
INTRODUCTION
Oral anticoagulant drugs
Oral anticoagulants (OAs) are recommended drugs to reduce the risk of stroke and systemic embolism in patients with non-valvular atrial fibrillation (NVAF), and to treat and reduce the risk of deep venous thrombosis (DVT) and pulmonary embolism (PE) [1, 2].
OAs can be classified as vitamin K antagonists (VKAs) (warfarin, acenocoumarol, fluindione and phenprocoumon) [3] and direct oral anticoagulants (DOACs): dabigatran, apixaban, rivaroxaban and edoxaban [4, 5].
Pharmacogenetics of OAs
Genome Wide Association Studies (GWAs) have been very successful in finding genetic risk factors associated with complex diseases or with drug response (pharmacogenomics). This technique uses a general approach which allow a systematic agnostic research of common genetic factors across the whole genome [6, 7].
Furthermore, different GWAs analysis have been published describing variants associated with the interindividual and inter-ethnic variation of VKAs response [6–11] and, recently, also for dabigatran response [12]).
The aim of this review is to describe the genetic factors associated with the response of OAs drugs used in stroke and other cardiovascular diseases prevention and to discuss the future use of OAs pharmacogenomics in the clinical practice.
MATERIALS AND METHODS
Literature search
An extensive literature search was performed, up to December 2017, on PubMed with the following key words: ‘GWAs and warfarin’, ‘GWAs and acenocoumarol’, ‘GWAs and fluindione’, ‘GWAs and phenprocoumon’, ‘GWAs and dabigatran’, ‘GWAs and edoxaban’, ‘GWAs and rivaroxaban’ and ‘GWAs and apixaban’. Thirty-eight results were obtained for ‘GWAs and warfarin’, 6 of which were GWAs studies. For ‘GWAs and acenocoumarol’, 4 results were obtained. Only 1 of them was a GWAs study performed on acenocoumarol. One result was obtained for ‘GWAs and phenprocoumon’ which corresponded to a GWAs analysis. Two results were found for ‘GWAs and dabigatran’; only one was a GWAs analysis in patients treated with dabigatran. For the other searches, no results were obtained (Figure 1, Supplementary Table 1).
Figure 1: Flow diagram in the selection of articles included in the review.
In the case of drugs for which we did not find GWAS analyses, we searched in PubMed for other genetic studies, based on candidate genes, using the following terms: ‘Genetics and fluindione’, ‘Genetics and edoxaban’, ‘Genetics and rivaroxaban’ and ‘Genetics and apixaban’. We included three candidate gene studies for fluindione, one candidate gene study for edoxaban and one in vitro study for apixaban (Figure 1). We included all the original studies written in English including candidate gene analyses in more than 20 patients or in vitro studies analyzing the influence of one or more SNPs.
eQTLs analysis
We searched in GTEx portal (https://www.gtexportal.org/) the first and second most significant eQTLs genes for the SNPs from the published GWAs cited in this review (Supplementary Table 1), in two cases: 1-intragenic SNPs attributed to regulate other genes in the referenced paper; 2-intergenic SNPs (Table 1). We indicated the tissue in which the most significant eQTL for a gene is expressed and also if it is expressed in relevant tissues for the disease.
Table 1: Most significant eQTLs for significant intergenic SNPs in AOs GWAs
SNP from GWAS (Supplementary Table 1) |
Gene |
Near gene (cited in the GWAs) |
Most significant eQTLs |
p-value |
Tissue |
|---|---|---|---|---|---|
rs10871454 |
STX4 |
VKORC1 |
KAT8 |
1.1e-38 |
Skina |
VKORC1 |
1.5e-30 |
Liverb |
|||
rs9923231 |
VKORC1 |
KAT8 |
1.2e-38 |
Skina |
|
VKORC1 |
2.0e-33 |
Liverb |
|||
rs12777823 |
CYP2C18 |
CYP2C19 |
6.9e-31 |
Esophagus - Mucosa |
|
MTND4P19 |
3.5e-14 |
Skin |
|||
rs2104162 |
CYP2C9 |
CYP2C19 |
1.1e-10 |
Esophagus- Mucosa |
|
MTDN4P19 |
0.0000069 |
Esophagus- Mucosa |
|||
rs749671 |
ZNF646 |
VKORC1 |
KAT8 |
1.9e-39 |
Skina |
VKORC1 |
3.0e-32 |
Liverb |
|||
rs4918798 |
CYP2C9 |
C10orf129 |
1.2e-12 |
Adipose - Subcutaneous |
|
CYP2C8 |
0.000077 |
Breast - Mammary Tissue |
|||
rs17126068 |
DDHD1 |
RP11-547D23.1 |
1.4e-9 |
Testis |
|
DDHD1 |
7.0e-7 |
Muscle - Skeletal |
|||
rs889548 |
MYST1 |
VKORC1 |
KAT8 |
7.1e-43 |
Skin |
VKORC1 |
1.2e-30 |
Liverb |
|||
rs1978487 |
MYST1 |
VKORC1 |
KAT8 |
1.1e-39 |
Skina |
VKORC1 |
7.0e-26 |
Liverb |
|||
rs749767 |
BCKDK |
VKORC1 |
KAT8 |
5.7e-40 |
Skina |
VKORC1 |
3.9e-27 |
Liverb |
|||
rs3862009 |
CYP2C19 |
C10orf129 |
1.7e-40 |
Adipose-subcutaneousa |
|
CYP2C9 |
7.2e-9 |
Adipose-subcutaneous |
|||
rs12610189 |
CYP4F2 |
AC004791.2 |
1.3e-40 |
Lunga,b |
|
CYP4F2 |
9.3e-11 |
Skin |
|||
rs1998591 |
CYP2C18 |
CYP2C19 |
1.1e-17 |
Esophagus - Mucosa |
|
C10orf129 |
4.1e-10 |
Nerve - Tibiala |
|||
rs2104543 |
CYP2C18 |
CYP2C19 |
1.1e-15 |
Esophagus - Mucosa |
|
C10orf129 |
3.8e-10 |
Nerve - Tibiala |
|||
rs12772169 |
CYP2C18 |
CYP2C19 |
2.1e-17 |
Esophagus - Mucosa |
|
C10orf129 |
3.1e-10 |
Esophagus - Mucosaa |
|||
rs11150604 |
ZNF646 |
VKORC1 |
KAT8 |
1.1e-38 |
Skina |
VKORC1 |
1.5e-30 |
Liverb |
|||
rs1980889 |
CKS2 |
SEMA4D |
1.3e-7 |
Thyroid |
|
CKS2 |
0.0000091 |
Liver |
|||
rs746357 |
SHC3 |
SEMA4D |
1.3e-7 |
Thyroid |
|
CKS2 |
0.0000091 |
Liver |
Enumeration of the most significant eQTLs for SNPs associated with AOs response located in 1) intragenic regions attributed to regulate other genes in the referenced paper; 2-intergenic regions. Column one: SNP ID (from Supplementary Table 1); Column two: gene, for intragenic SNPs; Column three: near gene described in the original GWAs to be the effector gene; Column four: most significant eQTLs from GTEx project. P-value indicated for the most significant eQTL in the gene; Column five: p-values for the most significant eQTL for each gene; Column six: tissues where the most significant eQTLs are expressed.
a Significant eQTL also in arteries (p<10-5); b Significant eQTL also in whole blood (p<10-5).
eQTL= expression quantitative trait loci; GTEx project= Genotype-Tissue Expression project.
VITAMIN K ANTAGONIST (VKA)
The first oral anticoagulant drug was discovered in 1941 through its identification as the cause of fatal bleeding in cattle [13]. These animals had eaten spoiled hay made from sweet clover which contained dicoumarol, a type of coumarin [14]. Since then, coumarins have long been used in the pharmaceutical industry to synthetize anticoagulant drugs due to their antagonistic effect on vitamin K [15].
VKAs are used worldwide. Warfarin is extensively used in North America, Scandinavia, the UK and Asian countries. Acenocoumarol and phenprocoumon are used in continental European countries [14, 16, 17]. Fluindione is widely used in France [18].
Warfarin and acenocoumarol are 4-hydroxycoumarins, which are vitamin K epoxide reductase (VKOR) inhibitors [14]. They inhibit recycling of the inactive oxidized to the active reduced form of vitamin K, a cofactor involved in activation of coagulation factors II, VII, IX and X [19] (Figure 2A).
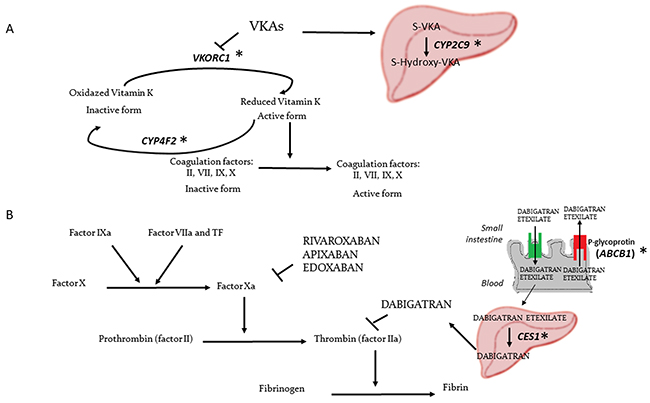
Figure 2: Mechanism of action of the different OAs and polymorphic genes associated with OAs response. (A) Mechanism of action of VKAs. (B) Mechanism of action of DOAC. *Genes with polymorphisms associated with OAs variation in GWAs.
VKAs have demonstrated to be effective, preventing 15 deaths and 15 non-fatal strokes per 1,000 patients with non-valvular AF. However, they caused 8 non-fatal major extracranial bleeds [20]. VKAs have a narrow therapeutic/toxic ratio [7, 11, 16, 21, 22], and their daily dose varies between individuals and ethnic groups [23].
Normally, during the first days to weeks of treatment, the correct dose is not achieved as measured by the international normalized ratio (INR). INR values above 3 or 4 increase the bleeding risk, whereas INR values below 2 are associated with low efficacy of VKAs [24]. Patients may therefore be undertreated, increasing the risk of cardioembolism, or overtreated, leading to bleeding, as the therapeutic range is very small [6, 7, 21].
WARFARIN
Warfarin is a racemic mixture comprised by S and R enantiomers. The S enantiomer has a half-life of 24-33h, whereas R-warfarin has a half-life of 35-58h [14]. S-warfarin is the most potent enantiomer and is almost completely responsible for the anticoagulant effect. This enantiomer is metabolized almost exclusively by the hepatic cytochrome CYP2C9 enzyme [25, 26]. R-warfarin is metabolized by CYP1A1, CYP1A2 and CYP3A4 [27].
The daily dose of warfarin varies between individuals. Doses range from 2 to 10 mg per day [23]. Furthermore, different ethnicities often require different dose of warfarin: Asian populations often have a lower daily dose requirement (3 mg per day) compared to European and African populations (5 mg and 6.5 mg per day, respectively) [23].
Relevant non-genetic factors affecting warfarin dose variance include age, weight, body surface, gender, drug interactions, diseases and the quantity of vitamin K intake [28].
Pharmacogenetics
The main genetic factors affecting warfarin dose variance found and replicated in different retrospective and prospective studies are SNPs in vitamin K epoxide reductase complex 1 (VKORC1) gene, in cytochrome P450 2C9 (CYP2C9) gene and in cytochrome P450 4F2 (CYP4F2) gene [3, 6, 7, 16, 21, 23, 26, 28–34].
Several GWAs analyses were performed in different populations to analyze the association of genetics with warfarin dose variations [6–10] (Supplementary Table 1). The largest GWAs analysis included 1,508 Japanese patients with the aim of finding associations with warfarin maintenance dose [8]. They found SNPs in VKORC1 gene to be the most significantly associated with warfarin maintenance dose, followed by polymorphisms in CYP2C9 and CYP4F2 genes [8].
VKORC1
Most studies highlight VKORC1 as the most important gene in warfarin pharmacogenetics [6–9, 35]. VKORC1 encodes the catalytic subunit of the vitamin K epoxide reductase complex, which reduces the inactive vitamin K to recycle it to the active form in the endoplasmic reticulum membrane [19, 36]. In the first reported systemic review and meta-analysis on the impact of polymorphisms in or near VKORC1 gene on warfarin dosage requirement, which included 19 studies and a total of 4,621 patients, the C allele in rs9934438, A allele in rs7294 and G allele in rs9923231 were associated with an increase in the daily warfarin dose requirement [28]. The heterozygous rs7294 CT and rs9923231 GA carriers required a 50% higher dose than the homozygous rs9934438 TT and rs9923231 AA. The homozygous rs9934438 CC and rs9923231 GG carriers required approximately double the dose of rs9934438 TT and rs9923231 AA. The effect of rs7294 SNP on the warfarin dose requirement was less clear. However, rs7294 A carriers required a dose approximately 30% higher than rs7294 GG carriers [28].
In three different GWAs studies, in Swedish, Japanese and African-American populations, the SNP rs9923231 upstream VKORC1 gene was also found to be significantly associated with the mean warfarin dose (Supplementary Table 1) [7–9]. The Swedish GWAs also found this SNP to be associated with over-anticoagulation (INR > 4) during the first 5 weeks of treatment (Supplementary Table 1) [7]. rs9923231 was linked to rs10871454 (associated with daily maintenance dose in the Caucasian GWAs analyses) [6, 35]. In the GWAs in African-American population [9], rs9934438 in the VKORC1 gene was also associated with a stable maintenance warfarin dose (Supplementary Table 1), considering the maintenance dose as a stable dose of three or more clinical visits. SNPs in linkage disequilibrium (LD) with rs9923231 on chromosome 16 were also significantly associated with stable warfarin dose [9].
The SNP rs749671, downstream the VKORC1 gene reached statistical significance in a GWAs study in a Brazilian population (Supplementary Table 1) [10]. The G allele of this SNP was strongly associated with a high warfarin dose requirement [10]. Other SNPs significantly associated with warfarin dose in the VKORC1 region in this GWAs were rs749670 and rs14235 [10] (Supplementary Table 1).
We have searched the most significant eQTLs of the intragenic and intergenic SNPs attributed to regulate VKORC1 in the referenced paper. We have found that all these SNPs have eQTLs in VKORC1 expressed in whole blood, among other tissues (Table 1). These SNPs described to regulate VKORC1 have also eQTLs in KAT8, expressed in arteries (Table 1).
CYP2C9
The CYP2C9 gene encodes a member of the superfamily of the cytochrome P450 enzymes important in drug metabolism and lipids synthesis. CYP2C9 enzyme is relevant in the metabolism of almost all the S-warfarin in liver [26, 37]. The most described variants functionally associated with warfarin dosage are CYP2C9*2 (rs1799853, R144C) and CYP2C9*3 (rs1057910, I359L), which are associated with lower warfarin dose requirements [26]. In a systemic review and meta-analysis including almost 8,000 genotyped individuals, it was reported that the heterozygous genotypes *1*2, *1*3, *2*3 required 19,6%, 33,7% and 56,7%, respectively, lower doses compared to the wild type (WT) genotype *1*1. The homozygous genotypes *2*2 and *3*3 required 36% and 78,1% lower doses compared to *1*1 genotype to achieve a stable INR [26]. These genotypes were more influential in individuals without interactions with other drugs [26]. CYP2C9 variants have an impact on different aspects in warfarin pharmacokinetics. Patients with CYP2C9*2 and CYP2C9*3 variants require a lower maintenance dose, more time to reach a stabilized dose (mean of 95 days) and have a higher risk of major bleeding compared to patients without these variants. Moreover, these variants are associated with the ratio of above-range INRs [23, 33].
Different GWAs have found other significant variants in CYP2C9 associated with mean warfarin dose. A GWAs study in a Swedish population confirmed previous results [38] about the significant association between the SNP rs4917639 in CYP2C9 and the mean warfarin dose given to a patient (Supplementary Table 1). This SNP was in almost complete LD with the composite of CYP2C9*2 and CYP2C9*3 variants associated with lower warfarin dose requirements [7].
Another GWAs study in Japanese population identified the SNP rs10509680 in the CYP2C9 gene significantly associated with maintenance dosage of warfarin. The GG genotype of this polymorphism was associated with higher therapeutic dose requirement [8].
Finally, in a GWAs study performed in Brazil, rs4918798 and rs9332238 (the G allele) in or near CYP2C9 were also associated with high warfarin dose. Rs9332238 and rs4918798, the latter to a lesser degree, were in almost complete LD with CYP2C9*2 and CYP2C9*3 [10] (Supplementary Table 1).
CYP4F2
CYP4F2 is another member of the cytochrome P450 enzymes superfamily. This enzyme is a vitamin K1 oxidase important in vitamin K metabolism [19, 30, 39].
In two GWAs, the variant rs2108622 CYP4F2*3 (1297G>A) in the CYP4F2 gene was associated with warfarin dose [7, 8] (Supplementary Table 1). This variant was associated with a higher warfarin dose requirement. This mutation affects the metabolizing function of CYP4F2 over vitamin K [30]. The cytochrome encoded by the gene CYP4F2 inhibits vitamin E by hydroxylation of its tocopherol phytyl side chain. This side chain is similar to the side chain of vitamins from the vitamin K group. Thus, it is possible that this cytochrome also inhibits vitamin K, and thereby the activation of vitamin K dependent coagulation factors is reduced when the enzyme works normally [11, 40]. Patients with the CYP4F2*3 allele had higher levels of vitamin K1 in the liver and needed an increase of 1–2.5 mg/day in the warfarin dose to achieve proper anticoagulant effect [30].
In relation with ethnic origin, it has been observed that 50% of African-Americans have one of the above described variants in CYP2C9, VKORC1 or CYP4F2, whereas this percentage increases up to 90% in Asians, Caucasians, Hispanics and Ashkenazi Jewish populations [30].
Another GWAs has recently identified two SNPs in NEDD4 and 2kb downstream DDHD (rs2288344 and rs17126068, respectively) associated with warfarin dose. In this GWAs, two SNPs in ASPH (rs4379440 and rs17791091) were associated with time in therapeutic range [35] (Supplementary Table 1).
Implications of warfarin pharmacogenetics
As a consequence of the studies published in relation to the pharmacogenomics of warfarin, in August 2007 the US Food and Drug Administration (FDA) updated the warfarin labelling to incorporate information on the genotyping of VKORC1 and CYP2C9 to better adjust the warfarin dose [7, 28]. However, in April 2009, the Centers for Medicare and Medicaid Services (CMS) decided not to routinely pay for the genotyping because they considered there had not been enough evidence supporting the improvement of this genetic test in patient’s health. CMS decided to pay only the genetic test in patients enrolled in specifically designed post-marketing clinical trials [41].
Another consequence of the different pharmacogenetic studies on warfarin was the creation of the International Warfarin Pharmacogenetics Consortium (IWPC) to combine the efforts of warfarin pharmacogenetics researchers. The IWPC included 22 research groups from 11 different countries with total data from more than 5,700 patients treated with warfarin [42].
The described variants in VKORC1 and CYP2C9 and the non-genetic factors explain roughly 50% of the interindividual variance in warfarin dose requirement [30]. Thus, around 50% of the warfarin dose variance remains unexplained. Specifically, VKORC1 polymorphisms explain about 27% (ranging from 15% to 34%) of the variance in the stabilized warfarin dose, whereas the CYP2C9 polymorphisms contribute on average 12% (ranging from 4 to 20%) [29]. Furthermore, polymorphism in CYP4F2 (rs2108622) explains roughly 1.5% of warfarin dose variation [7].
Many algorithms have been designed including genetic and non-genetic factors to attempt to predict the appropriate initial or daily stable warfarin dose [16, 21, 42–52].
Gage et al. designed a pharmacogenetic algorithm (PA) using stepwise regression in a multicenter study cohort. They used an additive genetic model, numbering the variant alleles at each locus from 0 to 2. The variables included were weighted according to their ratio of variance on the therapeutic warfarin dose. They tested the effect of several clinical and demographic variables and only retained variables that were significantly independent predictors of warfarin dose in the derivation cohort (N=1,015 patients). The formula was analyzed prospectively in 292 patients from the validation cohort [21]. The pharmacogenetic equation included the 1639G>A VKORC1 polymorphism, CYP2C9 *2 and *3 polymorphisms and physiological and clinical factors such as body surface area, age, target INR, amiodarone use, smoker status, African-American race and current thrombosis. This equation explained 53-54% of the warfarin dose variance. When the genetic factors were not included, the equation explained 17-21% of the warfarin dose variability and had higher prediction error [21]. In patients from the validation cohort, the warfarin dose was prescribed prospectively using the PA or an almost equal preliminary version of this algorithm. These patients took a first warfarin dose without accounting for CYP2C9*2 and CYP2C9*3 alleles. During the 30-day follow-up, 2 patients had a major hemorrhage or PE and 3 patients had symptomatic DVT. They demonstrated the feasibility and safety of the pharmacogenetics algorithm [21].
Recently, two prospective randomized trials were designed to compare patients with initial warfarin dose based on PA or clinical decision [45, 46]. In one of the studies, patients were randomized receiving warfarin by a validated algorithm (96 patients) [53] or CYP2C9 genotype-adjusted algorithm (95 patients) [46]. They found that the inclusion of CYP2C9 polymorphisms information decreased minor bleedings and improved the INR control, reaching the first therapeutic INR and stable anticoagulation earlier [46]. In the other study, 200 patients were randomized (101 patients treated using PA and 99 using standard clinical decision). They received warfarin and had at least 1 follow-up INR to prospectively validate the pharmacogenetics-guided dosing formula (determined by a regression equation) [45]. The primary endpoint was the percentage of out-of-range INRs. They did not find significant differences in the ratio of patients with out-of-range INR. However, PA more accurately predicted the individual dose when the patients were divided into subsets: patients without allele variants (WT), patients with a single allele variant and patients with multiple allele variants. The PA was better than the standard decision for predicting the higher average warfarin dose in WT and the lower dose in patients with multiple allele variants. Despite not being statistically significant, total adverse events were lower in patients with dose predicted based on PA than on standard decision [45].
Pharmacogenetic refinement algorithms have been developed to validate whether genotype can refine maintenance dose of warfarin after some days of therapy. One study derived clinical and pharmacogenetics refinement algorithms using INR values on day 4 or 5, clinical factors quantified using stepwise selection and genotype [16]. Variables which achieved statistical significance in the multivariable linear regression model and thereby were maintained were: INR, A allele of the 1639G>A polymorphism in VKORC1, CYP2C9*2 and CYP2C9*3 alleles, prior warfarin dose, age, BSA, stroke, diabetes, race, target INR and use of amiodarone or fluvastatin. The clinical refinement algorithm was similar to the pharmacogenetics algorithm but without the inclusion of genotype and race [16]. In the derivation cohort (N=969), the pharmacogenetics algorithm explained 63% of variation (R2), whereas the clinical algorithm had an R2 of 48%. The PA was tested in 204 patients of the internal validation cohort who had INR available on day 4th of therapy. The R2 was 58%, whereas the R2 for clinical algorithm was 43% [16]. Algorithms were also tested in another validation cohort of 105 patients with INR values measured on day 5th. In this case, the R2 for the PA was 60%, whereas the R2 for the clinical algorithm was 44% [16]. Similar results were obtained when final algorithms were validated in an external validation cohort (N=517 patients with INR measurements on day 4th of therapy; N=438 patients with INR measurements on day 5th of therapy). The authors concluded that PAs were more accurate than clinical algorithms for predicting the maintenance dose of warfarin [16].
PAs developed to predict the initial therapeutic dose of warfarin have shortcomings [21, 45, 46]. They do not indicate how to dose warfarin when the INR response to the therapy is known. Furthermore, in some occasions the time to obtain the genotyping results is too longer. Some experts have argued that when the genotyping of VKORC1 and CYP2C9 will be available in practice they may be not relevant or cost-effective. The initiation algorithms have been developed in small populations, with some exception. Thus, the predictive value of these algorithms in large populations could be different [16]. Hence, a large controlled, multicentre and randomized trial seems necessary to quantify the effect of pharmacogenetics algorithms in INR control and in the occurrence of adverse events [21]. In addition, for PA to be used in clinical practice, it is necessary to include the pharmacogenetics technology and knowledge in the clinical infrastructure [34].
The International Warfarin Dose-Refinement (Warfarin DR) Collaboration was created to develop and validate a pharmacogenetics refinement algorithm in an international cohort of patients and determine whether the genotyping predicts the therapeutic dose, also when the INR value is available in the 4th or 5th day after the therapy initiation [16].
ACENOCOUMAROL
Acenocoumarol is a racemic mixture comprised by S and R enantiomers. S-acenocoumarol is the most potent enantiomer and it is metabolized by the CYP2C9 enzyme, while R-acenocoumarol is metabolized by CYP1A2, CYP3A4, CYP2C9 and CYP2C19. The half-life of S-acenocoumarol is 1.8 hours and the half-life of R-acenocoumarol is 6.6 hours [14]. The slower elimination of the R-enantiomer makes R-acenocoumarol responsible for the anticoagulant effect of this drug. R-acenocoumarol is the clinically most important enantiomer due to the short half-life of S-acenocoumarol [27].
Administration of acenocoumarol is also limited by the narrow therapeutic/toxic profile of this drug and the interindividual and interethnic dose variation. Environmental and genetics factors influence in acenocoumarol dose variation [54].
Pharmacogenetics
Variations in VKORC1, CYP2C9 and CYP4F2 are associated with changes on stabilized acenocoumarol dosage. VKORC1 and CYP2C9 genetic variants also correlate with the maintenance dose, the first INR after the initial standard dose, the time until the stable dose is achieved, the time until the correct therapeutic rank is achieved and the ratio of bleeding events of acenocoumarol [11].
Only one GWAs study has been performed on acenocoumarol pharmacogenetics. A large population-based cohort of 1,451 Caucasian patients from the Rotterdam study and 287 individuals from the extended Rotterdam cohort in the replication were analyzed [11].
Near VKORC1 gene, the SNP rs10871454 in the Syntaxin-4 (STX4) gene was significantly associated with acenocoumarol dose variance in this GWAs and subsequently, replicated [11] (Supplementary Table 1). The authors suggested this polymorphism might decrease the hepatic expression of VKORC1 mRNA, decreasing the amount of the drug target [11]. Furthermore, this SNP was in complete LD with the rs9934438 SNP on the VKORC1 gene that is one of the polymorphisms associated with the dose variation in warfarin treated patients [11]. However, other roles are plausible to explain the association of the SNP in STX4 and the acenocoumarol dose variance. The protein encoded by STX4 is a SNARE molecule and SNAREs are involved in endothelial and other cells, such as platelets, secretion. However, its role in platelet exocytosis varies depending on secretary granules types, but appears to be essential for lysosomal release in platelets [55, 56]. Endothelial exocytosis is relevant in thrombosis, hemostasis and inflammation [57, 58].
Regarding CYP2C9 gene, in this GWAs, the SNP rs4086116 was associated with interindividual variation on stabilized acenocoumarol dosage.
In the same study, the polymorphism rs2108622 in CYP4F2 was also associated with the acenocoumarol dose when the significant SNPs of VKORC1 and CYP2C9 were included in the analysis as covariates [11] (Supplementary Table 1). It has been postulated that the T allele of the rs2108622 variant decreased the enzyme activity, preventing vitamin K inactivation. Consequently, patients with one CYP4F2 allele variant need an increment of the dose of about 1mg/week per allele [40].
Polymorphisms within and flanking CYP2C18 and CYP2C19 gene were also associated with acenocoumarol maintenance dose and replicated in the replication stage [11].
The combination of the CYP2C9*3 genotype and the polymorphisms in 1639G>A or 1173C>T in the VKORC1 gene explained around 50% of the interindividual variability on the anticoagulation effect of acenocoumarol [54]. In addition, the combination of variants in CYP2C9 and VKORC1 was strongly associated with severe over-anticoagulation [27]. The addition of the polymorphism rs2108622 (in CYP4F2) increased the r2 adjusted of the clinical and genetic (CYP2C9 and VKORC1) model for acenocoumarol dosage variation by 1.3%, while the addition of the polymorphism in CYP2C18 (rs1998591) increased the r2 adjusted by 1.2% [11].
Implications of acenocoumarol pharmacogenetics
Different acenocoumarol pharmacogenetic-guided dosing algorithms have been derived in different cohorts from different populations [17, 59–67]. An observational retrospective study analyzed 8 different acenocoumarol pharmacogenetic algorithms in a cohort of 189 patients [65]. The algorithm which achieved similar doses to the real stable doses was the EU-PACT algorithm [17]. However, considering the patients with over- or under-estimation of the dose and patients correctly classified (deviation from the actual stable dose of ≤20%), the algorithm which classified most patients correctly was the Borobia algorithm [59]. This algorithm correctly classified 40.7% of the 189 patients included [65]. However, an algorithm correctly classifying this percentage of patients remains far from being used in clinical practice.
PHENPROCOUMON
Phenprocoumon is another coumarin derivate from the VKA family which is also found as a racemic mixture. The S-phenprocoumon is the most potent enantiomer. The half-life of phenprocoumon is 110-130h for the S-enantiomer and 110-125h for the R-enantiomer [14]. Given the long half-life of phenprocoumon, over-anticoagulated patients under this treatment have higher risk of major bleeding [27].
Phenprocoumon is metabolized by CYP3A4 enzyme, which is a non-polymorphic cytochrome [14]. The main role of CYP3A4 in the metabolism of phenprocoumon makes it safer than acenocoumarol and warfarin. 60% of oral phenprocoumon is metabolized, whereas 40% is excreted unchanged. Thus, phenprocoumon could be a better option for those patients that metabolise coumarin poorly [27].
Despite having a narrow therapeutic/toxic profile, patients treated with phenprocoumon achieve more stable INR measurements and require less monitoring [27].
Pharmacogenetics
It has been suggested than phenprocoumon metabolism is less influenced by the CYP2C9 genotype, with VKORC1 having greater relevance. It has been shown that patients with a CYP2C9 variant, and without VKORC1 variant alleles, required a 30% lower dose than WT patients. Patients with CYP2C9 and VKORC1 allele variants have an increased risk of over-anticoagulation. CYP2C9 genotype is also associated with delayed phenprocoumon stabilization [27].
A gene candidate study was performed with VKORC1, CYP2C9 and CYP4F2 genes. The authors found that an allele variant in VKORC1 decreased the maintenance dose of phenprocoumon by 4.8 mg/week. An allele variant in CYP2C9 also decreased the dose by 2.2 mg/week and an allele variant in CYP4F2 increased the dose by 1.5 mg/week. They generated a clinical-genetic model including age, sex, BMI, target INR, VKORC1, CYP2C9 and CYP4F2 genotypes, explaining the 46% of the maintenance dose [68].
There is a GWAs study with 202 patients treated with phenprocoumon. The authors found 32 SNPs in chromosome 16 (within or flanking VKORC1 gene) significantly associated with phenprocoumon dose. The stronger associations were for the SNPs rs10871454 and rs11150604 (Supplementary Table 1). Both SNPs were in complete LD and were associated with a decrease in the dose. Both SNPs were found replicated in the validation cohort (n=42). Four additional SNPs nominally associated with phenprocoumon dose in chromosome 9 were found. However, they were not replicated [68] (Supplementary Table 1).
FLUINDIONE
Fluindione is an inandione derivate widely used in France (about the 80% of OA prescription in this country) [69]. Unlike coumarin derivates, fluindione is not found as a racemic mixture [70].
Knowledge of the pharmacokinetics of fluindione is scarce. An intermediate half-life has been described for fluindione, similar to the half-life of the more potent enantiomer of warfarin [18]. One study described a fluindione half-life of 31h [71], while another found a median half-life of 69h [70]. Fluindione is less often displaced by other drugs in their receptors and it has higher affinity to albumin than warfarin [70]. The role of CYP2C9 is unknown in the fluindione metabolism [72].
The efficacy and, thereby, the dose of fluindione, varies among patients depending on environmental and genetic factors. The individual fluindione dose varies between 5 and 40 mg per day [18, 73, 74]. The interindividual variability of this drug is associated with the same adverse events as with the other VKAs, mainly thromboembolic events and bleedings.
Pharmacogenetics
No GWAs study was performed in fluindione treated patients. Only some candidate gene analyses have evaluated the genes that could be implicated in the interindividual variability of fluindione. However, it would be necessary to perform GWAs in fluindione to find reliable associations.
In a study including 465 patients with a venous thromboembolic event, several polymorphisms in VKORC1, CYP2C9, CYP4F2 and EPHX1 genes were analyzed [72]. The C1173T polymorphism in VKORC1 gene was associated with different outcomes: the risk of a first INR measure ≥ 2, the mean time to achieve an INR measure in therapeutic range (2-3), the time to have a first INR > 4 (over-anticoagulation). The target dose of fluindione was also associated with the C1173T polymorphism. Patients with a T allele in this polymorphism were more sensitive to fluindione, achieving the first INR in therapeutic range earlier, with an increased risk of over-anticoagulation (achieving a INR value > 4 before) and with a decrease of more than a half the dose of patients with CC genotype [72]. However, these results were not replicated in a validation cohort.
Another study analyzed different variables which could be influencing the fluindione clearance in 24 healthy white patients. The CYP2C9*2 and *3 genotype, VKORC1 1173 C>T genotype, CYP1A2 phenotype and body weight were found to be predictors of the fluindione pharmacodynamics and pharmacokinetics [18].
To predict the fluindione dose in an elderly population, one study analyzed 13 polymorphisms in 7 genes (VKORC1, CYP4F2, EPHX1, CYP2C9, CYP2C19, CYP3A5 and ABCB1) potentially involved in the pharmacological effect or in the metabolism/transport of fluindione in 156 patients. The variables body weight, amiodarone intake, VKORC1, CYP4F2 and ABCB1 genotypes were included in a prediction model. This model explained 31,5% of the dose variability and the accuracy in the prediction of the dose within 5mg per day of fluindione was 89,7%. The model was validated in 74 patients, obtaining the correct dosing within 5mg per day in 83,3% of the patients. Patients with a variant allele 2kb upstream VKORC1 (rs9923231) and patients with a variant allele in ABCB1 2677 G>T/A required lower doses than WT patients. Interestingly, comparing with coumarin derivates, CYP2C9 was not associated with fluindione maintenance dose, suggesting the possible use of fluindione in patients hypersensitive to coumarins [74].
There are some differences among the different VKAs. They have different pharmacokinetics, such as different half-life [14]. In pharmacogenetics, there are also some differences in the variants associated with dose variation of the different VKAs. Only in the case of acenocoumarol, polymorphisms flanking CYP2C18 were identified associated with the acenocoumarol variation dose [27]. Furthermore, CYP2C9*2 has different effect in patients treated with acenocoumarol or warfarin. While CYP2C9*2 allele decreases the warfarin maintenance dose, this variant does not affect the acenocoumarol dose [27]. In addition, the CYP2C9 genotype seems to be less relevant for the phenprocoumon and fluindione dose variation than for the other VKAs [27, 74]. Moreover, CYP1A2 appears to have a more important role for fluindione than for the other VKAs [18].
DIRECT ORAL ANTICOAGULANTS (DOACS)
Nowadays, 4 DOACs have been developed to try to overcome the drawbacks of VKAs. Rivaroxaban, apixaban and edoxaban are direct oral factor Xa inhibitors, whereas dabigatran is a direct oral thrombin inhibitor [75] (Figure 2B).
DOACs are taken in fixed doses and the coagulation status does not need to be monitored periodically [12]. Furthermore, it has been observed that DOACs have fewer interactions with other drugs such as digoxin, aspirin or nonsteroidal anti-inflammatory drugs or with food [76].
All DOACs have been shown to be safe and effective alternatives to warfarin [77].
Different reviews and meta-analysis have analyzed data from real-world studies, registries and databases in patients with non-valvular AF. In general, they concluded that rivaroxaban is associated with lower rates of ischemic stroke or systemic embolism compared to warfarin [77]. The initiation of rivaroxaban is associated with higher risk of major bleeding compared to apixaban [78] but with lower risk of intracranial bleeding compared to warfarin [79].
Apixaban and dabigatran are associated with lower ratio of major bleeding compared to warfarin [77] and both, apixaban and dabigatran, have similar major bleeding rates [78]. They are also associated with lower ratio of intracranial bleeding compared to warfarin [79, 80]. Both apixaban and dabigatran are associated with lower death rates compared to warfarin [77].
In general, DOACs are similar to warfarin preventing ischemic stroke [77, 80], however with a lower risk of bleeding [77].
DABIGATRAN
Dabigatran is approved by the FDA and European Medicines Agency (EMA) and indicated for reducing the risk of stroke and systemic embolism in patients with NVAF [1, 2].
Dabigatran is a reversible direct thrombin inhibitor effectively completely converted by the liver esterase CES1 from the oral prodrug dabigatran etexilate to the active drug [12, 81, 82]. Dabigatran etexilate is a substrate of the P-glycoprotein intestinal efflux transporter (encoded by ABCB1 gene), an efflux pump for xenobiotics. Dabigatran is excreted predominantly by renal pathway (80%) [83]. Maximum plasma concentrations of dabigatran are achieved at 1 to 3 hours after the dose intake [12].
Recently, idarucizumab, a reversal agent for dabigatran action was approved by the FDA and the EMA to treat dabigatran patients with any bleeding or requiring emergency surgery [84].
Several real-world studies with dabigatran have been performed to determine its effectiveness in clinical practice. A meta-analysis of 20 observational real-world studies comparing patients with NVAF treated with dabigatran and with VKA found the incidence of ischemic stroke, major bleeding and mortality was lower for dabigatran than for VKA. However, the risk of gastrointestinal (GI) bleeding was higher for the patients treated with dabigatran (Supplementary Table 2) [85].
Another meta-analysis including 7 observational real-world studies found similar results (Supplementary Table 2). However, they observed a similar rate of stroke between patients treated with dabigatran and patients treated with warfarin. Furthermore, the higher ratio for GI bleeding was potentiated in the elderly subgroup (≥75 years). They included two studies which analyzed the GI risk in patients using dabigatran 110 mg. One of these studies found a similar risk compared with warfarin in patients < 75 years and a higher risk for dabigatran in patients ≥75 years. The other study found lower risk of GI for dabigatran 110 mg compared to warfarin in patients < 75 years. In general, the bleeding outcomes from this meta-analysis were in concordance with the ones in the RE-LY phase III clinical trial [80].
Other datasets included patients with NVAF treated with dabigatran. All of them observed lower risk for major bleeding, intracranial bleeding and death for patients treated with dabigatran compared to patients treated with warfarin (Supplementary Table 2). These associations were stronger for dabigatran 150 mg. Risk of major GI bleeding was similar or higher for patients with dabigatran treatment compared to warfarin. Risk of ischemic stroke was lower or similar between both drugs (Supplementary Table 2) [86–90]. The Danish registry compared the bleeding rates of VKA naïve patients and patients in treatment with VKA switched to dabigatran. They observed that warfarin starters had the highest bleeding rates. Dabigatran 110 mg switchers from VKA had higher rates of bleeding compared to patients continuing warfarin treatment [88–90].
Dabigatran 75 mg was approved in the USA for patients with renal impairment. In an observational study, it was observed that the ratio for stroke, mortality and bleeding was similar in patients treated with dabigatran 75 mg and in patients treated with warfarin. However, the ratio for intracranial bleeding was lower for dabigatran patients. However, most of patients included in this study treated with dabigatran 75 mg did not have renal impairment, so the prescription of this drug was off-label [91, 92].
Dabigatran, like the other DOACs, is taken in a fixed dose. However, there are interindividual differences between patients in blood concentration (estimation of 30% variation for systemic exposure) [12].
The unique GWAs with patients treated with DOACs was developed to identify variants associated with interindividual variability in dabigatran etexilate blood concentration. 1,694 patients of white European ancestry from the RE-LY study treated with dabigatran were analyzed. The polymorphism rs2244613 in the esterase gene CES1 was associated with trough concentrations (each minor allele was associated with 15% decrease in trough concentrations) and with lower risk of any bleeding. The polymorphisms rs4148738 in the ABCB1 gene and rs8192935 in CES1 gene were associated with peak concentrations but not with clinical outcome [12]. However, the results were not analyzed in a replication cohort. Importantly, both genes (ABCB1 and CES1) encode for proteins related to dabigatran pharmacokinetics, which means that variants in genes related to dabigatran absorption and metabolism explain part of the variations in dabigatran concentrations between individuals [12].
Another study in 92 patients with AF has recently evaluated the effect of the three polymorphisms found in the dabigatran GWAs analysis (rs2244613 and rs8192935 in CES1 gene and rs4148738 in ABCB1 gene) to analyze the influence of these polymorphisms on the interindividual variation in dabigatran concentration. They found significant association of the SNP rs8192935 in CES1 gene with dabigatran trough concentration [93].
It is relevant that VKAs and dabigatran have different metabolic pathways. These differences are also observed in pharmacogenetics studies: different polymorphisms in different genes are associated with metabolic variance for each drug. Consequently, future drug decision could be performed in those patients based on their genetic background.
RIVAROXABAN
Rivaroxaban is a direct oral inhibitor of the Xa factor approved by EMA and FDA, indicated for prevention of stroke and systemic embolism in patients with non-valvular AF [1, 2].
Rivaroxaban does not require coagulation monitoring. The pharmacokinetics of rivaroxaban is dose-proportional and it has high oral bioavailability and maximum plasma concentrations at 3-4 hours after drug intake, which can be affected by the co-administration of food [83]. Rivaroxaban is not recommended in patients with moderate or severe hepatic impairment, as the clearance of rivaroxaban is decreased [83]. Rivaroxaban exhibits little interindividual variability based on age, gender and body weight [94].
This drug is a substrate of the P glycoprotein and is metabolized through CYP3A4/5 and CYP2J2 (two-thirds of rivaroxaban) and CYP independent mechanisms [82, 83, 94].
Approximately 1/3 of unchanged rivaroxaban is eliminated by the kidneys. The other 2/3 of rivaroxaban is metabolized by the liver [95].
There are different studies, registries and databases describing the use of rivaroxaban in real-world for patients with AF [96–103].
In a meta-analysis of 9 studies [96], the results obtained (Supplementary Table 2) were in concordance with the results from the ROCKET-AF clinical trial, confirming the benefit-risk profile of rivaroxaban [97, 99].
The Xantus study, a prospective, observational, phase IV analysis, observed that patients with higher CHADS2 and CHA2DS2-VASc (a scale for the risk of stroke and systemic embolism) had higher rates of stroke and systemic embolism, major bleeding and death [99, 100].
Different registries and datasets have found results which were similar to the ROCKET-AF phase III clinical trial observations [98, 99, 101] (Supplementary Table 2). In general, the ratio of intracranial bleeding and ischemic stroke were lower for rivaroxaban compared to VKAs [99, 102, 103] (Supplementary Table 2).
There are no GWAs studies performed in patients treated with rivaroxaban. It is possible that polymorphisms in genes involved in the metabolism of the drug could be associated with rivaroxaban blood levels, similar to the results found with warfarin, acenocoumarol and dabigatran. Taking into consideration the proteins associated with the absorption (P-glycoprotein) and metabolism (CYP enzymes) of rivaroxaban, polymorphisms in ABCB1 gene or CYP genes could be associated with rivaroxaban concentration in blood.
APIXABAN
Apixaban, like rivaroxaban, is a direct oral inhibitor of the Xa factor. Apixaban is indicated by the EMA and FDA for reducing the risk of stroke and systemic embolism in patients with non-valvular AF. It is used in patients with risk factors (previous stroke, high blood pressure, diabetes, heart failure or being ≥75 years old) [1, 2].
The bioavailability of apixaban is around 50% and it reaches the maximum concentration 1-3 hours after intake. Its half-life is between 8 and 15 hours. Roughly 25% of the drug is excreted by the kidneys [104]. Apixaban is also a substrate for the P-glycoprotein transporter and mainly for CYP3A4/5 [83] but also CYP 1A2, 2C8, 2C9, 2C19, 2J2 [94].
A genetic study on the effect of rs4148738 polymorphism in ABCB1 gene in apixaban concentrations (the same polymorphism of the dabigatran GWAs) showed an association with apixaban peak concentrations [105]. However, it would be necessary to perform GWAs to really assess which genes are important for the interindividual variability.
Currently, there are no GWAs studies in patients treated with apixaban. Taking into account the proteins associated with the metabolism of rivaroxaban, it is possible that polymorphisms in the ABCB1 gene or CYP genes which metabolize apixaban (CYP3A4/5, CYP 1A2, 2C8, 2C9, 2C19 and 2J2) could be associated with rivaroxaban blood levels, similar to the results found with warfarin, acenocoumarol or dabigatran.
EDOXABAN
Edoxaban, such as rivaroxaban and apixaban, is a direct oral inhibitor of factor Xa indicated by the FDA for reducing the risk of stroke and systemic embolism in patients with non-valvular AF [2]. EMA has also approved edoxaban to prevent stroke and systemic embolism in patients with non-valvular AF, but in this case only in patients who have one or more risk factors (previous stroke, high blood pressure, diabetes, heart failure or being ≥ 75 years old) [1].
Edoxaban achieves maximum plasmatic concentrations within 1 to 2 hours after being taken. It has predictable pharmacokinetic profile and 62% oral bioavailability. Edoxaban, as the other DOACs, is also a substrate of the P-glycoprotein. It is metabolized by hydrolysis through a carboxylesterase (CES) and oxidized by the CYP enzyme CYP3A4 [82]. Some 50% of edoxaban is excreted by kidneys [106].
A meta-analysis of 24 real-world studies included patients with AF to analyzed patients treated with a daily dose of edoxaban 30 mg or 60 mg versus placebo (no treatment), aspirin and aspirin plus clopidogrel. They observed how treatment with edoxaban 30 mg decreased the risk for all stroke, ischemic stroke and mortality. Patients treated with edoxaban 30 mg versus patients treated with aspirin plus clopidogrel had a lower risk of intracranial hemorrhage. Edoxaban 60 mg reduced the risk of any stroke and systemic embolism versus placebo, aspirin and aspirin plus clopidogrel. Thus, both edoxaban doses had a positive net clinical benefit compared to antiplatelet treatment or no treatment in the real world [107].
Another study with patients from the Danish nationwide cohort tested the hypothesis that edoxaban had a net clinical benefit (NCB), which is the balance between systemic embolism and intracerebral bleeding, superior to warfarin. Comparing without treatment, warfarin had a NCB of 0.26 prevented events per 100 patients-years, edoxaban 60 mg had a NCB of 0.71 per 100 patients-years and edoxaban 30 mg had a NCB of 0.71 per 100 patients-years. At all CHADS2 and CHA2DS2-VASc score, both edoxaban doses had higher NCB than warfarin. At CHA2DS2-VASc scores 0 and 1, warfarin had no positive NCB compared to no treatment. In patients with CHADS2 and CHA2DS2-VASc score ≥ 2, edoxaban 60 mg had a better NCB than edoxaban 30 mg and warfarin. At CHA2DS2-VASc scores 0 and 1, edoxaban 30 mg had superior NCB than edoxaban 60 mg. In patients with HAS-BLED score ≤ 2 (a scale for the risk of major bleeding), both warfarin doses had higher NCB compared to warfarin independently of the CHADS2 and CHA2DS2-VASc scores. In patients with a HAS-BLED score ≥ 3, both edoxaban doses had positive NCB compared to warfarin, in patients with CHADS2 and CHA2DS2-VASc score ≥ 2 [108].
One in vitro study was performed with the purpose of determining if two polymorphisms in factor Xa (Ala152Thr and Gly192Arg) were affecting the edoxaban activity. The authors concluded that these mutations do not account for the interindividual variability of edoxaban [109].
There are no GWAs studies performed in patients treated with edoxaban. Studies analyzing the role of ABCB1 gene, or genes involved in the edoxaban metabolism (CYP3A4 or CES such as dabigatran), and the response to edoxaban could be interesting.
CONCLUSIONS
Variants in CYP2C9, VKORC1 and CYP4F2 are associated with VKAs dose variation between individuals. However, these variants together with non-genetic factors explain about 50% of the interindividual dose variation [7, 30]. Further analyses with more patients are necessary to find other genetic variations associated with dose variations or with vascular events and dosing treatment. However, other mechanisms could explain the remaining percentage of variation, such as rare mutations, epigenetics mechanisms and environmental factors. To find rare mutations, it would be necessary to perform exome sequencing or whole genome sequencing as was done for statin drugs [7]. For epigenetics, some mechanisms such as DNA methylation could be implicated in OAs dose variation. Some studies have found how DNA methylation modifications could be influencing the effects of antiplatelet drugs [110, 111].
Diverse pharmacogenetics algorithms have been developed, including polymorphisms in CYP2C9 and VKORC1 together with clinical variables to predict the initial dose of warfarin. The different PAs improved the accuracy and efficiency of warfarin dose initiation. However, the results are controversial in the reduction of out-of-range INRs. Some studies observed that genotyping variants in CYP2C9 improves INR control [46], whereas other studies did not find the usefulness of these algorithms to control INR [21, 45]. Although the percentage of out-of-range INRs is not statistically significant among patients treated according to PA or clinical/standard decision in these studies, PA more accurately predicted the individual dose depending on the subset of patients (WT, with one single allele variant, with multiple allele variants). The percentage of each of these subsets of patients could differ between studies and cause the mentioned differences among studies. Moreover, VKORC1 have not been genotyped in all studies and could modify the results and conclusions. Probably larger number of patients are necessary to achieve higher statistically power to observe differences among PA and clinical algorithms. Moreover, the genetic variants identified to date explain only a proportion of the drug response, thus more genetic variants can contribute to increasing the percentage of drug variability and may be included in pharmacogenetics algorithms [34].
To improve the main shortcomings of VKA, mainly the frequent monitoring, DOACs have been approved for the prevention of stroke recurrence and systemic embolism in patients having suffered a previous stroke with non-valvular AF, to treat DVT or PE. DOACs have a better efficacy-safety profile compared to warfarin (Daiichi Sankyo Europe GmbH 2014). Patients treated with DOACs have a lower ratio of bleeding events (rivaroxaban, apixaban and edoxaban) and life-threatening bleeding (dabigatran). Furthermore, the use of DOACs reduces intracranial bleeding compared to warfarin. In patients with AF, the results from the different pivotal clinical trials suggest that DOACs improve the outcomes of these patients [15]. Altogether, the information on the four DOACs from the clinical trials shows that they are not inferior to warfarin in terms of efficacy, although they seem to reduce bleeding events, improving their safety. However, DOACs have greater acquisition costs. Thus, greater cost versus greater efficacy/safety profile and avoidance of monitoring have to be balanced [112]. A review of the use of DOACs in an elderly population with non-valvular AF demonstrated that they are beneficial and lead to a reduction of stroke risk in this population [76].
Due to the lack of GWAs with DOACs (only one) [12], it would be important to perform pharmacogenetic studies based on a GWAs approach. It is possible that some genes could be associated with the metabolism of all or almost all DOACs, such as ABCB1, which is involved in the transport of all DOACs, or CYP3A4, involved in the metabolism of all DOACs with the exception of dabigatran [82, 94]. Furthermore, it could be interesting to assess whether there are polymorphisms associated with the response to the different DOACs in terms of efficacy (risk of stroke or systemic embolism) and safety (risk of bleeding).
Pharmacogenetics could be useful to identify the more appropriate anticoagulant for each specific patient, avoiding adverse events such as any bleeding or stroke recurrence. VKAs and DOACs do not share the same mechanism of action. This fact distinguishes both kinds of anticoagulant, meaning that VKAs could be more suitable for one individual but DOACs for another. Moreover, VKAs and dabigatran have different metabolic pathways and as different polymorphisms affect metabolism of both kind of drugs, knowing the specific genotype of each individual before starting anticoagulant treatment could help avoid adverse events. For instance, a sub-study of the ENGAGE-AF-TIMI 48 trial demonstrated that patients with the CYP2C9 and VKORC1 variants can benefit more from edoxaban compared to warfarin [113, 114]. Therefore, a personalized medicine for oral anticoagulants use is possible based on the fact that DOACs and VKAs involve different metabolic pathways and GWA studies confirm that different genetic risk factors are associated with the response of both types of drug.
FUTURE
It is expected that around year 2020 the results of large randomized clinical trials on the incorporation of pharmacogenetics algorithms to guide VKAs dosage will become available. Furthermore, by this time, it is likely that genotyping will be quickly and more cost-effective. It is expected that the use of pharmacogenetics algorithms for vitamin K inhibitors will reduce adverse reactions to these drugs [3].
Pharmacogenetic studies with DOACs would help to choose the most appropriate anticoagulant treatment for each patient. Thus, pharmacogenetics studies have to be translational, to include genotyping in clinical practice and achieve a more personalized medicine, as happens with other kind of treatments, such as in cancer [115, 116].
The applicability of pharmacogenetics in clinical practice has been mainly demonstrated with cancer treatments. Nowadays, there are cancer drugs which are dosed following pharmacogenetic tests. The genotyping of thiopurine S-methyltransferase (TPMT) in acute lymphoblastic leukemia (ALL) patients before treatment with 6-mercaptopurine is useful to select the correct dose based on genetics before the first drug intake. After changing the label for 6-mercaptopurine in 2004, some hospitals routinely started to order TPMT genotyping before initiation of the treatment [117].
In advanced colorectal cancer, irinotecan is the most widely-used drug. However, polymorphisms in diphosphate-glucuronosyltransferase 1A1 (UGT1A1) have been found to be associated with the risk of developing hematological and/or digestive toxicities. Thus, dose reduction was recommended in patients with the genotype *28/*28. In United States, the FDA recommended genotyped the UGT1A1 *28 variant before irinotecan prescription. In Europe, a Dutch workgroup and the French National Thesaurus of Digestive Oncology recommended dose reduction in *28/*28 patients [118]. A genotyping test has been generated to facilitate the detection of this genetic variation UGT1A1 (Mayo Medical Laboratory) [117, 119].
Other genes associated with adverse drug events with potential benefits for use in clinical practice could be: CYP2D6 and tamoxifen for breast cancer, DPYD and fluoropyrimidine antimetabolite 5-fluorouracil (5-FU) for colon cancer, SLCO1B1 and Simvastatin for the reduction of cholesterol levels, HLA-A*33:03 and ticlopidine for secondary prevention of atherothrombosis [117, 120].
Current medicine could incorporate pharmacogenetics to offer patients an effective personalized medicine by adjusting treatment and dosage in those cases with a proven genetic association based on GWAs analysis and subsequent approval by the FDA or the European authorities. This way, adverse events related with lack or excess of drug effect could be decreased.
LIMITATIONS
This review has some limitations. First, the idea was to revise and analyze all the GWAS published for the different OAs. However, for some of them (fluindione, edoxaban, rivaroxaban and apixaban), there are no studies that have performed GWAS analysis with these drugs. In these cases, other genetic studies based on gene candidates were reviewed. Nevertheless, these kind of studies are biased, as they are based on a previous hypothesis and do not replicate the results in independent populations. Thus, their reliability is limited. It would be necessary to perform a GWAs approach to really identify genes involved in the secondary response to these drugs.
Second, for many polymorphisms found associated with the effect of different OAs is assumed that the effector gene is the closest to the mutations. However, this is not always true and in vitro or in silico functional analysis have to be performed to demonstrate the effector gene for each specific mutation.
Abbreviations
OAs: Oral anticoagulants
AF: Atrial fibrillation
GWAs: Genome Wide Association Studies
VKAs: vitamin K antagonists
DOACs: direct oral anticoagulants
NVAF: nonvalvular atrial fibrillation
DVT: deep venous thrombosis
PE: pulmonary embolism
VKORC1: vitamin K epoxide reductase complex 1
CYP2C9: cytochrome P450 2C9
CYP4F2: cytochrome P450 4F2
FDA: US Food and Drug Administration
CMS: Centers for Medicare and Medicaid Services
IWPC: International Warfarin Pharmacogenetics Consortium
PA: pharmacogenetic algorithm
WT: wild type
Warfarin DR: international Warfarin Dose-Refinement
STX4: Syntaxin-4
EMA: European Medicines Agency
GI: gastrointestinal
CES: carboxylesterase
NCB: net clinical benefit
BMI: body mass index
TPMT: thiopurine S-methyltransferase
ALL: acute lymphoblastic leukemia
TCL1A: T-cell leukemia 1A.
Author contributions
Natalia Cullell: bibliography revision and manuscript writing
Caty Carrera, Elena Muiño, Núria Torres: revision of the manuscript
Jerzy Krupinski, Israel Fernández-Cadenas: orientation and revision of the manuscript.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by a grant from the Carlos III Institute of Health (PI15/01978) and by Boeringher Ingelheim funding (SEDMAN Project). We thank the International Stroke Genetics Consortium (ISGC), the Spanish Stroke Genetics Consortium (www.genestroke.com) and the RETICS Network INVICTUS, of which The Neurovascular Pharmacogenomics and Genetics Laboratory forms part.
CONFLICTS OF INTEREST
Nothing to disclose.
REFERENCES
1. European Medicines Agency - Find medicine - European public assessment reports [Internet]. [cited 2017 Jun 19]. Available 2017 Jun 19, from http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/epar_search.jsp&mid=WC0b01ac058001d124.
2. Drugs@FDA: FDA Approved Drug Products [Internet]. [cited 2017 Jun 19]. Available 2017 Jun 19, from https://www.accessdata.fda.gov/scripts/cder/daf/.
3. Manolopoulos VG, Ragia G, Tavridou A. Pharmacogenetics of coumarinic oral anticoagulants. Pharmacogenomics. 2010; 11:493–96. https://doi.org/10.2217/pgs.10.31.
4. Amin H, Nowak RJ, Schindler JL. Cardioembolic stroke: practical considerations for patient risk management and secondary prevention. Postgrad Med. 2014; 126:55–65. https://doi.org/10.3810/pgm.2014.01.2725.
5. Zirlik A, Bode C. Vitamin K antagonists: relative strengths and weaknesses vs. direct oral anticoagulants for stroke prevention in patients with atrial fibrillation. J Thromb Thrombolysis. 2017; 43:365–79. https://doi.org/10.1007/s11239-016-1446-0.
6. Cooper GM, Johnson JA, Langaee TY, Feng H, Stanaway IB, Schwarz UI, Ritchie MD, Stein CM, Roden DM, Smith JD, Veenstra DL, Rettie AE, Rieder MJ. A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood. 2008; 112:1022–27. https://doi.org/10.1182/blood-2008-01-134247.
7. Takeuchi F, McGinnis R, Bourgeois S, Barnes C, Eriksson N, Soranzo N, Whittaker P, Ranganath V, Kumanduri V, McLaren W, Holm L, Lindh J, Rane A, et al. A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS Genet. 2009; 5:e1000433. https://doi.org/10.1371/journal.pgen.1000433.
8. Cha PC, Mushiroda T, Takahashi A, Kubo M, Minami S, Kamatani N, Nakamura Y. Genome-wide association study identifies genetic determinants of warfarin responsiveness for Japanese. Hum Mol Genet. 2010; 19:4735–44. https://doi.org/10.1093/hmg/ddq389.
9. Perera MA, Cavallari LH, Limdi NA, Gamazon ER, Konkashbaev A, Daneshjou R, Pluzhnikov A, Crawford DC, Wang J, Liu N, Tatonetti N, Bourgeois S, Takahashi H, et al. Genetic variants associated with warfarin dose in African-American individuals: a genome-wide association study. Lancet. 2013; 382:790–96. https://doi.org/10.1016/S0140-6736(13)60681-9.
10. Parra EJ, Botton MR, Perini JA, Krithika S, Bourgeois S, Johnson TA, Tsunoda T, Pirmohamed M, Wadelius M, Limdi NA, Cavallari LH, Burmester JK, Rettie AE, et al. Genome-wide association study of warfarin maintenance dose in a Brazilian sample. Pharmacogenomics. 2015; 16:1253–63. https://doi.org/10.2217/pgs.15.73.
11. Teichert M, Eijgelsheim M, Rivadeneira F, Uitterlinden AG, van Schaik RH, Hofman A, De Smet PA, van Gelder T, Visser LE, Stricker BH. A genome-wide association study of acenocoumarol maintenance dosage. Hum Mol Genet. 2009; 18:3758–68. https://doi.org/10.1093/hmg/ddp309.
12. Paré G, Eriksson N, Lehr T, Connolly S, Eikelboom J, Ezekowitz MD, Axelsson T, Haertter S, Oldgren J, Reilly P, Siegbahn A, Syvanen AC, Wadelius C, et al. Genetic determinants of dabigatran plasma levels and their relation to bleeding. Circulation. 2013; 127:1404–12. https://doi.org/10.1161/CIRCULATIONAHA.112.001233.
13. Butt HR, Allen EV, Bollman JL. A preparation from spoiled sweet clover. 3, 3’-methylene-bis-(4-hydroxy-coumarin) which prolongs coagulation and prothrombin time of the blood: preliminary report of experimental and clinical studies. Proc Staff Meet Mayo Clin. 1941; 16:388–95. https://www.cabdirect.org/cabdirect/abstract/19411401648.
14. Ufer M. Comparative pharmacokinetics of vitamin K antagonists: warfarin, phenprocoumon and acenocoumarol. Clin Pharmacokinet. 2005; 44:1227–46. https://doi.org/10.2165/00003088-200544120-00003.
15. Morais J, De Caterina R. Stroke Prevention in Atrial fibrillation: a clinical perspective on trials of the novel oral anticoagulants. Cardiovasc Drugs Ther. 2016; 30:201–14. https://doi.org/10.1007/s10557-015-6632-3.
16. Lenzini P, Wadelius M, Kimmel S, Anderson JL, Jorgensen AL, Pirmohamed M, Caldwell MD, Limdi N, Burmester JK, Dowd MB, Angchaisuksiri P, Bass AR, Chen J, et al. Integration of genetic, clinical, and INR data to refine warfarin dosing. Clin Pharmacol Ther. 2010; 87:572–78. https://doi.org/10.1038/clpt.2010.13.
17. van Schie RM, Wessels JA, le Cessie S, de Boer A, Schalekamp T, van der Meer FJ, Verhoef TI, van Meegen E, Rosendaal FR, Maitland-van der Zee AH, and EU-PACT Study Group. Loading and maintenance dose algorithms for phenprocoumon and acenocoumarol using patient characteristics and pharmacogenetic data. Eur Heart J. 2011; 32:1909–17. https://doi.org/10.1093/eurheartj/ehr116.
18. Verstuyft C, Delavenne X, Rousseau A, Robert A, Tod M, Diquet B, Lebot M, Jaillon P, Becquemont L. A pharmacokinetic-pharmacodynamic model for predicting the impact of CYP2C9 and VKORC1 polymorphisms on fluindione and acenocoumarol during induction therapy. Clin Pharmacokinet. 2012; 51:41–53. https://doi.org/10.2165/11595560-000000000-00000.
19. Donohue MM, Tirschwell DL. Implications of pharmacogenetic testing for patients taking warfarin or clopidogrel. Curr Neurol Neurosci Rep. 2011; 11:52–60. https://doi.org/10.1007/s11910-010-0157-8.
20. Baker WL, Chamberlin KW. New oral anticoagulants vs. warfarin treatment: no need for pharmacogenomics? Clin Pharmacol Ther. 2014; 96:17–19. https://doi.org/10.1038/clpt.2014.48.
21. Gage BF, Eby C, Johnson JA, Deych E, Rieder MJ, Ridker PM, Milligan PE, Grice G, Lenzini P, Rettie AE, Aquilante CL, Grosso L, Marsh S, et al. Use of pharmacogenetic and clinical factors to predict the therapeutic dose of warfarin. Clin Pharmacol Ther. 2008; 84:326–31. https://doi.org/10.1038/clpt.2008.10.
22. Giri AK, Khan NM, Grover S, Kaur I, Basu A, Tandon N, Scaria V, Kukreti R, Brahmachari SK, Bharadwaj D; Consortium IGV; INDICO. Genetic epidemiology of pharmacogenetic variations in CYP2C9, CYP4F2 and VKORC1 genes associated with warfarin dosage in the Indian population. Pharmacogenomics. 2014; 15:1337–54. https://doi.org/10.2217/pgs.14.88.
23. Rieder MJ, Reiner AP, Gage BF, Nickerson DA, Eby CS, McLeod HL, Blough DK, Thummel KE, Veenstra DL, Rettie AE. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N Engl J Med. 2005; 352:2285–93. https://doi.org/10.1056/NEJMoa044503.
24. Odén A, Fahlén M, Hart RG. Optimal INR for prevention of stroke and death in atrial fibrillation: a critical appraisal. Thromb Res. 2006; 117:493–99. https://doi.org/10.1016/j.thromres.2004.11.025.
25. Wadelius M, Pirmohamed M. Pharmacogenetics of warfarin: current status and future challenges. Pharmacogenomics J. 2007; 7:99–111. https://doi.org/10.1038/sj.tpj.6500417.
26. Lindh JD, Holm L, Andersson ML, Rane A. Influence of CYP2C9 genotype on warfarin dose requirements—a systematic review and meta-analysis. Eur J Clin Pharmacol. 2009; 65:365–75. https://doi.org/10.1007/s00228-008-0584-5.
27. Beinema M, Brouwers JR, Schalekamp T, Wilffert B. Pharmacogenetic differences between warfarin, acenocoumarol and phenprocoumon. Thromb Haemost. 2008; 100:1052–57. https://doi.org/10.1160/TH08-04-0116.
28. Yang L, Ge W, Yu F, Zhu H. Impact of VKORC1 gene polymorphism on interindividual and interethnic warfarin dosage requirement—a systematic review and meta analysis. Thromb Res. 2010; 125:e159–66. https://doi.org/10.1016/j.thromres.2009.10.017.
29. Wadelius M, Chen LY, Lindh JD, Eriksson N, Ghori MJ, Bumpstead S, Holm L, McGinnis R, Rane A, Deloukas P. The largest prospective warfarin-treated cohort supports genetic forecasting. Blood. 2009; 113:784–92. https://doi.org/10.1182/blood-2008-04-149070.
30. Scott SA, Khasawneh R, Peter I, Kornreich R, Desnick RJ. Combined CYP2C9, VKORC1 and CYP4F2 frequencies among racial and ethnic groups. Pharmacogenomics. 2010; 11:781–91. https://doi.org/10.2217/pgs.10.49.
31. Voora D, Koboldt DC, King CR, Lenzini PA, Eby CS, Porche-Sorbet R, Deych E, Crankshaw M, Milligan PE, McLeod HL, Patel SR, Cavallari LH, Ridker PM, et al. A polymorphism in the VKORC1 regulator calumenin predicts higher warfarin dose requirements in African Americans. Clin Pharmacol Ther. 2010; 87:445–51. https://doi.org/10.1038/clpt.2009.291.
32. Rieder MJ, Reiner AP, Rettie AE. Gamma-glutamyl carboxylase (GGCX) tagSNPs have limited utility for predicting warfarin maintenance dose. J Thromb Haemost. 2007; 5:2227–34. https://doi.org/10.1111/j.1538-7836.2007.02744.x.
33. Higashi MK, Veenstra DL, Kondo LM, Wittkowsky AK, Srinouanprachanh SL, Farin FM, Rettie AE. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA. 2002; 287:1690–98. https://doi.org/10.1001/jama.287.13.1690.
34. Ross S, Paré G. Pharmacogenetics of antiplatelets and anticoagulants: a report on clopidogrel, warfarin and dabigatran. Pharmacogenomics. 2013; 14:1565–72. https://doi.org/10.2217/pgs.13.149.
35. Eriksson N, Wallentin L, Berglund L, Axelsson T, Connolly S, Eikelboom J, Ezekowitz M, Oldgren J, Paré G, Reilly P, Siegbahn A, Syvanen AC, Wadelius C, et al. Genetic determinants of warfarin maintenance dose and time in therapeutic treatment range: a RE-LY genomics substudy. Pharmacogenomics. 2016; 17:1425–39. https://doi.org/10.2217/pgs-2016-0061.
36. VKORC1. vitamin K epoxide reductase complex subunit 1 [Homo sapiens (human)]. 2018. https://www.ncbi.nlm.nih.gov/gene/?term=79001.
37. CYP2C9 cytochrome P450 family 2 subfamily C member 9 [Homo sapiens (human)]. 2018. https://www.ncbi.nlm.nih.gov/gene/?term=1559.
38. Wadelius M, Chen LY, Eriksson N, Bumpstead S, Ghori J, Wadelius C, Bentley D, McGinnis R, Deloukas P. Association of warfarin dose with genes involved in its action and metabolism. Hum Genet. 2007; 121:23–34. https://doi.org/10.1007/s00439-006-0260-8.
39. CYP4F2 cytochrome P450 family 4 subfamily F member 2 [Homo sapiens (human)]. 2018. https://www.ncbi.nlm.nih.gov/gene/?term=8529.
40. Caldwell MD, Awad T, Johnson JA, Gage BF, Falkowski M, Gardina P, Hubbard J, Turpaz Y, Langaee TY, Eby C, King CR, Brower A, Schmelzer JR, et al. CYP4F2 genetic variant alters required warfarin dose. Blood. 2008; 111:4106–12. https://doi.org/10.1182/blood-2007-11-122010.
41. Cohen J, Wilson A, Manzolillo K. Clinical and economic challenges facing pharmacogenomics. Pharmacogenomics J. 2013; 13:378–88. https://doi.org/10.1038/tpj.2011.63.
42. Cavallari LH, Shin J, Perera MA. Role of pharmacogenomics in the management of traditional and novel oral anticoagulants. Pharmacotherapy. 2011; 31:1192–207. https://doi.org/10.1592/phco.31.12.1192.
43. Sconce EA, Khan TI, Wynne HA, Avery P, Monkhouse L, King BP, Wood P, Kesteven P, Daly AK, Kamali F. The impact of CYP2C9 and VKORC1 genetic polymorphism and patient characteristics upon warfarin dose requirements: proposal for a new dosing regimen. Blood. 2005; 106:2329–33. https://doi.org/10.1182/blood-2005-03-1108.
44. Tham LS, Goh BC, Nafziger A, Guo JY, Wang LZ, Soong R, Lee SC. A warfarin-dosing model in Asians that uses single-nucleotide polymorphisms in vitamin K epoxide reductase complex and cytochrome P450 2C9. Clin Pharmacol Ther. 2006; 80:346–55. https://doi.org/10.1016/j.clpt.2006.06.009.
45. Anderson JL, Horne BD, Stevens SM, Grove AS, Barton S, Nicholas ZP, Kahn SF, May HT, Samuelson KM, Muhlestein JB, Carlquist JF, and Couma-Gen Investigators. Randomized trial of genotype-guided versus standard warfarin dosing in patients initiating oral anticoagulation. Circulation. 2007; 116:2563–70. https://doi.org/10.1161/CIRCULATIONAHA.107.737312.
46. Caraco Y, Blotnick S, Muszkat M. CYP2C9 genotype-guided warfarin prescribing enhances the efficacy and safety of anticoagulation: a prospective randomized controlled study. Clin Pharmacol Ther. 2008; 83:460–70. https://doi.org/10.1038/sj.clpt.6100316.
47. Schelleman H, Chen J, Chen Z, Christie J, Newcomb CW, Brensinger CM, Price M, Whitehead AS, Kealey C, Thorn CF, Samaha FF, Kimmel SE. Dosing algorithms to predict warfarin maintenance dose in Caucasians and African Americans. Clin Pharmacol Ther. 2008; 84:332–39. https://doi.org/10.1038/clpt.2008.101.
48. Klein TE, Altman RB, Eriksson N, Gage BF, Kimmel SE, Lee MT, Limdi NA, Page D, Roden DM, Wagner MJ, Caldwell MD, Johnson JA, Chen YT, et al, and International Warfarin Pharmacogenetics Consortium. Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med. 2009; 360:753–64. https://doi.org/10.1056/NEJMoa0809329.
49. Huang SW, Chen HS, Wang XQ, Huang L, Xu DL, Hu XJ, Huang ZH, He Y, Chen KM, Xiang DK, Zou XM, Li Q, Ma LQ, et al. Validation of VKORC1 and CYP2C9 genotypes on interindividual warfarin maintenance dose: a prospective study in Chinese patients. Pharmacogenet Genomics. 2009; 19:226–34. https://doi.org/10.1097/FPC.0b013e328326e0c7.
50. Pathare A, Al Khabori M, Alkindi S, Al Zadjali S, Misquith R, Khan H, Lapoumeroulie C, Paldi A, Krishnamoorthy R. Warfarin pharmacogenetics: development of a dosing algorithm for Omani patients. J Hum Genet. 2012; 57:665–69. https://doi.org/10.1038/jhg.2012.94.
51. Wei M, Ye F, Xie D, Zhu Y, Zhu J, Tao Y, Yu F. A new algorithm to predict warfarin dose from polymorphisms of CYP4F2, CYP2C9 and VKORC1 and clinical variables: derivation in Han Chinese patients with non valvular atrial fibrillation. Thromb Haemost. 2012; 107:1083–91. https://doi.org/10.1160/TH11-12-0848.
52. Mazzaccara C, Conti V, Liguori R, Simeon V, Toriello M, Severini A, Perricone C, Meccariello A, Meccariello P, Vitale DF, Filippelli A, Sacchetti L. Warfarin anticoagulant therapy: a Southern Italy pharmacogenetics-based dosing model. PLoS One. 2013; 8:e71505. https://doi.org/10.1371/journal.pone.0071505.
53. Ageno W, Johnson J, Nowacki B, Turpie AG. A computer generated induction system for hospitalized patients starting on oral anticoagulant therapy. Thromb Haemost. 2000; 83:849–52. https://doi.org/10.1055/s-0037-1613932.
54. Bodin L, Verstuyft C, Tregouet DA, Robert A, Dubert L, Funck-Brentano C, Jaillon P, Beaune P, Laurent-Puig P, Becquemont L, Loriot MA. Cytochrome P450 2C9 (CYP2C9) and vitamin K epoxide reductase (VKORC1) genotypes as determinants of acenocoumarol sensitivity. Blood. 2005; 106:135–40. https://doi.org/10.1182/blood-2005-01-0341.
55. Ye S, Karim ZA, Al Hawas R, Pessin JE, Filipovich AH, Whiteheart SW. Syntaxin-11, but not syntaxin-2 or syntaxin-4, is required for platelet secretion. Blood. 2012; 120: 2484-92. https://doi.org/10.1182/blood-2012-05-430603.
56. Chen D, Lemons PP, Schraw T, Whiteheart SW. Molecular mechanisms of platelet exocytosis: role of SNAP-23 and syntaxin 2 and 4 in lysosome release. Blood. 2000; 96:1782–88.
57. Tang BL. A unique SNARE machinery for exocytosis of cytotoxic granules and platelets granules. Mol Membr Biol. 2015; 32:120–26. https://doi.org/10.3109/09687688.2015.1079934.
58. Zhu Q, Yamakuchi M, Lowenstein CJ. SNAP23 Regulates Endothelial Exocytosis of von Willebrand Factor. PLoS One. 2015; 10: e0118737. https://doi.org/10.1371/journal.pone.0118737.
59. Borobia AM, Lubomirov R, Ramírez E, Lorenzo A, Campos A, Muñoz-Romo R, Fernández-Capitán C, Frías J, Carcas AJ. An acenocoumarol dosing algorithm using clinical and pharmacogenetic data in Spanish patients with thromboembolic disease. PLoS One. 2012; 7:e41360. https://doi.org/10.1371/journal.pone.0041360.
60. Rathore SS, Agarwal SK, Pande S, Singh SK, Mittal T, Mittal B. Therapeutic dosing of acenocoumarol: proposal of a population specific pharmacogenetic dosing algorithm and its validation in north Indians. PLoS One. 2012; 7:e37844. https://doi.org/10.1371/journal.pone.0037844.
61. Cerezo-Manchado JJ, Rosafalco M, Antón AI, Pérez-Andreu V, Garcia-Barberá N, Martínez AB, Corral J, Vicente V, González-Conejero R, Roldán V. Creating a genotype-based dosing algorithm for acenocoumarol steady dose. Thromb Haemost. 2013; 109:146–53. https://doi.org/10.1160/TH12-08-0631.
62. Pop TR, Vesa Ş, Trifa AP, Crişan S, Buzoianu AD. An acenocoumarol dose algorithm based on a South-Eastern European population. Eur J Clin Pharmacol. 2013; 69:1901–07. https://doi.org/10.1007/s00228-013-1551-3.
63. Wolkanin-Bartnik J, Pogorzelska H, Szperl M, Bartnik A, Koziarek J, Bilinska ZT. Impact of genetic and clinical factors on dose requirements and quality of anticoagulation therapy in Polish patients receiving acenocoumarol: dosing calculation algorithm. Pharmacogenet Genomics. 2013; 23:611–18. https://doi.org/10.1097/FPC.0000000000000004.
64. Jiménez-Varo E, Cañadas-Garre M, Gutiérrez-Pimentel MJ, Calleja-Hernández MA. Prediction of stable acenocoumarol dose by a pharmacogenetic algorithm. Pharmacogenet Genomics. 2014; 24:501–13. https://doi.org/10.1097/FPC.0000000000000082.
65. Jiménez-Varo E, Cañadas-Garre M, Garcés-Robles V, Gutiérrez-Pimentel MJ, Calleja-Hernández MÁ. Extrapolation of acenocoumarol pharmacogenetic algorithms. Vascul Pharmacol. 2015; 74:151–57. https://doi.org/10.1016/j.vph.2015.06.010.
66. Dimitrova-Karamfilova A, Tzveova R, Chilingirova N, Goranova T, Nachev G, Mitev V, Kaneva R. Acenocoumarol pharmacogenetic dosing algorithms and their application in two bulgarian patients with low anticoagulant requirements. Biochem Genet. 2015; 53:334–50. https://doi.org/10.1007/s10528-015-9695-8.
67. Tong HY, Dávila-Fajardo CL, Borobia AM, Martínez-González LJ, Lubomirov R, Perea León LM, Blanco Bañares MJ, Díaz-Villamarín X, Fernández-Capitán C, Cabeza Barrera J, Carcas AJ, and PGX-ACE Investigators Group. A new pharmacogenetic algorithm to predict the most appropriate dosage of acenocoumarol for stable anticoagulation in a mixed Spanish population. PLoS One. 2016; 11:e0150456. https://doi.org/10.1371/journal.pone.0150456.
68. Teichert M, Eijgelsheim M, Uitterlinden AG, Buhre PN, Hofman A, De Smet PA, Visser LE, Stricker BH. Dependency of phenprocoumon dosage on polymorphisms in the VKORC1, CYP2C9, and CYP4F2 genes. Pharmacogenet Genomics. 2011; 21:26–34. https://doi.org/10.1097/FPC.0b013e32834154fb.
69. Peoc’h K, Pruvot S, Gourmel C, dit Sollier CB, Drouet L. A new VKORC1 mutation leading to an isolated resistance to fluindione. Br J Haematol. 2009; 145:841–43. https://doi.org/10.1111/j.1365-2141.2009.07687.x.
70. Mentré F, Pousset F, Comets E, Plaud B, Diquet B, Montalescot G, Ankri A, Mallet A, Lechat P. Population pharmacokinetic-pharmacodynamic analysis of fluindione in patients. Clin Pharmacol Ther. 1998; 63:64–78. https://doi.org/10.1016/S0009-9236(98)90122-9.
71. Tillement JP, Thébault JJ, Mattei C, d’Athis P, Blatrix C. Anticoagulant effect and plasma kinetics of fluorophenindione after a single dose in man. Eur J Clin Pharmacol. 1975; 8:271–75. https://doi.org/10.1007/BF00567127.
72. Lacut K, Ayme-Dietrich E, Gourhant L, Poulhazan E, Andro M, Becquemont L, Mottier D, Le Gal G, Verstuyft C. Impact of genetic factors (VKORC1, CYP2C9, CYP4F2 and EPHX1) on the anticoagulation response to fluindione. Br J Clin Pharmacol. 2012; 73:428–36. https://doi.org/10.1111/j.1365-2125.2011.04095.x.
73. Comets E, Pousset F, Mentré F, Diquet B, Ankri A, Mallet A, Lechat P. Prediction of fluindione maintenance dosage hampered by large intraindividual variability. Ther Drug Monit. 2000; 22:668–75. https://doi.org/10.1097/00007691-200012000-00005.
74. Moreau C, Pautas E, Duverlie C, Berndt C, Andro M, Mahé I, Emmerich J, Lacut K, Le Gal G, Peyron I, Gouin-Thibault I, Golmard JL, Loriot MA, Siguret V. A model predicting fluindione dose requirement in elderly inpatients including genotypes, body weight, and amiodarone. Thromb Haemost. 2014; 111:705–12. https://doi.org/10.1160/TH13-07-0555.
75. Königsbrügge O, Simon A, Domanovits H, Pabinger I, Ay C. Thromboembolic events, bleeding, and drug discontinuation in patients with atrial fibrillation on anticoagulation: a prospective hospital-based registry. BMC Cardiovasc Disord. 2016; 16:254. https://doi.org/10.1186/s12872-016-0438-5.
76. Yates SW. Novel oral anticoagulants for stroke prevention in atrial fibrillation: a focus on the older patient. Int J Gen Med. 2013; 6:167–80. https://doi.org/10.2147/IJGM.S39379.
77. Larsen TB, Skjøth F, Nielsen PB, Kjældgaard JN, Lip GY. Comparative effectiveness and safety of non-vitamin K antagonist oral anticoagulants and warfarin in patients with atrial fibrillation: propensity weighted nationwide cohort study. BMJ. 2016; 353:i3189. https://doi.org/10.1136/bmj.i3189.
78. Lip GY, Pan X, Kamble S, Kawabata H, Mardekian J, Masseria C, Bruno A, Phatak H. Major bleeding risk among non-valvular atrial fibrillation patients initiated on apixaban, dabigatran, rivaroxaban or warfarin: a “real-world” observational study in the United States. Int J Clin Pract. 2016; 70:752–63. https://doi.org/10.1111/ijcp.12863.
79. Coleman CI, Antz M, Bowrin K, Evers T, Simard EP, Bonnemeier H, Cappato R. Real-world evidence of stroke prevention in patients with nonvalvular atrial fibrillation in the United States: the REVISIT-US study. Curr Med Res Opin. 2016; 32:2047–53. https://doi.org/10.1080/03007995.2016.1237937.
80. Romanelli RJ, Nolting L, Dolginsky M, Kym E, Orrico KB. Dabigatran versus warfarin for atrial fibrillation in real-world clinical practice: a systematic review and meta-analysis. Circ Cardiovasc Qual Outcomes. 2016; 9:126–34. https://doi.org/10.1161/CIRCOUTCOMES.115.002369.
81. Merali Z, Ross S, Paré G. The pharmacogenetics of carboxylesterases: CES1 and CES2 genetic variants and their clinical effect. Drug Metabol Drug Interact. 2014; 29:143–51. https://doi.org/10.1515/dmdi-2014-0009.
82. Chin PK. Which patients may benefit from dose adjustment of non-vitamin K antagonist oral anticoagulants? Semin Thromb Hemost. 2015; 41:195–207. https://doi.org/10.1055/s-0035-1546465.
83. Gong IY, Kim RB. Importance of pharmacokinetic profile and variability as determinants of dose and response to dabigatran, rivaroxaban, and apixaban. Can J Cardiol. 2013; 29:S24–33. https://doi.org/10.1016/j.cjca.2013.04.002.
84. Reilly PA, van Ryn J, Grottke O, Glund S, Stangier J. Idarucizumab, a specific reversal agent for dabigatran: mode of action, pharmacokinetics and pharmacodynamics, and safety and efficacy in phase 1 subjects. Am J Emerg Med. 2016; 34:26–32. https://doi.org/10.1016/j.ajem.2016.09.050.
85. Carmo J, Moscoso Costa F, Ferreira J, Mendes M. Dabigatran in real-world atrial fibrillation. Meta-analysis of observational comparison studies with vitamin K antagonists. Thromb Haemost. 2016; 116:754–63. https://doi.org/10.1160/TH16-03-0203.
86. Eikelboom JW, Wallentin L, Connolly SJ, Ezekowitz M, Healey JS, Oldgren J, Yang S, Alings M, Kaatz S, Hohnloser SH, Diener HC, Franzosi MG, Huber K, et al. Risk of bleeding with 2 doses of dabigatran compared with warfarin in older and younger patients with atrial fibrillation: an analysis of the randomized evaluation of long-term anticoagulant therapy (RE-LY) trial. Circulation. 2011; 123:2363-72. https://doi.org/10.1161/CIRCULATIONAHA.110.004747.
87. Villines TC, Schnee J, Fraeman K, Siu K, Reynolds MW, Collins J, Schwartzman E. A comparison of the safety and effectiveness of dabigatran and warfarin in non-valvular atrial fibrillation patients in a large healthcare system. Thromb Haemost. 2015; 114:1290–98. https://doi.org/10.1160/TH15-06-0453.
88. Larsen TB, Gorst-Rasmussen A, Rasmussen LH, Skjøth F, Rosenzweig M, Lip GY. Bleeding events among new starters and switchers to dabigatran compared with warfarin in atrial fibrillation. Am J Med. 2014; 127:650–656.e5. https://doi.org/10.1016/j.amjmed.2014.01.031.
89. Potpara TS. Dabigatran in ‘real-world’ clinical practice for stroke prevention in patients with non-valvular atrial fibrillation. Thromb Haemost. 2015; 114:1093–98. https://doi.org/10.1160/TH15-10-0825.
90. Ageno W, Eikelboom J, Lip GY. Dabigatran in clinical practice: contemporary overview of the evidence. Int J Cardiol. 2016; 220:417–28. https://doi.org/10.1016/j.ijcard.2016.06.078.
91. Graham DJ, Reichman ME, Wernecke M, Zhang R, Southworth MR, Levenson M, Sheu TC, Mott K, Goulding MR, Houstoun M, MaCurdy TE, Worrall C, Kelman JA. Cardiovascular, bleeding, and mortality risks in elderly Medicare patients treated with dabigatran or warfarin for nonvalvular atrial fibrillation. Circulation. 2015; 131:157–64. https://doi.org/10.1161/CIRCULATIONAHA.114.012061.
92. Lee LH. DOACs - advances and limitations in real world. Thromb J. 2016; 14:S17. https://doi.org/10.1186/s12959-016-0111-3.
93. Dimatteo C, D’Andrea G, Vecchione G, Paoletti O, Cappucci F, Tiscia GL, Buono M, Grandone E, Testa S, Margaglione M. Pharmacogenetics of dabigatran etexilate interindividual variability. Thromb Res. 2016; 144:1–5. https://doi.org/10.1016/j.thromres.2016.05.025.
94. Tran H, Joseph J, Young L, McRae S, Curnow J, Nandurkar H, Wood P, McLintock C, and Australasian Society of Thrombosis and Haemostasis. New oral anticoagulants: a practical guide on prescription, laboratory testing and peri-procedural/bleeding management. Intern Med J. 2014; 44:525–36. https://doi.org/10.1111/imj.12448.
95. ROCKET AF Study Investigators. Rivaroxaban-once daily, oral, direct factor Xa inhibition compared with vitamin K antagonism for prevention of stroke and Embolism Trial in Atrial Fibrillation: rationale and design of the ROCKET AF study. Am Heart J. 2010; 159:340–347.e1. https://doi.org/10.1016/j.ahj.2009.11.025.
96. Weeda ER, White CM, Peacock WF, Coleman CI. Rates of major bleeding with rivaroxaban in real-world studies of nonvalvular atrial fibrillation patients: a meta-analysis. Curr Med Res Opin. 2016; 32:1117–20. https://doi.org/10.1185/03007995.2016.1161610.
97. Patel MR, Mahaffey KW, Garg J, Pan G, Singer DE, Hacke W, Breithardt G, Halperin JL, Hankey GJ, Piccini JP, Becker RC, Nessel CC, Paolini JF, et al, and ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011; 365:883–91. https://doi.org/10.1056/NEJMoa1009638.
98. Beyer-Westendorf J, Förster K, Pannach S, Ebertz F, Gelbricht V, Thieme C, Michalski F, Köhler C, Werth S, Sahin K, Tittl L, Hänsel U, Weiss N. Rates, management, and outcome of rivaroxaban bleeding in daily care: results from the Dresden NOAC registry. Blood. 2014; 124:955–62. https://doi.org/10.1182/blood-2014-03-563577.
99. Beyer-Westendorf J, Camm AJ, Coleman CI, Tamayo S. Rivaroxaban real-world evidence: validating safety and effectiveness in clinical practice. Thromb Haemost. 2016; 116:S13–23. https://doi.org/10.1160/TH16-06-0485.
100. Camm AJ, Amarenco P, Haas S, Hess S, Kirchhof P, Kuhls S, van Eickels M, Turpie AG, and XANTUS Investigators. XANTUS: a real-world, prospective, observational study of patients treated with rivaroxaban for stroke prevention in atrial fibrillation. Eur Heart J. 2016; 37: 1145-53. https://doi.org/10.1093/eurheartj/ehv466.
101. Hecker J, Marten S, Keller L, Helmert S, Michalski F, Werth S, Sahin K, Tittl L, Beyer-Westendorf J. Effectiveness and safety of rivaroxaban therapy in daily-care patients with atrial fibrillation. Results from the Dresden NOAC Registry. Thromb Haemost. 2016; 115:939–49. https://doi.org/10.1160/TH15-10-0840.
102. Tamayo S, Frank Peacock W, Patel M, Sicignano N, Hopf KP, Fields LE, Sarich T, Wu S, Yannicelli D, Yuan Z. Characterizing major bleeding in patients with nonvalvular atrial fibrillation: a pharmacovigilance study of 27 467 patients taking rivaroxaban. Clin Cardiol. 2015; 38:63–68. https://doi.org/10.1002/clc.22373.
103. Coleman CI, Antz M, Ehlken B, Evers T. REal-LIfe Evidence of stroke prevention in patients with atrial Fibrillation—the RELIEF study. Int J Cardiol. 2016; 203:882–84. https://doi.org/10.1016/j.ijcard.2015.09.037.
104. Connolly SJ, Ezekowitz MD, Yusuf S, Eikelboom J, Oldgren J, Parekh A, Pogue J, Reilly PA, Themeles E, Varrone J, Wang S, Alings M, Xavier D, et al, and RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009; 361:1139–51. https://doi.org/10.1056/NEJMoa0905561.
105. Dimatteo C, D’Andrea G, Vecchione G, Paoletti O, Tiscia GL, Santacroce R, Correale M, Brunetti N, Grandone E, Testa S, Margaglione M. ABCB1 SNP rs4148738 modulation of apixaban interindividual variability. Thromb Res. 2016; 145: 24-6. https://doi.org/10.1016/j.thromres.2016.07.005.
106. Giugliano RP, Ruff CT, Braunwald E, Murphy SA, Wiviott SD, Halperin JL, Waldo AL, Ezekowitz MD, Weitz JI, Špinar J, Ruzyllo W, Ruda M, Koretsune Y, et al, and ENGAGE AF-TIMI 48 Investigators. Edoxaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2013; 369:2093–104. https://doi.org/10.1056/NEJMoa1310907.
107. Blann AD, Skjøth F, Rasmussen LH, Larsen TB, Lip GY. Edoxaban versus placebo, aspirin, or aspirin plus clopidogrel for stroke prevention in atrial fibrillation. An indirect comparison analysis. Thromb Haemost. 2015; 114:403–09. https://doi.org/10.1160/TH15-05-0383.
108. Blann AD, Banerjee A, Lane DA, Torp-Pedersen C, Lip GY. Net clinical benefit of edoxaban versus no treatment in a ‘real world’ atrial fibrillation population: A modelling analysis based on a nationwide cohort study. Int J Cardiol. 2015; 201:693–98. https://doi.org/10.1016/j.ijcard.2015.08.074.
109. Noguchi K, Morishima Y, Takahashi S, Ishihara H, Shibano T, Murata M. Impact of nonsynonymous mutations of factor X on the functions of factor X and anticoagulant activity of edoxaban. Blood Coagul Fibrinolysis. 2015; 26:117–22. https://doi.org/10.1097/MBC.0000000000000147.
110. Gallego-Fabrega C, Carrera C, Reny JL, Fontana P, Slowik A, Pera J, Pezzini A, Serrano-Heras G, Segura T, Martí-Fàbregas J, Muiño E, Cullell N, Montaner J, et al. TRAF3 epigenetic regulation is associated with vascular recurrence in patients with ischemic stroke. Stroke. 2016; 47:1180–86. https://doi.org/10.1161/STROKEAHA.115.012237.
111. Gallego-Fabrega C, Carrera C, Reny JL, Fontana P, Slowik A, Pera J, Pezzini A, Serrano-Heras G, Segura T, Bin Dukhyil AA, Martí-Fàbregas J, Muiño E, Cullell N, et al. PPM1A methylation is associated with vascular recurrence in aspirin-treated patients. Stroke. 2016; 47:1926–29. https://doi.org/10.1161/STROKEAHA.116.013340.
112. Kumana CR, Cheung BM, Siu DC, Tse HF, Lauder IJ. Non Vitamin K oral anticoagulants versus warfarin for patients with atrial fibrillation: absolute benefit and harm assessments yield novel insights. Cardiovasc Ther. 2016; 34:100–06. https://doi.org/10.1111/1755-5922.12173.
113. Mega JL, Walker JR, Ruff CT, Vandell AG, Nordio F, Deenadayalu N, Murphy SA, Lee J, Mercuri MF, Giugliano RP, Antman EM, Braunwald E, Sabatine MS. Genetics and the clinical response to warfarin and edoxaban: findings from the randomised, double-blind ENGAGE AF-TIMI 48 trial. Lancet. 2015; 385:2280–87. https://doi.org/10.1016/S0140-6736(14)61994-2.
114. Huynh K. Anticoagulation therapy. Optimal dosages and genotypes for edoxaban therapy. Nat Rev Cardiol. 2015; 12:256. https://doi.org/10.1038/nrcardio.2015.44.
115. Westwood M, Joore M, Whiting P, van Asselt T, Ramaekers B, Armstrong N, Misso K, Severens J, Kleijnen J. Epidermal growth factor receptor tyrosine kinase (EGFR-TK) mutation testing in adults with locally advanced or metastatic non-small cell lung cancer: a systematic review and cost-effectiveness analysis. Health Technol Assess. 2014; 18:1–166. https://doi.org/10.3310/hta18620.
116. Meleth S, Reeder-Hayes K, Ashok M, Clark R, Funkhouser W, Wines R, Hill C, Shanahan E, McClure E, Burson K, Coker-Schwimmer M, Garge N, Jonas DE. Technology Assessment of Molecular Pathology Testing for the Estimation of Prognosis for Common Cancers [Internet]. AHRQ Technology Assessments. 2014. http://europepmc.org/books/NBK285410.
117. Huang RS, Ratain MJ. Pharmacogenetics and pharmacogenomics of anticancer agents. CA Cancer J Clin. 2009; 59:42–55. https://doi.org/10.3322/caac.20002.
118. Etienne-Grimaldi MC, Boyer JC, Thomas F, Quaranta S, Picard N, Loriot MA, Narjoz C, Poncet D, Gagnieu MC, Ged C, Broly F, Le Morvan V, Bouquié R, et al, and Collective work by Groupe de Pharmacologie Clinique Oncologique (GPCO-Unicancer), and French Réseau National de Pharmacogénétique Hospitalière (RNPGx). UGT1A1 genotype and irinotecan therapy: general review and implementation in routine practice. Fundam Clin Pharmacol. 2015; 29:219–37. https://doi.org/10.1111/fcp.12117.
119. Ratain MJ. From bedside to bench to bedside to clinical practice: an odyssey with irinotecan. Clin Cancer Res. 2006; 12:1658–60. https://doi.org/10.1158/1078-0432.CCR-06-0159.
120. Lauschke VM, Milani L, Ingelman-Sundberg M. Pharmacogenomic biomarkers for improved drug therapy—recent progress and future developments. AAPS J. 2017; 20:4. https://doi.org/10.1208/s12248-017-0161-x.