A alone (18% versus 3% p = 0.003, Figure 6F). Both results indicated that the ability of rA3A to induce mutations and DSBs in genomic DNA was curtailed by rTRIB3.
Cytidine deamination in vivo
The above data showed that only rA3A was a functional as an efficient ssDNA cytidine deaminase in experimental settings (Figure 4 and Figure 5). As rA3A was widely expressed in vivo (Figure 2B), we sought edited cytoplasmic mtDNA (cymtDNA) in rabbit tissues. Using a nested PCR/3D-PCR protocol [38] focused on rabbit mtDNA CYTC, we found PCR products below the limiting Td = 83.9° C from lymph node and skin derived total DNA (Figure 7A). By contrast, nothing was recovered from brain derived DNA where A3A transcripts were not detected (Figure 2B). Cloning and sequencing of lymph nodes and skin 3D-PCR products obtained at 82.8° C (see asterisks, Figure 7A) confirmed multiple C->T substitutions (Figure 7B) with a preference for editing in a 5ʹTpC dinucleotide context, typical of A3A enzymes (Figure 7C). This suggests that only the A3A enzyme is active in vivo.
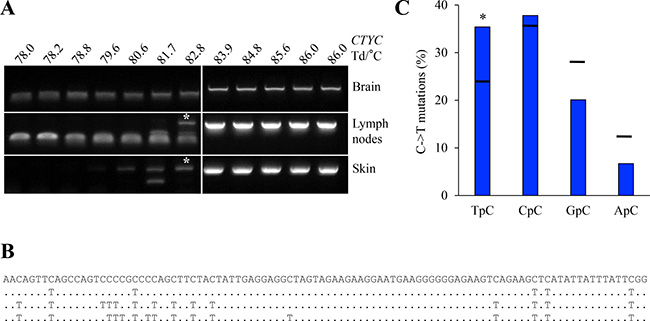
Figure 7: A3 edited mtDNA in vivo. (A) Rabbit cytochrome c mtDNA 3D-PCR gels using total DNA from brain, lymph node and skin tissues. Numbers indicates the Td for each well. White bar indicates restrictive Td. White asterisks indicate cloned PCR products for sequences analysis. If rA3A is orthologous to hA3A then the edited mtDNA is derived from mtDNA leaked to the cytoplasm. (B) Representative mutated mtDNA sequences harboring C->T substitutions. Mutations are noted compared to reference sequence. (C) Analysis of the 5′ nucleotide context of C->T transitions identified edited cytoplasmic mtDNA. Horizontal bars represent expected values. Asterisks show a statistically significant difference (p < 0.05).
DISCUSSION
The APOBEC3 locus is conserved among most placental mammals and usually encodes at least one Z1, Z2 and Z3 zinc-finger containing enzyme [5]. The notable exception is the entire order Rodentia that lacks a Z1 containing domain typified by the human A3A enzyme. The pig and cat genomes are also lacking a Z1 domain enzyme whereas the sheep and dog genomes do [28, 33]. The rabbit genome encoded two APOBEC3 genes in a head-to-head manner, reminiscent of the Carnivora APOBEC3 locus (Figure 1). The rAZ1 (rA3A) enzyme proved a bona fide member as it encodes the signature I128 residue (Supplementary Figure 2A) [28, 42, 43]. To date only the Z1 domain enzymes, notably human A3A and A3B, have been linked to cancers [3, 20, 22, 44, 45]. It has been suggested that one of the human A3H haplotypes might also be an endogenous mutagen [46] although A3H is a Z3 monodomain cytidine deaminase [5]. As the rA3A, A3 enzyme proved to be most closely related to primates, A3A enzyme opens up the perspective of using the rabbit as a small animal model, especially as rA3A was functional (Figures 4, 5 and 6). By contrast, the rabbit A3Z2Z3 protein was stable but functionally inactive (Figure 4A and 4C), using highly sensitive 3D-PCR, a technique that allows recovery of a single hypermutated sequence down to frequencies of 10–4 [37, 38]. Catalytically inactive enzymes that are nonetheless expressed in vivo have already been described [47, 48]. Human A3DE is an example as the protein is stably expressed, yet non-functional due to a single inactivating amino acid substitution [49]. Alternatively, it could result from an inactive polymorphism. No ssDNA hyperediting activity was detected with rA1 despite reports showing that rat A1 targets HIV DNA albeit much less so than for RNA [50]. This contrasts with murine A1, which can hypermutate both dsRNA and ssDNA [36]. 3D-PCR is excellent for detecting hyperedited ssDNA but is unable to recover DNA with a handful of edited cytidine bases per 100 bp.
Exploration of the cellular localization demonstrated a nucleo-cytoplasmic expression for the three rabbit proteins (rA1, rA3A, rAZ2Z3, Figure 3B). The expression of rA3A is the same for all mammalian A3A studied to date [28, 43]. By contrast, the nucleo-cytoplasmic localization of rA3Z2Z3 makes it different from murine A3 that was strictly cytoplasmic [36, 51, 52]. The rA1 expression pattern was consistent with a previous report [51].
The rabbit A3A enzyme was strictly orthologous to its human counterpart. Its localization was nucleo-cytoplasmic, able to hyperedit cytidine and 5-methylcytidine residues in foreign DNA substrates (Figure 5A), as well as able to access and deaminate nuDNA and induce DSBs (Figure 5C and 5E). Indeed, these are features of all mammalian A3A [25–30]. In addition, rA3A appeared to be similarly regulated. The ability of A3A to form homodimers is conserved in rabbits and the enzyme directly interacts with rTRIB3, the rabbit homolog of human TRIB3 that is the only A3A interactor identified so far [32]. As for the hA3A/hTRIB3 pair, rTRIB3 enhances rA3A degradation, this being followed by a decrease in rA3A editing activity (Figure 6).
The lemur and rabbit genomes are among the very few species that encode vestiges of endogenous lentiviruses [53, 54]. All but one of the exogenous lentiviruses encode a vif gene that neutralizes several, but not necessarily all, APOBEC3 enzymes. While the lemur endogenous retroviruses encode a vif gene, the rabbit elements referred to as RELIKs do not encode a vif gene [53, 54]. The present work suggests that the infectious counterpart was not vulnerable to restriction by the only functional rA3A enzyme, although other features could allow a virus to replicate despite the presence of APOBEC3 enzymes; for example human hepatitis B virus is vulnerable to editing by human APOBEC3 enzymes yet does not encode an APOBEC3 or interferon antagonist [14, 16, 55–57] given that several human APOBEC3 genes are interferon stimulated genes [32, 58–61].
The results presented here showed that the rabbit genome encodes an A3A enzyme that is strictly homologous to the human A3A in terms of substrate specificity and negative interaction with TRIB3. Moreover, the conservation of the relationship between A3A and TRIB3 over 100 million years, the time between the divergence of Glires and Primates, illustrates the importance of regulating genomic DNA editing. As all suborders in Rodentia do not encode an A3A gene, the rabbit thus appears as the only small available animal model for studying the role of A3A during human oncogenesis. However, rA3A is widely expressed (Figure 2B). By contrast the hA3A is preferentially expressed in hematopoietic cells and is massively up regulated by type I interferons [32, 59, 61]. This suggests that although rA3A is strictly orthologous to its human counterpart, and indeed to other mammalian A3A enzymes, it is differently regulated.
MATERIALS AND METHODS
Plasmids
Rabbit A3A, A3Z2Z3 and TRIB3 (NC_013672) cDNAs were synthetized (GeneCust) and subsequently cloned into pcDNA3.1/V5-His-TOPO vector (Invitrogen). All constructs were C-tagged by the V5 epitope excepting for the rabbit TRIB3 construct which was tagged with the 3XFLAG epitope in N-terminal position. The rabbit A3A catalytic mutant rA3A C98S (rA3A*) was generated using the GENEART® site-directed mutagenesis system. The hA3A, hA3A catalytic mutant, hTRIB3-3XFLAG and UGI plasmid have been previously described [20, 32]. The Cherry-GFP fusion protein encoding plasmid was from Addgene (#32643)
DNA extraction, 3D-PCR amplification and cloning
Total DNA from transfected cells was extracted using the MasterPure™ complete DNA and RNA purification kit (Epicentre) and suspended in 30 μL of sterile water. All amplifications were performed using first-round standard PCR with 5 μL of DNA extract followed by nested 3D-PCR with 5 μL of 1/50 dilution of the first PCR round. PCR was performed in 50 μL with 1 U Taq DNA polymerase (Eurobio) per reaction. 3D-PCR primer sequences are indicated in Supplementary Table 1. PCR conditions for the first round of amplification were 5 min. of denaturation at 95° C then 40 cycles of amplification (30 sec. 95° C, 30 sec. 58° C, 30 sec. 72° C), followed by 7 min. at 72° C. PCR conditions for the second round of amplification were 5 min of denaturation with dT° gradient of 80–90° C then 40 cycles of amplification (30 sec. 95° C, 30 sec. 80–90° C, 30 sec. 72° C), followed by 7 min. at 72° C. For methylated DNA 3D-PCR, primer sequences and PCR conditions were previously documented [26]. For human CMYC DNA 3D-PCR, primer sequences and PCR conditions were previously described [20]. After purification, PCR products were cloned into TOPO 2.1 vector (Invitrogen) and sequencing was outsourced to Eurofin.
Real time PCR quantification
Rabbit tissues were incubated in RNA later (RNA stabilization reagent, Qiagen) and mechanically disrupted before extraction of total RNA using RNeasy® lipid tissue mini kit (Qiagen) according to the manufacturer’s protocol. Corresponding cDNAs were synthetized using QuantiTect reverse transcription kit (Qiagen). Quantification was performed by TaqMan using Takyon Rox probe mastermix dTTP blue (Eurogentec). Sequences of specific primers and probes used are detailed in Supplementary Table 1. Cycling conditions were as follows: first step of denaturation at 95° C during 3 min., followed by 40 cycles of amplification (95° C 15 sec, 58° C 15 sec and 68° C 15 sec). Fluorescence was measured during the 68° C step incubation using a Realplex2 Mastercycler (Eppendorf). The specificity of the PCR products was verified by sequencing. Messenger RNA expression levels were normalized based on the EF1α reporter gene.
Cell culture
Japanese quail embryonic fibroblast QT6 cells (ATCC CRL 1708) were maintained in Ham’s medium supplemented with 1% chicken serum, 10% fetal bovine serum, 5% tryptose phosphate, 2 mM L-glutamine, 100 U/ml penicillin and 100 mg/ml streptomycin. HeLa, HEK-293T and HEK-293T-UGI cells (HEK-293T cells stably expressing Bacillus subtilis phage uracil-DNA glycosylase inhibitor or UGI) were maintained in DMEM glutamax medium (Invitrogen) supplemented with 10% FCS, 100 U/ml penicillin and 100 mg/ml streptomycin.
Cell transfection
Approximately 800,000 cells were seeded into 6-well plates and transfected with 2 μg of plasmid using the Jetprime transfection kit (Polypus Transfection™) according to manufacturer’s instructions. For cotransfections, a plasmid ratio of 1:1 was used except for rTRIB/rA3A cotransfection experiments where a ratio of 4:1 was used.
Deamination assay
At 48 hours post-transfection, A3-transfected HEK-293T cells were extensively washed with PBS and mechanically harvested. Total proteins were extracted using specific lysis buffer (25 mM HEPES [pH 7.4], 10% glycerol, 150 mM NaCl, 0.5% Triton X-100, 1 mM EDTA, 1 mM MgCl2, 1mM ZnCl2) supplemented with protease inhibitors. Deaminase activity was assessed by incubating whole cell lysates with 1 pmole DNA oligonucleotide 5′FAM-AAATTCTAATAGATAATGTGA-TAMRA in the presence of 0.4 U UDG (New England Biolabs) in a 20 mM Tris-HCl pH7, 1 mM dithiothreitol, and 1 mM EDTA reaction buffer. After 2 hours of incubation at 37° C, abasic sites were cleaved by heating for 2 min at 95° C, and endpoint fluorescence were measured using a Realplex 2 mastercycler (Bio Rad) with FAM setting. Results are normalized to the quantity of protein using Pierce BCA protein assay kit (Thermo Scientific).
Immunofluorescence
Approximately 50,000 HeLa cells were seeded in Nunc™ Lab-Tek™ II Chamber Slide™ System Thermo Scientific™ and transfected 24 hours later with 1 μg of plasmid DNA according to the Fugene® protocol. Two days after transfection, coverslip grown transfected HeLa cells were washed three times with PBS and fixed with 4% PFA for 15 min. Cells were then washed five times and permeabilized with a 50% methanol/acetone mix for 10 min. After five PBS washings, permeabilized cells were incubated for 1 hour at room temperature, first with 1% bovine serum albumin (BSA) PBS 1/200 mouse monoclonal anti-V5 antibody (Invitrogen) and then with 1% BSA PBS 1/750 anti mouse Alexa Fluor 488 conjugated antibody. After several PBS washings, coverslips were mounted with Mounting medium for immunofluorescence (Interchim). Imaging was performed using a Leica SP5 confocal microscope. Colocalization analyses were performed with Huygens Essential for Mac version 4.3.1p3 (Scientific Volume Imaging B.V.). Images were first deconvoluted then analyzed using the Costes method.
FACS analysis
For restriction of GFP expression plasmid experiments, transfected cells were trypsinized, washed with PBS, fixed in 2% ice-cold paraformaldehyde (Electron Microscopy Sciences) for 10–20 min. After one PBS washing, cells were permeabilized in 90% ice-cold methanol (Sigma) for 30 min. Following the second PBS washing, cells were incubated for one hour in ice with 1:100 PBS-0.5% BSA diluted mouse anti V5-Tag Alexa Fluor® 647 antibody (AbD Serotec). For apoptosis experiments, cells were first stained using an Annexin V Staining with Fixable Viability Dyes kit (eBioscience) using fixable Viability Dye eFluor®780 according to manufacturer’s instructions, then washed, permeabilized and incubated for one hour in ice with 1:100 PBS-0.5% BSA diluted mouse anti V5-Tag Alexa Fluor® 488 antibody (AbD Serotec). For DSBs experiments, fixed and permeabilized cells were incubated 1 hour in ice with 1:100 PBS-0.5% BSA diluted mouse anti V5-Tag Alexa Fluor® 488 antibody (AbD Serotec) and 1:100 diluted mouse anti human γH2AX and 1:100 Alexa Fluor® 647 antibody. For the FRET FACS assay, cells were simply trypsinized and washed with PBS. The data was acquired on a MACSQuant® analyzer harboring violet, blue, and either a red laser (measure of dsDNA breaks and apoptosis) or a yellow laser for the FRET FACS assay. The data were analyzed using the FlowJo® software (Tree Star Inc., version 10.1r5 for Mac).
Western blotting
Cells were recovered 24 hours after transfection. Protein extraction and Western blot analysis were carried out according to standard procedures. After blocking, membranes were probed with either a 1:5000 dilution of anti V5-tag horseradish peroxidase-coupled antibody, a 1:5000 dilution of anti 3x-FLAG peroxidase-coupled antibody (Sigma), or a 1:50000 dilution of anti β-actin. The membrane was subjected to detection by SuperSignal™ West Pico chemiluminescent substrate (ThermoFisher Scientific).
Author contributions
HCL, VC, MSB, XXL, FJ, MH, RS performed research, HCL, VC, MSB, XXL, FJ, MH, RS, SWH and JPV analyzed data, HCL, SWH and JPV designed research and wrote the paper.
ACKNOWLEDGMENTS
We thank Dr. Robin Lombard from Miltenyi Biotec for his help in setting up the FRET-FACS assay.
CONFLICTS OF INTEREST
No conflicts of interest
FUNDING
This work was supported by funds from the Institut Pasteur and Centre National de la Recherche Scientifique (CNRS). Hélène Laude, Vincent Caval and Mohamed Salah Bouzidi were supported by the “Assistance Publique des Hôpitaux de Paris”, OSEO (FUI AAP12), and the Ligue Nationale contre le Cancer (GB/MA/CD-11283) respectively, Xiong Xiong Li was supported by a stipend from Lanzhou Institute of Biological Products Co., Ltd (LIBP), subsidiary company of China National Biotec Group Company Limited (CNBG) and by a stipend from the Pasteur - Paris University (PPU) International PhD program.
REFERENCES
1. Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, Buck G, Chen L, Beare D, Latimer C, Widaa S, Hinton J, Fahey C, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010; 463:893–8.
2. Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordonez GR, Bignell GR, Ye K, Alipaz J, Bauer MJ, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010; 463:191–6.
3. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, et al. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–21.
4. Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, Stebbings LA, Leroy C, Edkins S, Mudie LJ, Greenman CD, Jia M, Latimer C, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009; 462:1005–10.
5. Larue RS, Andresdottir V, Blanchard Y, Conticello SG, Derse D, Emerman M, Greene WC, Jonsson SR, Landau NR, Lochelt M, Malik HS, Malim MH, Munk C, et al. Guidelines for Naming Non-Primate APOBEC3 Genes and Proteins. J Virol. 2008; 83:494–7.
6. Nabel CS, Lee JW, Wang LC, Kohli RM. Nucleic acid determinants for selective deamination of DNA over RNA by activation-induced deaminase. Proc Natl Acad Sci U S A. 2013; 110:14225–30.
7. Baumert TF, Rosler C, Malim MH, von Weizsacker F. Hepatitis B virus DNA is subject to extensive editing by the human deaminase APOBEC3C. Hepatology. 2007; 46:682–9.
8. Delebecque F, Suspene R, Calattini S, Casartelli N, Saib A, Froment A, Wain-Hobson S, Gessain A, Vartanian JP, Schwartz O. Restriction of foamy viruses by APOBEC cytidine deaminases. J Virol. 2006; 80:605–14.
9. Fan J, Wang J, Bensadoun A, Lauer SJ, Dang Q, Mahley RW, Taylor JM. Overexpression of hepatic lipase in transgenic rabbits leads to a marked reduction of plasma high density lipoproteins and intermediate density lipoproteins. Proc Natl Acad Sci U S A. 1994; 91:8724–8.
10. Lecossier D, Bouchonnet F, Clavel F, Hance AJ. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science. 2003; 300: 1112.
11. Mahieux R, Suspene R, Delebecque F, Henry M, Schwartz O, Wain-Hobson S, Vartanian JP. Extensive editing of a small fraction of human T-cell leukemia virus type 1 genomes by four APOBEC3 cytidine deaminases. J Gen Virol. 2005; 86:2489–94.
12. Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002; 418:646–50.
13. Suspène R, Aynaud MM, Koch S, Pasdeloup D, Labetoulle M, Gaertner B, Vartanian JP, Meyerhans A, Wain-Hobson S. Genetic editing of herpes simplex virus 1 and Epstein-Barr herpesvirus genomes by human APOBEC3 cytidine deaminases in culture and in vivo. J Virol. 2011; 85:7594–602.
14. Suspène R, Guétard D, Henry M, Sommer P, Wain-Hobson S, Vartanian JP. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc Natl Acad Sci U S A. 2005; 102:8321–6.
15. Vartanian JP, Guétard D, Henry M, Wain-Hobson S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science. 2008; 320:230–3.
16. Vartanian JP, Henry M, Marchio A, Suspène R, Aynaud MM, Guétard D, Cervantes-Gonzalez M, Battiston C, Mazzaferro V, Pineau P, Dejean A, Wain-Hobson S. Massive APOBEC3 editing of hepatitis B viral DNA in cirrhosis. PLoS Pathog. 2010; 6:e1000928.
17. Bogerd HP, Wiegand HL, Hulme AE, Garcia-Perez JL, O’Shea KS, Moran JV, Cullen BR. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc Natl Acad Sci U S A. 2006; 103:8780–5.
18. Esnault C, Heidmann O, Delebecque F, Dewannieux M, Ribet D, Hance AJ, Heidmann T, Schwartz O. APOBEC3G cytidine deaminase inhibits retrotransposition of endogenous retroviruses. Nature. 2005; 433:430–3.
19. Muckenfuss H, Hamdorf M, Held U, Perkovic M, Lower J, Cichutek K, Flory E, Schumann GG, Munk C. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J Biol Chem. 2006; 281:22161–72.
20. Suspène R, Aynaud M, Guétard D, Henry M, Eckhoff G, Marchio A, Pineau P, Dejean A, Vartanian JP, Wain-Hobson S. Somatic hypermutation of human mitochondrial and nuclear DNA by APOBEC3 cytidine deaminases, a pathway for DNA catabolism. Proc Natl Acad Sci U S A. 2011; 108:4858–63.
21. Caval V, Bouzidi MS, Suspène R, Laude H, Dumargne MC, Bashamboo A, Krey T, Vartanian JP, Wain-Hobson S. Molecular basis of the attenuated phenotype of human APOBEC3B DNA mutator enzyme. Nucleic Acids Res. 2015; 43:9340–9.
22. Caval V, Suspène R, Shapira M, Vartanian JP, Wain-Hobson S. A prevalent cancer susceptibility APOBEC3A hybrid allele bearing APOBEC3B 3′UTR enhances chromosomal DNA damage. Nat Commun. 2014; 5:5129.
23. Hoopes JI, Cortez LM, Mertz TM, Malc EP, Mieczkowski PA, Roberts SA. APOBEC3A and APOBEC3B Preferentially Deaminate the Lagging Strand Template during DNA Replication. Cell Rep. 2016; 14:1273–82.
24. Seplyarskiy VB, Soldatov RA, Popadin KY, Antonarakis SE, Bazykin GA, Nikolaev SI. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 2016; 26:174–82.
25. Carpenter MA, Li M, Rathore A, Lackey L, Law EK, Land AM, Leonard B, Shandilya SM, Bohn MF, Schiffer CA, Brown WL, Harris RS. Methylcytosine and Normal Cytosine Deamination by the Foreign DNA Restriction Enzyme APOBEC3A. J Biol Chem. 2012; 287:34801–8.
26. Suspène R, Aynaud MM, Vartanian JP, Wain-Hobson S. Efficient deamination of 5-methylcytidine and 5-substituted cytidine residues in DNA by human APOBEC3A cytidine deaminase. PLoS One. 2013; 8:e63461.
27. Wijesinghe P, Bhagwat AS. Efficient deamination of 5-methylcytosines in DNA by human APOBEC3A, but not by AID or APOBEC3G. Nucleic Acids Res. 2012; 40:9206–17.
28. Caval V, Suspène R, Vartanian JP, Wain-Hobson S. Orthologous mammalian APOBEC3A cytidine deaminases hypermutate nuclear DNA. Mol Biol Evol. 2014; 31:330–40.
29. Landry S, Narvaiza I, Linfesty DC, Weitzman MD. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 2011; 12:444–50.
30. Mussil B, Suspène R, Aynaud MM, Gauvrit A, Vartanian JP, Wain-Hobson S. Human APOBEC3A isoforms translocate to the nucleus and induce DNA double strand breaks leading to cell stress and death. PLoS One. 2013; 8:e73641.
31. Kidd JM, Newman TL, Tuzun E, Kaul R, Eichler EE. Population stratification of a common APOBEC gene deletion polymorphism. PLoS Genet. 2007; 3:e63.
32. Aynaud MM, Suspène R, Vidalain PO, Mussil B, Guétard D, Tangy F, Wain-Hobson S, Vartanian JP. Human Tribbles 3 protects nuclear DNA from cytidine deamination by APOBEC3A. J Biol Chem. 2012; 287:39182–92.
33. LaRue RS, Jonsson SR, Silverstein KA, Lajoie M, Bertrand D, El-Mabrouk N, Hotzel I, Andresdottir V, Smith TP, Harris RS. The artiodactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol Biol. 2008; 9:104.
34. Thielen BK, McNevin JP, McElrath MJ, Hunt BV, Klein KC, Lingappa JR. Innate immune signaling induces high levels of TC-specific deaminase activity in primary monocyte-derived cells through expression of APOBEC3A isoforms. J Biol Chem. 2010; 285:27753–66.
35. Yamanaka S, Balestra ME, Ferrell LD, Fan J, Arnold KS, Taylor S, Taylor JM, Innerarity TL. Apolipoprotein B mRNA-editing protein induces hepatocellular carcinoma and dysplasia in transgenic animals. Proc Natl Acad Sci U S A. 1995; 92:8483–7.
36. Petit V, Guétard D, Renard M, Keriel A, Sitbon M, Wain-Hobson S, Vartanian JP. Murine APOBEC1 is a powerful mutator of retroviral and cellular RNA in vitro and in vivo. J Mol Biol. 2009; 385:65–78.
37. Henry M, Guetard D, Suspene R, Rusniok C, Wain-Hobson S, Vartanian JP. Genetic editing of HBV DNA by monodomain human APOBEC3 cytidine deaminases and the recombinant nature of APOBEC3G. PLoS One. 2009; 4:e4277.
38. Suspène R, Henry M, Guillot S, Wain-Hobson S, Vartanian JP. Recovery of APOBEC3-edited human immunodeficiency virus G≥A hypermutants by differential DNA denaturation PCR. J Gen Virol. 2005; 86:125–9.
39. Bohn MF, Shandilya SM, Silvas TV, Nalivaika EA, Kouno T, Kelch BA, Ryder SP, Kurt-Yilmaz N, Somasundaran M, Schiffer CA. The ssDNA Mutator APOBEC3A Is Regulated by Cooperative Dimerization. Structure. 2015; 23:903–11.
40. Friew YN, Boyko V, Hu WS, Pathak VK. Intracellular interactions between APOBEC3G, RNA, and HIV-1 Gag: APOBEC3G multimerization is dependent on its association with RNA. Retrovirology. 2009; 6:56.
41. Huthoff H, Autore F, Gallois-Montbrun S, Fraternali F, Malim MH. RNA-dependent oligomerization of APOBEC3G is required for restriction of HIV-1. PLoS Pathog. 2009; 5:e1000330.
42. Bulliard Y, Narvaiza I, Bertero A, Peddi S, Rohrig UF, Ortiz M, Zoete V, Castro-Diaz N, Turelli P, Telenti A, Michielin O, Weitzman MD, Trono D. Structure-function analyses point to a polynucleotide-accommodating groove essential for APOBEC3A restriction activities. J Virol. 2011; 85:1765–76.
43. Henry M, Terzian C, Peeters M, Wain-Hobson S, Vartanian JP. Evolution of the primate APOBEC3A cytidine deaminase gene and identification of related coding regions. PLoS One. 2012; 7:e30036.
44. Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, Yee D, Temiz NA, Donohue DE, et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013; 494:366–70.
45. Middlebrooks CD, Banday AR, Matsuda K, Udquim KI, Onabajo OO, Paquin A, Figueroa JD, Zhu B, Koutros S, Kubo M, Shuin T, Freedman ND, Kogevinas M, et al. Association of germline variants in the APOBEC3 region with cancer risk and enrichment with APOBEC-signature mutations in tumors. Nat Genet. 2016; 48:1330–8.
46. Starrett GJ, Luengas EM, McCann JL, Ebrahimi D, Temiz NA, Love RP, Feng Y, Adolph MB, Chelico L, Law EK, Carpenter MA, Harris RS. The DNA cytosine deaminase APOBEC3H haplotype I likely contributes to breast and lung cancer mutagenesis. Nat Commun. 2016; 7:12918.
47. Bouzidi MS, Caval V, Suspene R, Hallez C, Pineau P, Wain-Hobson S, Vartanian JP. APOBEC3DE Antagonizes Hepatitis B Virus Restriction Factors APOBEC3F and APOBEC3G. J Mol Biol. 2016; 428:3514–28.
48. Dang Y, Wang X, Esselman WJ, Zheng YH. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J Virol. 2006; 80:10522–33.
49. Dang Y, Abudu A, Son S, Harjes E, Spearman P, Matsuo H, Zheng YH. Identification of a single amino acid required for APOBEC3 antiretroviral cytidine deaminase activity. J Virol. 2011; 85:5691–5.
50. Bishop KN, Holmes RK, Sheehy AM, Malim MH. APOBEC-mediated editing of viral RNA. Science. 2004; 305:645.
51. Ikeda T, Ohsugi T, Kimura T, Matsushita S, Maeda Y, Harada S, Koito A. The antiretroviral potency of APOBEC1 deaminase from small animal species. Nucleic Acids Res. 2008; 36:6859–71.
52. Renard M, Henry M, Guétard D, Vartanian JP, Wain-Hobson S. APOBEC1 and APOBEC3 cytidine deaminases as restriction factors for hepadnaviral genomes in non-humans in vivo. J Mol Biol. 2010; 400:323–34.
53. Gifford RJ, Katzourakis A, Tristem M, Pybus OG, Winters M, Shafer RW. A transitional endogenous lentivirus from the genome of a basal primate and implications for lentivirus evolution. Proc Natl Acad Sci U S A. 2008; 105:20362–7.
54. Katzourakis A, Tristem M, Pybus OG, Gifford RJ. Discovery and analysis of the first endogenous lentivirus. Proc Natl Acad Sci U S A. 2007; 104:6261–5.
55. Noguchi C, Ishino H, Tsuge M, Fujimoto Y, Imamura M, Takahashi S, Chayama K. G to A hypermutation of hepatitis B virus. Hepatology. 2005; 41:626–33.
56. Rosler C, Kock J, Kann M, Malim MH, Blum HE, Baumert TF, von Weizsacker F. APOBEC-mediated interference with hepadnavirus production. Hepatology. 2005; 42:301–9.
57. Turelli P, Mangeat B, Jost S, Vianin S, Trono D. Inhibition of hepatitis B virus replication by APOBEC3G. Science. 2004; 303:1829.
58. Bonvin M, Achermann F, Greeve I, Stroka D, Keogh A, Inderbitzin D, Candinas D, Sommer P, Wain-Hobson S, Vartanian JP, Greeve J. Interferon-inducible expression of APOBEC3 editing enzymes in human hepatocytes and inhibition of hepatitis B virus replication. Hepatology. 2006; 43:1364–74.
59. Koning FA, Newman EN, Kim EY, Kunstman KJ, Wolinsky SM, Malim MH. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J Virol. 2009; 83:9474–85.
60. Peng G, Lei KJ, Jin W, Greenwell-Wild T, Wahl SM. Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. J Exp Med. 2006; 203:41–6.
61. Stenglein MD, Burns MB, Li M, Lengyel J, Harris RS. APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat Struct Mol Biol. 2010; 17:222–9.