
INTRODUCTION
Unlimited proliferation is one of the hallmarks of cancer [1, 2]; however, to realize their capacity for unlimited growth in vivo tumors overcome multiple constraints posed by normal host tissues. To grow beyond the threshold size of several mm in diameter, tumors require autonomous blood supply, which is generated through neovascularization (angiogenesis), another hallmark of cancer [2]. Endogenous anti-angiogenic proteins are present at high levels in normal adult tissues and their expression is often decreased in the course of tumor progression, an event permissive for tumor angiogenesis [3]. Thrombospondin-1 (TSP1) was the first endogenous angiogenesis inhibitor to be identified [4] and its loss is among the critical events in progression of multiple cancers including carcinomas of the breast and colon as well as skin cancers [5, 6]. Previous studies identified TSP1 as a critical angiogenesis inhibitor in human and mouse skin, whose expression is blocked by ultraviolet B (UVB) [7, 8]. Ectopic TSP1 delays the growth of xenografted cutaneous tumors in mice and TSP1 transgene mitigates acute and long-term UVB damage in the skin [9, 10]. Thus TSP1 is an important therapy target in multiple cancer types, including non-melanoma skin cancers and melanoma [11, 12]. Here we report that plant flavonoid apigenin effectively restores TSP1 expression in UVB-irradiated skin.
Apigenin, is a potent chemopreventive agent and inhibitor of UVB-induced skin carcinogenesis [13]. We have shown that apigenin causes G1 and G2/M growth arrest by targeting cyclins B1 and D1 [14, 15] and by promoting p53-dependent transcription [16] in epidermal keratinocytes exposed to UVB. In colon and prostate cancers, apigenin causes growth arrest and cell death via DR5 [17, 18] and E-cadherin [19]. Importantly, apigenin blocks cyclooxygenase (Cox)-2 and its target Prostaglandin E2 (PGE2) [20, 21] via complex regulatory mechanism, whereby two RNA-binding protein HuR and translational repressor T-cell specific antigen 1-related protein inhibit Cox-2 induction by UVB [22].
In addition to its cytotoxic and cytostatic effects, apigenin inhibits angiogenesis. In non-skin tissues, this has been attributed to the suppression HIF-1α and VEGF [23–26]. Other in vitro studies indicate the role of nitric oxide (NO) and IL6/STAT pathways [27, 28]. Here, we show that apigenin inhibits cutaneous angiogenesis, at least in part, by maintaining high levels of anti-angiogenic TSP1. Apigenin mitigated the TSP1 loss due to UVB exposure in the epidermal keratinocytes in culture and in mouse skin. Moreover, it alleviated angiogenic and proliferative responses to UVB radiation in the skin in vivo. The regulation of TSP1 by apigenin was post-transcriptional. TSP1 mRNA was strongly decreased in UVB-treated cells and remained low in the presence of apigenin. In contrast, apigenin increased translational activity of existing TSP1 transcripts. Specifically, it increased cytosolic localization of the RNA-binding protein Human antigen R (HuR) and HuR association with TSP1 mRNA, causing enhanced translation and high TSP1 protein levels.
Thrombospondin-1 is a large multi-functional protein whose anti-angiogenic activity was mapped to the Type 1 malarial repeats, with CD36 as one of the signaling receptors on the vascular endothelium (reviewed in [29]). Importantly, peptide mimetic of TSP1, ABT-898, which represents anti-angiogenic thrombospondin repeats (TSR2) and blocks angiogenesis in CD36-dependent manner [29], was sufficient to reproduce the anti-angiogenic and cytostatic effects of apigenin in the skin suggesting a key functional role of TSP1 in apigenin’s chemoprevention of UVB-induced oncogenic events. To our knowledge, this is the first demonstration of the regulation of an endogenous anti-angiogenic factor by apigenin, with dramatic effect on cutaneous angiogenesis.
RESULTS
Apigenin restores TSP1 expression in UVB-treated skin keratinocytes
Seeking mechanisms underlying apigenin’s effects in the skin, we assessed TSP1 levels in mouse and human keratinocytes exposed to UVB and treated with apigenin. In immortalized 308 mouse keratinocytes (Fig. 1a) and in primary normal human epidermal keratinocytes (NHEKs, Fig. 1b), TSP1 expression was dramatically decreased 12 hr following UVB exposure. Importantly, both pre-treatment with apigenin (1-2 hrs prior to UVB) Fig. 1a, b and d) and treatment immediately after UVB irradiation (Fig. 1c) similarly increased TSP1 protein levels. In sham-irradiated cells, apigenin caused a moderate increase of already high TSP1 levels, compared to a robust increase in UVB-irradiated cells, where TSP1 was barely detectable (Fig. 1d).
Importantly, apigenin restored TSP1 expression in the UVB-treated skin in vivo. As was shown previously, TSP1 expression (detected by immunohistochemistry, IHC) was vanishingly low in the skins of mice after UVB irradiation. In contrast, in animals treated with topical apigenin, TSP1 staining in the epidermis was similar to that of untreated controls (Fig. 1e). Interestingly, one of the TSP1 anti-angiogenic receptors, CD36, was similarly downregulated by UVB and restored by apigenin (Supplementary Fig. 1).
Apigenin and TSP1 peptide mimetic blocks UVB-induced epidermal thickening
TSP1 active regions have been extensively analyzed and its anti-angiogenic activity mapped to the Type 1 malarial repeats (TSR), which yielded series of highly active TSP1 peptide mimetics. One of them, ABT-898, derived from TSR2, is especially potent in multiple models of angiogenesis and tumor growth and reproduces a full spectrum of TSP1 angioinhibitory effects [30–32]. We therefore tested its ability to reproduce apigenin’s effects in the UVB-treated skin. Epidermal thickening was noted as early as 12 hrs after UVB exposure (Fig. 2a, c) and lasted up to 48 hours (Supplementary Fig. 2). The thickening was largely due to hyperproliferation, as was evidenced by increased number of the keratinocyte layers (Fig. 2a, d). Apigenin significantly decreased both the thickness and the number of layers in the epidermis of irradiated mice (Fig. 2a, c, d). Importantly, subcutaneous injections of ABT-898 had a similar effect and the combination of apigenin and ABT-898 caused even greater decrease in epidermal thickness and the number of keratinocyte layers (Fig. 2c, d). In agreement, UVB caused an approximately 4-fold increase in proliferation, as was measured by IHC for Ki-67 (Fig. 2b, e). Treatment with either apigenin or ABT-898 caused a 2-fold reduction in the number of Ki-67 positive nuclei per linear 100μm of epidermis. The combined effect of apigenin and ABT-898 exceeded the effects of each compound alone and this difference was statistically significant (Fig. 2e).
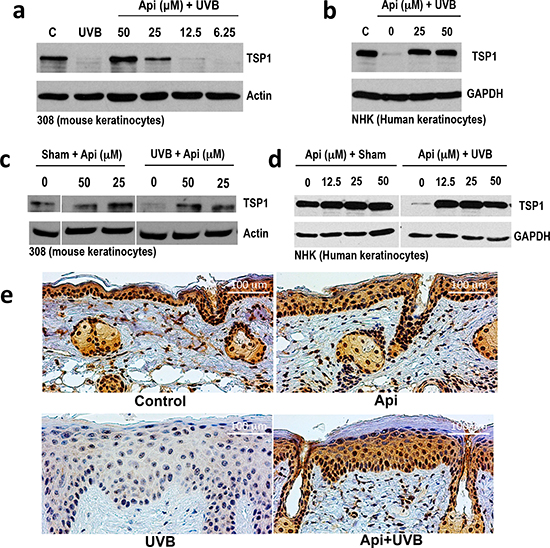
Figure 1: The effect of UVB and apigenin on TSP1 expression in epidermal keratinocytes in vitro and in vivo. (a, b) Mouse 308 keratinocytes (a) and normal human epidermal keratinocytes (NHEKs, b) were pre-treated with indicated concentrations of apigenin (Api) for 1 hr before UVB exposure (1000J/m2), and TSP1 was detected by Western Blot in cell extracts collected 12 h after UVB irradiation. (c) 308 cells were subjected to sham or UVB irradiation and apigenin added to cell cultures at indicated concentrations immediately after irradiation. The extracts were harvested after additional 12 hrs and analyzed by Western blot. (d) NHEKs were pre-treated with increasing concentrations of apigenin, where indicated and subjected to sham or UVB irradiation. Cell extracts were collected 16 hr post-irradiation and TSP1 expression analyzed by Western blot. Actin and GAPDH were used to assess loading. (e) SKH-1 mice were treated with UVB (5 consecutive days, 1300 J/m2 /day). Where indicated, the animals were pre-treated with topical apigenin (5 μMol in DMSO/acetone). Dorsal epidermis was harvested, sectioned and stained for TSP1.
ABT-898 recapitulates the anti-angiogenic effect of apigenin in the skin
The anti-angiogenic effect of apigenin in the non-skin tissues has been shown previously [23, 33, 34] and attributed to the decreased production of VEGF-A, due to the blockade of hypoxic and growth factor signaling. However, apigenin’s effect on cutaneous angiogenesis and TSP1 has not been documented. We therefore analyzed apigenin’s anti-angiogenic effects in the skin and tested the ability of TSP1 anti-angiogenic peptide mimetic to reproduce these actions. UVB significantly increased microvascular density (MVD) in the dermis, as was measured using IHC for the endothelial marker, CD31 (Fig. 3a, b), and in the underlying adipose tissue (data not shown). In contrast, treatment with apigenin and/or ABT-898 inhibited UVB-induced angiogenesis by at least 60%, suggesting a critical role for TSP1 in the anti-angiogenic action of apigenin.
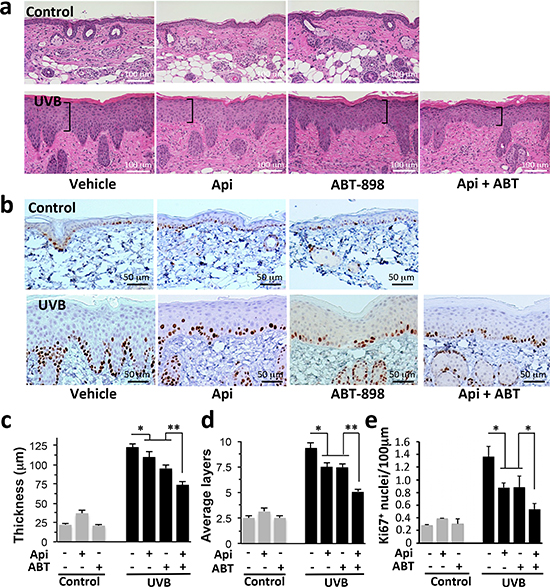
Figure 2: The effect of apigenin and TSP1 peptide mimetic (ABT-898) on UVB-induced skin thickening and proliferation. Mice were subjected to sham or UVB radiation (1300 J/m2 daily, 5 days). Where indicated, topical Apigenin (5 μMol) and subcutaneous ABT-898 (100 ng/ml in PBS, 30 mg/kg) were given 2 h prior to each UVB exposure. Mice were sacrificed at 12, 24 and 48 h after final exposure and dorsal skins harvested, fixed in formalin and paraffin-embedded. Five μm sections were stained with Hematoxylin and Eosin (H&E) for morphometric analysis (see Methods). (a, c, d) Dorsal skins harvested at 12 hrs post irradiation were used for H&E staining and subsequent morphometry analysis. Note the visibly decreased thickness of the UVB-irradiated epidermis in mice treated with apigenin and/or ABT-898. (b, e) IHC for proliferation marker Ki-67 at 24 hrs post irradiation. (c) Measurement of epidermal thickness. At least 3 sections per animal and 3 animals per data point were evaluated. (d) The number of epidermal cell layers was determined in 3 random measurements per section, a minimum of 2 sections per animal, in 3 animals per data point. **P value <0.0001; *P <0.003 as determined by one-way ANOVA. (e) Quantitative analysis of the experiment in (b) was performed using ImageJ64 software on thresholded images, using “Particle Analysis” function. A minimum of three 10x fields from 3 independent sections were examined and Ki-67 positive nuclei calculated per linear 100 μm of epidermis. Hair follicles were not included in analysis. *P<0.01 as was determined by two-tailed Student’s T-test using pairwise comparisons.
UVB is known to increase expression of Cox-2 [35, 36], a critical mediator of inflammation and angiogenesis [37, 38]. We have earlier shown that apigenin inhibits the induction of Cox-2 by UVB [20, 39]. ABT-898 alone caused a minor increase in CD31 staining (Fig. 3a, b) and Cox-2 expression (Fig. 3c, d), however this trend did not reach significance. In contrast, ABT-898 significantly reduced UVB-induced angiogenesis (Fig. 3b, P<0.001) and Cox-2 expression (Fig. 3c, d, P<0.009).
ABT-898 blocks the stimulation of HIF pathway by UVB
Apigenin inhibits HIF pathway and VEGF production in non-skin tissues. We used ABT-898 to determine whether these effects of apigenin could be related to TSP1.
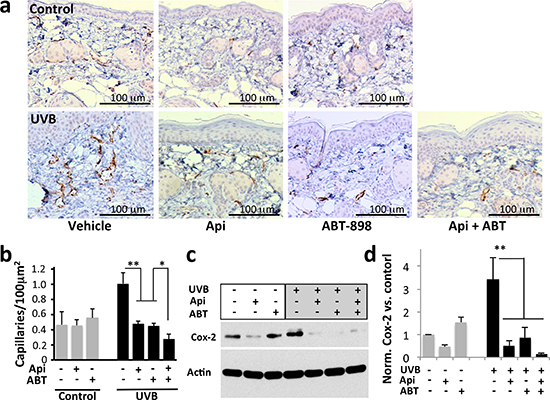
Figure 3: The effect of apigenin and ABT-898 on UVB-induced angiogenesis. Mice were subjected to sham or UVB radiation (1300 J/m2 daily, 5 consecutive days). Topical Apigenin (5 μMol) and subcutaneous ABT-898 (100 ng/ml in PBS, 30 mg/kg) were given daily 1–2 h prior to UVB exposure. Mice were sacrificed 48 hr after final exposure and dorsal skins were harvested, formalin fixed and paraffin embedded. (a) IHC for an endothelial cell marker CD31. Note a visible increase in microvascular density (MVD) caused by UVB. (b) Quantitative analysis of the IHC was performed using “Object Count” function in ImageJ64 software. A minimum of 10x fields from 3 independent skin sections were examined and MVD calculated per 100 μm2 area; *P>0.1 **P<0.001 by two-tailed Student’s T-test. (c, d) TSP1 and apigenin inhibit Cox-2 induction by UVB. Control and UVB-irradiated 308 keratinocytes were treated with apigenin (50μM) and ABT-898 (100nM) where indicated and Cox-2 in cell extracts was measured by Western blot (c) and quantified by densitometric analysis (ImageJ64 software) (d). The graph represents average of three independent experiments, all normalized to untreated control. **P<0.009
IHC for HIF-1α showed positive staining in control skin, predominantly in basal layer, which was not significantly altered by apigenin or ABT-898 (Fig. 4a). The expression of HIF-1α (staining intensity) increased 24 hrs post-irradiation (Fig. 4a, red arrow). This increase was abolished by apigenin and/or ABT-898 (Fig. 4a).
VEGF is the main pro-angiogenic target of the HIF pathway. Accordingly VEGF staining of epidermal keratinocytes was also increased by UVB 24 hrs post-irradiation (Fig. 4b, red arrow) and this increase was abolished by apigenin and by ABT-898, alone or in combination (Fig. 4b). In sub-epidermal skin layers, VEGF staining was localized mainly to the intercellular spaces (Fig. 4b, black arrow), presumably due to its binding to the extracellular matrix.
Apigenin regulates TSP1 via the RNA-binding protein HuR
Pre-treatment with apigenin strongly increased TSP1 levels in the UVB-irradiated skin and epidermal keratinocytes (Fig. 1). Surprisingly, apigenin failed to restore TSP1 mRNA levels in UVB-irradiated keratinocytes (Fig. 5a), suggesting regulation at post-transcriptional level. We therefore investigated its potential regulatory effect on TSP1 translation. We focused on ELAV-like 1 human antigen R (HuR) because it is one of apigenin’s molecular targets [20] and is involved in control of mRNA stability and translation. HuR can shift the ratio between translated and untranslated mRNA towards the active fraction [40] and has been reported to promote de novo TSP1 synthesis [41]. We used siRNA knockdown to test the role of HuR role in TSP1 regulation by apigenin. Indeed, HuR silencing diminished the ability of apigenin to rescue TSP1 expression in UVB-treated cells (Fig. 5b). Moreover, real time RT-PCR of mRNA from HuR complexes isolated by immunoprecipitation, showed increased recruitment of TSP1 mRNA in apigenin-treated keratinocytes (Fig. 5c). Importantly, metabolic labeling with [35S]-methionine showed higher de novo TSP1 synthesis in UVB irradiated cells treated with apigenin, which was abolished in proportion with the extent of HuR knockdown (Fig. 5d). Analysis of whole cell extracts was performed to show that equal amount of [35S]methionine/cysteine was incorporated into the cells and that protein translation in general was not affected by the different treatments.
Apigenin regulates HuR subcellular localization
In the cell, HuR activity is determined by its localization. Inactive HuR is sequestered in the nuclei and cytoplasmic HuR becomes active and controls mRNA stability and translation [42]. In sham- and UVB-irradiated 308 keratinocytes, apigenin treatment significantly increased the levels of HuR in the cytoplasm 4 hrs post irradiation (Fig. 5e). Importantly, apigenin increased cytoplasmic HuR in UV-irradiated mouse skin in vivo 24 hours post irradiation (Fig. 5f).
HuR localization can be controlled by multiple kinases including Erk1/2, p38, JNK-1 and Chk2 (reviewed in [43]). All of these are perturbed by apigenin in diverse cell types [44–46]. To identify the player(s) involved in apigenin regulation of HuR in the skin, we have measured TSP1 expression and assessed HuR cytoplasmic localization in the NHEK cells pre-treated with specific kinase inhibitors. The ability of apigenin to restore TSP1 expression in the UVB-irradiated keratinocytes was impaired when JNK-1 and Chk2 were blocked (Fig. 6a). Chk2 inhibitor caused a marked decrease in HuR cytoplasmic localization (Fig. 6b) suggesting that Chk2 is a critical mediator of apigenin’s regulation of HuR and TSP1. In addition, JNK-1 inhibitor markedly altered HuR staining in the cytoplasm, with striking punctate pattern. A similar, although less pronounced pattern was observed in the presence of Chk2 inhibitor (Fig. 6b).
However, JNK-1 blockade, but not Chk2 blockade, decreased TSP-1 levels regardless of apigenin and/or UVB exposure. (Supplementary Figure 3).
Together, our results indicate that apigenin prevents oncogenic UVB action in the skin, including proliferation and angiogenesis, through activation of CHK2 and possibly JNK-1, causing HuR translocation to the cytoplasm, where it binds TSP1 mRNA and increases its translational activity. In addition, TSP1 inhibits Cox-2 and poses additional restrictions on dermal angiogenesis and proliferation (Fig. 7).
DISCUSSION
Long-term sun exposure is widespread and significantly increases the risk of epithelial cell cancers; even a single UVB exposure causes skin alterations including erythema, epidermal hyperplasia, vascular dilation and hyperpermeability [47]. Vascular changes due to UVB are especially prominent in the skin and have been attributed to the increased pro-angiogenic cytokines and chemokines, which include VEGF, basic fibroblast growth factor (bFGF), and interleukin-8 (reviewed in [48]). On the other hand, neoangiogenesis and vascular leakage are tightly controlled by TSP1, an endogenous angiogenesis inhibitor expressed at high levels by epidermal keratinocytes. Previous studies showed that acute vascular reactions to UVB are more pronounced in the skin of TSP1-null mice [7, 8]. Moreover, mice with TSP1 overexpression targeted to dermal keratinocytes exhibit weakened vascular responses to UVB irradiation, including cutaneous angiogenesis, increased endothelial cell proliferation, and apoptosis [49]. Furthermore, TSP1 transgene significantly mitigates the longer-term UVB damage in the skin, such as photo-damage and wrinkling [49]. These results stress the importance of the cutaneous vasculature in UVB-induced skin damage. They point to anti-angiogenesis as a target in preventing chronic photo-damage and skin carcinogenesis and suggest TSP1 as a means for skin cancer prevention and therapy.
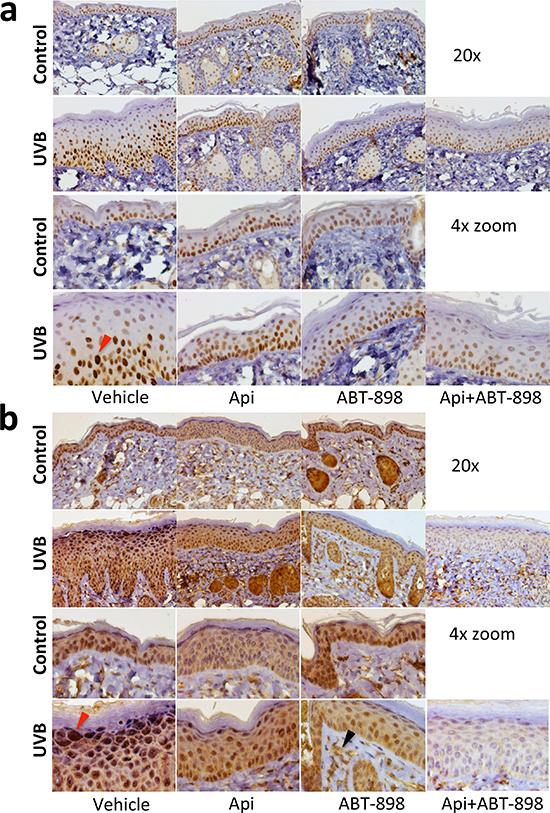
Figure 4: ABT-898 inhibits the induction of HIF pathway by UVB in epidermal keratinocytes. Mice were subjected to sham or UVB radiation (1300 J/m2 daily, 5 days). Where indicated, topical Apigenin (5 μMol) and subcutaneous ABT-898 (100 ng/ml in PBS, 30 mg/kg) were given 2 h prior to each UVB exposure. Mice were sacrificed at 24 h after final exposure to UVB and dorsal skins harvested, fixed in formalin and paraffin-embedded. Five μm sections were stained with antibodes agains HIF-1α and VEGF (see Methods) and countesrtained with hematoxylin. (a) Staining for HIF-1α. Images were taken at 20x magnification. A 4x zoom is shown below. Red arrow points to the nucleus with increased HIF-1α after UVB irradiation. (b) VEGF stiaining. Note a significant increase in VEGF staining in the uppermost keratinocyte layers after UVB exposure (red arrow). Also note the lack of VEGF regulation by apigenin and ABT-898 in non-irradiated skin and visible decrease in VEGF staining in the skin that was exposed to UVB. Sub-epidermal VEGF staining (black arrow) is localized to intercellular spaces; it appears unaltered and serves as an internal control.
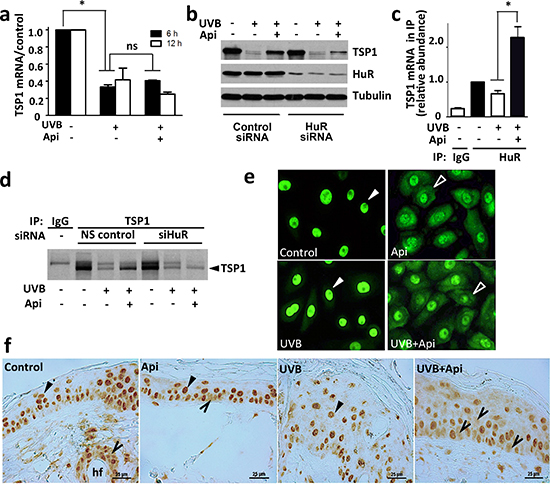
Figure 5: Apigenin regulates TSP1 translation via HuR. (a) Apigenin did not restore TSP1 mRNA level in UVB-irradiated cells. Control and UVB-irradiated 308 mouse keratinocytes were treated with apigenin where indicated and TSP1 mRNA measured by real-time RT-PCR. Statistical significance was determined in pairwise comparison by two-tailed Student’s T-test. *P<0.002; ns, not significant (P>0.4). (b) HuR was required for apigenin to maintain TSP1 expression. HuR knockdown in cells treated with control and HuR-specific siRNA was ascertained by Western blot (middle panel). The cells were then subjected to UVB irradiation and treated with apigenin or control vehicle. Note diminished TSP1 levels (top panel) upon HuR knockdown. The membrane was re-probed for alpha-tubulin to ascertain equal loading. (c) Apigenin increases HuR binding to TSP1 mRNA. The cells were treated with UVB and apigenin or control vehicle. The lysates were harvested and subjected to immunoprecipitation with HuR specific antibodies or control IgG and subsequent real-time PCR with primers for TSP1 mRNA. Note significant increase in the TSP1 mRNA in HuR protein complexes from the cells treated with the combination of UVB and apigenin. *P<0.008. (d) Apigenin controls de novo TSP1 protein synthesis via HuR. Mouse 308 keratinocytes were pre-incubated in methionine and cysteine-free medium and exposed to UVB in the absence and in the presence of apigenin. After 12 h the cells were given 15-min pulse of [35S] L-methionine and [35S] L-cysteine. The lysates were subjected to IP with anti-TSP1 antibody or normal mouse IgG (control). (e, f) Apigenin promotes cytoplasmic localization of HuR in UVB-irradiated keratinocytes and in the skin. (e) The cells were treated as indicated and HuR localization assessed by immunofluorescence (green). (f) Mice were subjected to UVB irradiation, with and without apigenin pre-treatment as above. Dorsal skins were harvested, sectioned and HuR localization assessed by IHC. Note nuclear HuR localization (filled arrows) in the absence of apigenin and cytoplasmic presence of HuR (empty arrowheads) in apigenin-treated cells/skins. Note a more pronounced cytoplasmic HuR localization in skins treated with UVB and apigenin combination.
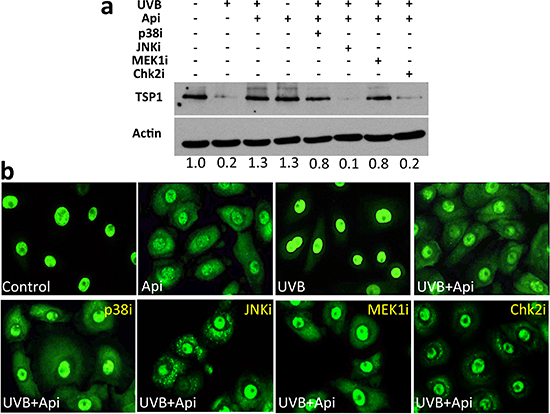
Figure 6: HuR-translocation and recovery of TSP1 expression by apigenin requires Chk2 kinase. NHEKs were treated with UVB and apigenin. Specific inhibitors of p38 (SB203580), MEK1 in the Erk1/2 pathway (U0126) and Chk2 inhibitor II, were added at 10 μM and JNK-1 inhibitor SP600125, was added at 50 μM. (a) TSP1 in whole cell extracts was measured by Western blot. Note that apigenin failed to restore TSP1 expression in the presence of JNK-1 and Chk-1 inhibitors. Numbers under the panel indicate values determined by densitometry (ImageJ) adjusted for loading (Actin) and normalized to untreated control. (b) NHEKs grown on coverslips were fixed and immunofluorescence performed to visualize HuR. Note decreased cytoplasmic staining in the presence of Chk2 inhibitor.
Apigenin is a natural chemopreventive agent with clear anti-angiogenic effects [23, 33]. Studies in cancer cell lines linked its anti-angiogenic potential to the decreased HIF-1 or STAT-3 signaling and resultant down-regulation of pro-angiogenic VEGF [23, 50]. Other studies show that apigenin decreases production and activity of proteases including MMP-1, uPA and MMP-2 and thus attenuates angiogenesis [34]. Our recent findings in prostate cancer cells and epidermal keratinocytes demonstrate the inhibition of Cox-2 and PGE2 synthesis by apigenin, which could potentially hinder angiogenesis [20, 21, 39].
Here, we show for the first time that apigenin inhibits UVB-induced cutaneous angiogenesis by maintaining normal high levels of endogenous TSP1. As was shown previously, UVB irradiation decreases TSP1 protein and mRNA in cultured mouse and human keratinocytes and in mouse skin. Importantly, we showed that pre-treatment with apigenin restored high TSP1 protein levels in vitro and in vivo. Moreover, in our study apigenin strongly attenuated neoangiogenesis, proliferation and epidermal thickening in mice exposed to UVB irradiation. Furthermore, TSP1 bioactive peptide mimetic, ABT-898, restored normal angiogenesis and skin thickness and reduced proliferation in the UVB-irradiated mouse skin, suggesting that TSP1 induction by apigenin is sufficient to alleviate the acute UVB damage. Full-length TSP-1 is a large molecule with multiple functional domains (Type-2 repeats, EGF-like domain, and N-terminal region) whose effects are distinct from these of the anti-angiogenic TSR represented by ABT-898. Because it reproduces a large portion of apigenin’s effects in UVB-irradiated skin and keratinocytes, we conclude that the main effects of TSP1 in this setting are mediated trough its Type I (TSR) repeats.
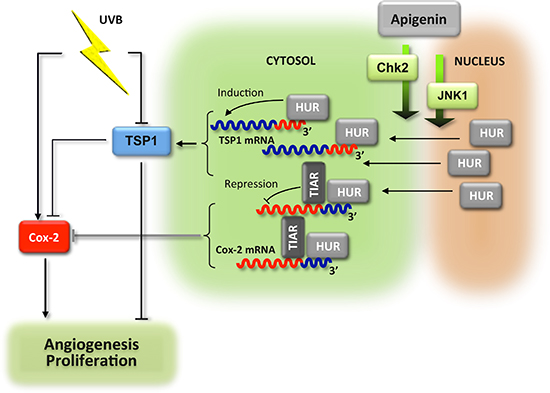
Figure 7: Schema of the anti-angiogenic cascade induced by apigenin in the UVB-irradiated skin. UVB exposure increases proliferation of the epidermal keratinocytes and elevated angiogenesis in sub-epidermal layers. These events are dependent on the increased production of Cox-2 and reduced levels of mRNA encoding anti-angiogenic TSP1. In the presence of apigenin, HuR enters cytoplasm of the epidermal keratinocytes where it binds and stabilizes TSP1 mRNA and increases de novo translation. In contrast, Cox-2 mRNA becomes destabilized. The accumulation of TSP1 protein results in attenuated proliferation and angiogenesis. In addition, TSP1 decreases Cox-2 production via unknown mechanism, and further augments anti-proliferative and anti-angiogenic action of apigenin.
In contrast with apigenin, TSP1 has no documented effects on HIF-1α. However, TSP1 peptide ABT-898 was shown to decrease VEGF production in mice with orthotopic ovarian cancers [51]. Here we show, the same TSP1 peptide mimetic decreased HIF-1α levels and inhibited VEGF production in the epidermal keratinocytes caused by UVB exposure. Moreover, the same peptide reproduced another important effect of apigenin in UVB-treated cells, the reduction of Cox-2 expression. Apparently apigenin controls Cox-2 via HuR, as was shown previously [20], and this pathway involves TSP1. Cox-2 is one of the mediators of VEGF-induced angiogenesis [52]. Thus the inhibition of Cox-2 induction could be a consequence of VEGF repression by ABT-898.
Interestingly, apigenin had opposing effects on the two modifiers of angiogenesis, Cox-2 and TSP1. We have recently discovered that apigenin causes cytoplasmic localization of the RNA-binding protein HuR in UVB-treated keratinocytes where it binds the AU-rich motif in the 3’ untranslated region (UTR) of the Cox-2 mRNA and engages another RNA-binding protein, TIAR, causing translational repression [20]. Here, we found that similar to Cox-2, apigenin increased the binding of HuR to the TSP1 mRNA. However, unlike Cox-2, HuR binding increased de novo TSP1 synthesis. This divergent effect could be due to the differences in the molecular landscape of the 3’UTR of the Cox-2 and TSP1 genes. Previous studies demonstrate that HuR binding to the 3’UTR and stabilization of the target mRNA can lead to multiple outcomes. In some cases, HuR partners with translational repressors, such as Smaug1 and thereby blocks protein synthesis [53]. In others, HuR interferes with Argonaut (Ago) binding and formation of RISC complexes containing miRNA, and thus lifts translational repression [54]. The miRNA response to UVB is not well understood, however several miRNA have been implicated [55]. The 3’UTR of human and mouse TSP1, unlike that of Cox-2, contains multiple binding sites for the UVB-responsive miRNA including miR-141, miR-125 and miR-495, adjacent to the putative HuR binding site(s) (Supplementary Tables 1a, b and 2a, b). Thus it is possible that inhibition of RISC underlies the increased translation of the TSP1 message via apigenin - HuR axis in the UVB-treated keratinocytes and skin. Our results also indicate that Chk2 (MAPKAPK2) and possibly JNK-1 are likely mediators of HuR cytoplasmic translocation due to apigenin exposure. Interestingly, blocking JNK and Chk2 resulted in characteristic punctate patterns of HuR staining. Similar patterns were noted previously in HeLa cells subjected to heat shock and ascribed to stress granule assembly [56]. Moreover, similar granules were identified as the site where HuR target mRNAs are degraded [57].
Previous studies identify TSP1 as a p53 target in some tissues, including fibroblasts and epithelial cells of the mammary gland [58–60]. However, the mechanisms that govern TSP1 expression are tissue-specific: distinct regulatory pathways have been identified in fibroblasts and epithelial cells [61]. Moreover, p53 regulation of TSP1 may be indirect, as in colon cancer, where p53 inhibits TSP1 expression through miR-194 [62]. Similar mechanisms may be at work in cutaneous keratinocytes, where of TSP1 levels rapidly decline despite stabilization and accumulation of p53 that occurs in response to UVB-induced DNA damage.
In conclusion, our results demonstrate a previously unknown effect of apigenin in the skin, where it counters UVB-induced angiogenesis by maintaining normal high levels of the endogenous angiogenesis inhibitor, TSP1. This finding has important therapeutic implications. Apigenin is orally bioavailable and non-toxic [63] and thus presents a cheap and effective alternative to the bioactive peptides, which remain the only available type of TSP1-derived therapy [29]. This is particularly relevant because of the general importance of TSP1 in blocking acute and long-term UVB skin damage and carcinogenesis [7–9, 64–66]. TSP1 inhibits angiogenic activity by multiple growth factors [67, 68], which is especially important because in addition to VEGF, UVB induces multiple pro-angiogenic cytokines including IL-6, IL-8 and bFGF [48]. Thus maintaining sufficient levels of endogenous TSP1 is more likely to have profound effect on angiogenesis than VEGF inhibitors alone.
Our study also yields important mechanistic insight into context-dependent dichotomy of translational regulation by the RNA-binding protein HuR in the skin. We show that cytoplasmic translocation of HuR caused by apigenin and subsequent binding to the 3’ regions on the target mRNA can cause either repression or activation of translation, as is the case for Cox-2 and TSP1, respectively, the events that culminate in the inhibition of angiogenesis and proliferation in the UVB-irradiated skin.
MATERIALS AND METHODS
Cells and reagents
All chemicals were from Sigma–Aldrich Co (St. Louis, MO) unless otherwise specified. For detailed procedures and antibodes, see Supplementary materials section Supplementary Methods and Supplementary Table 3. The mouse keratinocyte cell line 308 was derived from Balb/c mouse skin; primary normal human epidermal keratinocytes (NHEKs) were isolated at the Skin Disease Research Center Core (Northwestern University) as described previously [69]. Specific protein kinase inhibitors for MEK1 (U0126) in the Erk1/2 pathway and Chk2 (Chk2II) were from Millipore (Billerica, MA) and used at 10 μM working concentration. Inhibitors of p38 (SB203580) and JNK-1 kinases (SP600125) from InvivoGen (San Diego, CA) were used at 10 and 50 μM, respectively.
Treatment of cells
The cells were irradiated with FS40T12 lamps (National Biological, Twinsburg, OH, emission peak 313 nm) with Kodacel filter (Eastman Kodak, Rochester, NY) to eliminate UVC. Apigenin stock (50 mM in dimethyl sulfoxide, DMSO) was diluted in cell media. ABT-898 (Ac-GV-DalloIle-SQIRP-ethylamide), an octapeptide derived from TSP1 active internal peptide GVITRIR [30], was made to order (CPC Scientific, Sunnyvale, CA), reconstituted in water to 10 mg/ml and diluted in cell media as desired.
Treatment of mice
We have performed only the procedures approved by Northwestern University Animal Care and Use Committee, in strict adherence to the National Institutes of Health guidelines. Six to 8-week old female SKH-1 hairless mice (Charles River, Wilmington, MA) in groups of five were treated with sham or UVB radiation (1300 J/m2 daily, 5 days), topical Apigenin (5 μMol) in DMSO/acetone (1:9 v/v) and subcutaneous ABT-898 (100 ng/ml in PBS, 30 mg/kg) were given 1-2 h prior to each radiation. Mice were sacrificed at 12, 24 and 48 h after final UVB exposure to harvest dorsal skins.
Immunostaining
Immunostaining was performed as described elsewhere [70]. Stainings for CD31, Ki-67, HIF-1α and VEGF were performed at Northwestern University Mouse Histology and Phenotyping Core.
Immunofluorescence
The cells were grown on chamber slides (Nalgene Nunc International), fixed in paraformaldehyde and permeabilized with 0.1% Triton X-100. After blocking 1h in 5% goat serum the slides were incubated overnight with HuR antibody in 5% goat serum at 4°C followed by fluorescence-labeled secondary antibodies (2h at room temperature). The cells were counterstained with diaminophenylindole (DAPI).
Immunoblotting
The cells lysates were resolved on SDS PAGE and protein transferred onto nitrocellulose membranes, which were blocked in 5% dry milk in Tris buffered saline with 0.1% Tween 20 and incubated overnight with primary antibodies at 4°C followed by secondary antibodies (2 h, room temperature). The membranes were developed with Enhanced Chemoluminescence reagent (Amersham, Piscataway, NJ) and the signal detected by autoradiography.
RNA isolation
Total RNA was extracted using TRIzol reagent (Life Technologies, Carlsbad, CA), following manufacturer’s instructions.
RNA interference
For HuR knockdown, siRNA duplex targeting mouse HuR and non-silencing control (Santa Cruz, Santa Cruz, CA) were introduced using Lipofectamine RNAiMAX reagent (Life Technologies, Carlsbad, CA), following manufacturer’s instructions.
Immunoprecipitation and analysis of mRNP complexes
Cytoplasmic extracts collected as described [20] were incubated 2 h on ice with HuR antibody or control IgG (15 μg/sample) followed by slow rocking with Protein A/G Plus agarose beads (30 μL/sample, 1.5 h at 4°C). The beads were precipitated by centrifugation (2000g, 2 min at 4°C), and RNA extracted using TRIzol reagent. The RNA was treated with DNAse I (DNA-free kit, Ambion, Austin, TX) and reverse transcribed using SuperScript III with random hexamer primers (Life Technologies, Carlsbad, CA). Real-time PCR was performed using TaqMan Gene Express with TSP1 primers (Applied Biosystems, Foster City, CA) in ABI Prism 7900HT system, the product normalized to GAPDH and calculations performed using ΔΔCt method.
Analysis of Nascent TSP1 Protein
For metabolic labeling, 308 keratinocytes were pre-incubated 1 h in methionine and cysteine free DMEM (Sigma). Twelve h post-treatment, the cells were given a 15 min pulse of with 500 μCi [35S]L-methionine and [35S]L-cysteine (GE Healthcare, Piscataway, NJ) and harvested in lysis buffer as described previously [20]. Immunoprecipitation with anti-TSP1 or control antibody was carried out overnight at 4°C. Following extensive washes in 50 mM Tris-HCl (pH 7.5), 250 mM NaCl, 5 mM EDTA and 0.5% NP-40, the immunoprecipitates were resolved by SDS-PAGE and assessed by autoradiography.
Quantitative and statistical analyses
1. Measurements of epidermal thickness: the number of cell layers in the epidermis was determined in H&E-stained slides by counting the number of nucleated epithelial cells along a vertical line from the stratum basale to the stratum granulosum, away from hair follicles, in 10 representative fields at 400x magnification. The epidermal thickness (μm) was similarly measured in the same areas and included the full thickness from the stratum basale to the stratum corneum.
2. Microvascular density and Ki-67 positive nuclei were quantified using “Object Count” function of the ImageJ64 software (National Institutes of Health). CD31-positive structures (reported as “capillaries per area”) or Ki-67 positive nuclei in the epidermis, outside of the hair follicles, were quantified in at least 10 random fields, on three sections from three individual animals. The numbers of Ki67 positive nuclei was calculated per 100 linear μm of epidermal layer and the number of CD31-positive vascular structures per 100 μm2 of epidermal area.
3. Statistical significance was calculated by pairwise comparison, with 2-tailed Student’s T-test, and by one-way analysis of variance (ANOVA, Graph Pad Prism). P values below 0.05 were set as significant.
ACKNOWLEDGEMENTS
This work was supported in part by NIH CA172669 (JP, OV and XT), CA161181 (XT), resources provided by the Northwestern University Skin Disease Research Center (NIH/NIAMS P30AR057216) and H Foundation, via Robert H Lurie Cancer Center (JP and OV).
Conflict of interest
The authors state no conflict of interest.
ABBREVIATIONS
UVB, ultraviolet B; COX-2, cyclooxygenase 2; VEGF, Vascular Endothelial Growth Factor; NO, nitric oxide; TSP1, thrombospondin 1; PGE2, prostaglandin E2; NHEKs, normal human epidermal keratinocytes
REFERENCES
1. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000; 100:57–70.
2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674.
3. Folkman J. Tumor angiogenesis. Adv Cancer Res. 1985; 43:175–203.
4. Good DJ, Polverini PJ, Rastinejad F, Le Beau MM, Lemons RS, Frazier WA, Bouck NP. A tumor suppressordependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc Natl Acad Sci U S A. 1990; 87:6624–6628.
5. Roberts DD. Regulation of tumor growth and metastasis by thrombospondin-1. FASEB J. 1996; 10:1183–1191.
6. Kazerounian S, Yee KO, Lawler J. Thrombospondins in cancer. Cell Mol Life Sci. 2008; 65:700–712.
7. Yano K, Kadoya K, Kajiya K, Hong YK, Detmar M. Ultraviolet B irradiation of human skin induces an angiogenic switch that is mediated by upregulation of vascular endothelial growth factor and by downregulation of thrombospondin-1. Br J Dermatol. 2005; 152:115–121.
8. Yano K, Kajiya K, Ishiwata M, Hong YK, Miyakawa T, Detmar M. Ultraviolet B-induced skin angiogenesis is associated with a switch in the balance of vascular endothelial growth factor and thrombospondin-1 expression. J Invest Dermatol. 2004; 122:201–208.
9. Hawighorst T, Oura H, Streit M, Janes L, Nguyen L, Brown LF, Oliver G, Jackson DG, Detmar M. Thrombospondin-1 selectively inhibits early-stage carcinogenesis and angiogenesis but not tumor lymphangiogenesis and lymphatic metastasis in transgenic mice. Oncogene. 2002; 21:7945–7956.
10. Streit M, Velasco P, Brown LF, Skobe M, Richard L, Riccardi L, Lawler J, Detmar M. Overexpression of thrombospondin-1 decreases angiogenesis and inhibits the growth of human cutaneous squamous cell carcinomas. Am J Pathol. 1999; 155:441–452.
11. Rofstad EK, Galappathi K, Mathiesen B. Thrombospondin-1 treatment prevents growth of dormant lung micrometastases after surgical resection and curative radiation therapy of the primary tumor in human melanoma xenografts. Int J Radiat Oncol Biol Phys. 2004; 58:493–499.
12. Kalas W, Gilpin S, Yu JL, May L, Krchnakova H, Bornstein P, Rak J. Restoration of thrombospondin 1 expression in tumor cells harbouring mutant ras oncogene by treatment with low doses of doxycycline. Biochem Biophys Res Commun. 2003; 310:109–114.
13. Birt DF, Mitchell D, Gold B, Pour P, Pinch HC. Inhibition of ultraviolet light induced skin carcinogenesis in SKH-1 mice by apigenin, a plant flavonoid. Anticancer Res. 1997; 17:85–91.
14. Lepley DM, Pelling JC. Induction of p21/WAF1 and G1 cell-cycle arrest by the chemopreventive agent apigenin. Mol Carcinog. 1997; 19:74–82.
15. Lepley DM, Li B, Birt DF, Pelling JC. The chemopreventive flavonoid apigenin induces G2/M arrest in keratinocytes. Carcinogenesis. 1996; 17:2367–2375.
16. McVean M, Xiao H, Isobe K, Pelling JC. Increase in wild-type p53 stability and transactivational activity by the chemopreventive agent apigenin in keratinocytes. Carcinogenesis. 2000; 21:633–639.
17. Oishi M, Iizumi Y, Taniguchi T, Goi W, Miki T, Sakai T. Apigenin sensitizes prostate cancer cells to Apo2L/TRAIL by targeting adenine nucleotide translocase-2. PLoS One. 2013; 8:e55922.
18. Kim EY, Yu JS, Yang M, Kim AK. Sub-toxic dose of apigenin sensitizes HepG2 cells to TRAIL through ERKdependent up-regulation of TRAIL receptor DR5. Mol Cells. 2013; 35:32–40.
19. Shukla S, MacLennan GT, Flask CA, Fu P, Mishra A, Resnick MI, Gupta S. Blockade of beta-catenin signaling by plant flavonoid apigenin suppresses prostate carcinogenesis in TRAMP mice. Cancer Res. 2007; 67:6925–6935.
20. Tong X, Van Dross RT, Abu-Yousif A, Morrison AR, Pelling JC. Apigenin prevents UVB-induced cyclooxygenase 2 expression: coupled mRNA stabilization, translational inhibition. Mol Cell Biol. 2007; 27:283–296.
21. Van Dross RT, Hong X, Pelling JC. Inhibition of TPAinduced cyclooxygenase-2 (COX-2) expression by apigenin through downregulation of Akt signal transduction in human keratinocytes. Mol Carcinog. 2005; 44:83–91.
22. Tong X, Pelling JC. Targeting the PI3K/Akt/mTOR axis by apigenin for cancer prevention. Anticancer Agents Med Chem. 2013; 13:971–978.
23. Fang J, Zhou Q, Liu LZ, Xia C, Hu X, Shi X, Jiang BH. Apigenin inhibits tumor angiogenesis through decreasing HIF-1alpha and VEGF expression. Carcinogenesis. 2007; 28:858–864.
24. Liu LZ, Fang J, Zhou Q, Hu X, Shi X, Jiang BH. Apigenin inhibits expression of vascular endothelial growth factor and angiogenesis in human lung cancer cells: implication of chemoprevention of lung cancer. Mol Pharmacol. 2005; 68:635–643.
25. Fang J, Xia C, Cao Z, Zheng JZ, Reed E, Jiang BH. Apigenin inhibits VEGF and HIF-1 expression via PI3K/ AKT/p70S6K1 and HDM2/p53 pathways. Faseb J. 2005; 19:342–353.
26. Mirzoeva S, Kim ND, Chiu K, Franzen CA, Bergan RC, Pelling JC. Inhibition of HIF-1 alpha and VEGF expression by the chemopreventive bioflavonoid apigenin is accompanied by Akt inhibition in human prostate carcinoma PC3-M cells. Mol Carcinog. 2008; 47:686–700.
27. Erdogan A, Most AK, Wienecke B, Fehsecke A, Leckband C, Voss R, Grebe MT, Tillmanns H, Schaefer CA, Kuhlmann CR. Apigenin-induced nitric oxide production involves calcium-activated potassium channels and is responsible for antiangiogenic effects. J Thromb Haemost. 2007; 5:1774–1781.
28. Lamy S, Akla N, Ouanouki A, Lord-Dufour S, Beliveau R. Diet-derived polyphenols inhibit angiogenesis by modulating the interleukin-6/STAT3 pathway. Exp Cell Res. 2012; 318:1586–1596.
29. Henkin J, Volpert OV. Therapies using anti-angiogenic peptide mimetics of thrombospondin-1. Expert Opin Ther Targets. 2011; 15:1369–1386.
30. Garside SA, Henkin J, Morris KD, Norvell SM, Thomas FH, Fraser HM. A thrombospondin-mimetic peptide, ABT-898, suppresses angiogenesis and promotes follicular atresia in pre- and early-antral follicles in vivo. Endocrinology. 2010; 151:5905–5915.
31. Nakamura DS, Edwards AK, Virani S, Thomas R, Tayade C. Thrombospondin-1 mimetic peptide ABT-898 affects neovascularization and survival of human endometriotic lesions in a mouse model. Am J Pathol. 2012; 181:570–582.
32. Sahora AI, Rusk AW, Henkin J, McKeegan EM, Shi Y, Khanna C. Prospective study of thrombospondin-1 mimetic peptides, ABT-510 and ABT-898, in dogs with soft tissue sarcoma. J Vet Intern Med. 2012; 26:1169–1176.
33. Zou Y, Chiou GC. Apigenin inhibits laser-induced choroidal neovascularization and regulates endothelial cell function. J Ocul Pharmacol Ther. 2006; 22:425–430.
34. Kim MH. Flavonoids inhibit VEGF/bFGF-induced angiogenesis in vitro by inhibiting the matrix-degrading proteases. J Cell Biochem. 2003; 89:529–538.
35. Chen W, Tang Q, Gonzales MS, Bowden GT. Role of p38 MAP kinases and ERK in mediating ultraviolet-B induced cyclooxygenase-2 gene expression in human keratinocytes. Oncogene. 2001; 20:3921–3926.
36. Tang Q, Gonzales M, Inoue H, Bowden GT. Roles of Akt and glycogen synthase kinase 3beta in the ultraviolet B induction of cyclooxygenase-2 transcription in human keratinocytes. Cancer Res. 2001; 61:4329–4332.
37. Bachelor MA, Bowden GT. UVA-mediated activation of signaling pathways involved in skin tumor promotion and progression. Semin Cancer Biol. 2004; 14:131–138.
38. Fosslien E. Molecular pathology of cyclooxygenase-2 in neoplasia. Ann Clin Lab Sci. 2000; 30:3–21.
39. Van Dross RT, Hong X, Essengue S, Fischer SM, Pelling JC. Modulation of UVB-induced and basal cyclooxygenase-2 (COX-2) expression by apigenin in mouse keratinocytes: role of USF transcription factors. Mol Carcinog. 2007; 46:303–314.
40. Simone LE, Keene JD. Mechanisms coordinating ELAV/ Hu mRNA regulons. Curr Opin Genet Dev. 2013; 23:35–43.
41. Mazan-Mamczarz K, Hagner PR, Corl S, Srikantan S, Wood WH, Becker KG, Gorospe M, Keene JD, Levenson AS, Gartenhaus RB. Post-transcriptional gene regulation by HuR promotes a more tumorigenic phenotype. Oncogene. 2008; 27:6151–6163.
42. Wang J, Guo Y, Chu H, Guan Y, Bi J, Wang B. Multiple Functions of the RNA-Binding Protein HuR in Cancer Progression, Treatment Responses and Prognosis. Int J Mol Sci. 2013; 14:10015–10041.
43. Tiedje C, Holtmann H, Gaestel M. The role of mammalian MAPK signaling in regulation of cytokine mRNA stability and translation. J Interferon Cytokine Res. 2014; 34:220–232.
44. Kroonen J, Artesi M, Capraro V, Nguyen-Khac MT, Willems M, Chakravarti A, Bours V, Robe PA. Casein kinase 2 inhibition modulates the DNA damage response but fails to radiosensitize malignant glioma cells. Int J Oncol. 2012; 41:776–782.
45. Gopalakrishnan A, Xu CJ, Nair SS, Chen C, Hebbar V, Kong AN. Modulation of activator protein-1 (AP-1) and MAPK pathway by flavonoids in human prostate cancer PC3 cells. Arch Pharm Res. 2006; 29:633–644.
46. Van Dross R, Xue Y, Knudson A, Pelling JC. The chemopreventive bioflavonoid apigenin modulates signal transduction pathways in keratinocyte and colon carcinoma cell lines. J Nutr. 2003; 133:3800S–3804S.
47. Chen AC, Halliday GM, Damian DL. Non-melanoma skin cancer: carcinogenesis and chemoprevention. Pathology. 2013; 45:331–341.
48. Sawane M, Kajiya K. Ultraviolet light-induced changes of lymphatic and blood vasculature in skin and their molecular mechanisms. Exp Dermatol. 2012; 21:22–25.
49. Yano K, Oura H, Detmar M. Targeted overexpression of the angiogenesis inhibitor thrombospondin-1 in the epidermis of transgenic mice prevents ultraviolet-Binduced angiogenesis and cutaneous photo-damage. J Invest Dermatol. 2002; 118:800–805.
50. Anso E, Zuazo A, Irigoyen M, Urdaci MC, Rouzaut A, Martinez-Irujo JJ. Flavonoids inhibit hypoxia-induced vascular endothelial growth factor expression by a HIF- 1 independent mechanism. Biochem Pharmacol. 2010; 79:1600–1609.
51. Campbell N, Greenaway J, Henkin J, Petrik J. ABT-898 induces tumor regression and prolongs survival in a mouse model of epithelial ovarian cancer. Mol Cancer Ther. 2011; 10:1876–1885.
52. Iniguez MA, Rodriguez A, Volpert OV, Fresno M, Redondo JM. Cyclooxygenase-2: a therapeutic target in angiogenesis. Trends Mol Med. 2003; 9:73–78.
53. Baez MV, Boccaccio GL. Mammalian Smaug is a translational repressor that forms cytoplasmic foci similar to stress granules. J Biol Chem. 2005; 280:43131–43140.
54. Meisner NC, Filipowicz W. Properties of the regulatory RNA-binding protein HuR and its role in controlling miRNA repression. Adv Exp Med Biol. 2010; 700:106–123.
55. Syed DN, Khan MI, Shabbir M, Mukhtar H. MicroRNAs in skin response to UV radiation. Curr Drug Targets. 2013; 14:1128–1134.
56. Papadopoulou C, Ganou V, Patrinou-Georgoula M, Guialis A. HuR-hnRNP interactions and the effect of cellular stress. Mol Cell Biochem. 2013; 372:137–147.
57. Yoon JH, Abdelmohsen K, Srikantan S, Guo R, Yang X, Martindale JL, Gorospe M. Tyrosine phosphorylation of HuR by JAK3 triggers dissociation and degradation of HuR target mRNAs. Nucleic Acids Res. 2014; 42:1196–1208.
58. Dameron KM, Volpert OV, Tainsky MA, Bouck N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science. 1994; 265:1582–1584.
59. Volpert OV, Stellmach V, Bouck N. The modulation of thrombospondin and other naturally occurring inhibitors of angiogenesis during tumor progression. Breast Cancer Res Treat. 1995; 36:119–126.
60. Su F, Pascal LE, Xiao W, Wang Z. Tumor suppressor U19/ EAF2 regulates thrombospondin-1 expression via p53. Oncogene. 2010; 29:421–431.
61. Watnick RS, Rodriguez RK, Wang S, Blois AL, Rangarajan A, Ince T, Weinberg RA. Thrombospondin-1 repression is mediated via distinct mechanisms in fibroblasts and epithelial cells. Oncogene. 2014.
62. Sundaram P, Hultine S, Smith LM, Dews M, Fox JL, Biyashev D, Schelter JM, Huang Q, Cleary MA, Volpert OV, Thomas-Tikhonenko A. p53-responsive miR-194 inhibits thrombospondin-1 and promotes angiogenesis in colon cancers. Cancer Res. 2011; 71:7490–7501.
63. Lopez-Jornet P, Camacho-Alonso F, Gomez-Garcia F, Molina Minano F, Canas X, Serafin A, Castillo J, Vicente- Ortega V. Effects of potassium apigenin and verbena extract on the wound healing process of SKH-1 mouse skin. Int Wound J. 2012.
64. Detmar M. The role of VEGF and thrombospondins in skin angiogenesis. J Dermatol Sci. 2000; 24:S78–84.
65. Streit M, Velasco P, Riccardi L, Spencer L, Brown LF, Janes L, Lange-Asschenfeldt B, Yano K, Hawighorst T, Iruela-Arispe L, Detmar M. Thrombospondin-1 suppresses wound healing and granulation tissue formation in the skin of transgenic mice. EMBO J. 2000; 19:3272–3282.
66. Velasco P, Huegel R, Brasch J, Schroder JM, Weichenthal M, Stockfleth E, Schwarz T, Lawler J, Detmar M, Lange-Asschenfeldt B. The angiogenesis inhibitor thrombospondin-1 inhibits acute cutaneous hypersensitivity reactions. J Invest Dermatol. 2009; 129:2022–2030.
67. Lawler J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J Cell Mol Med. 2002; 6:1–12.
68. Lawler J, Detmar M. Tumor progression: the effects of thrombospondin-1 and -2. Int J Biochem Cell Biol. 2004; 36:1038–1045.
69. Tong X, Smith KA, Pelling JC. Apigenin, a chemopreventive bioflavonoid, induces AMP-activated protein kinase activation in human keratinocytes. Mol Carcinog. 2012; 51:268–279.
70. Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, Boivin GP, Bouck N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998; 93:1159–1170.