
INTRODUCTION
Approximately 30 Members of the TGF-β superfamily, including TGF-βs, activins, bone morphogenic proteins (BMPs), anti-Mullerian hormone (MIS/AMH), and growth/differentiation factors (GDFs), such as myostatin, represent multifunctional cytokines secreted by various cell types. TGF-β superfamily ligands regulate a vast array of biological processes such as growth, differentiation, migration, extracellular matrix production, angiogenesis, cytokine secretion, apoptosis, setting up of the body plan and organogenesis during embryogenesis, tissue homeostasis and immune regulation in adults [1–7]. Disruption of TGF-β signalling has been implicated in the progression of diseases such as cancer, fibrosis and autoimmune disease [8].
The primary signalling pathway downstream of TGF-β superfamily receptor/ligand interactions (for instance, interaction of TGF-β1 with type I and type II TGF-β receptors [9], and of BMPs with type I and type II serine/threonine kinase receptors [10, 11]) involves activation of Smad transcription factors [12–16]. In response to receptor/ligand interactions, Smad2 or Smad3 proteins become phosphorylated and form homomeric and heteromeric complexes with co-Smads, after which the complexes translocate to the nucleus where they either directly bind DNA or co-operate with sequence specific transcription factors/DNA binding proteins to regulate transcription of genes either positively or negatively [17]. In opposition to Smad2, or Smad3 effector signalling, the vertebrate I-Smads, Smad6 and Smad7 act as potent antagonists of TGF-β signalling [18–23] through recruitment of either ubiquitin ligases (Smurf1/2), or protein phosphatase I, or by becoming part of the TGF-β receptor complex and thereby interfering in the formation of SMAD2/SMAD3/co-SMAD complexes.
In cancer, TGF-β1 plays a dual role, involving both tumor suppressor and oncogenic functions, depending on context [24]. For instance, in the early stages of tumorigenesis the TGF-β signalling pathway induces growth arrest and promotes apoptosis, and mutation or deletion of members of the TGF-β signalling pathway define a tumor suppressor role for TGF-β signalling [8]. In contrast, activation of the TGF-β signalling pathway occurs in the late stages of tumorigenesis, as tumor cells become more invasive and prone to metastases, which is accompanied by induction of epithelial to mesenchymal transition (EMT).
During kidney development, ectopic expression of Activin A, a member of the TGF-β superfamily, inhibits GDNF-induced ureteric bud outgrowth and cell proliferation [25], and reduces Pax2 expression [26]. In contrast, BMP-7 induces Pax2 expression during kidney development [2]. Furthermore, exogenous expression of TGF-β1 in dysplastic kidney epithelial-like cells, decreased expression of Pax2, which was associated with decreased proliferation and transition to a mesenchymal phenotype [27]. In addition, TGF-β1 treatment caused suppression of Pax2 expression in proximal renal tubule cells [28]. Taken together, these data suggest there is an interaction between TGF-β1 signalling and Pax2 in kidney development and disease.
PAX2 is the second member of the Paired box (Pax) gene family, which has critical roles during early patterning events of the embryo [29, 30]. PAX2 is expressed at a very early stage in the nephric lineage and is required for normal kidney development in both mice [31, 32] and humans [33]. In early kidney development, the induction and conversion of metanephric mesenchyme to nephrogenic epithelium has been shown to require Pax2 activity during mesenchymal to epithelial transition (MET) [32], and this mechanism is also thought to operate during kidney regeneration and following kidney injury [34]. While Pax2 is only transiently expressed in the nephrogenic mesenchyme of the kidney, unabated expression of Pax2 leads to abnormal kidney hyperplasia and cystogenesis [35].
PAX2 is expressed in multiple different cancer types, including in renal cell carcinoma (RCC), ovarian cancer, endometrial carcinoma, breast cancer, and prostate cancer, [30]. Of these various cancer types, some subtypes frequently express PAX2 at high levels, whereas in the surrounding normal adult tissue the PAX2 expression is repressed following cessation of development [30]. In serous ovarian cancer cells wild-type p53 was shown to activate PAX2 expression [36]. However, the mechanisms by which PAX2 expression is regulated in cancer remain relatively poorly understood, and in particular the mechanisms associated with repression of PAX2 expression. For example, PAX2 protein expression has been noted at relatively early stages of tumor formation in serous ovarian carcinoma, but at later stages of progression the acquisition of metastasis is accompanied by loss of PAX2 expression [36]. Concomitantly, the loss of epithelial differentiation is associated with increased levels of TGF-β1 and TGF-β signalling in higher grades of ovarian cancer [37]. In serous ovarian carcinoma, down-regulation of PAX2 expression during later stages of tumor development in secretory cell outgrowths (SCOUTs) has been identified, and members of the TGF-β downstream signalling pathway were expressed in the same cells [38, 39].
In RCC, loss of VHL and hypoxia has been shown to activate PAX2 expression [40], but the potential role of PAX2 expression in RCC at later stages of tumor development is less well understood. PAX2 expression is also associated with several other kidney abnormalities, such as renal interstitial fibrosis [41] and polycystic kidney disease [42]. Following kidney injury, generation of renal fibrosis or scar tissue is dependent on the expression of TGF-β [43] and eventually, EMT-dependent suppression of PAX2 expression following its transient activation [41, 44]. Moreover, PAX2 expression in RCC has been shown to promote the expression of ADAM10, which is a negative regulator of EMT, and suggests further an association between PAX2 and suppression of EMT in RCC [45].
In this study we investigated the potential role of TGF-β1 signalling in promoting down-regulation of PAX2 expression in RCC tissue. We have identified a direct role for SMAD proteins in binding to the PAX2 gene promoter to suppress PAX2 expression in human clear cell renal cell carcinoma (CC-RCC) cells, during canonical TGF-β signalling. This finding suggests there is a direct relationship between TGF-β1 signalling and PAX2 expression during RCC tumor progression, with the implication that direct suppression of PAX2 expression in some cancer cell types by TGF-β1 signalling is a corollary of the promotion of EMT, tumor invasion and metastasis.
RESULTS
Treatment of RCC cancer cell lines with TGF-β1 leads to suppression of PAX2 mRNA and protein levels
Treatment of PAX2 expressing RCC cell lines, 786-O and A498 (Figure 1A, 1B), with 10 ng/ml TGF-β1 for 24 h resulted in substantial suppression of PAX2 mRNA and protein levels (Figure 1C-1E), and further analysis revealed significant inhibition of PAX2 mRNA and protein expression in 786-O cells in response to increasing concentrations of TGF-β1, which was sustained from 6 h to more than 48 h after treatment (Figure 1E, Supplementary Figure 1). The results of these investigations are consistent with previous studies, suggesting that TGF-β signalling pathways inhibit Pax gene expression [46].
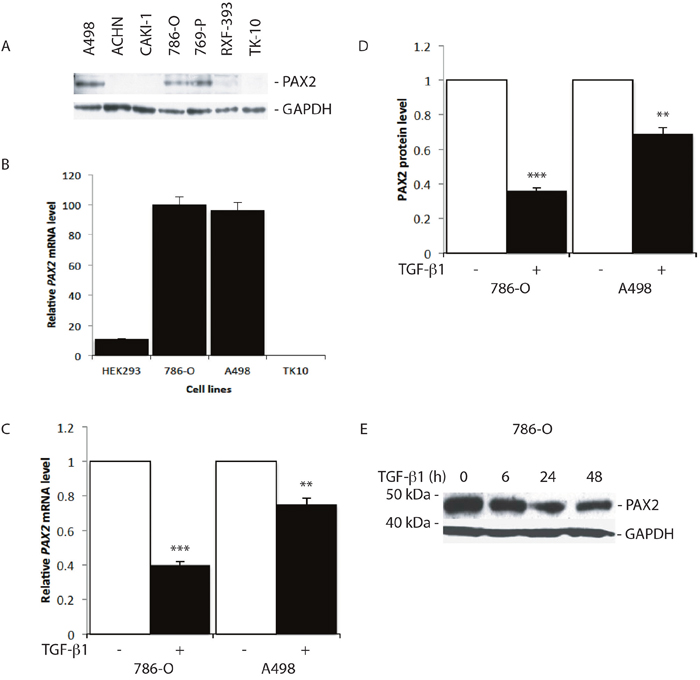
Figure 1: TGF-β1 treatment suppresses PAX2 mRNA and protein expression in CC-RCC cell lines. (A) Western blot analysis of PAX2 protein expression in RCC cell lines. GAPDH was used as a loading control. (B) QRT-PCR analysis of the relative level of endogenous PAX2 mRNA in HEK293 and three CC-RCC human cell lines. The data represent three separate experiments. (C) Relative PAX2 mRNA expression level, and (D) PAX2 protein following treatment of 786-O and A498 CC-RCC cells with 10 ng/ml TGF-β1 (labelled “+”, black columns) versus vehicle treated controls (labelled “-“, white columns). The results from three separate experiments after normalisation with GAPDH are shown. (E) Western blot of PAX2 protein expression relative to GAPDH following no treatment (0 h) of 786-O cells, or treatment for 6, 24 or 48 h with 10 ng/ml TGF-β1. Data are represented as mean ± S.E.M. The data were analysed by Student’s t test using GraphPad Prism 5.01, ** p < 0.01, *** p < 0.001 versus controls.
TGF-β1 treatment suppresses PAX2 promoter activity in RCC cells
To determine whether TGF-β treatment mediates inhibition of PAX2 expression directly or indirectly via regulation of the PAX2 promoter, we investigated whether TGF-β1 treatment could suppress transiently transfected human PAX2 promoter constructs in kidney and RCC cell lines. Previous studies have shown that there is potential for positive transcriptional auto-activation on PAX gene promoters by PAX proteins [46, 48]. We therefore reasoned that it would be important to minimize any potential confounding effects of auto-regulation on TGF-β1- mediated PAX2 suppression. HEK293 (human embryonic kidney), or TK10 (CC-RCC) cell lines have previously been shown to express little or no endogenous PAX2 mRNA or protein [47] (Figure 1). We found that PAX2 promoter-luciferase reporter constructs (pGPxp17 and pGPxp2, Figure 2A) were transcriptionally active in the HEK293 and TK10 cell lines (Figure 2), suggesting that these cells would be suitable to carry out the reporter assays. The pGPxp17 and pGPxp2 promoter-reporter constructs were transiently transfected into the HEK293 and TK-10 cells, which were then treated for 24 h with 10 ng/ml TGF-β1. Luciferase reporter activity was suppressed by the TGF-β1 treatment (Figure 2B, 2C), suggesting that TGF-β1 treatment caused transcriptional suppression of the PAX2 promoter construct activity in the transient transfection assays.
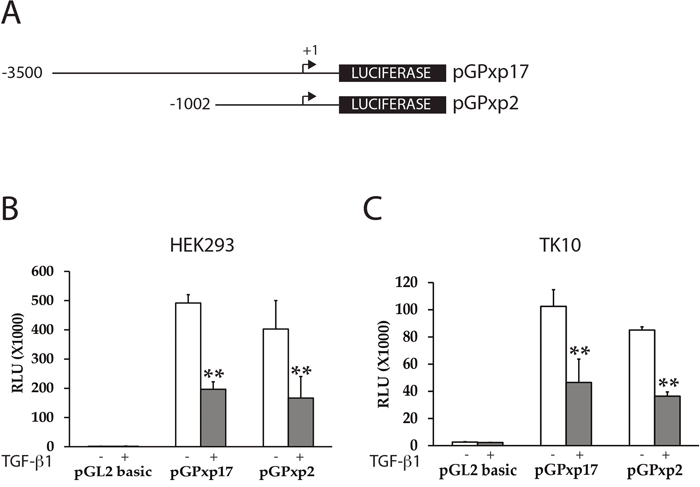
Figure 2: TGF-β1 treatment suppresses PAX2 promoter activity in CC-RCC cell lines. (A) The PAX2 promoter reporter constructs (pGPxp17 and pGPxp2) are shown. (B) HEK293 or (C) TK10 cells were transiently transfected with PAX2 promoter constructs (pGPxp17 or pGPxp2) and treated with 10 ng/ml TGF-β1 and the luciferase activity was assayed in TGF-β1 treated (grey columns) versus cells treated with vehicle control (white columns) The data represent the mean ± S.E.M of at least three experiments and were analyzed by Student’s t test (**, p < 0.01). HEK293 (n=5), TK10 (n=4).
Expression of SMAD-7 in RCC cells abolishes the inhibitory effect of TGF-β1 on PAX2 protein and mRNA expression
We next investigated whether signalling via SMAD proteins, which are known to mediate canonical TGF-β1 signalling, could be involved in the transcriptional suppression of PAX2 by TGF-β1. Exposure of 786-O cells to TGF-β1 for 6 h, 24 h or 48 h led to the strong phosphorylation of SMAD3 protein (Figure 3A), consistent with the expected effects of TGF-β1 treatment. Evidence for the involvement of SMAD proteins in the regulation of PAX2 expression was obtained by expressing an inhibitory SMAD (SMAD7), which we predicted would abolish the suppressive effect of TGF-β signalling on PAX2 expression levels if indeed SMAD signalling was involved in suppressing PAX2 transcription. To investigate this, 786-O cells were transiently transfected with an inhibitory SMAD7 expression construct, or an empty expression vector, then treated with either TGF-β1 or vehicle 24 h post-transfection. After transfection the cells were analyzed for relative levels of PAX2 protein and mRNA expression a further 48 h later. The treatment of 786-O cells with TGF-β1 suppressed PAX2 protein levels by 50 +/- 0.03% (p<0.001) and PAX2 mRNA by 60 +/- 0.03% (p<0.001) in the absence of SMAD7 (Figure 3B, lane 2), but in the presence of SMAD7, TGF-β1 treatment did not significantly suppress either levels of PAX2 protein (Figure 3B, lane 4 and Figure 3C), or PAX2 mRNA (Figure 3D). SMAD3 phosphorylation was observed following TGF-β1 treatment of 786-O cells, irrespective of the presence of SMAD7 expression (Figure 3B, lanes 2 and 4). These data suggest that SMAD proteins are directly associated with suppression of PAX2 expression, as the suppression of PAX2 by TGF-β1 treatment, which was associated with phosphorylation of SMAD3, was overcome by overexpression of the inhibitory SMAD (SMAD7) in 786-O cells.
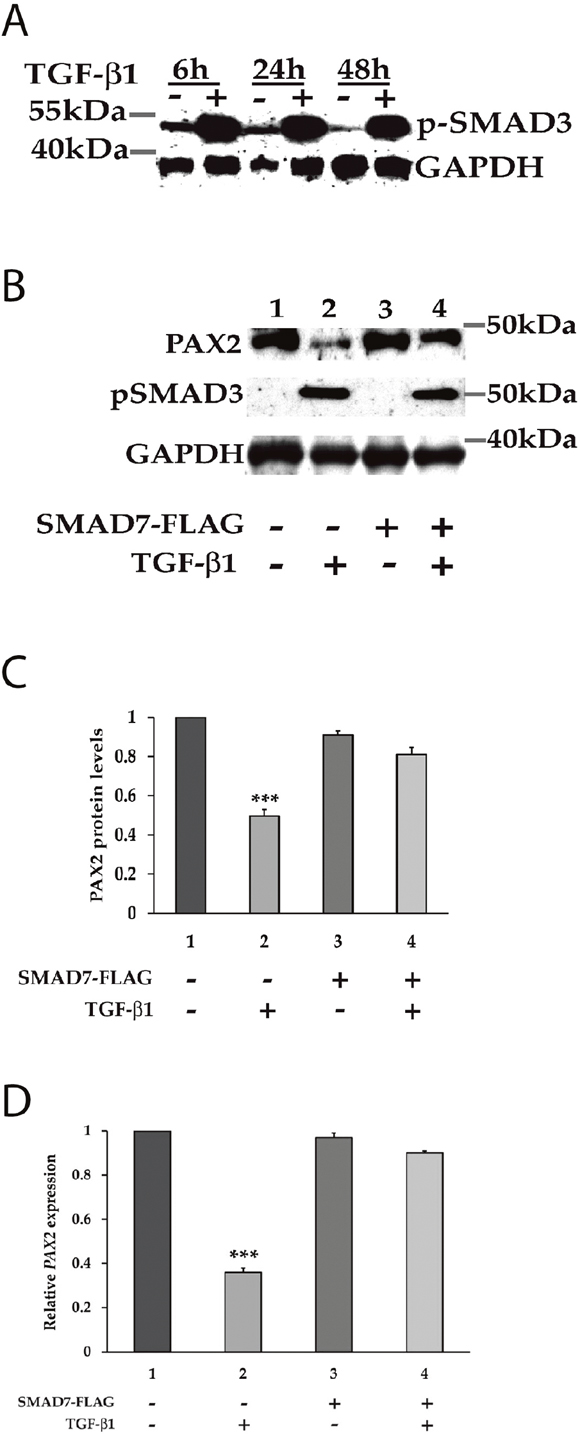
Figure 3: The canonical SMAD signalling pathway is involved in the regulation of PAX2 by TGF-β1 in 786-O cells. (A) Following treatment with TGF-β1, downstream SMAD3 becomes phosphorylated at carboxyl terminal serine residues. The phosphorylation of SMAD3 was determined in vehicle (-) and TGF-β1 (+) treated 786-O cells at 6 h, 24 h and 48 h post stimulation using phospho-Smad3 antibody, which detects endogenous levels of Smad3 when phosphorylated at Ser423/425. GAPDH served as a loading control. (B) Phosphorylation of SMAD3 upon treatment with TGF-β1 was associated with inhibition of PAX2 expression, and PAX2 suppression was abrogated upon expression of the iSMAD, SMAD7. (C) Quantitation of the PAX2 protein levels and (D) PAX2 mRNA levels in 786-O cell as treated in (B) in two separate experiments after normalization with GAPDH or with housekeeping genes relative to vehicle treated cells (with the PAX2 expression level set to 1). The data are represented as mean ± S.E.M. and were analyzed by one-way ANOVA followed by Tukey’s multiple comparison post-test (***, p < 0.001 versus control cells). The first column represents cells with no transfection and the dark grey (column 3) represents cells transiently transfected with the SMAD7-FLAG expression construct, while the lighter grey (columns 2 and 4) represent respective cells treated with TGF-β1.
Is there a physical interaction between PAX2 and SMAD2 or SMAD3?
SMAD proteins have been shown to physically interact with PAX6 protein, preventing PAX6 from auto-activating its own gene promoter [46, 48]. We therefore next determined whether SMAD proteins interact with the PAX2 protein, but we first sought to determine whether PAX2 auto-activates its own promoter. HEK293 cells were transiently co-transfected with 150 ng of PAX2 promoter-luciferase reporter construct and PAX2 expression constructs. We observed increased PAX2 promoter-luciferase activity (~444,000 to 449,000 RLU for PAX2b and PAX2c, respectively) in these co-transfections compared to the co-transfection of the equivalent amount of PAX2 promoter-luciferase reporter and empty vector expression constructs (~118,000 RLU, Figure 4A, 4B). When co-transfected promoter-luciferase reporter construct, pGPxp2, was increased to 425 ng, while maintaining PAX2b or PAX2c at 150 ng, this resulted in a further increase in luciferase activity (~620,000 to 670,000 RLU for PAX2b and PAX2c, respectively). In addition, when pGPxp2 was maintained at 150 ng, while increasing the PAX2b or PAX2c to 425 ng, this also resulted in increased luciferase activity (~300,000 RLU for either PAX2b and PAX2c). These results suggest that PAX2 auto-activates its own promoter. It is therefore possible that, like Pax6, SMAD proteins could interact directly with the PAX2 protein and thereby prevent auto-activation of the PAX2 promoter, as had been demonstrated for TGF-β1-mediated regulation of Pax6 expression [46, 48]. To investigate this, we carried out co-immunoprecipitation using an anti-SMAD2/3 antibody to determine whether a direct protein-protein interactions occurs between SMAD2 or SMAD3 proteins and the PAX2 protein. No evidence was found for an interaction between SMAD2/3 and PAX2 proteins (Figure 4B). In contrast, there was evidence of a potential interaction between SMAD2/3 and PAX6 protein (Supplementary Figure 3), as reported previously [48].
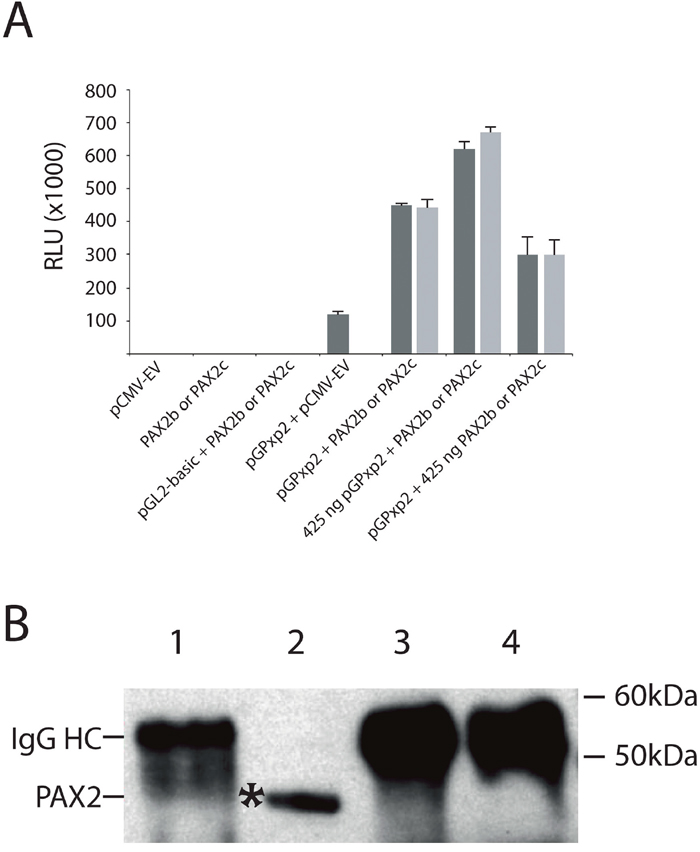
Figure 4: The PAX2 protein positively auto-regulates its own gene promoter, but does not physically interact with SMAD2/SMAD3 proteins. (A) Demonstration of auto-regulation of the PAX2 promoter in HEK293 cells. HEK293 cells were co-transfected with 150 ng of the PAX2 promoter plasmid (pGPxp2) together with pCMV-empty vector (“pGPxp2 + pCMV-EV”), which resulted in moderate luciferase activity (118,300 RLU). However, when HEK293 cells were co-transfected with 150 ng of PAX2 promoter reporter construct, pGPxp2, together with 150 ng of PAX2 expression construct, either pCMV-PAX2b (PAX2b, dark grey bar; 449,375 RLU), or pCMV-PAX2c (PAX2c, light grey bar; 444,159 RLU), there was 3.75-3.8-fold increased luciferase activity induced compared to pGPxp2 + pCMV-EV co-transfection. The graph summarizes three separate experiments for each co-transfection. The amounts of all PAX2 promoter-reporter or PAX2 expression construct plasmid that were used in the graph were 150 ng, unless otherwise specified. Error bars show the standard error of the mean. (B) Investigation of protein-protein interactions between PAX2 and SMAD2 or SMAD3. Lane 1: Eluate from beads used for pre-clearing the lysate. Lane 2: Input. Lane 3: Eluate from beads, which captured the protein complexes incubated with anti-IgG antibody. Lane 4: Eluate from beads, which captured the protein complexes incubated with anti-SMAD2/3 antibody. The figure shows that in lane 4, PAX2 was not immunoprecipitated with anti-SMAD2/3 antibody, suggesting there was no functional interaction between PAX2 and SMAD2 or SMAD3. The experiment was repeated twice. IgG HC: IgG heavy chain.
SMAD3 directly binds to the PAX2 promoter in RCC cells in response to TGF-β1 treatment
We next investigated whether SMAD3 could bind directly to the PAX2 promoter. Using the binding site prediction tool, ConTra v.2, at least seven SMAD (SMAD1 or SMAD3) binding sites were predicted in the ~1Kb of the PAX2 promoter upstream of the transcription start site (TSS), or in the 5’ untranslated region (5’UTR) (Figure 5A). Promoter-ChIP-PCR binding assays were used to investigate the binding of SMAD2 or SMAD3 proteins directly to the PAX2 promoter following TGF-β1 treatment in both 786-O and HEK293 cells, and an approximately 4 to 5-fold enrichment of SMAD2/3 binding to PAX2 promoter fragments was observed (p<0.001) (Figure 5B, 5D). Although the individual binding sites could not be resolved by the binding of SMAD proteins in the PAX2 promoter in ChIP analysis, since individual sonicated fragments were between 200-800 bp in length and potentially encompassed multiple SMAD binding sites, the results nevertheless suggested that significantly increased binding of SMAD proteins to the PAX2 promoter occurred in cells treated with TGF-β1. As a negative control the GNB2L1 gene promoter was investigated, which contains no SMAD binding site in its promoter, and no enrichment of SMAD2 or SMAD3 binding was observed following TGF-β1 treatment in either 786-O or HEK293 cells (Figure 5B, 5D). We then investigated the effects of the inhibitory SMAD, SMAD7 on binding of SMAD2/3 to the PAX2 promoter. The 786-O or HEK293 cells were transiently transfected with a SMAD7 expression construct, and the promoter-ChIP-PCR binding assays were again carried out. Following expression of SMAD7, the SMAD2/3 binding was inhibited (Figure 5C, 5E). These ChIP-PCR results suggest that SMAD3 protein is able to bind directly to DNA fragments within a 1Kb region of the PAX2 promoter in ChIP-PCR binding assays, and that this binding is inhibited upon overexpression of SMAD7.
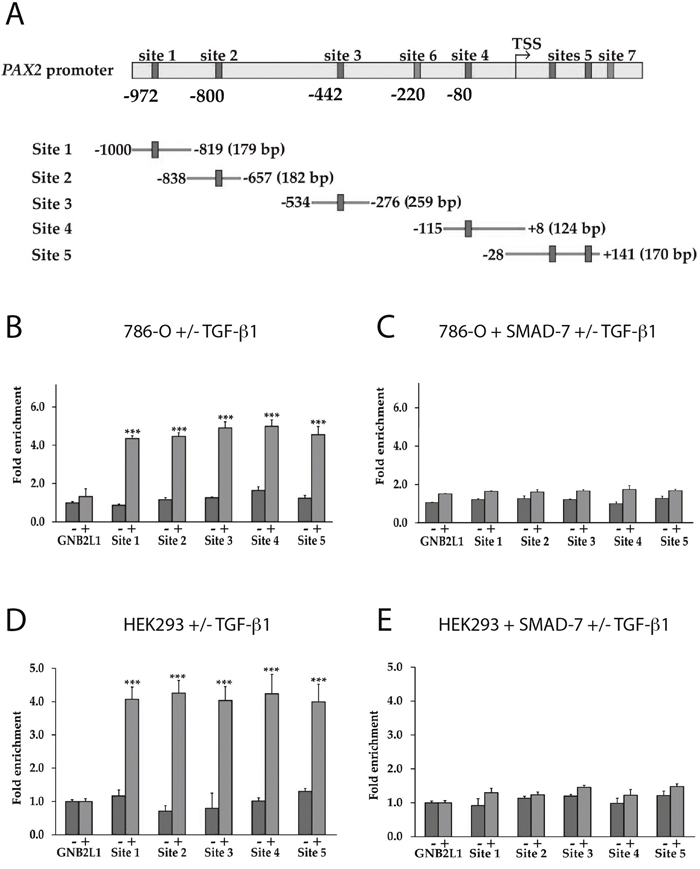
Figure 5: The binding of SMAD3 to the PAX2 promoter in response to TGF-β1 treatment is inhibited in the presence of SMAD7 overexpression. (A) Representation of the human PAX2 promoter (not drawn to scale) showing SMAD1 or SMAD3 binding sites predicted using ConTra v2, and PCR fragments used for PCR-ChIP assays. Putative SMAD3 binding sites were identified at sites 4 and 6, while the remaining sites predicted by the ConTra binding site prediction tool were putative SMAD1 binding sites. The “TSS” shows the transcription start site. In (B) to (E) ChIP assays were carried out in 786-O and HEK293 cells following TGF-β1 treatment to show binding of SMAD3 to PCR-amplified fragments the PAX2 promoter. Sites 6 and 7 in the PAX2 promoter were unable to be amplified and were not analysed, but all other sites (sites 1-5) were investigated. In parallel experiments ChIP assays were carried out in cells transfected with a SMAD7-FLAG expression construct (see Supplementary Figure 2 for confirmation of SMAD7 overexpression in 786-O and HEK293 cells). A region of the GNB2L1 promoter without any putative SMAD3 binding site was used as a negative control. Dark grey columns represent fold enrichment (over no antibody controls) in cells stimulated with vehicle and lighter grey columns represent fold enrichment in cells stimulated with TGF-β1. The data represent the mean ± S.E.M of three independent experiments and were analyzed by one-way ANOVA followed by Tukey’s multiple comparison post-test (***, p < 0.001 versus negative control and vehicle treated cells).
DISCUSSION
Here we show that TGF-β1 treatment of renal cell carcinoma cells is associated with transcriptional supression of PAX2, through the direct binding of SMAD proteins to the PAX2 promoter. Although our ChIP data demonstrate binding of SMAD3 to the PAX2 promoter, these results do not distinguish which sites are functional, and further work will be required to determine this. Nevertheless, these findings represent the first evidence for direct promoter-mediated inhibition of PAX2 expression by TGF-β1 signalling in tumor cells. With respect to several other Pax family members, it has previously been shown that TGF-β signalling regulates Pax6 or Pax8 gene expression through protein-protein interactions between SMAD2/3 and the Pax protein, resulting in inhibition of auto-activation of the respective Pax gene promoter [48, 49]. In contrast, our findings suggest that, in the case of PAX2 expression in CC-RCC, SMAD2/3 proteins directly bind to the gene promoter, resulting in PAX2 transcriptional supression.
Many previous studies have shown that SMAD3 binding to gene promoters is often associated with target gene activation. While the mechanism by which the binding of SMADs to the PAX2 promoter leads to transcriptional suppression in CC-RCC cells has not been elucidated, SMAD proteins have been shown to interact with, and are able to form transcriptionally inactive complexes with co-repressors in kidney cells [50]. It is therefore possible that in CC-RCC cells the binding of SMADs to the PAX2 promoter interferes with factors transcriptionally activating the promoter, which then leads to suppression of promoter activation, similar to the suppression of C/EBPβ- and STAT3-mediated transcriptional activation of the haptoglobin promoter by SMAD proteins, which has previously been reported [51]. However, it is presently unknown which factors are involved in generating the PAX2 promoter activity observed in vitro in RCC cells.
The findings reported here are of potential importance in understanding the role of PAX2 expression in cancer progression and metastasis. Dysgulated expression of PAX2 has been linked to several cancer types, including RCC, ovarian cancer, endometrial cancer, prostate cancer, breast cancer, melanoma, medulloblastoma and Wilms tumor [30]. While PAX2 expression has been shown to promote tumor cell proliferation and survival in multiple cancer types, paradoxically in serous ovarian cancer it has been shown that at advanced tumor stages, with onset of invasion and metastasis, PAX2 mRNA and protein expression decline, and eventually PAX2 expression is lost in the invading cells [36]. This apparent selection against PAX2 expression within subpopulations of tumor cells seems counter-intuitive to the notion that PAX2 plays an oncogenic role in tumorigenesis. However, it is possible that switching away from PAX2 expression might reflect characteristic physiological changes within the cells, associated with acquisition of invasive properties. In serous ovarian cancer, the down-regulation of PAX2 expression occurs in the secretory outgrowths (termed SCOUTs), in which aggressive changes have been documented to occur [38, 39, 52]. These outgrowths are associated with poorer patient prognosis and progression to metastasis. Interestingly, Pax2 is expressed normally in mouse adult oviduct tissue [47], and it has been shown that Pax2 expression maintains the differentiation of adult mouse oviduct epithelium and inhibits the transition to a stem cell-like state [53]. Similarly in breast cancer, expression of PAX2 has been associated with a low invasive phenotype in luminal breast cancer cells [54], while in endometrial cancer joint loss of PAX2 and PTEN expression was associated with progression to carcinoma [55].
PAX2 protein expression has been associated with 81-95% of CC-RCCs [56, 57], and we have also shown that in vivo tumorigenicity of RCC cells is reduced following siRNA-mediated knockdown of PAX2 expression [58]. Moreover, PAX2 protein is strongly expressed in primary CC-RCC in a high percentage (~66%) of tumor cells, and in CC-RCC metastases expression of PAX2 appears to be more intense and in a higher proportion (~80%) of tumor cells [57]. However, PAX2 expression has been shown to positively regulate expression of ADAM10 in CC-RCC [45], and the suppression of ADAM10 expression is associated with EMT, as well as up-regulation of Slug, and down-regulation of E-Cadherin expression [45]. In independent experiments we observed that, following siRNA-mediated silencing of PAX2 expression in A498 RCC cells, down-regulation of ADAM10 mRNA levels occurred (unpublished data), which is in agreement with previously published work [45]. Down-regulation of PAX2 expression as a result of enhanced TGF-β1 signalling in CC-RCC would thus be consistent with a role for PAX2 in facilitating EMT-related changes at least in part via reduced ADAM10 expression. Importantly, an increased level of TGF-β1 signalling has clearly been associated with increased invasion and metastatic changes in CC-RCC [59–63]. However, in contrast to ovarian cancer where PAX2 expression is reduced in SCOUTs, the high levels of PAX2 expression observed in metastatic sites of CC-RCC [57] seem to contradict the notion that decreased PAX2 expression is associated with EMT in CC-RCC. Furthermore, it has been observed that PAX2 expression levels are increased by hypoxia and VHL mutations in CC-RCC [40], and so further investigations will be required to determine how the TGF-β1/SMAD-mediated signalling pathway promotes invasion through modulation of PAX2 expression in CC-RCC.
We propose that pathways involving TGF-β1 signalling, and subsequent down-regulation of PAX2 expression facilitate the switching of tumor cells from PAX2-mediated epithelial cell differentiation to more stem cell-like mesenchymal programs. These are associated with EMT, and greater dependence on other signalling pathways, in which tumor cells have acquired increased motility, invasiveness and treatment resistance [36]. Suppression of PAX2 expression with concomintant EMT-related changes might be applicable to multiple cancer types in which PAX2 is expressed, including RCC, ovarian, breast, endometrial and prostate cancer. It is conceivable, however, that the therapeutic inhibition of PAX2 could induce tumor progression and metastasis in some cancer types, particularly if continued expression of PAX2 has a role in promoting epithelial differentiation and suppression of tumor metastasis in advanced stages of cancer. Although not investigated here, mechanisms involving TGF-β signalling, and suppressed expression of PAX2 might also be the case in renal fibrosis, where a role for PAX2 expression in kidney injury has also been identified [41]. In contrast, TGF-β–mediated suppression of PAX2 expression might not be universal to all cancer types. For example, increasing levels of PAX2 expression have been linked to metastatic progression in esophageal cancer [64].
In conclusion, our studies have demonstrated that TGF-β1 signalling in CC-RCC cells results in the direct inhibition of PAX2 expression through SMAD-mediated transcriptional suppression of the PAX2 gene promoter. These data therefore provide clearer understanding of the role of TGF-β1 signalling and PAX2 expression in cancer.
MATERIALS AND METHODS
Cell cultures and TGF-β1 treatment
Cell lines used in this study were HEK293 (human embryonic kidney immortalized with adenovirus 5 DNA), A498, ACHN, CAKI-1, 786-O, 769-P, RXF-393, and TK10 (renal cell carcinoma cell lines) and were obtained from the Developmental Therapeutics Program, NIH (as part of the NCI-60 cell line panel), and cultured either in Dulbecco’s Modified Eagle’s Media (DMEM) (Invitrogen, USA) or in Roswell Park Memorial Institute (RPMI) 1640 medium (Invitrogen, USA) supplemented with 10% fetal bovine serum (FBS) at 37°C in a humidified environment with 5% CO2. Cells were seeded at 50-60% confluence, and in some experiments were incubated with recombinant human TGF-β1 (R & D systems, Minneapolis, MN) at a concentration of 10 ng/ml (whenever the concentration was not specified). Control cells were treated with vehicle (4 mM HCl containing 1 mg/ml bovine serum albumin). Cells were harvested at 24 h post TGF-β1 treatment for RNA extraction and at 48 h post TGF-β1 treatment for Western blot analysis.
Plasmids and promoter reporter constructs
Luciferase reporter plasmids (pGPxp17 and pGPxp2) have been reported elsewhere [65], and contained the human PAX2 gene promoter fused to the luciferase reporter gene of pGL2 basic vector (Promega, USA). The pGPxp17 and pGPxp2 plasmids contain 4.2 kb and 1.7 kb of the human PAX2 promoter and 5’ – flanking sequences, respectively. Expression plasmids encoding PAX2 and inhibitory SMAD, SMAD7 (pSMAD7-FLAG) were described previously [66, 67]. A plasmid encoding β-galactosidase (βgal) under the CMV promoter (pCMV-βgal) was used as an internal control of transfection efficiency in all reporter assays. Plasmids used for transient transfections were prepared using Qiagen plasmid Midi/Maxi kits, according to the manufacturer’s instructions (Qiagen, Chatsworth, CA).
Luciferase reporter assays
Luciferase reporter assays were carried out following transient transfection of cells with promoter-luciferase reporter constructs as previously described [65]. Briefly, cells were plated in 24-well plates to give 30-50% confluence after 24 h, and transfections were carried out using FuGENE® 6 Transfection Reagent (Roche Applied Science, Germany) according to the manufacturer’s instructions. An optimal FuGENE (μl): DNA (μg) ratio of 6:1 was used for all cell types except TK10 cells for which the FuGENE: DNA ratio was 3:1. Control cells were transfected with an equal amount of pGL2 basic to measure background luciferase activity. Fifty nanograms of pCMV-βgal was co-transfected as an internal control for transfection efficiency and used for normalizing the data. Transfected cells were treated with 10 ng/ml TGF-β1 24 h after transfection, and cells were harvested at 48 h post TGF-β1 treatment. Luminescence was measured using the Synergy 2 Multi-Mode Microplate Reader (BioTek, USA) and Gen5 interface software (version 1.01.9).
β-galactosidase activity assays
β-galactosidase assays were carried out as described previously [67]. Briefly, 10 μl lysate was incubated with 100 μl βgal substrate solution (200 mM sodium phosphate buffer pH 7.3, 2 mM MgCl2, 100 mM β-mercapthoethanol, 1.33 mg/ml ONPG (Sigma-Aldrich). The samples were incubated at 37°C until the appearance of a bright yellow colour. The absorbance of the reaction was measured at 410 nm, using the Synergy 2 plate reader.
Total RNA purification and quantitative real-time PCR (qRT-PCR)
Total RNA for gene expression analysis was isolated as described previously [47]. Briefly, cells were lysed with 200 μl TRIzol reagent (Invitrogen, USA) per well of a 24-well plate. Total RNA was extracted using 1-bromo-3-chloropropane (BCP) (Molecular Research Centre, USA) and 70% ethanol (in DEPC-H2O) (ethanol, Sharlau, Spain; DEPC, Sigma-Aldrich, USA) then purified using the PureLink RNA Mini Kit (Invitrogen, USA) according to the manufacturer’s instructions. 200 ng of total RNA from each sample was used for generating cDNA in a 20-μl reaction. First-strand cDNA was synthesized using a SuperScript III Reverse Transcriptase Kit (Invitrogen, USA), primed with dNTPs and 250 ng random hexamers (Invitrogen, USA), according to the enzyme manufacturer’s instructions, and was performed at 25°C for 5min, 50°C for 1 hr, and 70°C for 15 min. Three microliters of first strand cDNA (diluted 1:10) and 0.2 μM gene-specific primers (Table 1) were used with the Platinum® SYBR® Green qPCR SuperMix UDG (Invitrogen, USA), to amplify and detect target genes. The qRT-PCR reactions were performed in an ABI-7300 Real time PCR system (Applied Biosystems, USA) in duplicates, using the following program: 50°C – 2 min, 95°C – 10 min, 45 cycles of 95°C – 15 sec and 60°C – 1 min, with an additional dissociation stage: 95°C – 15 sec, 60°C – 1 min, and 95°C – 15 sec. Relative gene expression data were normalised to three housekeeping genes, and represented relative to vehicle treated control cells. Gene-specific primers (Invitrogen) for qRT-PCR analysis were designed using Primer3 (version 0.4.0, http://frodo.wi.mit.edu/primer3/). In each QRT-PCR experiment, the three most stable housekeeping genes (also listed in Table 1) were identified.
Table 1: Oligonucleotide primers used for qRT-PCR
Target gene |
Forward primer 5’- 3’ |
Reverse primers 5’ – 3’ |
|---|---|---|
PAX Genes |
||
PAX2 |
CCTGGCCACACCATTGTTC |
TCACGTTTCCTCTTCTCACCAT |
SMAD Genes |
||
SMAD2 |
TATGGACACAGGCTCTCCAG |
CACCAAAATGCAGGTTCTGA |
SMAD3 |
GCAGGTTCTCCAAACCTATCC |
AGGAGATGGAGCACCAGAAG |
Housekeeping Genes |
||
GAPDH |
CTCAAGATCATCAGCAATGCC |
GGTCATGAGTCCTTCCACGATAC |
GNB2L1 |
CACAACGGGCACCACCAC |
CACACACCCAGGGTATTCCAT |
HPRT1 |
ATTATGGACAGGACTGAACGTCTTG |
TGAGCACACAGAGGGCTACAAT |
RPL32 |
AACGTCAAGGAGCTGGAAGTG |
GGCTTTGCGGTTCTTGGA |
RPL13a |
GGAAAGAGAAAGCCAAGATCCA |
GCTCAGACCAGGAGTCCGTG |
YWHAZ |
ACTTGACATTGTGGACATCGGATAC |
GTTGGAAGGCCGGTTAATTTTC |
PPIB |
ATGATCCAGGGCGGAGACTT |
CAGGCCCGTAGTGCTTCAG |
Total protein sample preparation, SDS-PAGE and western blotting
Protein isolation and Western blots were performed as previously described [68]. Briefly, cells were lysed in RIPA buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS supplemented with protease inhibitors (Complete, Roche; 1 mM PMSF, Sigma-Aldrich; 1mM sodium orthovandate, Sigma-Aldrich). Total protein in the extracts was quantitated using a colorimetric BCA Protein Assay Kit as per the manufacturer’s instructions. 40 μg of total cellular protein for each sample was boiled in Laemmli sample buffer and separated by SDS-PAGE under reducing conditions and transferred to nitrocellulose membrane (Hybond-C Extra). Immunoreactive bands were visualized by chemiluminescence using SuperSignal West Pico Chemiluminescent Substrate (containing equal parts of the Stable Peroxide Solution and the Luminol/Enhancer Solution) (Thermo Scientific, USA). Antibodies used for Western blotting were rabbit anti-PAX2 (1:500; Zymed), mouse anti-SMAD2/3 (1:1000; Santa Cruz Biotechnology), goat anti-GAPDH (1:2000, Santa Cruz Biotechnology), and anti-rabbit and anti-mouse horseradish peroxidase IgG (Sigma-Aldrich). The protein ladder used was MagicMark XP Standard (Invitrogen, USA).
Chromatin immunoprecipitation (ChIP)-PCR
ChIP-PCR assays were carried out as described in Sehgal et al (2009) [69] with several modifications. Briefly, approximately 1.0 x 108 cells, with or without TGF-β1 treatment, were crosslinked per ChIP analysis. Following washing, scraping and centrifugation of the cells in ice cold PBS, the cells were then lysed in 50 mM Tris-HCl pH 8.0, 10 mM EDTA, 1% SDS supplemented with protease inhibitors (Complete, Roche; 1 mM PMSF, Sigma-Aldrich; 1mM sodium orthovandate, Sigma-Aldrich). The lysates were then sonicated to obtain DNA fragments in the range of 200-800 bp using a Sonics Vibra-Cell sonicator (Sonics and Materials Inc., USA, Model: VCX 130) set to 25% amplitude. HEK293 cells were sonicated for 5 sets of 10-second pulses and 786-O cells for 5 sets of 20-second pulses. Following centrifugation (12,000 x g, 10 min at 4°C) supernatants were diluted 2.5 times with ChIP dilution buffer (0.5% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.0, 100 mM NaCl), and 5% of the diluted supernatant was saved as an Input control. The rest of the supernatant was precleared using Dynabeads® Protein G (Invitrogen) for 1 h at 4°C. For immunoprecipitation 4 μg of anti-SMAD2/3 (Santa Cruz Biotechnology) was added to precleared lysates and rotated at 4°C overnight. Lysates with no antibody served as a negative control. The remainder of the protocol was carried out essentially as described in Sehgal et al (2009). The qRT-PCR reactions were performed as described previously (ChIP PCR primers listed in Table 2) except an additional dissociation was performed at 99°C instead of 95°C. The data were analysed according to the SuperArray ChIP-qPCR Data Analysis Template (www.sabiosciences.com/manuals/chipqpcranalysis.xls). Individual binding sites were unable to be resolved by the binding of SMAD proteins in the PAX2 promoter in ChIP analysis, because individual sonicated fragments were between 200-800 bp in length and potentially contained multiple SMAD binding sites.
Table 2: Oligonucleotide primers used for ChIP-PCR analysis
Target Gene |
Accession no./Reference |
Promoter region |
Forward sequence 5’–3’ |
Reverse sequence 5’–3’ |
|---|---|---|---|---|
PAX2 |
4842, NM_000278 |
-997/-819, site 1 |
GGATCCACCGAGCTAGCAG |
GGGACTTGGTTTCTGAACCC |
PAX2 |
4842, NM_000278 |
-838/-657, site 2 |
GGGTTCAGAAACCAAGTCCC |
GTCTTGTCCCCTCCCGTTCC |
PAX2 |
4842, NM_000278 |
-534/-276, site 3 |
GCTGGGCGAGTTAGAACTGA |
GCCCGGGATTAAAACTACACTG |
PAX2 |
4842, NM_000278 |
-115/8, site 4 |
TGGCGAATCACAGAGTGGTGGAAT |
GCTCCCGGTGTGTGTCTCTCTAAAA |
PAX2 |
4842, NM_000278 |
-28/141, site 5 |
GGGCTTTGCAGCTTTTAGAGAG |
GGAAAAGGCAGGCGCACG |
GNB2L1 |
33871, NM_006098 |
-259/-134, GNB2L1 |
ACTTCACCTCTTTCGCTTCTCGCT |
ACAGTCCCGTCTTCCGTACAACAA |
SMAD2 or SMAD3 protein-protein co-immunoprecipitation
HEK293 cells were transiently transfected with the PAX2 expression construct, pCMV-PAX2b, using FuGENE. After 42h of transfection, the cells were treated with TGF-β1 for 6h to activate SMAD2 and SMAD3. The cells were then lysed in non-denaturing lysis buffer supplemented with protease inhibitors (Sigma-Aldrich). The protein lysates (500 μg per antibody) were precleared for 1h at 4°C using Dynabeads® Protein G (Invitrogen), followed by immunoprecipitation using mouse anti-SMAD2/3 (Santa Cruz Biotechnology) or control IgG antibodies (Sigma-Aldrich), which was carried out overnight at 4°C. This was followed by incubation with 50 μl Dynabeads for 4h at 4°C, and then the magnetically captured protein complexes on Dynabeads were washed twice with lysis buffer, and the protein complexes eluted by boiling in Laemmli sample buffer (0.24 M Tris-HCl pH 6.8, 10% SDS, 40% Glycerol, 20% β-mercaptoethanol, 0.02% Bromophenol blue) under reducing conditions. Proteins were then resolved by SDS-PAGE and analyzed by Western blot to investigate SMAD2- or SMAD3-PAX2 or PAX6 interactions. Following the detection of PAX2, the Western blot membrane was stripped and incubated with an anti-SMAD2/3 antibody (1:1000, Santa Cruz Biotechnology) to detect SMAD2/3 proteins, and then the membrane was directly re-probed with anti-PAX6 antibody (1:100, Developmental Studies Hybridoma Bank) to detect PAX6 protein.
PAX2 autoregulation assays
In co-transfection assays, HEK293 cells were transiently transfected with one of two PAX2 expression constructs (either CMV-PAX2b (PAX2b), or CMV-PAX2c (PAX2c) [67], together with the PAX2 promoter-reporter plasmid, pGPxp2. Cells were co-transfected with a total of 600 ng of plasmids, comprising 150 ng of either pCMV-PAX2b or pCMV-PAX2c, and 150 ng of pGPxp2 with the remainder of the transfected plasmid being made up to an amount of 575 ng using pCMV empty vector (pCMV-EV). In some transfections up to 425 ng of either PAX2 expression vector, or PAX2 promoter-reporter construct was transfected, while the concentrations of either the expression, or the reporter construct, respectively, were maintained at 150 ng, and the amount of pCMV-EV was adjusted accordingly to make the total plasmid up to 575 ng. In addition, pCMV- β-gal (25 ng) was also included in all transient transfections to normalize the data to the β-gal value. Control transfections included cells only, pGL2 basic vector alone, CMV-PAX2b or CMV-PAX2c alone, and PGL2-basic together with CMV-PAX2b or CMV-PAX2c, and these were included in each experiment. The luciferase activity from “cells only”, and “pGL2 basic vector alone” was undetectable and is not shown. Luciferase activity is represented in Relative Light Units (RLU) normalised to the β-gal value.
Online software and statistical analysis
For qRT-PCR analysis, the qBase software (version 1.3.5, http://medgen.ugent.be/qbase/) was used. For identification of SMAD binding sites in the PAX2 promoter, the online web-based tools ConTra v.2 and TRANSFAC were used. We used ConTra v.2 for visualization of SMAD1 or SMAD3 binding sites in a region 3.5Kb upstream, or the 5’UTR region of the human PAX2 gene on the sequence, NM_003987, with a core of 0.90, and similarity matrix of 0.75. Subsequently, we used ConTra v.3 for visualization of SMAD3 binding sites in the same 3.5Kb region of the same sequence (NM_003987), and a core of 0.95 and similarity matrix of 0.85, and this identified only the SMAD3 binding site previously identified at position -220 upstream of the TSS, which was conserved in species from humans to zebrafish.
Author contributions
GK, GL, AC, CS, JH and MRE designed the studies. GK and GL performed the experimental work. GK and GL analysed the data and prepared the tables and figures. AC, CS, JH and MRE oversaw the experiments. MRE wrote the manuscript, and all authors read, contributed to and approved the final manuscript.
ACKNOWLEDGMENTS
The authors thank Dr D Wynford Thomas for the providing K1 thyroid carcinoma cells, Drs Phil Daniel and Jenny Grant for technical advice.
CONFLICTS OF INTEREST
The authors declare they have no competing interests.
GRANT SUPPORT
GK was supported by a New Zealand International Doctoral Research Scholarship (NZIDRS), and AC was supported by an overseas travel fellowship from the Royal Society, and the Norfolk and Waveney BigC Cancer Charity. Funding for this research was from the Health Research Council of NZ (MRE).
REFERENCES
1. Dudley AT, Lyons KM, Robertson EJ. A requirement for bone morphogenetic protein-7 during development of the mammalian kidney and eye. Genes Dev. 1995; 9:2795–807. https://doi.org/10.1101/gad.9.22.2795.
2. Luo G, Hofmann C, Bronckers AL, Sohocki M, Bradley A, Karsenty G. BMP-7 is an inducer of nephrogenesis, and is also required for eye development and skeletal patterning. Genes Dev. 1995; 9:2808–20. https://doi.org/10.1101/gad.9.22.2808.
3. Miyazaki Y, Oshima K, Fogo A, Hogan BL, Ichikawa I. Bone morphogenetic protein 4 regulates the budding site and elongation of the mouse ureter. J Clin Invest. 2000; 105:863–73. https://doi.org/10.1172/JCI8256
4. Piscione TD, Yager TD, Gupta IR, Grinfeld B, Pei Y, Attisano L, Wrana JL, Rosenblum ND. BMP-2 and OP-1 exert direct and opposite effects on renal branching morphogenesis. Am J Physiol. 1997; 273:F961–75.
5. Ritvos O, Tuuri T, Erämaa M, Sainio K, Hildén K, Saxén L, Gilbert SF. Activin disrupts epithelial branching morphogenesis in developing glandular organs of the mouse. Mech Dev. 1995; 50:229–45. https://doi.org/10.1016/0925-4773(94)00342-K.
6. Rogers SA, Ryan G, Purchio AF, Hammerman MR. Metanephric transforming growth factor-beta 1 regulates nephrogenesis in vitro. Am J Physiol. 1993; 264:F996–1002.
7. Massagué J. TGF-beta signal transduction. Annu Rev Biochem. 1998; 67:753–91. https://doi.org/10.1146/annurev.biochem.67.1.753
8. Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000; 342:1350–58. https://doi.org/10.1056/NEJM200005043421807.
9. Wrana JL, Attisano L, Cárcamo J, Zentella A, Doody J, Laiho M, Wang XF, Massagué J. TGF beta signals through a heteromeric protein kinase receptor complex. Cell. 1992; 71:1003–14. https://doi.org/10.1016/0092-8674(92)90395-S.
10. Estevez M, Attisano L, Wrana JL, Albert PS, Massagué J, Riddle DL. The daf-4 gene encodes a bone morphogenetic protein receptor controlling C. elegans dauer larva development. Nature. 1993; 365:644–49. https://doi.org/10.1038/365644a0.
11. Nellen D, Affolter M, Basler K. Receptor serine/threonine kinases implicated in the control of Drosophila body pattern by decapentaplegic. Cell. 1994; 78:225–37. https://doi.org/10.1016/0092-8674(94)90293-3.
12. Wrana JL, Attisano L. The Smad pathway. Cytokine Growth Factor Rev. 2000; 11:5–13. https://doi.org/10.1016/S1359-6101(99)00024-6.
13. Attisano L, Wrana JL. Smads as transcriptional co-modulators. Curr Opin Cell Biol. 2000; 12:235–43. https://doi.org/10.1016/S0955-0674(99)00081-2.
14. Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003; 113:685–700. https://doi.org/10.1016/S0092-8674(03)00432-X.
15. Derynck R, Zhang YE. Smad-dependent and Smadindependent pathways in TGF-beta family signalling. Nature. 2003; 425:577–84. https://doi.org/10.1038/nature02006.
16. Attisano L, Wrana JL. Signal transduction by the TGFbeta superfamily. Science. 2002; 296:1646–47. https://doi.org/10.1126/science.1071809.
17. Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005; 21:659–93. https://doi.org/10.1146/annurev.cellbio.21.022404.142018.
18. Imamura T, Takase M, Nishihara A, Oeda E, Hanai J, Kawabata M, Miyazono K. Smad6 inhibits signalling by the TGF-beta superfamily. Nature. 1997; 389:622–26. https://doi.org/10.1038/39355.
19. Nakao A, Afrakhte M, Morén A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten Dijke P. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997; 389:631–35. https://doi.org/10.1038/39369.
20. Topper JN, Cai J, Qiu Y, Anderson KR, Xu YY, Deeds JD, Feeley R, Gimeno CJ, Woolf EA, Tayber O, Mays GG, Sampson BA, Schoen FJ, et al. Vascular MADs: two novel MAD-related genes selectively inducible by flow in human vascular endothelium. Proc Natl Acad Sci U S A. 1997; 94:9314–19. https://doi.org/10.1073/pnas.94.17.9314.
21. Hata A, Lagna G, Massagué J, Hemmati-Brivanlou A. Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev. 1998; 12:186–97. https://doi.org/10.1101/gad.12.2.186.
22. Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, Richardson MA, Topper JN, Gimbrone MA Jr, Wrana JL, Falb D. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell. 1997; 89:1165–73. https://doi.org/10.1016/S0092-8674(00)80303-7.
23. Tsuneizumi K, Nakayama T, Kamoshida Y, Kornberg TB, Christian JL, Tabata T. Daughters against dpp modulates dpp organizing activity in Drosophila wing development. Nature. 1997; 389:627–31. https://doi.org/10.1038/39362.
24. Elliott RL, Blobe GC. Role of transforming growth factor Beta in human cancer. J Clin Oncol. 2005; 23:2078–93. https://doi.org/10.1200/JCO.2005.02.047.
25. Maeshima A, Maeshima K, Nojima Y, Kojima I. Involvement of Pax-2 in the action of activin A on tubular cell regeneration. J Am Soc Nephrol. 2002; 13:2850–59. https://doi.org/10.1097/01.ASN.0000035086.93977.E9.
26. Maeshima A, Vaughn DA, Choi Y, Nigam SK. Activin A is an endogenous inhibitor of ureteric bud outgrowth from the Wolffian duct. Dev Biol. 2006; 295:473–85. https://doi.org/10.1016/j.ydbio.2006.03.011.
27. Yang SP, Woolf AS, Yuan HT, Scott RJ, Risdon RA, O’Hare MJ, Winyard PJ. Potential biological role of transforming growth factor-beta1 in human congenital kidney malformations. Am J Pathol. 2000; 157:1633–47. https://doi.org/10.1016/S0002-9440(10)64801-8.
28. Liu S, Cieslinski DA, Funke AJ, Humes HD. Transforming growth factor-beta 1 regulates the expression of Pax-2, a developmental control gene, in renal tubule cells. Exp Nephrol. 1997; 5:295–300.
29. Dahl E, Koseki H, Balling R. Pax genes and organogenesis. BioEssays. 1997; 19:755–65. https://doi.org/10.1002/bies.950190905.
30. Robson EJ, He SJ, Eccles MR. A PANorama of PAX genes in cancer and development. Nat Rev Cancer. 2006; 6:52–62. https://doi.org/10.1038/nrc1778.
31. Torres M, Gómez-Pardo E, Dressler GR, Gruss P. Pax-2 controls multiple steps of urogenital development. Development. 1995; 121:4057–65.
32. Rothenpieler UW, Dressler GR. Pax-2 is required for mesenchyme-to-epithelium conversion during kidney development. Development. 1993; 119:711–20.
33. Sanyanusin P, Schimmenti LA, McNoe LA, Ward TA, Pierpont ME, Sullivan MJ, Dobyns WB, Eccles MR. Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat Genet. 1995; 9:358–64. https://doi.org/10.1038/ng0495-358. Erratum in: Mutation of the gene in a family with optic nerve colobomas, renal anomolies and vesicoureteral reflux. [Nat Genet. 1996].
34. Lindoso RS, Verdoorn KS, Einicker-Lamas M. Renal recovery after injury: the role of Pax-2. Nephrol Dial Transplant. 2009; 24:2628–33. https://doi.org/10.1093/ndt/gfp307.
35. Dressler GR, Wilkinson JE, Rothenpieler UW, Patterson LT, Williams-Simons L, Westphal H. Deregulation of Pax-2 expression in transgenic mice generates severe kidney abnormalities. Nature. 1993; 362:65–67. https://doi.org/10.1038/362065a0.
36. Modi DA, Tagare RD, Karthikeyan S, Russo A, Dean M, Davis DA, Lantvit DD, Burdette JE. PAX2 function, regulation and targeting in fallopian tube-derived highgrade serous ovarian cancer. Oncogene. 2017; 36:3015–24. https://doi.org/10.1038/onc.2016.455.
37. Chen K, Wei H, Ling S, Yi C. Expression and significance of transforming growth factor-β1 in epithelial ovarian cancer and its extracellular matrix. Oncol Lett. 2014; 8:2171–74. https://doi.org/10.3892/ol.2014.2448.
38. Chen EY, Mehra K, Mehrad M, Ning G, Miron A, Mutter GL, Monte N, Quade BJ, McKeon FD, Yassin Y, Xian W, Crum CP. Secretory cell outgrowth, PAX2 and serous carcinogenesis in the Fallopian tube. J Pathol. 2010; 222:110–16.
39. Mehra K, Mehrad M, Ning G, Drapkin R, McKeon FD, Xian W, Crum CP. STICS, SCOUTs and p53 signatures; a new language for pelvic serous carcinogenesis. Front Biosci (Elite Ed). 2011; 3:625–34.
40. Luu VD, Boysen G, Struckmann K, Casagrande S, von Teichman A, Wild PJ, Sulser T, Schraml P, Moch H. Loss of VHL and hypoxia provokes PAX2 up-regulation in clear cell renal cell carcinoma. Clin Cancer Res. 2009; 15:3297–304. https://doi.org/10.1158/1078-0432.CCR-08-2779.
41. Zhou TB. Signaling pathways of PAX2 and its role in renal interstitial fibrosis and glomerulosclerosis. J Recept Signal Transduct Res. 2012; 32:298–303. https://doi.org/10.3109/10799893.2012.738231.
42. Stayner C, Iglesias DM, Goodyer PR, Ellis L, Germino G, Zhou J, Eccles MR. Pax2 gene dosage influences cystogenesis in autosomal dominant polycystic kidney disease. Hum Mol Genet. 2006; 15:3520–28. https://doi.org/10.1093/hmg/ddl428.
43. Vanhove T, Goldschmeding R, Kuypers D. Kidney Fibrosis: origins and Interventions. Transplantation. 2017; 101:713–26. https://doi.org/10.1097/TP.0000000000001608.
44. Li L, Wu Y, Yang Y. Paired box 2 induces epithelialmesenchymal transition in normal renal tubular epithelial cells of rats. Mol Med Rep. 2013; 7:1549–54. https://doi.org/10.3892/mmr.2013.1365.
45. Doberstein K, Pfeilschifter J, Gutwein P. The transcription factor PAX2 regulates ADAM10 expression in renal cell carcinoma. Carcinogenesis. 2011; 32:1713–23. https://doi.org/10.1093/carcin/bgr195.
46. Frost V, Grocott T, Eccles MR, Chantry A. Self-regulated Pax gene expression and modulation by the TGFbeta superfamily. Crit Rev Biochem Mol Biol. 2008; 43:371–91. https://doi.org/10.1080/10409230802486208.
47. Muratovska A, Zhou C, He S, Goodyer P, Eccles MR. Paired-Box genes are frequently expressed in cancer and often required for cancer cell survival. Oncogene. 2003; 22:7989–97. https://doi.org/10.1038/sj.onc.1206766.
48. Grocott T, Frost V, Maillard M, Johansen T, Wheeler GN, Dawes LJ, Wormstone IM, Chantry A. The MH1 domain of Smad3 interacts with Pax6 and represses autoregulation of the Pax6 P1 promoter. Nucleic Acids Res. 2007; 35:890–901. https://doi.org/10.1093/nar/gkl1105.
49. Costamagna E, García B, Santisteban P. The functional interaction between the paired domain transcription factor Pax8 and Smad3 is involved in transforming growth factorbeta repression of the sodium/iodide symporter gene. J Biol Chem. 2004; 279:3439–46. https://doi.org/10.1074/jbc.M307138200.
50. Yang J, Zhang X, Li Y, Liu Y. Downregulation of Smad transcriptional corepressors SnoN and Ski in the fibrotic kidney: an amplification mechanism for TGF-beta1 signaling. J Am Soc Nephrol. 2003; 14:3167–77. https://doi.org/10.1097/01.ASN.0000099373.33259.B2.
51. Zauberman A, Lapter S, Zipori D. Smad proteins suppress CCAAT/enhancer-binding protein (C/EBP) beta- and STAT3-mediated transcriptional activation of the haptoglobin promoter. J Biol Chem. 2001; 276:24719–25. https://doi.org/10.1074/jbc.M005813200.
52. Quick CM, Ning G, Bijron J, Laury A, Wei TS, Chen EY, Vargas SO, Betensky RA, McKeon FD, Xian W, Crum CP. PAX2-null secretory cell outgrowths in the oviduct and their relationship to pelvic serous cancer. Mod Pathol. 2012; 25:449–55. https://doi.org/10.1038/modpathol.2011.175.
53. Alwosaibai K, Abedini A, Al-Hujaily EM, Tang Y, Garson K, Collins O, Vanderhyden BC. PAX2 maintains the differentiation of mouse oviductal epithelium and inhibits the transition to a stem cell-like state. Oncotarget. 2017; 8:76881–97. https://doi.org/10.18632/oncotarget.20173.
54. Beauchemin D, Lacombe C, Van Themsche C. PAX2 is activated by estradiol in breast cancer cells of the luminal subgroup selectively, to confer a low invasive phenotype. Mol Cancer. 2011; 10:148. https://doi.org/10.1186/1476-4598-10-148.
55. Monte NM, Webster KA, Neuberg D, Dressler GR, Mutter GL. Joint loss of PAX2 and PTEN expression in endometrial precancers and cancer. Cancer Res. 2010; 70:6225–32. https://doi.org/10.1158/0008-5472.CAN-10-0149.
56. Mentrikoski MJ, Wendroth SM, Wick MR. Immunohistochemical distinction of renal cell carcinoma from other carcinomas with clear-cell histomorphology: utility of CD10 and CA-125 in addition to PAX-2, PAX-8, RCCma, and adipophilin. Appl Immunohistochem Mol Morphol. 2014; 22:635–41. https://doi.org/10.1097/PAI.0000000000000004.
57. Ozcan A, de la Roza G, Ro JY, Shen SS, Truong LD. PAX2 and PAX8 expression in primary and metastatic renal tumors: a comprehensive comparison. Arch Pathol Lab Med. 2012; 136:1541–51. https://doi.org/10.5858/arpa.2012-0072-OA.
58. Hueber PA, Waters P, Clark P, Eccles M, Goodyer P. PAX2 inactivation enhances cisplatin-induced apoptosis in renal carcinoma cells. Kidney Int. 2006; 69:1139–45. https://doi.org/10.1038/sj.ki.5000136. Erratum in: Kidney Int. 2006; 70:1666. Clarke, P [corrected to Clark, P].
59. Huang W, Cen S, Kang XL, Wang WF, Wang Y, Chen X. TGF-β1-induced Fascin1 promotes cell invasion and metastasis of human 786-0 renal carcinoma cells. Acta Histochem. 2016; 118:144–51. https://doi.org/10.1016/j.acthis.2015.12.005.
60. Kou B, Liu W, Tang X, Kou Q. HMGA2 facilitates epithelial-mesenchymal transition in renal cell carcinoma by regulating the TGF-β/Smad2 signaling pathway. Oncol Rep. 2018; 39:101–08.
61. Lebdai S, Verhoest G, Parikh H, Jacquet SF, Bensalah K, Chautard D, Rioux Leclercq N, Azzouzi AR, Bigot P. Identification and validation of TGFBI as a promising prognosis marker of clear cell renal cell carcinoma. Urol Oncol. 2015; 33:69.e11-8. https://doi.org/10.1016/j.urolonc.2014.06.005.
62. Shang D, Liu Y, Yang P, Chen Y, Tian Y. TGFBI-promoted adhesion, migration and invasion of human renal cell carcinoma depends on inactivation of von Hippel-Lindau tumor suppressor. Urology. 2012; 79:966.e1-7. https://doi.org/10.1016/j.urology.2011.12.011.
63. Zhu J, Liang C, Hua Y, Miao C, Zhang J, Xu A, Zhao K, Liu S, Tian Y, Dong H, Zhang C, Li P, Su S, et al. The metastasis suppressor CD82/KAI1 regulates cell migration and invasion via inhibiting TGF-β 1/Smad signaling in renal cell carcinoma. Oncotarget. 2017; 8:51559–68. https://doi.org/10.18632/oncotarget.18086.
64. Liu P, Gao Y, Huan J, Ge X, Tang Y, Shen W, Tian Y, Shen W, Zou S, Zhou J, Zhang S. Upregulation of PAX2 promotes the metastasis of esophageal cancer through interleukin-5. Cell Physiol Biochem. 2015; 35:740–54. https://doi.org/10.1159/000369734.
65. Stayner CK, Cunliffe HE, Ward TA, Eccles MR. Cloning and characterization of the human PAX2 promoter. J Biol Chem. 1998; 273:25472–79. https://doi.org/10.1074/jbc.273.39.25472.
66. Wicks SJ, Haros K, Maillard M, Song L, Cohen RE, Dijke PT, Chantry A. The deubiquitinating enzyme UCH37 interacts with Smads and regulates TGF-beta signalling. Oncogene. 2005; 24:8080–84. https://doi.org/10.1038/sj.onc.1208944.
67. McConnell MJ, Cunliffe HE, Chua LJ, Ward TA, Eccles MR. Differential regulation of the human Wilms tumour suppressor gene (WT1) promoter by two isoforms of PAX2. Oncogene. 1997; 14:2689–700. https://doi.org/10.1038/sj.onc.1201114.
68. Li CG, Nyman JE, Braithwaite AW, Eccles MR. PAX8 promotes tumor cell growth by transcriptionally regulating E2F1 and stabilizing RB protein. Oncogene. 2011; 30:4824–34. https://doi.org/10.1038/onc.2011.190.
69. Sehgal R, Sheibani N, Rhodes SJ, Belecky Adams TL. BMP7 and SHH regulate Pax2 in mouse retinal astrocytes by relieving TLX repression. Dev Biol. 2009; 332:429–43. https://doi.org/10.1016/j.ydbio.2009.05.579.