
INTRODUCTION
Androgen-receptor splice variant 7 (AR-V7) is a constitutively-active isoform of the androgen receptor that is associated with a particularly aggressive form of advanced prostate cancer [1]. Because AR-V7 lacks the androgen-receptor ligand-binding domain, AR-V7-positive prostate cancers are generally resistant to novel hormonal therapies including abiraterone and enzalutamide [2, 3]. In addition, prostate cancers expressing AR-V7 often show poor responses to taxane chemotherapies including docetaxel and cabazitaxel [4, 5]. To this end, patients with AR-V7-positive prostate cancer generally have a median progression-free survival (PFS) of only 3-4 months and a median overall survival (OS) of 7-9 months. Therefore, developing effective therapies for AR-V7-expressing advanced prostate cancer represents an urgent unmet need.
Immune-checkpoint blockade may be one potential strategy to treat such patients. In many cancer types, inhibition of cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) and/or the programmed death 1 (PD-1) receptor has resulted in meaningful antitumor responses [6]. In some settings, combined blockade of both PD-1 (mediating T-cell exhaustion in peripheral tissues) and CTLA-4 (involved in earlier phases of T-cell activation) has proven more efficacious than inhibition of either pathway alone [7, 8]. Furthermore, tumors harboring DNA mismatch-repair defects or those with hypermutation may be particularly sensitive to immune-checkpoint inhibition [9, 10]. While prostate cancer is generally regarded as a low–mutation-burden tumor [11] and immune-checkpoint blockade has resulted in only modest benefits as a monotherapy [12, 13], recent data have suggested that AR-V7-expressing prostate cancers may be associated with a greater number of DNA-repair gene mutations and a higher mutation load [14].
We hypothesized that metastatic castration-resistant prostate cancer patients with AR-V7-positive circulating tumor cells (CTCs) would be susceptible to treatment with combined immune-checkpoint blockade, and that this approach would be safe and tolerable. We also sought to determine (in an exploratory fashion) whether treatment efficacy was associated with presence of DNA-repair gene mutations. To test these hypotheses, we conducted a phase-2 clinical trial testing ipilimumab plus nivolumab in patients with AR-V7-positive advanced prostate cancer.
RESULTS
Patient characteristics
From March 2016 through December 2016, a total of 36 patients underwent clinical-grade AR-V7 testing for eligibility purposes, 26 (72%) had detectable CTCs, and 16 men (44%) were AR-V7-positive. One patient failed screening, leaving 15 patients that comprised our study cohort. Supplementary Table 1 summarizes the baseline characteristics of the study participants. Median age was 65 years, 47% had ECOG performance-status of 1, median PSA was 115 ng/mL, 67% had visceral (liver or lung) metastases, and 60% had received ≥4 prior regimens for metastatic castration-resistant prostate cancer (mCRPC). All patients received at least one dose of the study drugs. At the time of data cutoff (October 2017), median follow-up was 8.6 (range, 1.9–17.9) months, and two patients remained alive.
Overall clinical outcomes
All patients were evaluable for efficacy (summarized in Table 1, Supplementary Figure 1). Overall, 2 of 15 men (13.3%, 95%CI 3.7–37.9%) achieved a PSA response. Among the 8 patients with measurable soft-tissue disease, the objective response rate (ORR) was 25.0% (95%CI 7.2–59.1%). Median PSA-PFS was 3.0 (95%CI 2.1–NR) months, and median PFS was 3.7 (95%CI 2.8–7.5) months. Three of 15 patients (20.0%, 95%CI 7.1–45.2%) achieved a “durable PFS”. Median OS was 8.2 (95%CI 5.5–10.4) months.
Table 1: Overall outcomes for all patients, and according to DNA-repair deficiency (DRD) status
Overall |
DRD Negative |
DRD Positive |
HR (95%CI) |
P value |
|
|---|---|---|---|---|---|
PSA50, N (%) |
2/15 (13.3%) |
0/9 (0%) |
2/6 (33.3%) |
– |
0.14 |
ORR, N (%) |
2/8 (25.0%) |
0/3 (0%) |
2/5 (40.0%) |
– |
0.46 |
Durable PFS |
3/15 (20.0%) |
0/9 (0%) |
3/6 (50.0%) |
– |
0.044 |
PSA-PFS (mo),(95% CI) |
2.96 (2.07–NR) |
2.07 (1.74–NR) |
5.82 (4.24–NR) |
0.19 (0.06–0.62) |
0.0003 |
PFS (mo),(95% CI) |
3.68 (2.76–7.52) |
2.83(1.87–NR) |
6.51 (3.88–NR) |
0.31 (0.10–0.92) |
0.014 |
OS (mo),(95% CI) |
8.18 (5.52–10.41) |
7.23 (3.45–NR) |
9.04 (8.18–NR) |
0.41 (0.14–1.21) |
0.11 |
NR: upper 95% confidence limit of survival probability not reached.
DNA-repair defects and outcomes
Six of 15 patients (40%) harbored potentially deleterious somatic and/or germline mutations in a least one DNA-repair gene (Table 2, Supplementary Table 7C), and were considered DNA-repair deficient (DRD+). Patient 3 had a germline BRCA2 mutation, patient 4 had somatic mutations in both BRCA2 and MSH6, patient 6 had a somatic ATM mutation, patient 8 had a germline BRCA2 and a somatic FANCM mutation, patient 9 had a somatic ATM mutation, and patient 14 had a somatic ERCC4 mutation. Baseline characteristics and clinical outcomes of the DRD+ and DRD– patients are summarized in Supplementary Tables 1 and 2. Two patients (3 and 8) had germline mutations in BRCA2, and two patients (4 and 8) had biallelic BRCA2 alterations resulting from LOH of the wild-type allele. No patient demonstrated microsatellite instability. Mean tumor mutational load was estimated at 3.2 (range, 0.8–7.8) mutations/Mb in DRD+ patients and 1.6 (range, 0.8–3.1) mutations/Mb in DRD– patients.
Table 2: Summary of DNA-repair deficiency (DRD) status among the 15 patients treated with ipilimumab plus nivolumab
Patient no. |
DRD status |
DNA-repair gene |
Pathogenic DNA-repair mutations |
Germline vs. somatic |
Loss of heterozygosity (LOH) |
MSI markers shifted |
Mutational load (muts/Mb) |
Source of tumor DNA |
|---|---|---|---|---|---|---|---|---|
1 |
– |
- |
- |
- |
- |
N/A |
1.1 |
Plasma |
2 |
– |
- |
- |
- |
- |
N/A |
2.4 |
Prostate |
3 |
+ |
BRCA2 |
E1646Qfs*23 |
Germline |
No |
0/5 |
1.6 |
Liver mass |
4 |
+ |
BRCA2 |
P3189H |
Somatic |
Yes |
0/5 |
7.8 |
Lymph node |
5 |
– |
- |
- |
- |
- |
N/A |
3.1 |
Plasma |
6 |
+ |
ATM |
D2708N |
Somatic |
No |
0/5 |
1.6 |
Lymph node |
7 |
– |
- |
- |
- |
- |
0/5 |
1.4 |
Epidural mass |
8 |
+ |
BRCA2 |
D3095E |
Germline |
Yes |
0/5 |
0.8 |
Prostate |
9 |
+ |
ATM |
E2039X |
Somatic |
No |
0/5 |
1.1 |
Plasma |
10 |
– |
- |
- |
- |
- |
N/A |
1.1 |
Plasma |
11 |
– |
- |
- |
- |
- |
0/5 |
1.3 |
Prostate |
12 |
– |
- |
- |
- |
- |
0/5 |
0.8 |
Prostate |
13 |
– |
- |
- |
- |
- |
0/5 |
1.3 |
Lymph node |
14 |
+ |
ERCC4 |
D762V |
Somatic |
No |
0/5 |
5.6 |
Lymph node |
15 |
– |
- |
- |
- |
- |
0/5 |
1.8 |
Liver mass |
To further examine whether DNA-repair defects are enriched in AR-V7-positive patients, we interrogated the StandUp2Cancer (SU2C) database comprising whole-exome and transcriptome sequencing from 150 mCRPC biopsies [11], of which 143 had adequate RNA yields. Of these, 17.5% of cases (25/143) had AR-V7/AR-FL ratios on RNA sequencing of >10%, and were designated as AR-V7-high; while the remaining 82.5% (118/143) were designated as AR-V7-low. This threshold was set so that the prevalence of an AR-V7-positive tissue-based test would be broadly similar to that of a positive CTC-based AR-V7 test. To this end, pathogenic DRD mutations were found in 36.0% (9/25) of AR-V7-high cases but only in 18.6% (22/118) of AR-V7-low cases (P=0.056), suggesting a possible (but non-significant) association between AR-V7 and DNA-repair defects. In the AR-V7-high SU2C cohort, the altered DNA-repair genes were BRCA2 (x4), ATM (x2), CDK12 (x2) and MSH2 (x1).
We then compared clinical outcomes in DRD+ and DRD– patients from our trial (Table 1). Response measures appeared generally better in DRD+ versus DRD– cases (Figure 1) with respect to PSA responses (33% vs. 0%; P=0.14, nonsignificant), ORR (40% vs. 0%; P=0.46, nonsignificant) and “durable PFS” (50% vs. 0%; P=0.04, significant). Interestingly, both patients who achieved PSA responses (4 and 8) had biallelic BRCA2 alterations. Similarly, time-to-event outcomes also appeared better in DRD+ versus DRD– patients (Figure 2) with respect to PSA-PFS (HR 0.19, 95%CI 0.06–0.62; P<0.001, significant), PFS (HR 0.31, 95%CI 0.10–0.92; P=0.01, significant), and OS (HR 0.41, 95%CI 0.14–1.21; P=0.11, nonsignificant).
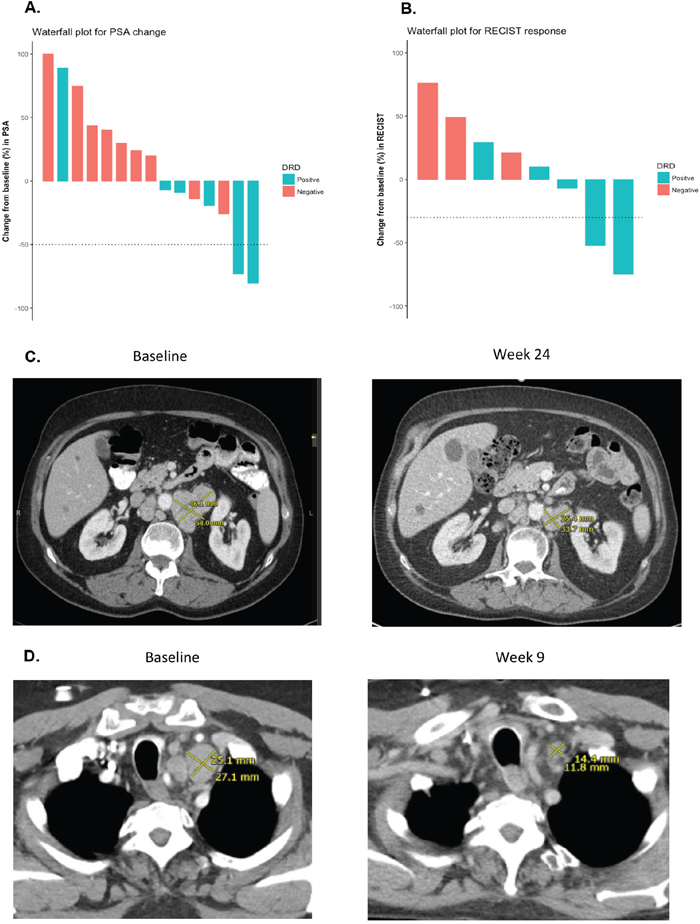
Figure 1: PSA responses and radiographic responses according to DRD status. (A) Waterfall plot showing PSA responses according to DRD status. The two patients with PSA50 responses (#4 and #8) both had biallelic BRCA2 gene mutations. Patient #4 had a mixed soft-tissue response (some measurable lesions decreased while others increased) and achieved a durable PFS. Patient #8 did not have any measurable disease, but also achieved a durable PFS, and experienced complete resolution of malignant bone pain (pain score 7/10 decreased to 0/10 after 12 weeks of therapy); he is still alive after 17.5+ months of follow-up. (B) Waterfall plot showing objective RECIST responses according to DRD status. The two patients with soft-tissue responses (#6 and #14) had mutations in ATM and ERCC4, respectively. Patient #6 achieved a durable PFS, and is still alive after 17.9+ months of follow-up. (C) CT scan of radiographic response for patient #6 (with somatic ATM mutation) at baseline and after 24 weeks of treatment. The sum diameter of his target lesions decreased by 52% at the time of his best response. (D). CT scan of radiographic response for patient #14 (with somatic ERCC4 mutation) at baseline and after 9 weeks of treatment. The sum diameter of his target lesions decreased by 75% at the time of his best response.
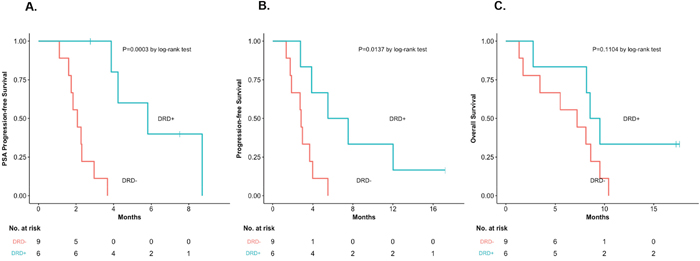
Figure 2: Time-to-event outcomes, according to DRD status. (A) PSA-PFS, according to DRD status [HR 0.19, 95%CI 0.06–0.62, P=0.0003]. (B) PFS, according to DRD status [HR 0.31, 95%CI 0.10–0.92, P=0.014]. (C) OS, according to DRD status [HR 0.41, 95%CI 0.14–1.21, P=0.11].
Other biomarkers and outcomes
To examine the prognostic impact of CTC phenotypic heterogeneity, we compared outcomes in patients with a high (≥1.5) versus low (<1.5) Shannon index (Supplementary Table 3). Five (33%) and 10 men (67%) were classified as Shannon-high and Shannon-low, respectively. There were numerically more Shannon-high cases among DRD+ compared to DRD– patients (50% [3/6] vs. 22% [2/9] respectively, P=0.26, nonsignificant). Outcomes appeared generally better in Shannon-high vs. Shannon-low patients with respect to PSA responses (20% vs. 10%; P=1.0, nonsignificant), ORR (100% vs. 0%; P=0.04, significant), “durable PFS” (40% vs. 10%; P=0.24, nonsignificant), PSA-PFS (HR 0.67, 95%CI 0.23–1.99; P=0.44, nonsignificant), PFS (HR 0.43, 95%CI 0.15–1.22; P=0.11, nonsignificant), and OS (HR 0.34, 95%CI 0.11–0.99; P=0.07, nonsignificant) (Figure 3). Interestingly, both men with RECIST-defined objective responses (6 and 14) had high Shannon indices. CTC pleomorphism (high vs. low) was also assessed in relation to clinical outcomes. No statistical trends were observed (Supplementary Table 4, Supplementary Figure 2), although both patients with PSA responses (4 and 8) were classified as pleomorphism-high.
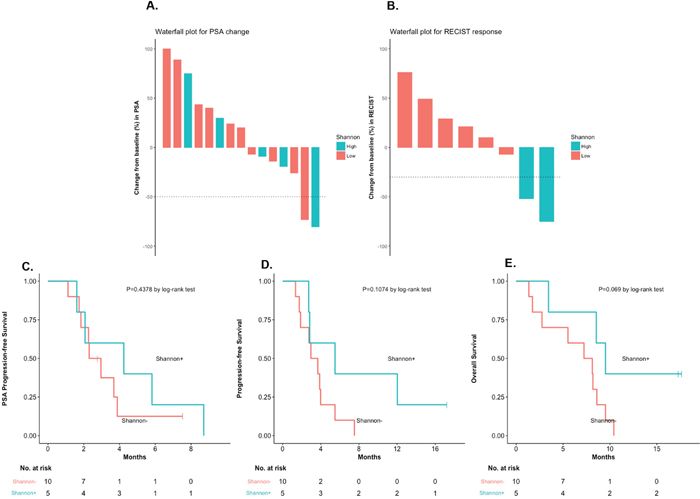
Figure 3: Clinical outcomes, according to Shannon index (low vs. high). (A) PSA responses, according to Shannon Index. (B) RECIST responses, according to Shannon index. (C) PSA-PFS, according to Shannon index [HR 0.67, 95%CI 0.23–1.99, P=0.44]. (D) PFS, according to Shannon index [HR 0.43, 95%CI 0.15–1.22, P=0.11]. (E) OS, according to Shannon index [HR 0.34, 95%CI 0.11–0.99, P=0.07].
Eight patients underwent new metastatic biopsies and were evaluable for PD-L1 status. Five (62%) and 3 men (38%) were PD-L1–positive and -negative, respectively. Representative immunostains are shown in Supplementary Figure 3. There were numerically more PD-L1–positive cases among DRD+ compared to DRD– tumors (80% [4/5] vs. 33% [1/3] respectively, P=0.19). No statistical trends between PD-L1 status and clinical outcomes were observed (Supplementary Table 5, Supplementary Figure 4), although both patients with objective responses (6 and 14) had PD-L1–expressing tumors.
Safety and adverse events
The most common toxicities that developed during or after treatment were fatigue, AST elevation, diarrhea and anorexia (Supplementary Table 6). Seventeen grade 3-4 adverse events occurred in 7 of 15 patients (46%). There were two cases of grade 3-4 fatigue, two cases of grade 3-4 diarrhea/colitis, and two cases of grade 3-4 elevated lipase. Immune-related adverse events were of particular interest. There were five events (affecting 33% of patients) that were possibly or probably related to autoimmune phenomena and that required treatment with corticosteroids: two episodes of colitis, two episodes of pneumonitis, and one episode of hepatitis; hypophysitis was not observed. There were no treatment-related deaths.
DISCUSSION
Prostate cancer expressing AR-V7 represents a lethal phenotype with inadequate treatment options. Here, we report data from the first trial specifically targeting AR-V7-positive disease and the first trial of ipilimumab plus nivolumab in prostate cancer. Although sufficient clinical activity was not observed in the overall study population (and the primary endpoint was not met), encouraging clinical activity using combined immune-checkpoint blockade was seen in the subset of patients harboring germline and/or somatic mutations in DNA-repair genes (and not restricted to mismatch-repair genes). Moreover, there appeared to be a positive correlation between AR-V7 detection and the presence of sequence alterations in DNA-repair genes, further supporting an immunotherapy approach in these patients.
It is now appreciated that approximately 20-25% of metastatic prostate cancers harbor somatic mutations involving DNA-repair genes, primarily homologous-recombination repair genes (e.g. BRCA2, ATM) and, to a lesser extent, mismatch-repair genes (e.g. MSH2, MSH6) [11, 21]. The current study, coupled with our secondary analysis of the StandUp2Cancer dataset, suggests that DNA-repair defects (DRD) may be further enriched in AR-V7-positive prostate cancers with a prevalence approaching 40%. These DRD+ patients may benefit from alternative treatment strategies including poly–ADP-ribose polymerase (PARP) inhibitors [22] or other genetically-targeted approaches [23, 24]. The potential association between AR-V7 detection and DRD mutations has also been suggested by a previous study,[14] but still requires further confirmation.
The correlation between DNA mismatch-repair deficiency (resulting in microsatellite instability) and responsiveness to PD-1 inhibitor therapy is now well established, although MMR mutations are only observed in 2-3% of advanced prostate cancers [9]. Our data suggest that sensitivity to immune-checkpoint inhibitors may perhaps be expanded to other types of DNA-repair alterations, particularly homologous-recombination deficiency (HRD) mutations. Among the six DRD+ patients in this study, five had HRD lesions (three in BRCA2, two in ATM) and one had a nucleotide-excision repair (ERCC4) lesion. Interestingly, mean tumor mutational burden was approximately 2-fold higher in DRD+ versus DRD– cancers, although none of these patients demonstrated microsatellite instability. These findings are consistent with two prior studies (including one in prostate cancer) that reported a modestly higher mutational load in BRCA2-mutant vs. wild-type tumors [25, 26]. Two other studies in BRCA1-deficient breast cancers and BRCA1/2-deficient ovarian cancers, respectively, demonstrated that these tumors may have higher predicted neoantigen loads, more tumor-infiltrating lymphocytes and increased expression of PD-1 and CTLA-4 as compared to their homologous-repair–proficient counterparts [27, 28]. Furthermore, a recent study combining durvalumab (a PD-L1 inhibitor) with olaparib (a PARP inhibitor) in mCRPC patients reported high response rates in men with HRD mutations [29]. Finally, a recent clinical study in advanced urothelial carcinoma suggested that outcomes to PD-1 or PD-L1 inhibitors were superior in patients with vs. without HRD mutations [30]. Taken together, these data imply that HRD alterations, not just MMR alterations, may sensitize patients to immune-checkpoint blockade. In addition, the current study is the first to suggest that defects in nucleotide-excision repair (e.g. ERCC4) may also be associated with immunotherapy sensitivity.
We also observed a trend between high phenotypic CTC heterogeneity (Shannon index) and favorable responses to combination immunotherapy. In addition, DRD+ patients demonstrated a trend towards higher CTC heterogeneity compared to DRD– patients. Previous studies showed that mCRPC patients with Shannon-high CTCs respond poorly to novel hormonal therapies and better to taxane chemotherapies [20]. Interestingly, the two patients with the highest Shannon indices (6 and 14) both had objective tumor responses, both harbored DRD alterations, and both expressed PD-L1. This suggests a theoretical model whereby DRD mutations result in greater genomic heterogeneity, manifesting as greater phenotypic CTC heterogeneity, and increasing the likelihood of a favorable response to immune-checkpoint inhibition. This hypothesis remains to be proven.
In conclusion, our data suggest that the combination of nivolumab plus ipilimumab demonstrates acceptable safety and encouraging efficacy in men with AR-V7-expressing advanced prostate cancer who also harbor DNA-repair alterations, but not in the overall study population. Moreover, the prevalence of these DNA-repair defects appears to be higher in AR-V7-positive patients. Both of these findings require large-scale prospective validation.
MATERIALS AND METHODS
Patient eligibility
Eligible patients had histologically confirmed, progressive, metastatic castration-resistant prostate cancer (mCRPC) with detectable AR-V7 transcripts using the Johns Hopkins CTC-based clinical-grade AR-V7 assay (see below) [15, 16]. Additional eligibility criteria included an ECOG performance-status of 0-1, at least 18 years of age, serum testosterone <50 ng/dL with ongoing androgen-deprivation therapy, adequate organ (liver, kidney, bone marrow) function, and availability of new or archival tumor tissue for biomarker analysis. Key exclusion criteria included a second active malignancy within 5 years, prior immune-checkpoint inhibitor therapy, active brain or meningeal metastases, history of autoimmune disease, or requirement for systemic corticosteroids. Complete eligibility criteria are available in the Supplementary Materials.
Study design
This was a single-institution one-arm open-label phase 2 study conducted at Johns Hopkins. Patients received treatment by intravenous infusion consisting of 3 mg per kilogram of nivolumab plus 1 mg per kilogram of ipilimumab every 3 weeks for 4 doses, followed by a maintenance regimen of 3 mg per kilogram of nivolumab every 2 weeks thereafter. Treatment continued until radiographic progression, unequivocal clinical progression, development of unacceptable toxicity, or withdrawal of consent. Suspected immune-related toxicities were managed using available guidelines. Patients were not permitted to receive nivolumab maintenance therapy unless they tolerated all four doses of combination immunotherapy.
The primary endpoint was the PSA response rate, defined as a ≥50% decline in PSA from baseline maintained for ≥4 weeks. Secondary endpoints included freedom-from-PSA-progression (PSA-progression-free-survival; PSA-PFS), freedom-from-clinical/radiographic-progression (progression-free-survival; PFS), objective response rate (ORR) according to RECIST1.1 criteria [17] in patients with measurable disease, PFS lasting >24 weeks (termed “durable PFS”), and overall survival (OS). PSA-progression was defined as a ≥25% increase in PSA from baseline or nadir, requiring confirmation ≥4 weeks later (PCWG2 criteria [18]). Clinical/radiologic-progression was defined as unequivocal symptomatic progression (worsening disease-related symptoms or new cancer-related complications), or radiographic progression (CT scan showing ≥20% enlargement in sum diameter of soft-tissue target lesions [RECIST1.1]; bone scan showing ≥2 new osseous lesions not related to bone flare) or death, whichever occurred first. Safety and adverse effects were also assessed.
Study assessments were prospectively defined. PSA measurements were obtained at baseline and every 4 weeks on study. Radiographic evaluations (CT of chest/abdomen/pelvis and technetium-99 bone scans) were performed at baseline and every 12 weeks. Physical examination, toxicity assessments, and laboratory studies (complete blood count, comprehensive metabolic panel, thyroid function) were performed every 4 weeks. Safety was assessed by collecting and grading adverse events according to CTCAE v4.0 criteria.
This was an investigator-initiated trial (NCT02601014) designed by the principal investigators (E.S.A. and C.G.D.) and funded by Bristol Myers-Squibb who also provided both study drugs free of cost. The study was approved by the Johns Hopkins University IRB, and was overseen by an independent scientific review committee and an independent data and safety monitoring committee. All patients provided written informed consent before participation.
DNA sequencing
All 15 patients underwent prospective tumor DNA sequencing. Details of targeted next-generation sequencing methods performed on pre-treatment tumor, matched normal and circulating-tumor (ct)DNA samples, and bioinformatic analyses, are provided in the Supplementary Materials. We performed targeted sequencing on 8 matched tumor-normal and 3 tumor-only cases (Supplementary Table 7A). In 4 patients, where tumor tissue was not available, we performed next-generation sequencing of cell-free ctDNA (Supplementary Table 7B). In addition to examining sequence alterations and microsatellite instability, we generated estimates of mutation burden for each tumor. We subsequently focused on sequence alterations in DNA-repair genes, identified somatic and germline variants and assessed allele-specific copy-number and loss-of-heterozygosity events for these loci. Putative pathogenic variants were determined by an ensemble of bioinformatic platforms, as described in the Supplementary Materials. To correlate genomic findings with clinical outcomes, patients were classified as “positive” or “negative” for potentially pathogenic mutations in DNA-repair genes. Patients were considered to be DNA repair-deficient (DRD-positive [DRD+]) if they had at least one pathogenic mutation in a gene involved in DNA-damage repair [22]; otherwise they were classified as DRD-negative (DRD–).
AR-V7 and CTC analyses
A modified AdnaTest assay (Qiagen, Hannover, Germany) conducted in our CLIA-certified laboratory was used to interrogate CTCs for AR-V7 mRNA detection [15], and a positive test was required for eligibility. Briefly, this employs EpCAM-based CTC capture followed by multiplexed reverse-transcription polymerase-chain-reaction (qRT-PCR) using custom primers to detect full-length androgen receptor (AR-FL) mRNA and AR-V7 mRNA, as previously described [2, 15]. In addition, all patients underwent collection of CTCs at baseline using the Epic Sciences platform (San Diego, CA) [19], and these cells were analyzed for phenotypic heterogeneity (Shannon index)[20] and degree of pleomorphism, as described in the Supplementary Materials. Clinical outcomes were compared among patients with high versus low CTC heterogeneity and high versus low pleomorphism.
PD-L1 analysis
In patients undergoing a new metastatic tumor biopsy, expression of PD-L1 protein was assessed using immunohistochemistry (rabbit monoclonal antibody, Ventana, Tucson, AZ), as described in the Supplementary Materials. A positive test was defined as any percentage of PD-L1 staining on tumor cells.
Statistical analyses
The primary endpoint was PSA response, and a response rate above 5% was considered clinically meaningful in this AR-V7-positive population. Accordingly, a sample size of 15 patients with ≥3 PSA responses would produce a 90% confidence interval of 6–44%, which would be above the 5% threshold. A positive study would therefore be defined as ≥3 of 15 patients achieving a PSA response.
Analyses of response endpoints (e.g. PSA response, ORR) were expressed as proportions with 2-sided Wilson binomial 95% confidence intervals. Time-to-event endpoints (e.g. PFS, OS) were analyzed using the Kaplan-Meier method and 95% confidence intervals were generated using the generalized Brookmeyer-Crowley method after log-transformation. Clinical outcomes were compared among patients who were DRD+ and DRD– (primary biomarker analysis), as well as according to other biomarker categories (CTC heterogeneity, CTC pleomorphism, tumor PD-L1 expression). To examine associations between clinical outcomes and biomarker status, response endpoints were compared using Fisher’s exact test, and time-to-event endpoints were compared using the log-rank test with Cox proportional-hazards models to derive hazard ratios. All tests were two-sided, and P values ≤0.05 were considered significant; we did not correct for multiple hypotheses. Statistical analyses were performed using R (version 3.4.3).
ACKNOWLEDGMENTS
We thank Arul Chinnaiyan (and the East Coast/International Dream Team investigators) for providing DNA and RNA sequencing data from the StandUp2Cancer metastatic tumor cohort for reanalysis. We are also grateful to all of the patients and their families who participated in this clinical trial.
CONFLICTS OF INTEREST
E.S.A. is a paid consultant/advisor to Janssen, Astellas, Sanofi, Dendreon, Medivation, ESSA, AstraZeneca, Clovis and Merck; has received research funding to his institution from Janssen, Johnson & Johnson, Sanofi, Dendreon, Genentech, Novartis, Tokai, Bristol Myers-Squibb, AstraZeneca, Clovis and Merck; and is the co-inventor of a biomarker technology that has been licensed to Qiagen. S.A.T. has served as a consultant and received honoraria from Roche/ Ventana Medical Systems, Almac Diagnostics, Janssen, AbbVie and Astellas/Medivation; he is also a co-founder of, consultant for and Laboratory Director of Strata Oncology. Y.W., A.J., R.P.G. and R.D. are employees of Epic Sciences. V.E.V. is a founder of Personal Genome Diagnostics (PGDx), is a member of its Scientific Advisory Board and Board of Directors, and owns PGDx stock, which is subject to certain restrictions under university policy; he is also on the Scientific Advisory Board for Ignyta; the terms of these arrangements are managed by the Johns Hopkins University in accordance with its conflict of interest policies. The remaining authors disclose no relevant conflicts of interest.
FUNDING
This research was sponsored by Bristol-Myers Squibb (Princeton, NJ) who also provided both study drugs free of cost. This research was also partially supported by National Institutes of Health grants P30 CA006973 (E.S.A) and R01 CA185297 (J.L. and E.S.A.), Department of Defense grants W81XWH-13-PCRP-CCA (M.A.C. and E.S.A) and W81XWH-15-2-0050 (J.L.), and the Bloomberg-Kimmel Institute for Cancer Immunotherapy (E.S.A.). Genomic studies were partially supported by National Institutes of Health grant R01 CA121113 (V.A. and V.E.V.), and the Commonwealth Foundation (V.E.V.).
REFERENCES
1. Antonarakis ES, Armstrong AJ, Dehm SM, Luo J. Androgen receptor variant-driven prostate cancer: clinical implications and therapeutic targeting. Prostate Cancer Prostatic Dis. 2016; 19:231–41. https://doi.org/10.1038/pcan.2016.17.
2. Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, Lotan TL, Zheng Q, De Marzo AM, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014; 371:1028–38. https://doi.org/10.1056/NEJMoa1315815.
3. Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, Zhu Y, Silberstein JL, Taylor MN, Maughan BL, Denmeade SR, Pienta KJ, Paller CJ, Carducci MA, et al. Clinical significance of androgen receptor splice variant-7 mRNA detection in circulating tumor cells of men with metastatic castration-resistant prostate cancer treated with first- and second-line abiraterone and enzalutamide. J Clin Oncol. 2017; 35:2149–56. https://doi.org/10.1200/JCO.2016.70.1961.
4. Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, Nakazawa M, Nadal R, Paller CJ, Denmeade SR, Carducci MA, Eisenberger MA, Luo J. Androgen receptor splice variant 7 and efficacy of taxane chemotherapy in patients with metastatic castration-resistant prostate cancer. JAMA Oncol. 2015; 1:582–91. https://doi.org/10.1001/jamaoncol.2015.1341.
5. Scher HI, Lu D, Schreiber NA, Louw J, Graf RP, Vargas HA, Johnson A, Jendrisak A, Bambury R, Danila D, McLaughlin B, Wahl J, Greene SB, et al. Association of AR-V7 on circulating tumor cells as a treatment-specific biomarker with outcomes and survival in castration-resistant prostate cancer. JAMA Oncol. 2016; 2:1441–49. https://doi.org/10.1001/jamaoncol.2016.1828.
6. Topalian SL. Targeting immune checkpoints in cancer therapy. JAMA. 2017; 318:1647–48. https://doi.org/10.1001/jama.2017.14155.
7. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015; 373:23–34. https://doi.org/10.1056/NEJMoa1504030.
8. Hammers HJ, Plimack ER, Infante JR, Rini BI, McDermott DF, Lewis LD, Voss MH, Sharma P, Pal SK, Razak AR, Kollmannsberger C, Heng DY, Spratlin J, et al. Safety and efficacy of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma: the CheckMate 016 study. J Clin Oncol. 2017; 35:3851–58. https://doi.org/10.1200/JCO.2016.72.1985.
9. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, Wong F, Azad NS, Rucki AA, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017; 357:409–13. https://doi.org/10.1126/science.aan6733.
10. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, Watkins TB, Shafi S, Murugaesu N, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016; 351:1463–69. https://doi.org/10.1126/science.aaf1490.
11. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, Beltran H, Abida W, Bradley RK, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015; 161:1215–28. https://doi.org/10.1016/j.cell.2015.05.001. Erratum in: Cell. 2015; 162:454.
12. Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G, Ganju V, Polikoff J, Saad F, Humanski P, Piulats JM, Gonzalez Mella P, Ng SS, et al. Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J Clin Oncol. 2017; 35:40–47. https://doi.org/10.1200/JCO.2016.69.1584.
13. Hansen A, Massard C, Ott PA, Haas N, Lopez J, Ejadi S, Wallmark J, Keam B, Delord JP, Aggarwal R, Gould M, Qiu P, Saraf S, et al. Pembrolizumab for patients with advanced prostate adenocarcinoma: preliminary results from the KEYNOTE-028 study. Ann Oncol. 2016 (suppl_6); 27:vi243–65. https://doi.org/10.1093/annonc/mdw372.09.
14. Joshi H, Pinski JK. Association of AR-V7 expression with molecular and clinical characteristics in prostate cancer. J Clin Oncol. 2016; 34:109.
15. Lokhandwala PM, Riel SL, Haley L, Lu C, Chen Y, Silberstein J, Zhu Y, Zheng G, Lin MT, Gocke CD, Partin AW, Antonarakis ES, Luo J, Eshleman JR. Analytical validation of androgen receptor splice variant 7 detection in a Clinical Laboratory Improvement Amendments (CLIA) laboratory setting. J Mol Diagn. 2017; 19:115–25. https://doi.org/10.1016/j.jmoldx.2016.08.003.
16. Markowski MC, Silberstein JL, Eshleman JR, Eisenberger MA, Luo J, Antonarakis ES. Clinical utility of CLIA-grade AR-V7 testing in patients with metastatic castration-resistant prostate cancer. JCO Precis Oncol. 2017; 1–9. https://doi.org/10.1200/PO.17.00127.
17. Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000; 92:205–16. https://doi.org/10.1093/jnci/92.3.205.
18. Scher HI, Halabi S, Tannock I, Morris M, Sternberg CN, Carducci MA, Eisenberger MA, Higano C, Bubley GJ, Dreicer R, Petrylak D, Kantoff P, Basch E, et al, and Prostate Cancer Clinical Trials Working Group. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008; 26:1148–59. https://doi.org/10.1200/JCO.2007.12.4487.
19. Werner SL, Graf RP, Landers M, Valenta DT, Schroeder M, Greene SB, Bales N, Dittamore R, Marrinucci D. Analytical validation and capabilities of the Epic CTC platform: enrichment-free circulating tumour cell detection and characterization. J Circ Biomark. 2015; 4:3. https://doi.org/10.5772/60725.
20. Scher HI, Graf RP, Schreiber NA, McLaughlin B, Jendrisak A, Wang Y, Lee J, Greene S, Krupa R, Lu D, Bamford P, Louw JE, Dugan L, et al. Phenotypic heterogeneity of circulating tumor cells informs clinical decisions between AR signaling inhibitors and taxanes in metastatic prostate cancer. Cancer Res. 2017; 77:5687–98. https://doi.org/10.1158/0008-5472.CAN-17-1353.
21. Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, Danila D, Rathkopf D, Morris M, Slovin S, McLaughlin B, Curtis K, Hyman DM, et al. Prospective genomic profiling of prostate cancer across disease states reveals germline and somatic alterations that may affect clinical decision making. JCO Precis Oncol. 2017; 1–16. https://doi.org/10.1200/PO.17.00029.
22. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, Boysen G, Porta N, Flohr P, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015; 373:1697–708. https://doi.org/10.1056/NEJMoa1506859.
23. Teply BA, Antonarakis ES. Treatment strategies for DNA repair-deficient prostate cancer. Expert Rev Clin Pharmacol. 2017; 10:889–98. https://doi.org/10.1080/17512433.2017.1338138.
24. Schweizer MT, Antonarakis ES. Prognostic and therapeutic implications of DNA repair gene mutations in advanced prostate cancer. Clin Adv Hematol Oncol. 2017; 15:785–95.
25. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, Seja E, Lomeli S, Kong X, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016; 165:35–44. https://doi.org/10.1016/j.cell.2016.02.065. Erratum in: Cell. 2017; 168:542.
26. Taylor RA, Fraser M, Livingstone J, Espiritu SM, Thorne H, Huang V, Lo W, Shiah YJ, Yamaguchi TN, Sliwinski A, Horsburgh S, Meng A, Heisler LE, et al. Germline BRCA2 mutations drive prostate cancers with distinct evolutionary trajectories. Nat Commun. 2017; 8:13671. https://doi.org/10.1038/ncomms13671.
27. Nolan E, Savas P, Policheni AN, Darcy PK, Vaillant F, Mintoff CP, Dushyanthen S, Mansour M, Pang JB, Fox SB, Perou CM, Visvader JE, Gray DHD, and Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab). Combined im mune checkpoint blockade as a therapeutic strategy for BRCA1-mutated breast cancer. Sci Transl Med. 2017; 9. https://doi.org/10.1126/scitranslmed.aal4922.
28. Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, Garber JE, Chowdhury D, Wu CJ, D’Andrea AD, Matulonis UA, Konstantinopoulos PA. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget. 2016; 7:13587–98. https://doi.org/10.18632/oncotarget.7277.
29. Karzai F, Madan RA, Owens H, Couvillon A, Hankin A, Williams M, Bilusic M, Cordes LM, Trepel JB, Killian K, Meltzer PS, Gulley JL, Lee JM, Dahut WL. A phase 2 study of olaparib and durvalumab in metastatic castrate-resistant prostate cancer (mCRPC) in an unselected population. J Clin Oncol. 2018; 36:163. https://doi.org/10.1200/JCO.2018.36.6_suppl.163.
30. Teo MY, Seier K, Ostrovnaya I, Regazzi AM, Kania BE, Moran MM, Cipolla CK, Bluth MJ, Chaim J, Al-Ahmadie H, Snyder A, Carlo MI, Solit DB, et al. Alterations in DNA damage response and repair genes as potential marker of clinical benefit from PD-1/PD-L1 blockade in advanced urothelial cancers. J Clin Oncol. 2018; 36:1685-1694. https://doi.org/10.1200/JCO.2017.75.7740.