
INTRODUCTION
EGFR is a member of the ErbB family of transmembrane receptor tyrosine kinases (TKs) and upon ligand-binding and dimerization undergoes auto-phosphorylation of its intracellular domain, resulting in recruitment of different adaptors and signal-transducers and activation of downstream signaling-pathways. These especially include the RAS-RAF-MEK-MAPK, the PI3K-AKT-PTEN-mTOR, and the STAT pathways, ultimately resulting in cancer-promoting effects, such as increased cell proliferation, survival, protein synthesis, migration, and angiogenesis [1]. Common gain-of-function mutations of EGFR, such as most of microdeletions in exon 19 and the point-substitution p.L858R in exon 21, which together represent almost 90% of all EGFR-mutations, result in constitutive ligand-independent EGFR-TK activity and oncogenicity as well as increased affinity and sensitivity to the first- and second-generation EGFR-TK inhibitors (TKIs) gefitinib, erlotinib and afatinib. These drugs are currently the standard of care first-line treatment for patients with advanced EGFR-mutated NSCLC, with higher response rate (RR) observed for patients with exon 19-microdeletions [1, 2]. Despite initial response to first-line EGFR-TKIs, most patients inevitably become resistant with median progression-free survival (PFS) of 10-12 months. In up to 60% of cases, this is due to the emergence of the secondary p.T790M EGFR-mutation in exon 20, which also is an activating mutation, but possesses increased affinity for ATP, thereby competitively impeding the binding of reversible EGFR-TKIs to the EGFR ATP-binding pocket [3, 4]. The third-generation EGFR-TKI osimertinib is currently the standard of care for treating advanced EGFR-mutant NSCLC with p.T790M-positive acquired TKI-resistance.
Most of the activating EGFR-mutations occurring in non-small-cell lung cancer (NSCLC) before treatment are mutually exclusive with those in other oncogenic drivers. However, additional distinct driver alterations such as ALK-rearrangement, KRAS-mutations, PIK3CA-mutations, MET-amplification and others were recently reported to co-exist with EGFR-mutations in a small percentage of TKI therapy-naïve pulmonary adenocarcinomas (ADCs) [5–13]. A recent database study including 17664 lung cancer patients identified 2-3 concomitant driver mutations in almost 1% of these cases [6].
Importantly, the occurrence of co-mutations in EGFR itself or other cancer-drivers at diagnosis may potentially impair the efficacy of tyrosine-kinase-inhibitors (TKIs) and partly explain why approximately 10% of TKI-treated NSCLCs are intrinsically resistant [4].
Consequently, the evaluation of EGFR-mutations and ALK-rearrangements for selecting NSCLC patients treatable with first-line TKIs may not be sufficient to predict the response to these treatments. Thus, we examined the frequency of an extended panel of cancer-relevant mutations that could potentially reduce the initial response to TKIs in a cohort of newly diagnosed, EGFR-mutated, advanced NSCLC of primarily ADC subtype. Additionally, we evaluated the response to erlotinib of EGFR-mutated tumors with or without co-mutations.
RESULTS
EGFR-mutations and patients characteristics
Thirty-six (7%) of the 514 tested NSCLCs displayed EGFR-mutations, including two tumors with co-existing p.L858R and p.T790M mutations, one of which was from an operable patient. Of the 514 patients, 283 (55.1%) had advanced stage or were not amenable for definitive local treatment because of poor pulmonary function. Twenty-three of these patients (8.1%) had tumors harboring activating EGFR-mutations and were treated with erlotinib, their individual characteristics are shown in Table 1 and histological subtype and type of EGFR-mutations in Table 2. Eighteen (78%) of the patients with EGFR-mutated tumors were females and five (22%) males, whereas in terms of smoking habit, 18 (78%) were never/previous smokers and 5 (22%) current smokers. According to established criteria [14], the specimens from 20 of the EGFR-mutant cases (87%) were diagnosed as lung ADCs, based on the presence of acinar/papillary/micropapillary/solid epithelial structures, mucin production, and immunohistochemical markers (all were CK7+/TTF1+/CK5-/p40-), whereas one specimen (4%) was classified as SCC because of lack of mucin production and marker status (CK7-/TTF1-/CK5+/p40+). The specimens of the other two EGFR-mutant cases (9%) were classified on biopsies as most likely adenosquamous carcinomas (ADSC), based on the co-existence of cells with mucin production, keratinization and ADC/SCC immunohistochemical markers (CK7+/TTF1+/CK5+/p40+). Seven patients had a single p.L858R mutation, 14 an exon 19-microdeletion, one harbored the p.G719C/p.S768I double mutation, one p.L858R combined with the intrinsic erlotinib-resistant p.T790M mutation, and one the unusual p.E746_R748del/p.A750P combination (Figure 1A). Taken together, these results are consistent with the well-established notions that, regardless of ethnicity, the frequency of EGFR-mutations is higher among women, never/light smokers, and with NSCLC of ADC type, and that the exon 19-microdeletions and the p.L858R substitution in exon 21 are the most frequent EGFR-mutations in NSCLC [1].
Table 1: Individual characteristics and outcome of 23 EGFR-mutation positive, treatment naïve NSCLC patients treated with erlotinib
Patient no. |
Gender |
Age |
PS |
Smoking status |
Stage |
Objective response to erlotinib |
PFS (months) |
OS (months) |
|---|---|---|---|---|---|---|---|---|
1 |
Female |
75 |
1 |
Never |
IIIA |
PR |
24 |
24 |
2 |
Female |
58 |
1 |
Current |
IV |
PR |
19 |
41 |
3 |
Female |
61 |
0 |
Never |
IV |
PR |
5 |
7 |
4 |
Female |
62 |
0 |
Previous |
IV |
PR |
8 |
25 |
5 |
Female |
76 |
1 |
Current |
IV |
PR |
7 |
16 |
6 |
Female |
81 |
0 |
Previous |
IIIA |
PR |
20 |
50 (Alive) |
7 |
Male |
83 |
1 |
Never |
IV |
PR |
18 |
29 |
8 |
Female |
62 |
1 |
Never |
IV |
NC |
10 |
47 |
9 |
Female |
59 |
0 |
Previous |
IIIB |
PR |
17 |
34 |
10 |
Female |
50 |
0 |
Current |
IV |
NC |
20 |
35 |
11 |
Female |
83 |
1 |
Previous |
IV |
PR |
17 |
32 |
12 |
Female |
60 |
0 |
Never |
IV |
Mixed |
15 |
21 |
13 |
Female |
53 |
0 |
Never |
IV |
NC |
14 |
23 |
14 |
Female |
56 |
0 |
Current |
IV |
PR |
8 |
28 |
15 |
Female |
47 |
1 |
Previous |
IV |
PR |
6 |
9 |
16 |
Female |
82 |
2 |
Never |
IV |
PR |
8 |
15 |
17 |
Female |
81 |
1 |
Never |
IIIA |
PR |
9 |
22 |
18 |
Male |
67 |
1 |
Previous |
IV |
PR |
8 |
17 |
19 |
Male |
63 |
0 |
Previous |
IV |
NC |
9 |
23 |
20 |
Female |
63 |
0 |
Never |
IV |
CR |
13 |
33 |
21 |
Female |
59 |
1 |
Previous |
IV |
CR |
8 |
19 |
22 |
Male |
75 |
1 |
Previous |
IIIB |
PR |
8 |
16 |
23 |
Male |
49 |
1 |
Current |
IV |
Mixed |
1 |
10 |
PS: WHO Performance status; CR: Complete response; PR: Partial response; NC: No change.
Table 2: Histological subtype and biomarker status in 23 EGFR- mutation- positive NSCLC patients treated with erlotinib
Patient no. |
NSCLC histotype |
EGFR-mutations^ |
MET-amplification§ |
MET protein overexpression§§ |
TP53 mutation |
Other mutations |
|---|---|---|---|---|---|---|
1 |
ADSC |
c.2573T>G, p.L858R |
c.473G>T, p.R158L (pathogenic) |
KRAS: c.35G>T, p.G12V (pathogenic) |
||
2 |
ADC |
c.2155G>T, p.G719C + c.2303G>T, p.S768I |
Intermediate-level of amplification |
3+ in 80% of tumor cells |
c.503A>G, p.H168R (likely-pathogenic) |
|
3 |
ADC |
c.2573T>G, p.L858R |
||||
4 |
ADSC |
c.2573T>G, p.L858R |
High-level amplification |
3+ in 100% of tumor cells |
||
5 |
ADC |
c.2573T>G, p.L858R |
High-level amplification |
3+ in 70% of tumor cells |
||
6 |
ADC |
c.2239_2253del15, p.L747_T751delLREAT |
Not enough cells for analysis |
Not enough cells for analysis |
||
7 |
ADC |
c.2239_2253del15, p.L747_T751delLREAT |
c.723Cdel, p.S241fs* (unknown) |
|||
8 |
ADC |
c.2573T>G, p.L858R + c.2369C>T, p.T790M |
||||
9 |
ADC |
c.2236_2244del9, p.E746_R748del + c.2248G>C, p.A750P |
MET: |
|||
10 |
ADC |
c.2213-2230dup, p.I744_K745insKIPVAI |
Low-level amplification |
3+ in 100% of tumor cells |
c.463A>C, p.T155P (neutral) |
|
11 |
SCC |
c.2235_2249del15, p.E746_A750delELREA |
Not sufficient DNA for NGS analysis |
|||
12 |
ADC |
c.2573T>G, p.L858R |
SMAD4: c.403C>T, p.R135* (stop) (pathogenic) |
|||
13 |
ADC |
Exon 19 del (by Cobas) |
Not sufficient DNA for NGS analysis |
|||
14 |
ADC |
c.2236_2250del15insTTA, |
c.734G>A, p.G245D (pathogenic) |
|||
15 |
ADC |
c.2235_2249del15, p.E746_A750delELREA |
c.818G>T, p.R273L (pathogenic) |
NRAS: |
||
16 |
ADC |
c.2573T>G, p.L858R |
c.216Cdel, p.P74fs* (unknown) |
PIK3CA: c.1633G>A, p.E545K (pathogenic) |
||
17 |
ADC |
c.2240_2257del18, p.L747_P753delinsS |
CTNNB1: c.98C>G, p.S33C (pathogenic) |
|||
18 |
ADC |
c.2235_2249del15, p.E746_A750delELREA |
3+ in 100% of tumor cells |
c.535C>T, p.H179Y (pathogenic) |
||
19 |
ADC |
c.2236_2250del15, p.E746_A750delELREA |
c.403T>G, p.C135G (pathogenic) |
|||
20 |
ADC |
c.2235_2249del15, p.E746_A750delELREA |
c.376-1G>T (Substitution - intronic) (pathogenic) |
FGFR1: c.373_374insTCA, p.S125-E126insS (unknown) |
||
21 |
ADC |
c.2235_2249del15, p.E746_A750delELREA |
||||
22 |
ADC |
c.2573T>G, p.L858R |
CTNNB1: c.110C>T, p.S37F (pathogenic) |
|||
23 |
ADC |
c.2235_2249del15, p.E746_A750delELREA |
. |
FGFR3: c.2349_2350delAG, p.D785fs*31 (unknown) |
ADC: Adenocarcinoma; SCC: Squamous cell carcinoma; ADSC: Adenosquamous carcinoma
In parenthesis the pathogenic prediction of the mutations according to the catalogue of somatic mutations in cancer (COSMIC) at http://cancer.sanger.ac.uk/cosmic. ^All identified EGFR-mutations were COSMIC-verified as being pathogenic.
§ For clarity only cases with some level of MET-amplification (high-, intermediate-, low-level) are shown; empty rows = no MET-amplification, as defined in Materials and methods.
§§ For clarity only cases with MET-overexpression (3+) and % of overexpressing cells are shown, empty rows = cases with MET-expression 2+, 1+ or 0, as defined in Materials and methods.
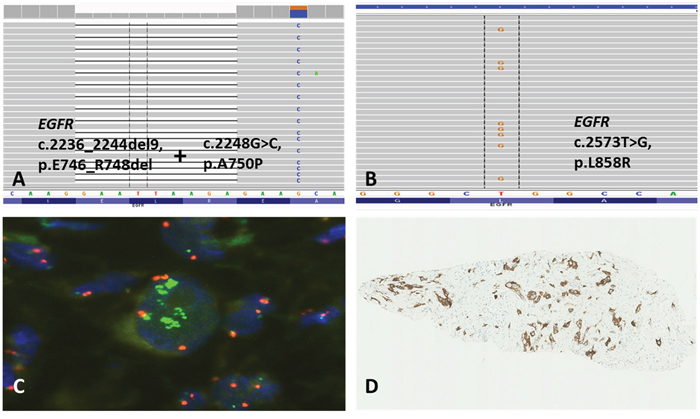
Figure 1: Unusual double p.E746_R748del/p.A750P EGFR-mutation identified by NGS in one NSCLC-case (A; patient no. 9 in Table 2). Biopsy from NSCLC of ADC subtype with activating p.L858R EGFR-mutation (B; patient no. 5 in Table 2) identified by NGS, high-level MET-amplification detected by FISH (C; green, x100), and overexpression of MET receptor assessed by IHC (D; x10).
NGS findings
DNA for NGS was available from 21 of the 23 EGFR-mutated cases (Table 2). Complete concordance was observed between EGFR-mutation-status by Cobas® EGFR Mutation Test and NGS. However, one sample identified by Cobas®Test as exon 19-microdeletion turned out to be an exon 19-insertion resulting in 6-amino-acid duplication (c.2213-2230dup, p.I744_K745insKIPVAI) by NGS analysis. Fifteen of the 21 NGS-analyzed cases displayed co-mutations in other cancer-relevant genes (Table 2), with TP53-mutations being the most represented (n=10, 67%, subdivided in 7 missense mutations, 1 intronic substitution, 2 previously unreported frameshift deletions). Other mutations were identified in CTNNB1 (n=2, 13%), KRAS, FGFR1, FGFR3, MET, SMAD4, PIK3CA, NRAS, DDR2, ERBB4 (all n=1, 7%), with the latter 3 gene-mutations concomitantly present in the same tumor (patient 15, Table 2). The spectrum and pathogenic prediction of these co-mutations according to the catalogue of somatic mutations in cancer (COSMIC, http://cancer.sanger.ac.uk/cosmic) are indicated in Table 2.
FISH and IHC
None of the 23 EGFR-mutated cases showed expression of ALK-fusion protein. FISH and IHC for MET were feasible in 22 of the cases. We detected significant variation concerning average MET GCN per tumor cell nucleus (range 1.62-13.74; mean 3.81) and MET/CEN7 ratio (range 0.74-4.58; mean 1.31), which is consistent with other recent results in treatment-naïve NSCLCs [11].
Two of the FISH-analyzed cases (9.1%) exhibited high-level MET gene-amplification concomitant with the p.L858R EGFR-mutation (Figure 1B-1D), while another case (4.5%) showed intermediate-level of MET GCN gain together with the above-mentioned p.G719C/p.S768I double mutation. Additionally, the sample (4.5%) harboring the above-mentioned EGFR exon 19-insertion p.I744_K745insKIPVAI had low-level of MET GCN gain (Table 2). The remaining 18 cases (81.8%) were negative for MET-amplification. The 4 cases with MET-amplification, regardless of the GCN increase level, showed MET-overexpression by IHC (Table 2, Figure 1D). In addition, one EGFR-mutated tumor exhibited MET-overexpression without MET-amplification (Table 2), suggesting other mechanisms increasing the receptor expression in this case.
Clinical outcomes
Among the 23 EGFR-mutated patients treated with erlotinib, 17 (74%) responded, 4 (17%) had no OR, and 2 (9%) harboring a SMAD4-mutation (p.R135*(stop)) and a FGFR3-mutation (p.D785fs*31), respectively, showed mixed response with simultaneously progressing and responding tumor lesions (Tables 1 and 2). The above-named tumor harboring the double p.L858R/p.T790M EGFR-mutation, did not respond to erlotinib, while another case with p.L858R did, despite concurrently harboring the p.G12V KRAS-mutation and p.R158L TP53-mutation. Median PFS was 8.8 months, 95%CI [7.3; 10.5] and OS was 23.3 months, 95%CI [20.1; 26.6] among all 23 patients harboring EGFR-mutations. No significant differences in PFS (8.5 months, 95%CI [7.7; 9.3] vs. 9.9 months, 95%CI [3.0; 16.7]) (p=0.56) or OS (23.3 months, 95%CI [20.5; 26.2] vs. 22.8 months, 95%CI [7.2; 38.3]) (p=0.18) were observed when comparing EGFR-mutated tumors with or without concomitant genetic aberrations (Figure 2). However, 2 tumors with insufficient DNA for NGS and 1 tumor with too few cells for FISH and IHC were included in the analysis as being without concomitant mutations.
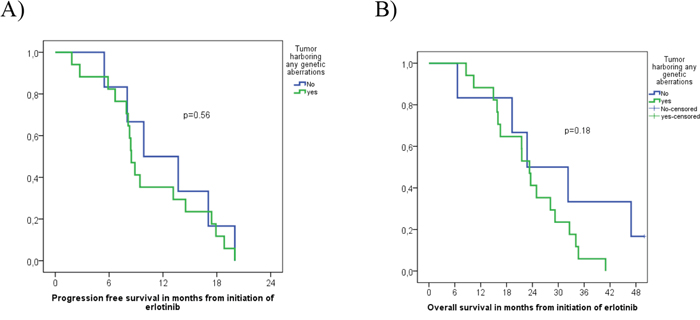
Figure 2: Progression free survival (A) and Overall survival (B) of 23 EGFR-mutation positive NSCLC patients with or without identified concomitant genetic aberrations.
DISCUSSION
Certain EGFR-mutated NSCLCs are inherently resistant to TKI-inhibitors and NSCLCs initially responding will ultimately become TKI-resistant and progress. In our study, 7% (36/514) of tested NSCLCs of all stages and 8% (23/283) of NSCLCs in advanced stage harbored EGFR-mutations, consistent with previous data concerning Danish patients [15].
Among the advanced erlotinib-treated NSCLCs, one simultaneously carried the p.L858R and the intrinsic erlotinib-resistant p.T790M EGFR-mutations, but no mutations in other analyzed genes. Known as the most common mechanism of acquired resistance to first-/second-generation EGFR-TKIs, p.T790M occurrence at baseline seems frequently associated with lack of OR and poor outcome in patients treated with these TKIs [3, 4]. Accordingly, our p.L858R/p.T790M case displayed no OR. Another erlotinib-treated case harbored the double p.G719C/p.S768I EGFR-mutation, each considered uncommon and reportedly less sensitive to gefitinib/erlotinib than more common EGFR-mutations [16, 17]. Yet, p.G719C and p.S768I may coexist more frequently than other rare mutations in NSCLC [16, 18]. Moreover, patients with EGFR co-mutations exhibit shorter PFS and lower RR than patients with single EGFR-mutations [19]. Although S768I alone or concomitant with exon 18 G719X does not necessarily appear to be sensitive to erlotinib [16, 17] it was shown to respond to afatinib [20, 21]. Our p.G719C/p.S768I case also showed intermediate-level of MET-amplification, MET-overexpression, and the fairly rare p.H168R TP53-mutation, nevertheless it did display an OR to erlotinib. Thus, given the rarity and variable response of TKI-treated cases with exon 20 S768I, the exact prognostic and predictive role of this mutation in EGFR-mutant NSCLC seems still unclear. A separate EGFR-mutant NSCLC carried the unusual combination of the two rare mutations p.E746_R748del and p.A750P in EGFR exon 19, together with the p.T1010I point-mutation in the MET-gene. The sensitivity of these two exon 19-mutations to EGFR-TKIs is poorly described, while the MET-substitution has been reported to be associated with compromised response to these drugs [22]. In any case, this patient did show a partial response to erlotinib with PFS longer than 17 months. Another case displayed an EGFR exon 19-insertion resulting in 6 amino acid duplication (p.I744_K745insKIPVAI) together with a missense TP53-mutation and low-level MET-amplification. EGFR exon 19-insertions have been reported in only 0.26% and 0.11% of large Caucasian and Asian NSCLC-cohorts, respectively [23, 24], thus their response to EGFR-TKIs is uncertain. A recent meta-analysis of the few reported patients with these mutations suggested that they had lower RR than patients with common EGFR-mutations, but incomplete PFS/OS data in this small cohort rendered difficult the comparison [24]. In this regard, our patient with exon 19-insertion showed no OR to erlotinib, but one cannot exclude that the concomitant TP53-mutation and possibly the low-level MET-amplification associated with MET-overexpression influenced this lack of response.
We also addressed whether possible co-mutations in alternative cancer-drivers could interfere with the response to erlotinib. In a previous exploratory study of 197 consecutive NSCLC cases with TKI-sensitive EGFR-mutations, 11 exhibited primary resistance to EGFR-TKIs, but apart from three of these cases exhibiting either concomitant p.T790M, MET-amplification, or ALK-fusion, no other co-mutations in driver genes that could explain the intrinsic TKI-resistance were identified by targeted NGS [25]. Of the 21 erlotinib-treated NSCLCs that we assessed by targeted NGS, 71% exhibited concomitant mutations in alternative cancer-drivers before TKI-therapy. The most frequent were TP53-mutations found in 67% of these cases, whereas 60% of them harbored co-mutations in either MET, KRAS, SMAD4, PIK3CA, CTNNB1, NRAS, DDR2, ERBB4, FGFR1, or FGFR3. Using the same targeted NGS platform as ours, two other groups recently detected cases of advanced EGFR-mutant NSCLC that prior to gefitinib-treatment displayed co-mutations very similar to those identified in our study and at comparable frequency. Notably, these cases responded significantly worse to gefitinib than those without co-mutations [26, 27].
Moreover, in a recent large database-study assessing characteristics and outcomes of patients harboring multiple molecular alterations, patients with EGFR/KRAS and EGFR/PIK3CA co-mutations experienced worse PFS during TKI-therapy than patients having only EGFR-mutations [6]. Collectively, these results indicate that the concurrent mutations we have detected are among the most frequent in advanced therapy-naïve EGFR-mutant NSCLC and could be involved in primary resistance to gefitinib and erlotinib.
Mutant TP53 occurs in over 50% of pulmonary ADCs [8] and may interfere with TKI-induced cell-cycle arrest and apoptosis, thereby contributing to acquired TKI-resistance development [28–31]. Regarding intrinsic TKI-resistance, though, relatively small cohorts of gefitinib- or erlotinib-treated NSCLC-patients comparable to ours have shown only a marginal negative impact of coexisting TP53-mutations on the OR to TKIs [26, 27]. Similarly, we found no significant association between TP53-mutations and sensitivity to erlotinib. These findings may be due to stochastic variations related to relatively few observations and/or the type of identified TP53-mutations that may differently interfere with the effect of TKIs.
As for MET-status in the EGFR-mutated NSCLCs, we found 1 case with MET-mutation, four cases with different MET-amplification levels and MET-overexpression, and one case with MET-overexpression not associated with amplification. The overall frequency (22.2%) of MET GCN gain in our cohort of EGFR-mutated cases was similar to that reported by Schildhaus et al. in their overall NSCLC population and subgroup of EGFR-mutated tumors [11]. Also consistent with Schildhaus et al. [11], we found almost complete concordance between MET-amplification and MET-overexpression (4/5 cases). Furthermore, TP53-mutation occurred in 3/5 of patients with MET-amplification and/or MET-overexpression, suggesting possible growth advantages for NSCLCs that concomitantly have altered EGFR-, MET- and p53-dependent signaling. In preclinical models, MET-amplification has been shown to be a driver for NSCLC growth and survival [32]. In the clinical setting 5-20% of NSCLC patients with acquired resistance to EGFR-TKIs have MET-amplification likely due to clonal selection by TKI-treatment of preexisting MET-amplified cells, resulting in activated MET-signaling, which bypasses EGFR-blockade and induces cell proliferation and survival [3, 4, 33]. Although this role of MET in acquired TKI-resistance is well established, the potential impact on the intrinsic TKI-resistance is less clear [3, 4]. Single cases of NSCLC presenting with concomitant EGFR-mutation and de novo MET-amplification associated with primary resistance to erlotinib have been reported. Interestingly, these cases have shown response to a dual EGFR/MET blockade mediated by erlotinib/crizotinib combination [34, 35], thereby illustrating feasibility and therapeutic potential of combinatorial strategies in EGFR-mutant NSCLC with activation of bypass signaling pathways, such as MET-amplification. However, we found no indication in our cohort that MET-status at baseline inevitably hampered the efficacy of erlotinib-treatment. Only the case with EGFR exon 19-duplication (p.I744_K745insKIPVAI) that concomitantly harbored a TP53-mutation (p.T155P; classified as “neutral” in COSMIC) and low-level MET-amplification together with MET-overexpression, did not respond to erlotinib (Tables 1 and 2). The other four cases with MET-amplification and/or MET-overexpression, two of which also had TP53-mutations, as well as the case with MET-mutation, all partially responded to erlotinib (Tables 1 and 2). Although concurrent EGFR-mutation and ALK-rearrangement in lung ADC is more frequent than initially anticipated [7-10, 36], we detected no ALK fusion-protein expression, i.e. no sign of ALK-rearrangement, in any of the 23 EGFR-mutated patients. Consistent with the reported possibility of rare concomitance of EGFR- and KRAS-mutations in lung ADCs [7, 8, 12, 13], one of our EGFR-mutated cases harbored the p.G12V KRAS-mutant together with the p.R158L TP53-mutant, and somehow surprisingly it partially responded to erlotinib. KRAS-mutations are one of the most common genetic events in lung ADC and constitutively activate effectors downstream of EGFR, thus they can potentially cause TKI-resistance and be a negative predictive biomarker for response to EGFR-TKIs in NSCLC [37]. Nevertheless, the role of KRAS-mutations in intrinsic TKI-resistance remains unclear. Another of our cases partially responding to erlotinib displayed TP53- and PIK3CA-mutations concurrently with the activating p.L858R EGFR-mutant. Somatic mutations in the catalytic domain of PIK3CA have been implicated in acquired TKI-resistance and also found in 1%–3% of NSCLCs prior to TKI therapy [38]. These PIK3CA-mutations render EGFR-mutant NSCLC cell lines resistant to EGFR-TKIs by activating AKT-signaling and inhibiting TKI-induced apoptosis [38]. In a large, retrospectively analyzed cohort of patients with advanced EGFR-mutant lung ADC, the concomitant occurrence of a PIK3CA-mutation was a negative prognostic factor associated with decreased median OS, but did not impact benefit from EGFR-TKI monotherapy in terms of objective RR, PFS and duration of response [39]. As our erlotinib-treated PIK3CA-comutated case, these data suggest that PIK3CA-mutations despite being considered cancer-drivers do not necessarily represent a mechanism of primary resistance to erlotinib in EGFR-mutated NSCLC.
Two other patients partially responding to erlotinib displayed co-mutation of the CTNNB1 gene coding for β-catenin, the main effector in the Wnt/β-catenin signaling pathway that transactivates cell proliferation-related genes [40]. Interestingly, preclinical data show that β-catenin is activated by EGFR-mutants and contributes to the development of EGFR-mutated NSCLC, so that CTNNB1-mutations can potentially induce resistance to EGFR-TKIs [41–43]. Therefore, targeting the Wnt/β-catenin pathway might provide novel strategies to counteract TKI-resistance [41]. However, the role of CTNNB1-mutations in TKI-resistance awaits further clinical confirmation.
Partial response to erlotinib was also seen in a case concomitantly revealing an EGFR exon 19-microdeletion, “pathogenic” mutations in TP53 and ERBB4 and “unknown”/unreported mutations in NRAS and DDR2 (patient 15, Table 2). NRAS-mutations are detectable in ~1% of NSCLCs, particularly ADCs in current/former smokers [44], but their role in TKI-resistance, despite being hypothetically similar to that of KRAS-mutations, is poorly explored. Missense mutations of DDR2, which encodes the collagen receptor discoidin domain receptor 2, are present in 4% of pulmonary SCCs and approximately 1.5% of ADCs (http://cancer.sanger.ac.uk/cosmic). However, their frequency was reported increased to 16% in EGFR-mutated NSCLC, without significant impact on gefitinib-treatment [26]. ERBB4-mutations occur in ~1% of NSCLCs (http://cancer.sanger.ac.uk/cosmic). The observed p.S239P mutation resides in ERBB4 extracellular dimerizing domain, has formerly been reported in esophageal cancer as ERBB4-activating mutation [45] and might represent a bypass-mechanism for erlotinib-resistance, but the role of ERBB4-mutants in TKI-resistance needs further clarification. Finally, three of the 23 EGFR-mutant NSCLCs harbored a concomitant mutation in either SMAD4, FGFR1, or FGFR3. The former encodes the SMAD4 transcriptional co-factor that mediates TGF-β tumor-suppressive function by inducing growth arrest and apoptosis [46], but the incidence and function of inactivating SMAD4-mutations in TKI-resistance are scantly known. Recently SMAD4 co-mutations were observed in EGFR-mutated NSCLCs especially occurring in gefitinib-sensitive patients [26, 27]. However, our patient with SMAD4 co-mutation exhibited mixed response to erlotinib. Thus, the significance of SMAD4-mutations in TKI-resistance remains unclear.
Constitutive FGFR1-activation by FGFR1-amplification, -translocation or -mutation is associated with various malignancies. FGFR1-amplification has been observed in up to 20% of pulmonary SCCs and less frequently in ADCs and small-cell carcinomas [47]. Single preclinical and clinical studies suggest that activated FGFR1-signaling may represent a mechanism of acquired resistance to EGFR-TKIs [48–50]. Lim et al. reported that 2 of 20 EGFR-mutant NSCLCs not responding to gefitinib carried a concomitant FGFR1-mutation [26]. Our FGR1-mutated case concurrently displayed an intronic substitution in TP53 predicted to be “pathogenic” in COSMIC and nonetheless did show OR to TKI-treatment.
Activating FGR3-mutations have been reported in small fractions of advanced pulmonary SCCs and ADCs [51–53], including cases with co-existing EGFR-mutations. Additionally, lung ADC may contain oncogenic FGFR3-TACC3 fusions that may function as bypass-mechanism associated with intrinsic/acquired resistance to EGFR-TKIs reversible by FGFR-inhibitors [54–56]. As we recently described elsewhere, our advanced EGFR-mutant ADC-case with concomitant FGFR3-mutation displayed mixed response, with pleural metastasis progressing already 7 weeks after initiating first-line erlotinib-treatment, whereas other metastatic sites progressed only 6 months later after acquiring additional p.T790M EGFR-mutation [57].
Somehow surprisingly, among our 23 EGFR-mutated patients treated with erlotinib, 17 showed OR (complete/partial) regardless of the presence of cancer-relevant co-mutations that can potentially represent mechanisms of primary resistance to EGFR-TKIs, because they occurred in genes coding for proteins that either are directly downstream the EGFR or belong to alternative by-pass pathways, as discussed above. No statistically significant difference in PFS or OS was observed when stratifying according to concomitant driver status, potentially due to the low number of patients included (Figure 2). Although concurrent aberrations in other driver genes did not seem to significantly impair erlotinib efficacy in most of our cases, they could potentially decrease the time to progression by serving as alternative driver-pathways. Once treated with erlotinib, NSCLCs may become resistant either by selecting pre-existing subclones carrying resistance-mutations such as p.T790M or possessing the ability to depend on alternative oncogenic pathways for survival [3, 58]. Thus, acquired TKI-resistance may already exist in subclones at baseline and targeting a single activating driver mutation will eventually lead to treatment failure, while combining different pathway-targeted drugs based on pre-treatment molecular analyses could potentially be applied in NSCLC to avert or postpone the appearance of resistant tumor cells. In this regard, our study presents limitations, such as the fairly low number of investigated EGFR-mutated cases and the fact that the utilized techniques (NGS, FISH, IHC) covered only a specified number of driver genes. In contrast, the optimal implementation of combination targeted therapy for NSCLC in the future will also require broader knowledge of other genetic or epigenetic events that potentially can represent additional targets or resistance mechanisms and thereby serve as predictive biomarkers as well. This will obviously add to the complexity of combinatorial therapeutic strategies, nevertheless, implementing broad genomic sequencing panels, such as the recently reported MSK-IMPACT assay, can detect multiple potentially actionable targets coexisting within individual tumors in a significant amount of patients, further underlining the future need of combination therapies against NSCLC [5].
In conclusion, the vast majority of advanced EGFR-mutated NSCLCs in our cohort showed co-mutations in other cancer driver genes before receiving first-line erlotinib-treatment. These concurrent alternative mutations may not necessarily lead to initial resistance to erlotinib but they may impact the time to progression to first-line TKI-treatment and represent potential therapeutic targets for combined targeted therapy.
MATERIALS AND METHODS
Tumor samples and mutation analysis
Five hundred and fourteen consecutive NSCLCs of all stages diagnosed at our institution from July 2013 to August 2015 were included. All tumors of non-squamous cell type were examined, while squamous cell carcinomas (SCCs) were only tested on specific clinical indication (patients younger than 50 years or never smokers/light smokers with less than 2 pack-years). DNA was extracted from two 5-μm-thick formalin-fixed paraffin-embedded (FFPE) tissue sections of diagnostic biopsies or from smears of cytological samples obtained from the primary tumor or metastases in lymph nodes or distant organs. Samples diagnosed as ADCs and judged suitable (minimal relative tumor cell nuclei content of 20%) were routinely tested for EGFR-mutations using the Cobas® EGFR Mutation Test (Roche Diagnostics) on the Cobas z480 analyzer (analytical sensitivity of 5%) according to the supplier´s instructions. This fully automated analysis covers 41 EGFR-mutations, including G719X (G719A, G719C, or G719S) in exon 18, 29 variable exon 19-microdeletions, S768I and T790M in exon 20, five different exon 20-insertions, and L858R in exon 21 (two variants).
Specimens below the 20% cut-off were enriched for tumor content using manual micro-dissection.
Samples from EGFR-mutated NSCLC patients that were candidate for first-line TKI treatment (advanced stage, PS 0-2) were further tested by next generation sequencing (NGS) for other possible simultaneous cancer-relevant gene mutations and for confirming and specifying the type of EGFR-mutations detected by Cobas® Test, whereas cases not eligible to TKI therapy (low stage/operable or receiving chemo-radiotherapy, PS 3, or deceased before therapy start) were not further analyzed. For each FFPE specimen, 10 ng of genomic DNA, purified by the QIAamp DNA Minikit (Qiagen) and quantified by the Qubit® dsDNA HS assay on a Qubit® 2.0 Flourometer (Thermo Fisher Scientific), were used for library preparation with the Ion AmpliSeq Library Kit 2.0 (LifeTechnologies) and the Ion AmpliSeq Colon-Lung Cancer Research Panel v2 (Ion Torrent, Thermo Fisher Scientific). This panel yields 92 amplicons covering 504 mutational hot-spot regions and 1825 hot-spot mutations in the following 22 lung/colon cancer-associated genes: AKT1 (NCBI reference sequence: NM_005163), ALK (NM_004304), BRAF (NM_004333), CTNNB1 (NM_001904), DDR2 (NM_001014796), EGFR (NM_005228), ERBB2 (NM_004448), ERBB4 (NM_005235), FBXW7 (NM_033632), FGFR1 (NM_023110), FGFR2 (NM_022970), FGFR3 (NM_000142), KRAS (NM_033360), MAP2K1 (NM_002755), MET (NM_001127500), NOTCH1 (NM_017617), NRAS (NM_002524), PIK3CA (NM_006218), PTEN (NM_000314), SMAD4 (NM_005359), STK11 (NM_000455) and TP53 (NM_000546). Library concentration was determined by the Ion Ampliseq™ TaqMan Quantification Kit.
Preparation of sequencing templates and Ion Spheres followed by loading on Ion 316 chips v2 was automatically performed on the Ion Chef System (Thermo Fisher Scientific). Sequencing was carried out on an Ion Torrent Personal Genome™ (PGM) sequencer using the Ion PGM Sequencing 200 Kit v2 according to the manufacturer’s instructions. Data analysis, including alignment to the hg19 human reference genome and variant calling, was performed by the Torrent Suite Software v.4.4 (Thermo Fisher Scientific) and visually verified with the Integrative Genomics Viewer; IGV v.2.1 (Broad Institute)
Fluorescence in-situ hybridization (FISH) and immunohistochemistry (IHC)
FISH was utilized to detect possible MET-amplification, which can represent a mechanism of TKI-resistance [59].
Two-μm-thick FFPE tissue sections from biopsies or cell-blocks of cytological samples were hybridized overnight with the Zyto-Light SPEC MET/CEN7 Dual Color Probe (ZytoVision) that detects the MET-gene and the centromeric portion of the MET-harboring chromosome 7, as previously described with minor modifications [11].
Briefly, slides were scanned using a X63 objective and appropriate filter sets (DM5500 fluorescent microscope; Leica), using normal fibroblasts, leukocytes, endothelial cells or non-neoplastic lung tissue as internal controls and individually analyzing 100 tumor cell nuclei (20 neighboring tumor cell nuclei from five random areas of homogenous distribution of MET signals) with the X100 objective counting MET (green) and CEN7 (orange) signals. FISH was assessed by one reader (ES-R) without knowing the IHC data. The tumor samples were classified into the following four groups of MET-amplification status [11].
A) High-level amplification when they displayed a MET/CEN7 ratio ≥2.0 or an average MET gene copy number (GCN) per cell of ≥6.0 or ≥10% of tumor cells containing ≥15 MET signals. B) Intermediate-level of GCN gain when ≥50% of cells contained ≥5 MET signals and the criteria for high-level amplification were not fulfilled. C) Low-level of GCN gain when ≥40% of tumor cells showed ≥4 MET signals and criteria for high-/intermediate-level amplification were not fulfilled. D) Negative (no amplification/GCN gain) when none of the above criteria were fulfilled.
IHC for membranous and cytoplasmic expression of MET-receptor was performed on FFPE 2.5-μm-thick tissue sections using a BenchMark ULTRA automated slide immunostainer (Ventana Medical Systems Inc.; item no. N750-BMKU-FS), Ultra Cell Conditioning solution (CC1) pretreatment (Ventana) for 8 min at 95°C, four CC1 treatments (20, 36, 52, and 64 min), and incubation with the pre-diluted CONFIRM anti-Total c-MET (SP44) Rabbit mAb (Ventana Medical Systems, Inc.; Cat. # 790-4430) for 16 min, as described [11]. The reaction was visualized using ultraView DAB Detection Kit and hematoxylin counterstaining (Ventana). MET expression was scored in a blinded manner (without knowing the FISH results) by one observer (ES-R), assessing staining intensity (negative, weak, moderate, or strong) and the percentage of cells with these intensities using bronchial/alveolar epithelial cells as internal controls for weak-moderate intensity. Thereby, 4 diagnostic “immunoscores” were defined: 3+/overexpression (strong intensity in ≥50% of tumor cells); 2+ (moderate intensity in ≥50% of tumor cells); 1+ (weak intensity in ≥50% of tumor cells); and 0 (no staining or <50% of tumor cells stained with any intensity) [11].
For ALK immunostaining, as indicator of ALK-rearrangement [60], the fully automated IHC assay using the pre-diluted VENTANA anti-ALK (D5F3) Rabbit mAb (Ventana Medical Systems Inc.; Cat. # 790-4794) was used together with the Optiview DAB IHC detection kit and Optiview Amplification kit on the Benchmark XT stainer (Ventana Medical Systems Inc.), implementing a binary scoring system (present/absent moderate/strong granular cytoplasmic staining in any percentage of tumor cells) according to manufacturer’s instructions [11, 60].
Each case was also stained with an unrelated matched rabbit IgG mAb used as negative control. Samples with known MET-amplification and MET-overexpression or ALK--rearrangement and ALK-overexpression were used as positive controls [61].
Assessment of tumor response and definition of lack of response/intrinsic resistance
Objective response (OR) evaluation was done by CT-scan at baseline (maximum 1 month prior to TKI-treatment start), followed by CT-scans every 9 weeks. RECIST v1.1 criteria for definitions of response and progression were used [62]. Patients who progressed within 18 weeks (4 months) were classified as having intrinsic resistance.
Statistical analysis
Overall survival (OS) was defined as the time from onset of treatment to the time of death from any cause or last follow-up. PFS was defined as the time from onset of treatment with erlotinib to a documented progression or death from any cause. For patients without any progression at the time of analysis, the date of last follow-up was considered right-censored. Kaplan-Meier curves were used to estimate curves for OS and PFS and chi-square test was used for comparison of proportions. A p-value < 0.05 was considered statistically significant.
The study was approved by the institutions’ local ethical committee and was with data collection on pre-defined case report forms.
ACKNOWLEDGMENTS AND FUNDING
We are very grateful to Camilla C. Mortensen, Sanni C. Pedersen and Tine W. Adelfest for their excellent technical assistance. The study was partly funded by a grant from Roche.
CONFLICTS OF INTEREST
Eric Santoni-Rugiu and Jens B. Sørensen have received lecture honoraria from Roche, Pfizer, and Boehringer Ingelheim. Jan N. Jakobsen, Morten Grauslund and Linea C. Melchior have no potential conflicts of interest to disclose.
REFERENCES
1. Mitsudomi T, Yatabe Y. Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci. 2007; 98:1817–24. https://doi.org/10.1111/j.1349-7006.2007.00607.x.
2. Lee CK, Wu YL, Ding PN, Lord SJ, Inoue A, Zhou C, Mitsudomi T, Rosell R, Pavlakis N, Links M, Gebski V, Gralla RJ, Yang JC. Impact of Specific Epidermal Growth Factor Receptor (EGFR) Mutations and Clinical Characteristics on Outcomes After Treatment With EGFR Tyrosine Kinase Inhibitors Versus Chemotherapy in EGFRMutant Lung Cancer: A Meta-Analysis. J Clin Oncol. 2015; 33:1958–65. https://doi.org/10.1200/JCO.2014.58.1736.
3. Morgillo F, Della Corte CM, Fasano M, Ciardiello F. Mechanisms of resistance to EGFR-targeted drugs: lung cancer. ESMO Open. 2016; 1:e000060. https://doi.org/10.1136/esmoopen-2016-000060.
4. Tetsu O, Hangauer MJ, Phuchareon J, Eisele DW, McCormick F. Drug Resistance to EGFR Inhibitors in Lung Cancer. Chemotherapy. 2016; 61:223–35. https://doi.org/10.1159/000443368.
5. Jordan EJ, Kim HR, Arcila ME, Barron D, Chakravarty D, Gao J, Chang MT, Ni A, Kundra R, Jonsson P, Jayakumaran G, Gao SP, Johnsen HC, et al. Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov. 2017; 7:596–609. https://doi.org/10.1158/2159-8290.CD-16-1337.
6. Guibert N, Barlesi F, Descourt R, Lena H, Besse B, Beau-Faller M, Mosser J, Pichon E, Merlio JP, Ouafik L, Guichard F, Mastroianni B, Moreau L, et al. Characteristics and Outcomes of Patients with Lung Cancer Harboring Multiple Molecular Alterations: Results from the IFCT Study Biomarkers France. J Thorac Oncol. 2017; 12:963–973. https://doi.org/10.1016/j.jtho.2017.02.001.
7. Gainor JF, Varghese AM, Ou SH, Kabraji S, Awad MM, Katayama R, Pawlak A, Mino-Kenudson M, Yeap BY, Riely GJ, Iafrate AJ, Arcila ME, Ladanyi M, et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res. 2013; 19:4273–81. https://doi.org/10.1158/1078-0432.CCR-13-0318.
8. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511:543–50. https://doi.org/10.1038/nature13385
9. Baldi L, Mengoli MC, Bisagni A, Banzi MC, Boni C, Rossi G. Concomitant EGFR mutation and ALK rearrangement in lung adenocarcinoma is more frequent than expected: report of a case and review of the literature with demonstration of genes alteration into the same tumor cells. Lung Cancer. 2014; 86:291–95. https://doi.org/10.1016/j.lungcan.2014.09.011.
10. Won JK, Keam B, Koh J, Cho HJ, Jeon YK, Kim TM, Lee SH, Lee DS, Kim DW, Chung DH. Concomitant ALK translocation and EGFR mutation in lung cancer: a comparison of direct sequencing and sensitive assays and the impact on responsiveness to tyrosine kinase inhibitor. Ann Oncol. 2015; 26:348–54. https://doi.org/10.1093/annonc/mdu530.
11. Schildhaus HU, Schultheis AM, Rüschoff J, Binot E, Merkelbach-Bruse S, Fassunke J, Schulte W, Ko YD, Schlesinger A, Bos M, Gardizi M, Engel-Riedel W, Brockmann M, et al. MET amplification status in therapy-naïve adeno- and squamous cell carcinomas of the lung. Clin Cancer Res. 2015; 21:907–15. https://doi.org/10.1158/1078-0432.CCR-14-0450.
12. Sholl LM, Aisner DL, Varella-Garcia M, Berry LD, Dias-Santagata D, Wistuba II, Chen H, Fujimoto J, Kugler K, Franklin WA, Iafrate AJ, Ladanyi M, Kris MG, et al, and LCMC Investigators. Multi-institutional Oncogenic Driver Mutation Analysis in Lung Adenocarcinoma: The Lung Cancer Mutation Consortium Experience. J Thorac Oncol. 2015; 10:768–77. https://doi.org/10.1097/JTO.0000000000000516.
13. Lee T, Lee B, Choi YL, Han J, Ahn MJ, Um SW. Non-small Cell Lung Cancer with Concomitant EGFR, KRAS, and ALK Mutation: Clinicopathologic Features of 12 Cases. J Pathol Transl Med. 2016; 50:197–203. https://doi.org/10.4132/jptm.2016.03.09.
14. Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JH, Beasley MB, Chirieac LR, Dacic S, Duhig E, Flieder DB, Geisinger K, Hirsch FR, Ishikawa Y, et al, and WHO Panel. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J Thorac Oncol. 2015; 10:1243–60. https://doi.org/10.1097/JTO.0000000000000630.
15. Skov BG, Høgdall E, Clementsen P, Krasnik M, Larsen KR, Sørensen JB, Skov T, Mellemgaard A. The prevalence of EGFR mutations in non-small cell lung cancer in an unselected Caucasian population. APMIS. 2015; 123:108–15. https://doi.org/10.1111/apm.12328.
16. Leventakos K, Kipp BR, Rumilla KM, Winters JL, Yi ES, Mansfield AS. S768I Mutation in EGFR in Patients with Lung Cancer. J Thorac Oncol. 2016; 11:1798–801. https://doi.org/10.1016/j.jtho.2016.05.007.
17. Chiu CH, Yang CT, Shih JY, Huang MS, Su WC, Lai RS, Wang CC, Hsiao SH, Lin YC, Ho CL, Hsia TC, Wu MF, Lai CL, et al. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Treatment Response in Advanced Lung Adenocarcinomas with G719X/L861Q/S768I Mutations. J Thorac Oncol. 2015; 10:793–99. https://doi.org/10.1097/JTO.0000000000000504.
18. Krawczyk P, Reszka K, Ramlau R, Powrózek T, Pankowski J, Wojas-Krawczyk K, Kalinka-Warzocha E, Szczęsna A, Nicoś M, Jarosz B, Szyszka-Barth K, Bryl M, Adamowicz K, et al. Prevalence of rare EGFR gene mutations in nonsmall-cell lung cancer: a multicenter study on 3856 Polish Caucasian patients. Ann Oncol. 2016; 27:358–59. https://doi.org/10.1093/annonc/mdv553.
19. Barnet MB, O’Toole S, Horvath LG, Selinger C, Yu B, Ng CC, Boyer M, Cooper WA, Kao S. EGFR-Co-Mutated Advanced NSCLC and Response to EGFR Tyrosine Kinase Inhibitors. J Thorac Oncol. 2017; 12:585–90. https://doi.org/10.1016/j.jtho.2016.09.001.
20. Yang JC, Sequist LV, Geater SL, Tsai CM, Mok TS, Schuler M, Yamamoto N, Yu CJ, Ou SH, Zhou C, Massey D, Zazulina V, Wu YL. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol. 2015; 16:830–38. https://doi.org/10.1016/S1470-2045(15)00026-1.
21. Russo A, Franchina T, Ricciardi GR, Adamo V. Rapid Acquisition of T790M Mutation after Treatment with Afatinib in an NSCLC Patient Harboring EGFR Exon 20 S768I Mutation. J Thorac Oncol. 2017; 12:e6–8. https://doi.org/10.1016/j.jtho.2016.09.132.
22. Ludovini V, Bianconi F, Pistola L, Pistola V, Chiari R, Colella R, Bellezza G, Tofanetti FR, Siggillino A, Baldelli E, Flacco A, Giuffrida D, Sidoni A, Crinò L. Optimization of patient selection for EGFR-TKIs in advanced non-small cell lung cancer by combined analysis of KRAS, PIK3CA, MET, and non-sensitizing EGFR mutations. Cancer Chemother Pharmacol. 2012; 69:1289–99. https://doi.org/10.1007/s00280-012-1829-7.
23. He M, Capelletti M, Nafa K, Yun CH, Arcila ME, Miller VA, Ginsberg MS, Zhao B, Kris MG, Eck MJ, Jänne PA, Ladanyi M, Oxnard GR. EGFR exon 19 insertions: a new family of sensitizing EGFR mutations in lung adenocarcinoma. Clin Cancer Res. 2012; 18:1790–97. https://doi.org/10.1158/1078-0432.CCR-11-2361.
24. Lin YT, Liu YN, Wu SG, Yang JC, Shih JY. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor-sensitive Exon 19 Insertion and Exon 20 Insertion in Patients With Advanced Non-Small-cell Lung Cancer. Clin Lung Cancer. 2017; 18:324–332.e1. https://doi.org/10.1016/j.cllc.2016.12.014.
25. Lee JK, Shin JY, Kim S, Lee S, Park C, Kim JY, Koh Y, Keam B, Min HS, Kim TM, Jeon YK, Kim DW, Chung DH, et al. Primary resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) in patients with non-small-cell lung cancer harboring TKI-sensitive EGFR mutations: an exploratory study. Ann Oncol. 2013; 24:2080–87. https://doi.org/10.1093/annonc/mdt127.
26. Lim SM, Kim HR, Cho EK, Min YJ, Ahn JS, Ahn MJ, Park K, Cho BC, Lee JH, Jeong HC, Kim EK, Kim JH. Targeted sequencing identifies genetic alterations that confer primary resistance to EGFR tyrosine kinase inhibitor (Korean Lung Cancer Consortium). Oncotarget. 2016; 7:36311–20. https://doi.org/10.18632/oncotarget.8904.
27. Bria E, Pilotto S, Amato E, Fassan M, Novello S, Peretti U, Vavalà T, Kinspergher S, Righi L, Santo A, Brunelli M, Corbo V, Giglioli E, et al. Molecular heterogeneity assessment by next-generation sequencing and response to gefitinib of EGFR mutant advanced lung adenocarcinoma. Oncotarget. 2015; 6:12783–95. https://doi.org/10.18632/oncotarget.3727.
28. VanderLaan PA, Rangachari D, Mockus SM, Spotlow V, Reddi HV, Malcolm J, Huberman MS, Joseph LJ, Kobayashi SS, Costa DB. Mutations in TP53, PIK3CA, PTEN and other genes in EGFR mutated lung cancers: correlation with clinical outcomes. Lung Cancer. 2017; 106:17–21. https://doi.org/10.1016/j.lungcan.2017.01.011.
29. Labbé C, Cabanero M, Korpanty GJ, Tomasini P, Doherty MK, Mascaux C, Jao K, Pitcher B, Wang R, Pintilie M, Leighl NB, Feld R, Liu G, et al. Prognostic and predictive effects of TP53 co-mutation in patients with EGFR-mutated non-small cell lung cancer (NSCLC). Lung Cancer. 2017; 111:23–29. https://doi.org/10.1016/j.lungcan.2017.06.014.
30. Canale M, Petracci E, Delmonte A, Chiadini E, Dazzi C, Papi M, Capelli L, Casanova C, De Luigi N, Mariotti M, Gamboni A, Chiari R, Bennati C, et al. Impact of TP53 Mutations on Outcome in EGFR-Mutated Patients Treated with First-Line Tyrosine Kinase Inhibitors. Clin Cancer Res. 2017; 23:2195–202. https://doi.org/10.1158/1078-0432.CCR-16-0966.
31. Huang S, Benavente S, Armstrong EA, Li C, Wheeler DL, Harari PM. p53 modulates acquired resistance to EGFR inhibitors and radiation. Cancer Res. 2011; 71:7071–79. https://doi.org/10.1158/0008-5472.CAN-11-0128.
32. Lutterbach B, Zeng Q, Davis LJ, Hatch H, Hang G, Kohl NE, Gibbs JB, Pan BS. Lung cancer cell lines harboring MET gene amplification are dependent on Met for growth and survival. Cancer Res. 2007; 67:2081–88. https://doi.org/10.1158/0008-5472.CAN-06-3495.
33. Beau-Faller M, Ruppert AM, Voegeli AC, Neuville A, Meyer N, Guerin E, Legrain M, Mennecier B, Wihlm JM, Massard G, Quoix E, Oudet P, Gaub MP. MET gene copy number in non-small cell lung cancer: molecular analysis in a targeted tyrosine kinase inhibitor naïve cohort. J Thorac Oncol. 2008; 3:331–39. https://doi.org/10.1097/JTO.0b013e318168d9d4.
34. Dietrich MF, Yan SX, Schiller JH. Response to Crizotinib/Erlotinib Combination in a Patient with a Primary EGFR-Mutant Adenocarcinoma and a Primary c-met-Amplified Adenocarcinoma of the Lung. J Thorac Oncol. 2015; 10:e23–25. https://doi.org/10.1097/JTO.0000000000000448.
35. Gainor JF, Niederst MJ, Lennerz JK, Dagogo-Jack I, Stevens S, Shaw AT, Sequist LV, Engelman JA. Dramatic Response to Combination Erlotinib and Crizotinib in a Patient with Advanced, EGFR-Mutant Lung Cancer Harboring De Novo MET Amplification. J Thorac Oncol. 2016; 11:e83–85. https://doi.org/10.1016/j.jtho.2016.02.021.
36. Ulivi P, Chiadini E, Dazzi C, Dubini A, Costantini M, Medri L, Puccetti M, Capelli L, Calistri D, Verlicchi A, Gamboni A, Papi M, Mariotti M, et al. Nonsquamous, Non-Small-Cell Lung Cancer Patients Who Carry a Double Mutation of EGFR, EML4-ALK or KRAS: Frequency, Clinical-Pathological Characteristics, and Response to Therapy. Clin Lung Cancer. 2016; 17:384–90. https://doi.org/10.1016/j.cllc.2015.11.004.
37. Martin P, Leighl NB, Tsao MS, Shepherd FA. KRAS mutations as prognostic and predictive markers in non-small cell lung cancer. J Thorac Oncol. 2013; 8:530–42. https://doi.org/10.1097/JTO.0b013e318283d958.
38. Wang J, Wang B, Chu H, Yao Y. Intrinsic resistance to EGFR tyrosine kinase inhibitors in advanced non-smallcell lung cancer with activating EGFR mutations. Onco Targets Ther. 2016; 9:3711–26. https://doi.org/10.2147/OTT.S106399.
39. Eng J, Woo KM, Sima CS, Plodkowski A, Hellmann MD, Chaft JE, Kris MG, Arcila ME, Ladanyi M, Drilon A. Impact of Concurrent PIK3CA Mutations on Response to EGFR Tyrosine Kinase Inhibition in EGFR-Mutant Lung Cancers and on Prognosis in Oncogene-Driven Lung Adenocarcinomas. J Thorac Oncol. 2015; 10:1713–19. https://doi.org/10.1097/JTO.0000000000000671.
40. Zhang X, Hao J. Development of anticancer agents targeting the Wnt/β-catenin signaling. Am J Cancer Res. 2015; 5:2344–60.
41. Nakayama S, Sng N, Carretero J, Welner R, Hayashi Y, Yamamoto M, Tan AJ, Yamaguchi N, Yasuda H, Li D, Soejima K, Soo RA, Costa DB, et al. β-catenin contributes to lung tumor development induced by EGFR mutations. Cancer Res. 2014; 74:5891–902. https://doi.org/10.1158/0008-5472.CAN-14-0184.
42. Li K, Mo C, Gong D, Chen Y, Huang Z, Li Y, Zhang J, Huang L, Li Y, Fuller-Pace FV, Lin P, Wei Y. DDX17 nucleocytoplasmic shuttling promotes acquired gefitinib resistance in non-small cell lung cancer cells via activation of β-catenin. Cancer Lett. 2017; 400:194–202. https://doi.org/10.1016/j.canlet.2017.02.029.
43. Togashi Y, Hayashi H, Terashima M, de Velasco MA, Sakai K, Fujita Y, Tomida S, Nakagawa K, Nishio K. Inhibition of β-Catenin enhances the anticancer effect of irreversible EGFR-TKI in EGFR-mutated non-small-cell lung cancer with a T790M mutation. J Thorac Oncol. 2015; 10:93–101. https://doi.org/10.1097/JTO.0000000000000353.
44. Ohashi K, Sequist LV, Arcila ME, Lovly CM, Chen X, Rudin CM, Moran T, Camidge DR, Vnencak-Jones CL, Berry L, Pan Y, Sasaki H, Engelman JA, et al. Characteristics of lung cancers harboring NRAS mutations. Clin Cancer Res. 2013; 19:2584–91. https://doi.org/10.1158/1078-0432.CCR-12-3173.
45. Lin DC, Hao JJ, Nagata Y, Xu L, Shang L, Meng X, Sato Y, Okuno Y, Varela AM, Ding LW, Garg M, Liu LZ, Yang H, et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat Genet. 2014; 46:467–73. https://doi.org/10.1038/ng.2935.
46. Massagué J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000; 103:295–309. https://doi.org/10.1016/S0092-8674(00)00121-5.
47. Schildhaus HU, Nogova L, Wolf J, Buettner R. FGFR1 amplifications in squamous cell carcinomas of the lung: diagnostic and therapeutic implications. Transl Lung Cancer Res. 2013; 2:92–100.
48. Terai H, Soejima K, Yasuda H, Nakayama S, Hamamoto J, Arai D, Ishioka K, Ohgino K, Ikemura S, Sato T, Yoda S, Satomi R, Naoki K, Betsuyaku T. Activation of the FGF2-FGFR1 autocrine pathway: a novel mechanism of acquired resistance to gefitinib in NSCLC. Mol Cancer Res. 2013; 11:759–67. https://doi.org/10.1158/1541-7786.MCR-12-0652.
49. Azuma K, Kawahara A, Sonoda K, Nakashima K, Tashiro K, Watari K, Izumi H, Kage M, Kuwano M, Ono M, Hoshino T. FGFR1 activation is an escape mechanism in human lung cancer cells resistant to afatinib, a pan-EGFR family kinase inhibitor. Oncotarget. 2014; 5:5908–19. https://doi.org/10.18632/oncotarget.1866.
50. Kim TM, Song A, Kim DW, Kim S, Ahn YO, Keam B, Jeon YK, Lee SH, Chung DH, Heo DS. Mechanisms of Acquired Resistance to AZD9291: A Mutation-Selective, Irreversible EGFR Inhibitor. J Thorac Oncol. 2015; 10:1736–44. https://doi.org/10.1097/JTO.0000000000000688.
51. Wang R, Zhang Y, Pan Y, Li Y, Hu H, Cai D, Li H, Ye T, Luo X, Zhang Y, Li B, Shen L, Sun Y, Chen H. Comprehensive investigation of oncogenic driver mutations in Chinese non-small cell lung cancer patients. Oncotarget. 2015; 6:34300–08. https://doi.org/10.18632/oncotarget.5549.
52. Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, Kurzrock R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin Cancer Res. 2016; 22:259–67. https://doi.org/10.1158/1078-0432.CCR-14-3212.
53. Chandrani P, Prabhash K, Prasad R, Sethunath V, Ranjan M, Iyer P, Aich J, Dhamne H, Iyer DN, Upadhyay P, Mohanty B, Chandna P, Kumar R, et al. Drug-sensitive FGFR3 mutations in lung adenocarcinoma. Ann Oncol. 2017; 28:597–603.
54. Ou SI, Horn L, Cruz M, Vafai D, Lovly CM, Spradlin A, Williamson MJ, Dagogo-Jack I, Johnson A, Miller VA, Gadgeel S, Ali SM, Schrock AB. Emergence of FGFR3-TACC3 fusions as a potential by-pass resistance mechanism to EGFR tyrosine kinase inhibitors in EGFR mutated NSCLC patients. Lung Cancer. 2017; 111:61–64. https://doi.org/10.1016/j.lungcan.2017.07.006.
55. Capelletti M, Dodge ME, Ercan D, Hammerman PS, Park SI, Kim J, Sasaki H, Jablons DM, Lipson D, Young L, Stephens PJ, Miller VA, Lindeman NI, et al. Identification of recurrent FGFR3-TACC3 fusion oncogenes from lung adenocarcinoma. Clin Cancer Res. 2014; 20:6551–58. https://doi.org/10.1158/1078-0432.CCR-14-1337.
56. Daly C, Castanaro C, Zhang W, Zhang Q, Wei Y, Ni M, Young TM, Zhang L, Burova E, Thurston G. FGFR3-TACC3 fusion proteins act as naturally occurring drivers of tumor resistance by functionally substituting for EGFR/ERK signaling. Oncogene. 2017; 36:471–81. https://doi.org/10.1038/onc.2016.216.
57. Santoni-Rugiu E, Grauslund M, Melchior LC, Costa JC, Sørensen JB, Urbanska EM. Heterogeneous resistance mechanisms in an EGFR exon 19-mutated non-small cell lung cancer patient treated with erlotinib: persistent FGFR3-mutation, localized transformation to EGFR-mutated SCLC, and acquired T790M EGFR-mutation. Lung Cancer. 2017; 113:14–17. https://doi.org/10.1016/j.lungcan.2017.08.024.
58. Ma C, Wei S, Song Y. T790M and acquired resistance of EGFR TKI: a literature review of clinical reports. J Thorac Dis. 2011; 3:10–18..
59. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007; 316:1039–43. https://doi.org/10.1126/science.1141478.
60. Ying J, Guo L, Qiu T, Shan L, Ling Y, Liu X, Lu N. Diagnostic value of a novel fully automated immunochemistry assay for detection of ALK rearrangement in primary lung adenocarcinoma. Ann Oncol. 2013; 24:2589–93. https://doi.org/10.1093/annonc/mdt295.
61. Rossing HH, Grauslund M, Urbanska EM, Melchior LC, Rask CK, Costa JC, Skov BG, Sørensen JB, Santoni-Rugiu E. Concomitant occurrence of EGFR (epidermal growth factor receptor) and KRAS (V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog) mutations in an ALK (anaplastic lymphoma kinase)-positive lung adenocarcinoma patient with acquired resistance to crizotinib: a case report. BMC Res Notes. 2013; 6:489. https://doi.org/10.1186/1756-0500-6-489.
62. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009; 45:228–47. https://doi.org/10.1016/j.ejca.2008.10.026.