
INTRODUCTION
Colorectal cancer (CRC) is the third most common and lethal cancer in the United States [1]. Diet is one of the strongest influential risk factors in the development of colorectal cancer. Thus, dietary intervention has been suggested to be an effective way to decrease colorectal cancer development and mortality [2–4]. Many studies have observed a beneficial or protective effect toward colorectal cancer, although other studies have reported contradicted these findings [5–8]. The fermentation of dietary fiber in the proximal colon produces bacterial derived-short chain fatty acids (SCFAs), which include acetate, propionate and butyrate. Among these SCFAs, butyrate has been considered a critical metabolite that mediates the tumor repressive effect of dietary fiber toward colorectal cancer [9–11].
Unlike other SCFAs, butyrate is primarily metabolized by colonocytes as an energy source and also plays a role in epigenetic modification where it increases histone acetylation through inhibiting histone deacetylases [12–14]. At physiologically relevant doses, butyrate decreases cell proliferation, increases cell differentiation and induces apoptosis in colorectal cancer cells [15–17]. Interestingly, butyrate’s effects on cell proliferation and apoptosis are associated with its metabolic fate in colonocytes [18, 19]. Several studies have shown that butyrate is primarily oxidized by isolated colonocytes, and that this oxidation can be influenced by exogenous factors [20–22]. Additionally, butyrate has been shown to decrease proliferation in cancerous colonocytes and increase proliferation in non-cancerous colonocytes [21, 23–25]. Thus, butyrate produces different effects on cancerous and normal colonocytes, which have different metabolic characteristics. In normal colonocytes, butyrate is oxidized through mitochondrial β-oxidation and is utilized to produce energy through the tricarboxylic acid (TCA) cycle or cytosolic acetyl-CoA. The acetyl-CoA produced in this process can also be used as a cofactor for histone acetyltransferases (HATs) or as a substrate for lipogenesis [18, 26]. However, cancerous colonocytes favor glucose over butyrate utilization as a result of a metabolic transformation called the Warburg effect. This glucose-preferred environment results in suppressed butyrate oxidation, and enhanced histone acetylation induced by butyrate [18, 19]. Therefore, to understand butyrate’s inhibitory and selective effects against colorectal cancer, it is essential to understand the mechanisms that contribute to decreased butyrate oxidation in cancerous colonocytes.
Short chain acyl-CoA dehydrogenase (SCAD) is an enzyme that catalyzes the first step in the oxidation of butyrate in the mitochondria [27]. Deletion of short chain acyl-CoA dehydrogenase in humans and mice decreases butyrate oxidation whiles simultaneously increasing butyryl-CoA accumulation and excretion, thereby illustrating the enzyme’s role in butyrate metabolism [28–30]. Additionally, loss of SCAD in isolated colonocytes results in a decrease in the oxidation of butyrate [31]. Previous studies have observed a reduction in SCAD expression at both gene and protein level in colorectal cancer [32–35]. However, there little is known about the mechanisms that regulate SCAD expression. In this study, we report that butyrate decreases its own metabolism in colorectal cancer cells. Pretreatment of colorectal cancer cells with butyrate suppresses SCAD expression. The mechanism as to how butyrate lowers SCAD levels in colorectal cancer cells is explored and is mediated through inhibition of a histone deacetylase. Importantly, non-cancerous colonocytes do not show this effect. This work identifies butyrate itself as part of the mechanism by which colorectal cancer cells shift away from utilizing butyrate.
RESULTS
Butyrate diminishes its own oxidation in colorectal cancer cells
In recent years, our lab has sought to identify factors or conditions that impact the oxidation of butyrate in colorectal cancer cells. Since butyrate modulates gene expression through inhibiting histone deacetylases, we postulated that butyrate could directly affect its own metabolic fate through changing the expression of proteins that oxidize short chain fatty acids. To begin to test this possibility, HCT116 colorectal cancer cells were pretreated with or without butyrate, and butyrate oxidation was measured as the percent change in the oxygen consumption rate (Figure 1A). In the assay, cells are maintained in a Fatty Acid Oxidation Media containing KHB buffer and only 2.5 mM glucose as the only exogenous carbon substrate. During the assay butyrate, 2-deoxyglucose (2DG), etomoxir, and sodium azide are injected into the media. The 2DG blocks glucose utilization, thus leaving butyrate as the sole exogenous energetic substrate. Etomoxir, inhibits carnitine palmitoyltransferase and the contribution of endogenous lipids to oxidative metabolism. Butyrate oxidation represents the total area under the curve following the addition of 2DG to the time when sodium azide is injected. In pretreated cells, butyrate repressed its own oxidation when compared to non-pretreated cells (Figure 1B). After 2DG injection, butyrate oxidation was significantly lower in pretreated cells as compared to non-pretreated control cells (Figure 1C). RKO cells were used to confirm whether butyrate pretreatment would decrease butyrate oxidation in an additional colorectal cancer line. Indeed, pretreatment with butyrate in RKO cells significantly diminished butyrate oxidation as compared to cells not pretreated with butyrate (Supplementary Figure 1). This suggested that the ability of butyrate to suppress its own oxidation is not limited to a single colorectal cancer cell line.
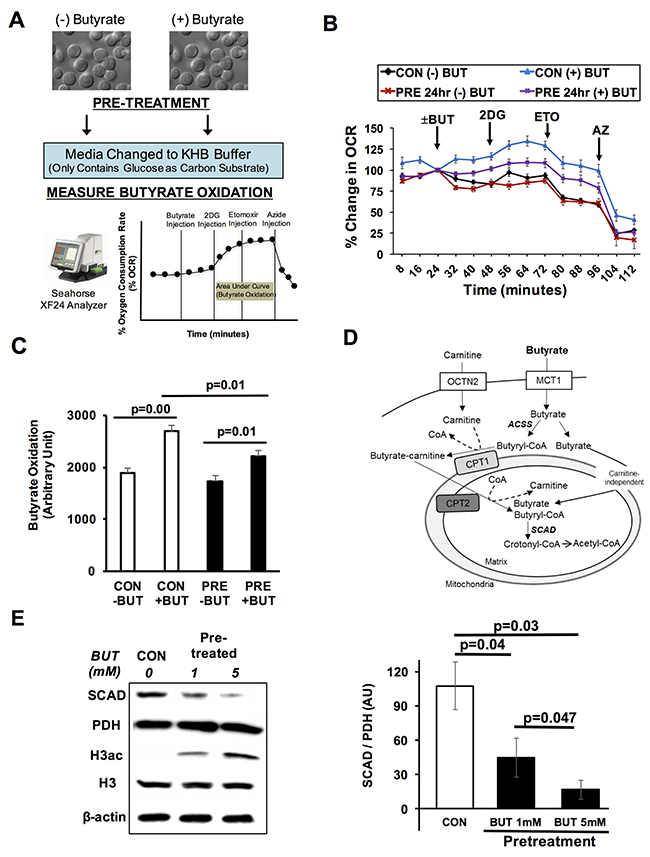
Figure 1: Butyrate suppresses its own oxidation in colorectal cancer cells. (A) Schematic shows butyrate pre-pretreatment and measurement of butyrate oxidation. (B) Percentage change in oxygen consumption rate (OCR) relative to baseline in pre-treated HCT116 cells with and without butyrate (5 mM, 24 hrs). (C) Area under the curve measurement from OCR analysis taken after 2-deoxyglucose (2DG) injection but before azide injection (56-104 min). (D) Diagram of butyrate oxidation in the cancerous colonocytes. Cancerous colonocytes oxidize butyrate in the mitochondria. SCAD plays a role in first step of the oxidation of butyrate. (E) Western blot confirming butyrate reduced the SCAD level. Quantification of western blots is shown in right panel. Data for western blots and butyrate oxidation represent the average of 3-5 replicates per condition. Error bars are ± SEM. MCT1; monocarboxylate transport protein 1, OCTN2; organic cation/carnitine transporter, ACSS; Acyl Co-A synthetases, CPT1/2; carnitine palmitoyltransferase 1/2, SCAD; short chain acyl-CoA dehydrogenase.
After butyrate moves into the mitochondria, short chain acyl-CoA dehydrogenase (SCAD) catalyzes the first dehydrogenation step where butyryl-CoA is metabolized to form acetyl-CoA [27] (Figure 1D). Western blot analysis of HCT116 colorectal cancer cells showed that butyrate treatment decreased the levels of SCAD (Figure 1E). Butyrate dose-dependently lowered the expression of SCAD. Since butyrate is known to increase global histone acetylation, H3 acetylation was also measured to confirm the cells were responding to butyrate as expected. Butyrate dose-dependently increasing histone H3 acetylation. Throughout this paper (unless otherwise mentioned), pyruvate dehydrogenase (PDH) is used as a loading control instead of β-Actin due to the fact that PDH is also a mitochondrial protein like SCAD. Moreover, by normalizing with PDH, it separates changes in SCAD expression from alterations in mitochondrial number.
Butyrate pretreatment does not affect its own oxidation in isolated colonocytes
Non-cancerous colonocytes efficiently oxidize butyrate, and it is unclear whether pretreatment with butyrate would affect this process. In addressing this issue, colonocytes where isolated from adult mice, and pretreated with and without butyrate. Butyrate oxidation was measured as before in colonocytes after pretreatment. Butyrate increased the oxygen consumption rate to the same level after 2DG addition in both experimental conditions, non-pretreated and pretreated (Figure 2A). As opposed to colorectal cancer cells, pretreatment with butyrate did not show decreased oxidation when compared to non-pretreated colonocytes (Figure 2B). Thus, the ability of butyrate to decrease its own oxidation appears to be selective to colorectal cancer cells. Although, these future studies are needed to confirm this is the case in primary human colonocytes.
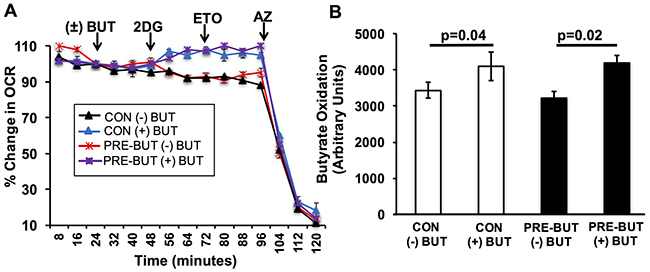
Figure 2: Butyrate has no effect on its oxidation in primary colonocytes. (A) Percentage change in oxygen consumption rate (OCR) relative to baseline in isolated colonocytes treated with and without butyrate. (B) Area under the curve measurement from OCR analysis taken after 2-deoxyglucose (2DG) injection but before azide injection. Data for butyrate oxidation represent the average of 5 replicates per condition. Error bars are ± SEM. p<0.05 indicates significant difference between groups.
Butyrate reduces SCAD expression only in colorectal cancer cells
Butyrate decreased SCAD levels in colorectal cancer cells; however, it wasn’t clear whether butyrate would have this same effect in non-cancerous colonocytes? Cancerous colonocytes have a reduced ability to oxidize butyrate compared to non-cancerous colonocytes, which is in part due to lower cellular carnitine levels, and decreased organic cation/carnitine transporter (OCTN2) and carnitine palmitoyltransferase (CPT1A) [36]. In addition, the diminished ability of cancerous colonocytes to oxidize butyrate may be due to the lowered SCAD levels compared to the non-cancerous colonocytes. Therefore, we compared SCAD levels in cancerous (HCT116 cells) and non-cancerous (FHC cells). colonocyte lines. To our surprise, SCAD expression was actually higher in HCT116 cells than FHC cells (Figure 3A). This may be due to the fact that the expression and activity of enzymes related to fatty acid metabolism (i.e. SCAD) are dramatically elevated after birth in tissues where strongly metabolize fatty acids [37]. Since FHC cells originated from a 13-week fetus, these cells may not be a good representative cell line for non-cancerous colonocytes. However, Kaiko et al. (2011) confirmed that primary colonocytes (non-cancerous) have high SCAD expression compared to the stem cells in the colonic crypts [31].
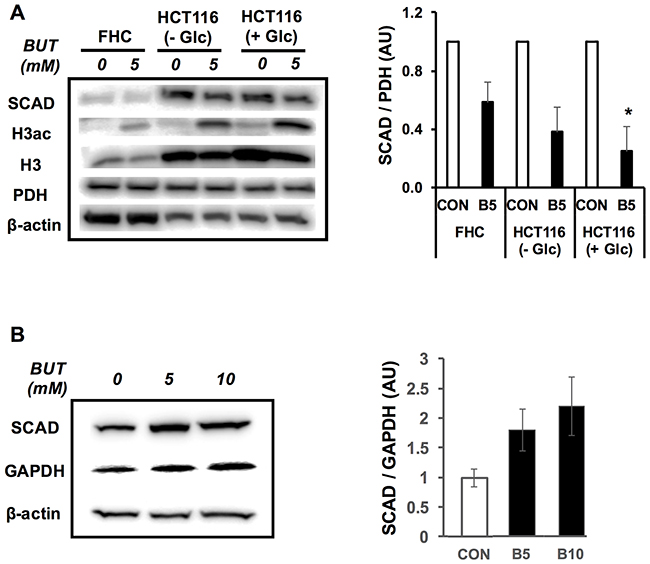
Figure 3: Butyrate decreases SCAD expression in cancerous but not primary colonocytes. (A) Western blot showing SCAD expression in FHC cells and HCT116 cells that were treated with (0 mM, CON) or without butyrate (5 mM, B5). HCT116 cells were grown under the absence (2.5 mM glucose) and presence (25 mM glucose) for the Warburg effect. Right panel shows quantification of SCAD levels relative to PDH levels. (B) Western blot showing SCAD levels in isolated normal colonocytes with butyrate (0 mM, CON; 5 mM, B5 and 10 mM. B10). Quantification of SCAD expression relative to GAPDH is shown in right panel. For statistical analysis, western blot was conducted 5 times per condition. Error bars are Mean ± SEM. *p<0.05 indicates significant difference between cells treated with butyrate vs. controls.
As opposed to non-cancerous colonocytes, cancerous colonocytes increase aerobic glycolysis by utilizing the Warburg effect, which may alter the expression of proteins such as SCAD and negatively impact the oxidation of butyrate. To directly test the importance of the Warburg effect in butyrate-mediated SCAD reduction, the glucose concentration in the media was manipulated to high or low. SCAD expression was observed in the HCT116 cells under the high and low glucose conditions treated with butyrate (Figure 3A and Supplementary Figure 2). Importantly, SCAD levels were only significantly decreased under with the high glucose condition. To test whether butyrate decreases SCAD levels in cells not undergoing the Warburg effect, non-cancerous colonocytes were isolated from mice and treated with butyrate. Non-cancerous isolated colonocytes treated with butyrate showed a trend toward increasing SCAD levels, although this effect was not significant (Figure 3B). SCAD expression was normalized to GAPDH. Butyrate levels in the colonic lumen can range from 0.5mM to as high as 20mM in the colonic lumen [38–40]. Since no decrease in SCAD levels was observed at 5 mM butyrate, the concentration was increased to 10 mM, which is still within the physiological range. Nevertheless, there was no significant effect on SCAD expression even at 10 mM butyrate.
To confirm whether butyrate suppressed SCAD at the mRNA level, we conducted qRT-PCR and measured Scad1, Scad2, and total Scad. Scad1 and Scad2 are products of alternative splicing of the Scad gene. Butyrate, at 5 mM, significantly decreased mRNA levels for Scad1, Scad2, and total Scad in HCT116 colorectal cancer cells (Figure 4). Additionally, at a low butyrate concentration (1 mM), Scad2 and total Scad mRNA were also significantly diminished. These data suggest that butyrate suppresses SCAD transcription and gene expression in colorectal cancer cells, which may involve inhibition of histone deacetylases.
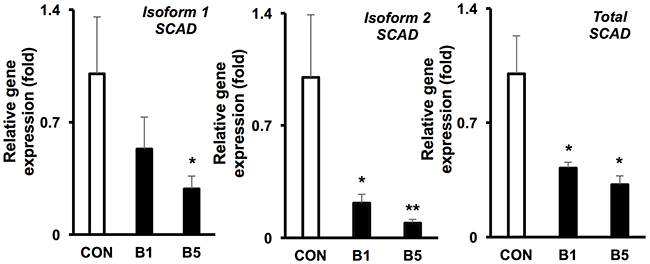
Figure 4: Butyrate reduces the mRNA levels of SCAD in colorectal cancer cell. mRNA expression of isoform 1, isoform 2 and total Scad was evaluated by semi-quantitative RT-PCR (0 mM, CON; 5 mM, B5 and 10 mM, B10). The relative mRNA level was normalized to 18S rRNA and shown as fold of the control value. For statistical analysis, qRT-PCR was conducted three times per condition. Error bars are Mean ± SEM. *p<0.05 and **p<0.01 indicates significant difference between cells treated with butyrate vs. controls.
Butyrate lowers SCAD expression as an HDAC inhibitor
The fermentation of dietary fiber in the colon produces additional SCFAs, which include acetate and propionate. Therefore, we wanted to investigate whether these other SCFAs reduced SCAD expression in colorectal cancer cells like butyrate. Both propionate and butyrate significantly decreased SCAD expression, while acetate did not impact SCAD expression in HCT116 colorectal cancer cells (Figure 5A). Similar to butyrate, propionate also inhibits histone deacetylases [41]. This alludes to HDAC inhibition as a key component in regulating SCAD expression in colorectal cancer cells.
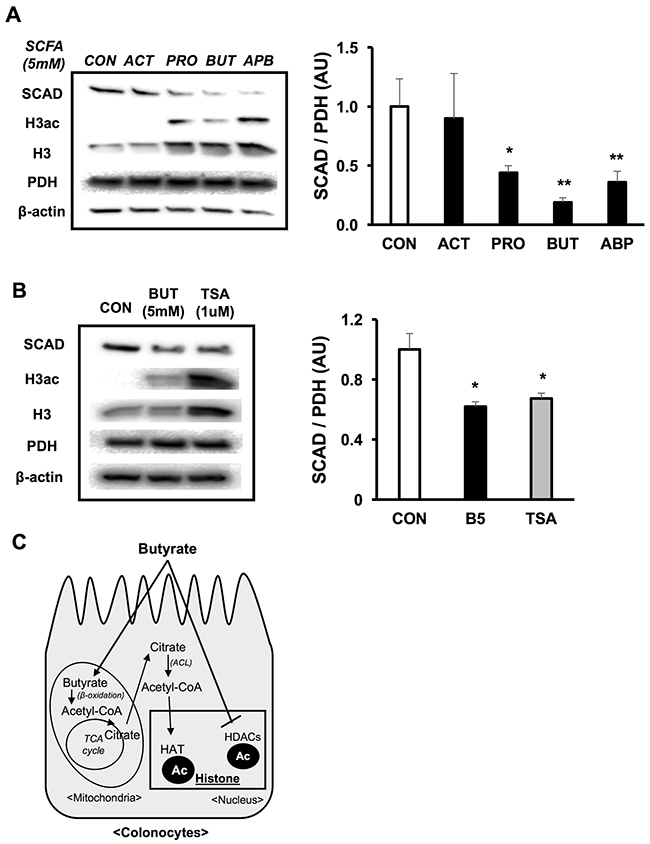
Figure 5: Butyrate decreases SCAD as an HDAC inhibitor. (A) Western blot showing SCAD expression in HCT116 cells that were treated with a set of SCFAs (ACT; acetate, PRO; propionate, BUT; butyrate and APB; all of SCFAs) at 5 mM. Right panel shows quantification of SCAD levels relative to PDH levels. (B) Western blot describing SCAD expression in HCT116 cells that were treated with butyrate (5 mM) or TSA (1 μM). Quantification of SCAD expression relative to PDH levels is shown in right panel. (C) Diagram showing epigenetic mechanisms as to how butyrate can modulate gene expression. Butyrate acts as a co-factor for histone acetyltransferases (HATs) through the involvement of ATP-citrate lyase (ACL). Also, butyrate directly inhibits histone deacetylases (HDACs). For statistical analysis, western blot was conducted 3 times per condition. Error bars are Mean ± SEM. *p<0.05 and **p<0.01 indicates significant difference between cells treated with butyrate vs. controls.
Butyrate is involved in epigenetic modifications through two mechanisms (Figure 5C) [18]. Mitochondrial butyrate oxidation results in the biogenesis of acetyl-CoA. Acetyl-CoA can be utilized as a cofactor for histone acetyltransferases (HATs) when regenerated in the cytosol via ATP-citrate lyase (ACL). In addition, butyrate directly goes into the nucleus where it functions as an HDAC inhibitor. First, to test the importance of HDAC inhibition, a structurally distinct HDAC inhibitor, trichostatin A (TSA), was used as a positive control. Both butyrate and TSA significantly reduced SCAD expression (Figure 5B). Next, a siRNA knockdown of ACL was performed and SCAD expression was evaluated with and without butyrate. Since ACL catalyzes the reaction that converts citrate into acetyl-CoA in the cytosol, an ACL knockdown would block butyrate’s involvement as a HAT cofactor. However, knockdown of ACL did not impact SCAD suppression induced by butyrate, indicating that this mechanism is unrelated to SCAD regulation. Moreover, there was no difference in the percentage change in OCR between siMock and siACL transfected cells (Supplementary Figure 3A and 3B). In addition, butyrate still significantly reduced SCAD expression in both siMock and siACL knockdown, suggesting that ACL was not an important mediator in the decreased SCAD levels (Supplementary Figure 3C). Taken together, this data point to inhibition of histone deacetyases as the major mechanism as to how butyrate diminishes SCAD expression in the colorectal cancer cells.
Butyrate decreases SCAD levels through selective inhibition of HDAC1
In colorectal cancer, HDAC 1, HDAC 2, and HDAC 3 are highly expressed in order to accelerate cell proliferation, growth and survival [42, 43]. Butyrate effectively inhibits most HDACs, which results in decreased cell proliferation and induction of cell apoptosis in the cancer cells [15, 18]. Since butyrate along with other HDAC inhibitors decreased SCAD levels, it was unclear whether a selective HDAC was responsible for the effect. Therefore, HDAC1, HDAC2, or HDAC3 were knocked down using RNAi and these cells were treated with butyrate to test whether SCAD reduction is augmented by a selective HDAC knockdown condition. While butyrate and TSA significantly decreased SCAD levels in siMock cells, siHDAC1 transfected cells did not show decreased SCAD levels after butyrate treatment (Figure 6A). Scad mRNA levels were also measured to confirm that knockdown of HDAC1 inhibited butyrate’s ability to reduce Scad gene expression. Butyrate was no longer able to decrease Scad mRNA levels when HDAC1 was knocked down (Figure 6B). RNAi knockdown of HDAC2 and HDAC3 in HCT116 cells did not impact SCAD levels like as HDAC1 (Supplementary Figure 4). These findings demonstrate that butyrate targets HDAC1 to decrease SCAD levels in the colorectal cancer cells. These data also suggest that although butyrate is a general HDAC inhibitor, its effects can be mediated through a specific HDAC.
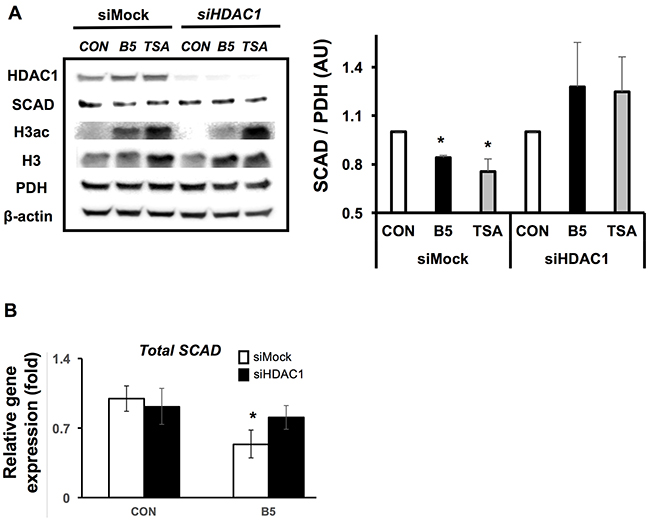
Figure 6: Butyrate inhibits HDAC 1 to reduce SCAD expression. (A) Western blot showing SCAD expression for siMock and siHDAC1 transfected HCT116 cells after treatment with or without butyrate (5 mM) or TSA (1 μM) for 6hrs. Right panel shows quantification of SCAD levels relative to PDH levels. (B) mRNA expression of total Scad was evaluated by semi-quantitative RT-PCR. The relative mRNA level was normalized to 18S rRNA and shown as fold of the control value. For statistical analysis, qRT-PCR was conducted three times per condition. Error bars are Mean ± SEM. *p<0.05 indicates significant difference between cells treated with butyrate vs. controls.
DISCUSSION
Normal colonocytes prefer to oxidize butyrate as a primary energy source whereas cancerous colonocytes increase glucose utilization. This is demonstrated through increased fluorodeoxyglucose (FDG) uptake as measured by positron emission tomography – computed tomography (PET-CT) in colorectal cancer patients [44–47]. In addition, colorectal cancer cells display a reduced capacity to oxidize butyrate compared to normal colonocytes, which could result from diminished intracellular carnitine and CPT1A levels [36]. These changes in metabolism significantly impact butyrate’s role as an inhibitor of histone deacetylases in colorectal cancer cells [18]. As an HDAC inhibitor, butyrate represses cell proliferation and induces cell death in cancer cells [16, 48, 49]. Thus, gaining knowledge toward the mechanisms that regulate butyrate oxidation in cancer cells is an important step in understanding butyrate’s role as an HDAC inhibitor, which is associated with the molecule’s protective action toward colorectal cancer. Here, it is demonstrated that butyrate suppresses its own oxidation in cancerous colonocytes. Butyrate diminishes SCAD levels, a critical enzyme involved in its oxidation. Specifically, butyrate regulates SCAD expression through inhibition of histone deacetylase 1 (HDAC1). The fact that butyrate increases histone acetylation, which is generally associated with upregulated gene expression might suggest that decreased SCAD levels observed after butyrate treatment most likely involves a secondary mechanism. An upregulation of a negative co-regulator of SCAD is a possibility.
Many studies have previously reported a role of SCAD in regulating butyrate oxidation [27, 28, 30]. SCAD contributes to energy metabolism in the colon via participating in SCFA oxidation [29]. Recently, Kaiko et al. reported that colonocytes isolated from SCAD-/- mice showed diminished butyrate oxidation compared to those from normal mice (SCAD+/+), which confirms the importance of SCAD in mediating butyrate oxidation in colonocytes [31]. In contrast to this study, a cancerous colonocyte cell line was used due to previous reports demonstrating diminished butyrate oxidation in this cell line [18, 36]. The fact that butyrate reduces its own oxidation in these cells reveals a potential mechanism as to why colorectal cancer cells are sensitive to butyrate’s HDAC inhibitory effects.
This mechanistic relationship between butyrate and SCAD in the colorectal cancer cells was significantly influenced by the Warburg effect. Previously, it was found that butyrate (1 mM) increased SCAD expression in a colorectal cancer cell line (HT15). However, similar to a non-cancerous cell line, these cells still preferentially utilized butyrate over glucose thereby negating the impact of the Warburg effect [50]. The colonic administration of butyrate in healthy subjects increases gene transcription relating to energy metabolism and fatty acid metabolism, while SCAD was not altered [51]. Germfree mice, which lack a microbiome and the capability to produce butyrate from fiber, show reduced SCAD expression compared to normal mice [12]. These findings are consistent with butyrate modulating SCAD levels differently in non-cancerous colonocytes. However, more studies will be needed to confirm whether this holds true in non-cancerous human colonocytes since the primary colonocytes used in this study were from mice. Butyrate infusion studies in humans, which also reported no change in SCAD mRNA levels in colon [52].
In general, butyrate inhibits most HDACs, except class 2 (HDAC6 and 10) and class 3, and specifically inhibits HDAC1 and 3 in colorectal cancer cells [15, 53]. As an HDAC inhibitor, butyrate effectively impedes cancer cell survival and growth [15, 54]. Butyrate suppresses intestinal inflammation and oxidative stress, while protecting the intestinal epithelial barrier via its HDAC inhibitor role; these effects likely help reduce CRC susceptibility [55–57]. Separate from the non-cell autonomous effects, butyrate availability can influence cancerous colonocyte metabolism and HDAC inhibition. The suppressive action of butyrate on SCAD expression and its own oxidation in the colorectal cancer cells is mediated by its function as an HDAC inhibitor. Butyrate specifically inhibits HDAC1 to have these regulatory actions as knockdown of this protein negates any change in SCAD caused by butyrate. These data also allude to butyrate promoting its own action as an HDAC inhibitor in colorectal cancer cells through decreasing its own metabolism. It will be interesting to conduct further research regarding why butyrate behaves in this way, and whether this mechanism helps mediate its specificity toward cancer cells. Colorectal cancer (all stages) has altered gene expression characterized by lower mitochondrial metabolism and a down-regulation in SCAD expression [32, 33]. Future studies are needed to investigate the outcome of reduced SCAD expression in cancerous colonocytes especially as it relates to the Warburg effect and diminished butyrate oxidation.
The oncological significance of this work relates to the fact that colorectal cancer is, in part, a metabolic disease, where alterations in metabolic pathways are key to tumor development and progression. One important metabolic shift involves the cancer cell’s switch from utilizing butyrate as its primary energetic substrate to adopting a more glycolytic phenotype characterized by enhanced glucose uptake. This work provides insight into the mechanisms that impact this metabolic shift. As demonstrated that butyrate suppresses its own oxidation in colorectal cancer cells, which is associated with HDAC inhibition regulating SCAD. Reversing the colorectal cell’s glycolytic phenotype and forcing metabolism back toward butyrate utilization represents an unexplored therapeutic strategy.
MATERIALS AND METHODS
Cell culture and siRNA transfection
HCT116 cell (ATCC, CCL-247) were grown in DMEM supplemented with 25mM glucose and 10% FBS. FHC cells (ATCC, CRL-1831) were grown in complete growth DMEM:F12 medium following the recommended recipe from ATCC with 20% FBS instead of 10%. Specifically, the following reagents were added to the DMEM:F12 medium; HEPES (final concentration of 25 mM), 10 ng/mL of cholera toxin, 0.005 mg/mL of insulin, 0.005 mg/mL of transferrin, and 100 ng/mL of hydrocortisone. RNAi transfection in HCT116 cells was performed as previously described [18], and siRNA pools for human ACL (Dharmacon, #L-004915-00), human HDAC1 (Dharmacon, #L-003493-00-0005), human HDAC2 (Dharmacon, #L-003495-02-0005), human HDCA3 (Dharmacon, #L-003496-00-0005) and non-targeting control (Dharmacon, D001810-01-05) were used at a 20nM final concentration. The optimized time for each siRNA transfection was confirmed with Western blotting.
Animal studies and colonocytes isolation
C57Bl/6J were purchased from Jackson Laboratories (Bar Harbor, ME) and were maintained on a 12-hour light-dark cycle with free access to water and standard laboratory chow (Purina lab chow, # 5001). Mouse studies were conducted according to federal regulations and were approved by the Institutional Animal Care and Use Committee at the University of Tennessee-Knoxville. Isolation of colonic epithelial cells from mice was performed from 8-12 weeks old male mice as previously described [12, 39]. Colons were washed several times with sterilized phosphate-buffered Saline (PBS). Then, the colon was incubated in PBS containing 5 mM EDTA (Fisher Scientific, Cat# S311-500) and 1% FBS, with or without butyrate (5 and 10mM), for 45 mins at 37°C. After 45 mins, the tissues were removed and isolated colonocytes were collected through centrifugation.
Biochemical assays
HDAC activity assay was performed according to manufacturer specifications (BioVision, Cat# K339-100). Briefly, HCT116 cells were seeded into 96 well plates and treated with butyrate (Sigma, B5887) and trichostatin A (Promega, G6560). Following treatment times, assay was performed. All values were normalized to total protein in each well.
Flux experiment
To measure percentage change of oxygen consumption rates (% OCR) in HCT116 cells, Seahorse XF24 Analyzer (Seahorse Bioscience) was used. All Seahorse assays were conducted according to the company guidelines, and the experimental design to measure butyrate oxidation in HCT116 cells were followed as stated [36]. HCT116 cells were seeded into XF24 cell culture microplates (Seahorse Bioscience, 100777-004) with an identical cell number per well. Cell plates are incubated with 1X KHB (2.5mM glucose and 50 μM carnitine) in non-CO2 incubator at 37°C for one hour before Seahorse assay. All Seahorse experiments were performed with identical conditions (unless otherwise stated). In brief, KHB media or sodium butyrate (Sigma, B5887) at 5mM final concentration were injected and the change in OCR was measured from baseline (% OCR). Then, 2-deoxyglucose (Sigma, D8375) was injected and % OCR was measured again. At last, 10% sodium azide was injected to block mitochondrial respiration by inhibiting complex IV.
Western blot
From FHC, HCT116 cells and isolated colonocytes, the proteins are extracted with RIPA buffer (Cell Signaling, #9806), 1mM PMSF (Cell Signaling, #8553) and phosphatase inhibitor cocktail (Cell Signaling, #5872). Quantifications of protein were measured by Bradford assay. Gel electrophoresis and transfer were performed using standard protocol for Western blotting. Antibodies that were used included pan-acetylated-histone H3 (Active motif, Cat# 39139), total Histone H3 (Active motif, Cat# 39763), total PDH (Abcam, Cat# ab110330), ACL (Cell Signaling, Cat # 4332), SCAD (Abcam, Cat# 154823), HDAC1 (Cell signaling, Cat# 34589), HDAC2 (Cell signaling, Cat# 57156), HDAC3 (Cell signaling, Cat# 85057) and β-actin (Sigma, Cat# A1978). Chemiluminescence detection was conducted with the Odyssey Fc and bands were quantified with Image Studio Software (LI-COR Biosciences, Lincoln, NE).
mRNA expression
Total RNA from (un) treated HCT116 cells were extracted using Trizol reagent (ambion, Cat# 15596-026). The concentration and integrity of RNA were measured by Nano-drop 1000. Reverse transcription was performed with RevertAid RT kit (Thermo Scientific, Cat# K1691) by following the company’s protocol. The amounts of product from RT-qPCR was measured by SYBR Green fluorescence (applied Biosystems, Cat# 4309155). SCAD primers for isoform 1 (Forward: GCGACTCATGGGTTCTGAAT and Reverse: TGCGACAGTCCTCAAAGATG), isoform 2 (Forward: GCCCGACTGGACCTATTTTT and Reverse: TGCGACAGTCCTCAAAGATG) and total (Forward: CAGGGATGGGCTTCAAGATA and Reverse: TGTCTGCCAACTTGAACTGG) were designed and their efficiency was confirmed by gel PCR. Relative gene expression levels were calculated through the ΔΔCt method and normalized to human 18S rRNA.
Statistical analysis
For biochemical assays, Seahorse experiments, and western blotting, the differences between experimental groups were determined by ANOVA followed by a Tukey post-hoc test. All data are expressed as mean ± SEM. Groups are considered significantly different at p < 0.05.
Abbreviations
HDAC: histone deacetylase; SCAD: acyl-CoA dehydrogenase – short chain; CRC: colorectal cancer; SCFAs: short-chain fatty acids; OCR: oxygen consumption rate; 2DG: 2-deoxyglucose; MCT1: monocarboxylate transporter-1; OCTN2: organic cation/carnitine transporter; CPT1A: carnitine palmitoyltransferase-1A; FHC: fetal human colonocytes; PDH: pyruvate dehydrogenase; TSA: trichostatin A; ACL: ATP-citrate lyase; KHB: krebs-HEPES buffer.
Author contributions
AH and DRD conceived and designed experiments. AH and NB performed experiments. AH, AB, JW, and DRD analyzed the data and reviewed the final version of the manuscript.
ACKNOWLEDGMENTS
The authors acknowledge the technical support of Beverly Samples.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by a University of Tennessee (Start-up fund to D.R.D).
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66:7–30.
2. Vargas AJ, Thompson PA. Diet and nutrient factors in colorectal cancer risk. Nutr Clin Pract. 2012; 27:613–23.
3. Haggar FA, Boushey RP. Colorectal cancer epidemiology: incidence, mortality, survival, and risk factors. Clin Colon Rectal Surg. 2009; 22:191–7.
4. Johnson CM, Wei C, Ensor JE, Smolenski DJ, Amos CI, Levin B, Berry DA. Meta-analyses of colorectal cancer risk factors. Cancer Causes Control. 2013; 24:1207–22.
5. Dahm CC, Keogh RH, Spencer EA, Greenwood DC, Key TJ, Fentiman IS, Shipley MJ, Brunner EJ, Cade JE, Burley VJ, Mishra G, Stephen AM, Kuh D, et al. Dietary fiber and colorectal cancer risk: a nested case-control study using food diaries. J Natl Cancer Inst. 2010; 102:614–26.
6. Bingham SA, Day NE, Luben R, Ferrari P, Slimani N, Norat T, Clavel-Chapelon F, Kesse E, Nieters A, Boeing H, Tjønneland A, Overvad K, Martinez C, et al, and European Prospective Investigation into Cancer and Nutrition. Dietary fibre in food and protection against colorectal cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC): an observational study. Lancet. 2003; 361:1496–501.
7. Romaneiro S, Parekh N. Dietary fiber intake and colorectal cancer risk: weighing the evidence from epidemiologic studies. Topics Clin Nutr. 2012; 27:41–7.
8. Michels KB, Edward Giovannucci, Joshipura KJ, Rosner BA, Stampfer MJ, Fuchs CS, Colditz GA, Speizer FE, Willett WC. Prospective study of fruit and vegetable consumption and incidence of colon and rectal cancers. J Natl Cancer Inst. 2000; 92:1740–52.
9. Blackwood AD, Salter J, Dettmar PW, Chaplin MF. Dietary fibre, physicochemical prope rties and their relationship to health. J R Soc Promot Health. 2000; 120:242–47.
10. Scharlau D, Borowicki A, Habermann N, Hofmann T, Klenow S, Miene C, Munjal U, Stein K, Glei M. Mechanisms of primary cancer prevention by butyrate and other products formed during gut flora-mediated fermentation of dietary fibre. Mutat Res. 2009; 682:39–53.
11. Macfarlane GT, Macfarlane S. Bacteria, colonic fermentation, and gastrointestinal health. J AOAC Int. 2012; 95:50–60.
12. Donohoe DR, Garge N, Zhang X, Sun W, O’Connell TM, Bunger MK, Bultman SJ. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011; 13:517–26.
13. Steliou K, Boosalis MS, Perrine SP, Sangerman J, Faller DV. Butyrate histone deacetylase inhibitors. Biores Open Access. 2012; 1:192–8.
14. Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost F, Brummer RJ. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther. 2008; 27:104–19.
15. Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr. 2003; 133:2485S–93S.
16. Archer SY, Johnson J, Kim HJ, Ma Q, Mou H, Daesety V, Meng S, Hodin RA. The histone deacetylase inhibitor butyrate downregulates cyclin B1 gene expression via a p21/WAF-1-dependent mechanism in human colon cancer cells. Am J Physiol Gastrointest Liver Physiol. 2005; 289:G696–G703.
17. Hinnebusch BF, Meng S, Wu JT, Archer SY, Hodin RA. The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. J Nutr. 2002; 132:1012–7.
18. Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell. 2012; 48:612–26.
19. Andriamihaja M, Chaumontet C, Tome D, Blachier F. Butyrate metabolism in human colon carcinoma cells: implications concerning its growth-inhibitory effect. J Cell Physiol. 2009; 218:58–65.
20. Ahmad MS, Krishnan S, Ramakrishna BS, Mathan M, Pulimood AB, Murthy SN. Butyrate and glucose metabolism by colonocytes in experimental colitis in mice. Gut. 2000; 46:493–9.
21. Donohoe DR, Holley D, Collins LB, Montgomery SA, Whitmore AC, Hillhouse A, Curry KP, Renner SW, Greenwalt A, Ryan EP, Godfrey V, Heise MT, Threadgill DS, et al. A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov. 2014; 4:1387–97. https://doi.org/10.1158/2159-8290.CD-14-0501.
22. Roediger WE, Nance S. Selective reduction of fatty acid oxidation in colonocytes: correlation with ulcerative colitis. Lipids. 1990; 25:646–52.
23. Gibson PR, Moeller I, Kagelari O, Folino M, Young GP. Contrasting effects of butyrate on the expression of phenotypic markers of differentiation in neoplastic and non-neoplastic colonic epithelial cells in vitro. J Gastroenterol Hepatol. 1992; 7:165–72.
24. Kripke SA, Fox AD, Berman JM, Settle RG, Rombeau JL. Stimulation of intestinal mucosal growth with intracolonic infusion of short-chain fatty acids. JPEN J Parenter Enteral Nutr. 1989; 13:109–16. https://doi.org/10.1177/0148607189013002109.
25. Sakata T. Stimulatory effect of short-chain fatty acids on epithelial cell proliferation in the rat intestine: a possible explanation for trophic effects of fermentable fibre, gut microbes and luminal trophic factors. Br J Nutr. 1987; 58:95–103.
26. Rémésy C, Demigne C, Morand C. Metabolism and utilisation of short chain fatty acids produced by colonic Dietary Fibre — A Component of Food. ILSI Human Nutrition Reviews. Springer, London. 1992: 137–50.
27. Astbury SM, Corfe BM. Uptake and metabolism of the short-chain fatty acid butyrate, a critical review of the literature. Curr Drug Metab. 2012; 13:815–21.
28. Wood PA, Amendt BA, Rhead WJ, Millington DS, Inoue F, Armstrong D. Short-chain acyl-coenzyme A dehydrogenase deficiency in mice. Pediatr Res. 1989; 25:38–43.
29. Augenlicht LH, Anthony GM, Church TL, Edelmann W, Kucherlapati R, Yang K, Lipkin M, Heerdt BG. Short-chain fatty acid metabolism, apoptosis, and Apc-initiated tumorigenesis in the mouse gastrointestinal mucosa. Cancer Res. 1999; 59:6005–9.
30. Bhala A, Willi SM, Rinaldo P, Bennett MJ, Schmidt-Sommerfeld E, Hale DE. Clinical and biochemical characterization of short-chain acyl-coenzyme A dehydrogenase deficiency. J Pediatr. 1995; 126:910–5.
31. Kaiko GE, Ryu SH, Koues OI, Collins PL, Solnica-Krezel L, Pearce EJ, Pearce EL, Oltz EM, Stappenbeck TS. The colonic crypt protects stem cells from microbiota-derived metabolites. Cell. 2016; 167:1137.
32. Birkenkamp-Demtroder K, Christensen LL, Olesen SH, Frederiksen CM, Laiho P, Aaltonen LA, Laurberg S, Sørensen FB, Hagemann R, Ørntoft TF. Gene expression in colorectal cancer. Cancer Res. 2002; 62:4352–63.
33. Kitahara O, Furukawa Y, Tanaka T, Kihara C, Ono K, Yanagawa R, Nita ME, Takagi T, Nakamura Y, Tsunoda T. Alterations of gene expression during colorectal carcinogenesis revealed by cDNA microarrays after laser-capture microdissection of tumor tissues and normal epithelia. Cancer Res. 2001; 61:3544–9.
34. Jankova L, Chan C, Fung CL, Song X, Kwun SY, Cowley MJ, Kaplan W, Dent OF, Bokey EL, Chapuis PH, Baker MS, Robertson GR, Clarke SJ, Molloy MP. Proteomic comparison of colorectal tumours and non-neoplastic mucosa from paired patient samples using iTRAQ mass spectrometry. Mol Biosyst. 2011; 7:2997–3005.
35. Kim H, Kang HJ, You KT, Kim SH, Lee KY, Kim TI, Kim C, Song SY, Kim HJ, Lee C. Suppression of human selenium-binding protein 1 is a late event in colorectal carcinogenesis and is associated with poor survival. Proteomics. 2006; 6:3466–76.
36. Han A, Bennett N, MacDonald A, Johnstone M, Whelan J, Donohoe DR. Cellular metabolism and dose reveal carnitine-dependent and-independent mechanisms of butyrate oxidation in colorectal cancer cells. J Cell Physiol. 2016; 231:1804–13.
37. Nagao M, Parimoo B, Tanaka K. Developmental, nutritional, and hormonal regulation of tissue-specific expression of the genes encoding various acyl-CoA dehydrogenases and alpha-subunit of electron transfer flavoprotein in rat. J Biol Chem. 1993; 268:24114–24.
38. Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut. 1987; 28:1221–7.
39. Donohoe DR, Wali A, Brylawski BP, Bultman SJ. Microbial regulation of glucose metabolism and cell-cycle progression in mammalian colonocytes. PLoS One. 2012; 7:e46589.
40. Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett. 2009; 294:1–8. https://doi.org/10.1111/j.1574-6968.2009.01514.x.
41. Aoyama M, Kotani J, Usami M. Butyrate and propionate induced activated or non-activated neutrophil apoptosis via HDAC inhibitor activity but without activating GPR-41/GPR-43 pathways. Nutrition. 2010; 26:653–61.
42. Weichert W, Röske A, Niesporek S, Noske A, Buckendahl AC, Dietel M, Gekeler V, Boehm M, Beckers T, Denkert C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clinical Cancer Research. 2008; 14:1669–77.
43. Wilson AJ, Byun DS, Popova N, Murray LB, L’Italien K, Sowa Y, Arango D, Velcich A, Augenlicht LH, Mariadason JM. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem. 2006; 281:13548–58.
44. Roediger W. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut. 1980; 21:793–8.
45. Miles K. FDG-PET and colon cancer. Cancer Imaging. 2015; 3:135.
46. Delbeke D, Martin WH. FDG PET and PET/CT for colorectal cancer. Methods Mol Biol. 2011; 727:77–103.
47. de Geus-Oei LF, Ruers TJ, Punt CJ, Leer J, Corstens FH, Oyen WJ. FDG-PET in colorectal cancer. Cancer Imaging. 2006; 6:S71–81.
48. Archer SY, Meng S, Shei A, Hodin RA. p21WAF1 is required for butyrate-mediated growth inhibition of human colon cancer cells. Proc Natl Acad Sci USA. 1998; 95:6791–6.
49. Medina V, Edmonds B, Young GP, James R, Appleton S, Zalewski PD. Induction of caspase-3 protease activity and apoptosis by butyrate and trichostatin A (inhibitors of histone deacetylase): dependence on protein synthesis and synergy with a mitochondrial/cytochrome c-dependent pathway. Cancer Research. 1997; 57:3697–707.
50. Serpa J, Caiado F, Carvalho T, Torre C, Gonçalves LG, Casalou C, Lamosa P, Rodrigues M, Zhu Z, Lam EW. Butyrate-rich colonic microenvironment is a relevant selection factor for metabolically adapted tumor cells. J Biol Chem. 2010; 285:39211–23.
51. Vanhoutvin SA, Troost FJ, Hamer HM, Lindsey PJ, Koek GH, Jonkers DM, Kodde A, Venema K, Brummer RJ. Butyrate-induced transcriptional changes in human colonic mucosa. PLoS One. 2009; 4:e6759.
52. Luceri C, Femia AP, Fazi M, Di Martino C, Zolfanelli F, Dolara P, Tonelli F. Effect of butyrate enemas on gene expression profiles and endoscopic/histopathological scores of diverted colorectal mucosa: a randomized trial. Dig Liver Dis. 2016; 48:27–33. https://doi.org/10.1016/j.dld.2015.09.005.
53. Thangaraju M, Carswell KN, Prasad PD, Ganapathy V. Colon cancer cells maintain low levels of pyruvate to avoid cell death caused by inhibition of HDAC1/HDAC3. Biochem J. 2009; 417:379–89.
54. Bultman SJ. Molecular pathways: gene–environment interactions regulating dietary fiber Induction of proliferation and apoptosis via butyrate for cancer prevention. Clin Cancer Res. 2014; 20:799–803.
55. Canani RB, Di Costanzo M, Leone L. The epigenetic effects of butyrate: potential therapeutic implications for clinical practice. Clinical Epigenetics. 2012; 4:4.
56. Leonel AJ, Alvarez-Leite JI. Butyrate: implications for intestinal function. Curr Opin Clin Nutr Metab Care. 2012; 15:474–9.
57. Plöger S, Stumpff F, Penner GB, Schulzke JD, Gäbel G, Martens H, Shen Z, Günzel D, Aschenbach JR. Microbial butyrate and its role for barrier function in the gastrointestinal tract. Ann N Y Acad Sci. 2012; 1258:52–9.