
Introduction
Multiple myeloma (MM) is a malignant disease that is characterized by clonal expansion of terminally differentiated B cells. Despite major advances in overall survival (OS) achieved by the introduction of novel agents within the last decade, MM remains an incurable malignancy in most cases [1].
Although very heterogeneous with respect to molecular characteristics, a common feature of MM cells is the relatively slow rate of cell proliferation in most newly diagnosed patients [2, 3]. Equally important for MM cell survival and therapeutic resistance are impaired mechanisms of apoptosis. The anti-apoptotic Bcl-2 family member Mcl-1, in particular, seems to be essential for MM cell survival [4-7].
Survivin is a member of the IAP (inhibitors of apoptosis) protein family, encoded by the BIRC5 gene, and is highly expressed in cancer cells while virtually absent in most differentiated normal tissues [8, 9]. Furthermore, expression levels of survivin have been found to correlate with poor prognosis in colorectal, non-small-cell lung, prostate, and breast cancer, as well as in MM [10-14]. Functionally, survivin has been shown to counteract apoptosis induction upstream of effector caspases [15] and to have an essential role in cell proliferation by regulating spindle assembly and microtubule attachment to the kinetochore as a member of the chromosomal passenger complex [16, 17]. In MM, survivin has been implicated in the mechanisms of action of several therapeutic approaches. Specifically, inhibition of AKT, STAT-3 and NFkB signaling is associated with reduced intracellular gene expression and protein levels of survivin [18-20]. Long-term knockdown of survivin resulted in moderate inhibition of MM cell growth and increased drug sensitivity [21].
Although survivin appears to be an attractive therapeutic target from this initial set of data, it has never been analyzed in a large cohort of patients for its prognostic and therapeutic value. The portfolio of clinical grade survivin specific antagonists has been surprisingly limited [9]. Recently, a novel imidazolium-based small molecule suppressant of survivin, YM155, has been described. This compound was identified in a cell-based promoter activity assay and was described to specifically abrogate survivin gene (BIRC5) expression, resulting in preclinical activity in several tumor models both in vitro and in vivo [22, 23]. YM155 is currently being evaluated in phase II clinical trials in lymphoma, melanoma, as well as in cancers of the breast, lung, and prostate.
Based on these data we sought to re-assess the prognostic significance of BIRC5 gene expression in CD138-purified MM cells from a cohort of 1093 previously untreated patients and to pre-clinically evaluate YM155 for its therapeutic potential in MM. BIRC5 expression proved to be a powerful prognostic marker for event-free (EFS) and OS in two independent cohorts of patients. YM155 potently abrogates MM cell growth associated with inhibition of survivin expression. Furthermore, delineation of molecular sequelae in MM cells showed that down regulation of Mcl-1 appears to be an even more important downstream effector mechanism in MM cells exposed to YM155. Mcl-1 is expressed in all 1093 MM cell samples and is likewise associated with an adverse clinical prognosis.
Taken together, YM155 displays great therapeutic potential on MM cells via inhibition of survivin and, more importantly, Mcl-1 expression. Further clinical evaluation of this compound in MM is strongly warranted.
Results
Aberrant expression of survivin gene transcripts in multiple myeloma
The survivin gene (BIRC5, probe set 202095_s_at) was not expressed in normal bone marrow plasma cells (n = 10, absent call 10/10), but was aberrantly expressed in 158/370 (42.7 %, present call) MM cell samples in our patient cohort. The mean expression in MM cells is 4-fold higher compared to that in normal plasma cells (P < 0.001) and 2-fold compared to plasma cells from patients with MGUS (p < 0.001), respectively (Figure 1A). In turn, BIRC5 is expressed in all samples of proliferating normal or malignant plasma cells, i.e. polyclonal plasmablastic cells (PPC, non-malignant, n = 10), or human myeloma cell lines (n = 32) (Figure 1A). In both cases, the expression is significantly higher compared to MM cell samples (p < 0.001, p < 0.001). BIRC5 expression in primary MM cells correlates significantly with proliferation as assessed by the gene expression profiling derived proliferation index, GPI (r = 0.81, p < 0.001) or propidium iodide staining [2] (r = 0.59, p < 0.001; n = 36), and the expression is significantly different between a low/median/high GPI (Figure 1A). BIRC5 gene expression also increases with higher Durie-Salmon stage (p < 0.001, Suppl. Figure S1A). BIRC5 expression was validated by qRT-PCR [14]. In agreement with microarray and qRT-PCR-data, survivin protein could be detected in 12/12 MM cell lines, being absent in 2/2 bone marrow stromal cell samples used as control (Figure 1B).
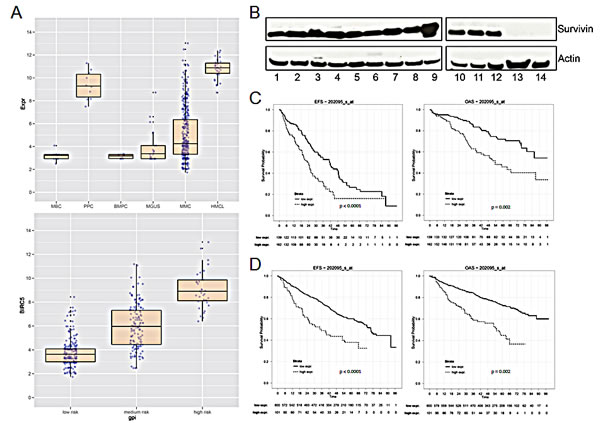
Figure 1: Survivin gene (BIRC5) expression and survival in multiple myeloma. (A) Upper panel: logarithmic scale of BIRC5 expression in MBC (memory B-cells), PPC (polyclonal plasmablastic cells), BMPC (bone marrow plasma cells), MGUS, MMC (multiple myeloma cells) and HMCL (human myeloma cell lines). Lower panel: logarithmic scale of BIRC5 expression in patient samples with low, medium or high risk GPI. (B) Survivin protein expression in 12 different MM cell lines and in primary BMSC. The indicated samples were: 1 L-363; 2 KARPAS-620; 3 AMO-1; 4 OPM-2; 5 JJN-3; 6 EJM; 7 U-266; 8 RPMI-8226; 9 LP-1; 10 KMS-12-BM; 11 MOLP-8; 12 NCI-H929; 13 BMSC Patient1; 14 BMSC Patient2. (C) Event-free and overall survival in 301 patients undergoing high-dose chemotherapy at our center in Heidelberg with low or high BIRC5 expression, respectively. (D) UAMS Arkansas validation group: event-free and overall survival in 701 patients treated within the total therapy 2 and 3 protocols.
Survivin expression, patient survival, and risk factors
In 301 patients undergoing high-dose chemotherapy, the presence of BIRC5 expression was associated with inferior EFS and OS (22.6 vs. 35.4 months, p < 0.001; 52.9 vs. not reached, p = 0.002) using an optimized cutoff defined by maximum logrank statistics (Figure 1C). This strategy was also applied to 701 patients of the UAMS Arkansas group treated within the total therapy 2 or therapy 3 protocols with EFS of 12.3 vs. 54.1 months (P < 0.001) and OS of 17.4 vs. n.r. (p = 0.002) (Figure 1D). BIRC5 expression is also significantly higher in patients harboring a del17p or gain of 1q21 (Suppl. Figure S1B). BIRC5 expression is likewise higher in patients designated as “high risk” by the gene expression based risk scores of the UAMS (UAMS 70 gene score [24] or the IFM-score (IFM 15 gene score) [25] (Suppl. Figure S1C and S1D).
YM155 specifically inhibits cell growth and induces apoptosis in myeloma cells
The small molecule survivin suppressant YM155 has been shown to effectively reduce cellular survivin expression at the mRNA and protein levels [22]. In our panel of 10 MM cell lines, increasing concentrations of YM155 reduced cell growth (i.e., viable cells after 48h) in all cell lines within a range of IC50 values of 2 nM to 50 nM (Figure 2A). Likewise, YM155 specifically inhibited proliferation in these cell lines already within 24 hours of exposure at similar IC50 values, assessed by [3H]-thymidine uptake (Figure 2B). Moreover, the viability of CD138-positive cells isolated from the bone marrow samples of three heavily pre-treated MM patients was also strongly reduced upon treatment with YM155, with only minor effects on corresponding primary bone marrow stromal cells, used as non-malignant controls (Figure 2C). In addition, YM155 induced apoptosis as detected by caspase 3/7 activation in the two most sensitive cell lines, U-266 and INA-6, as well as in primary myeloma cells, at concentrations comparable to those that inhibited proliferation (Figure 2D). Less sensitive cell lines, such as OPM-2, required a 10-fold higher concentration (500 nM) for induction of apoptosis (Suppl. Figure 2)
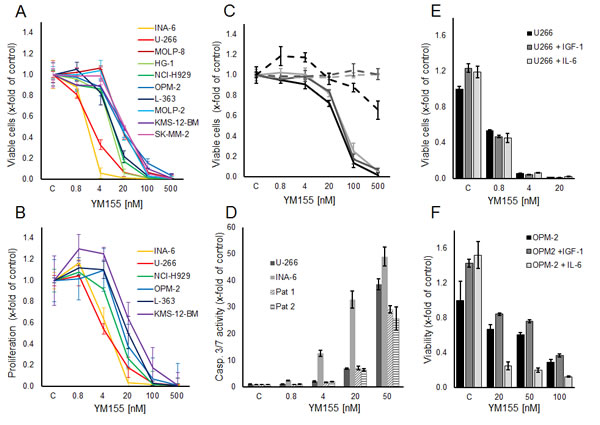
Figure 2: YM155 inhibits viability, proliferation and induces apoptosis in MM cell lines and primary MM cells. (A) Viable and (B) proliferating myeloma cells are shown as percentage of control after 48 and 24 hours of treatment with YM155 at indicated concentrations or vehicle only, respectively. (C) Primary MM cells (solid lines) and BMSCs (dashed lines) from three patients. Viable cells as percentage of control after 48 hours of treatment with YM155. (D) Caspase 3/7 activity in U-266 and INA-6 cell lines and primary cells from two myeloma patients at 24 hours after treatment with YM155 at indicated concentrations. (E, F) IL-6 sensitizes OPM-2 cells to YM155. OPM-2 and U-266 cells were grown in starvation medium with or without IL-6 (50 ng/ml) or IGF-I (100 ng/ml), respectively, and YM155 at indicated concentrations. All quantitative data are shown as the mean +/- SD of 3 independent experiments conducted in triplicates.
IL-6 sensitizes OPM-2 cells to YM155
As the growth factors IL-6 and IGF-1 are known to promote MM cell growth and survival and to confer drug resistance, we next examined whether YM155 can overcome these protective effects. While both cytokines triggered MM cell growth in serum-free media in untreated controls, neither IL-6 nor IGF-1 were able to attenuate the growth inhibitory effect of YM155 in U-266 and OPM-2 cells (Figure 2E,F). Moreover, IL-6 strikingly sensitized OPM-2 cells to treatment with YM155 (Figure 2F). U-266 cells have been reported to utilize a constitutively activated IL-6/Stat-3 pathway by the autocrine secretion of IL-6 [27]. Considering that U-266 cells as well as the IL-6-dependent INA-6 cells were highly sensitive to YM155 under normal growth conditions, the fact that IL-6 stimulation in serum-free conditions sensitized OPM-2 cells to YM155 treatment suggested that YM155 might interfere with the IL-6/JAK/STAT3 signaling pathway.
YM155 induces ER stress response signaling in myeloma cells
To gain further insight into the mechanism of action of YM155 in MM cells, we performed serial gene expression profiling after 6 (4 nM and 50 nM), 12 (4 nM and 50 nM), and 36 hours (4 nM) of YM155 treatment in U-266 cells. Among significantly differentially regulated genes (P < 0.01), 189 were up-regulated and 63 down-regulated > 1.5-fold. Pathway analysis using the Ingenuity Systems software revealed up-regulation of genes associated with the endoplasmic reticulum (ER) stress/ unfolded protein response (UPR) suggesting the involvement of this pathway in the anti-MM effects of YM155 (Figure 3). To confirm these data, we assessed key markers of the ER stress response on MM cells by immunoblot assay (Figure 4). PERK-dependent phosphorylation of eIF2a and induction of its downstream target CHOP together with the stress-related transcription factor ATF-3 were detected in U-266 cells within 24 hours at 4nM and within 6 to 12 hours (50 nM) of YM155 exposure. The up-regulation of CHOP, which has previously been shown to inhibit proliferation and to induce apoptosis in MM cells [29, 30], was closely associated with PARP cleavage, indicating the onset of apoptotic signaling. Similar results were obtained in INA-6 cells. Thapsigargin (TG) was used as a positive control for markers of ER stress response (Figure 4).
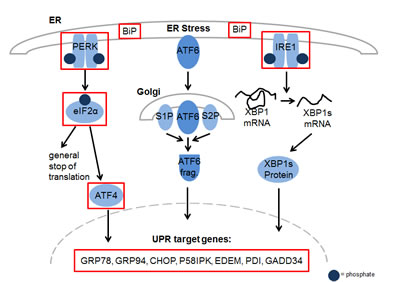
Figure 3: YM155 exposure regulates genes involved in ER stress in MM cells. Activation of key components of the Unfolded Protein Response (UPR) in U-266 cells upon YM155 treatment. The marked genes were significantly upregulated (p < 0.05) after 12 hours of incubation at concentrations of 4 nM and 50 nM.
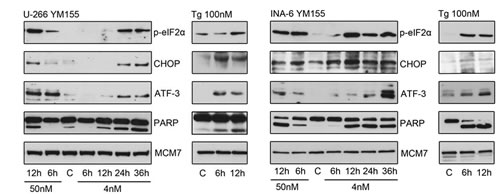
Figure 4: YM155 triggers ER stress signaling in MM cells. UPR signaling pathways were assessed by immunoblot of p-eIF2α, CHOP and ATF-3 protein levels in U-266 and INA-6 cells at indicated times and concentrations. Cleaved PARP indicated onset of apoptosis, MCM7 served as loading control. thapsigargin (Tg) treated cells served as positive control samples.
YM155 inhibits IL-6/STAT3-signaling
Gene expression profiling further suggested concordant down-regulation of genes that have been described to be regulated by the transcription factor STAT3 (Figure 5) [28]. While IL-6 signals via at least 3 pathways in multiple myeloma, namely STAT3, ERK1/2, and AKT [31, 32], we first biologically validated the impact of YM155 on IL-6/STAT3-signalling, finding a concentration-dependent decrease of phospho-STAT3 in both cell lines after 24 hours under normal growth conditions. This correlated closely with inhibition of survivin and Mcl-1 protein expression (Figure 6A). Phosphorylated STAT3 translocates from the cytoplasm to the nucleus. Immunofluorescence staining of STAT3 showed that nuclear STAT3 was depleted upon treatment with YM155 U-266 and INA-6 cells (Suppl. Figure 3).
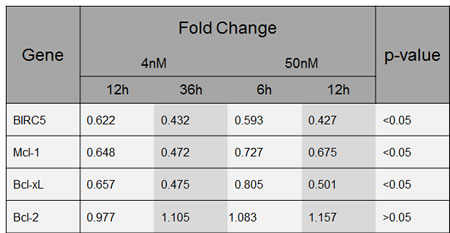
Figure 5:YM155 regulates gene expression of Bcl-2 family members. Fold change of BIRC5, MCL1, Bcl-xL and Bcl-2 gene expression in U-266 cells upon exposure to YM155 at indicated concentrations.
There was also a strong time-dependent correlation between the inhibition of STAT3 activation and the decrease of Mcl-1. Apoptosis induction as assessed by PARP cleavage was observed to have a close temporal correlation with the reduction of STAT3 phosphorylation and Mcl-1 suppression (Figure 6B).
In contrast, we found a transient increase in the phosphorylation of AKT in both U-266 and INA-6 cells upon YM155 treatment, in particular at 50 nM of YM155. Similarly, phosphorylation of ERK was markedly induced in INA-6, while it was less pronounced in U-266 cells, indicating the activation of alternative signaling pathways (Figure 6B).
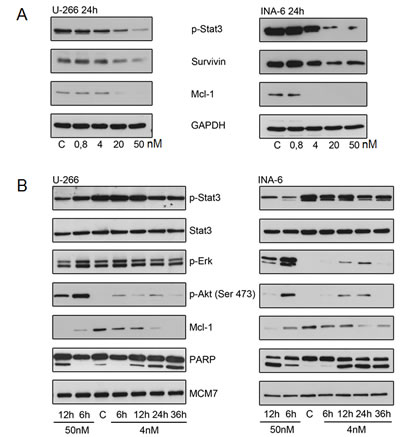
Figure 6: YM155 targets Mcl-1 in MM cells. (A) p-Stat3, survivin and Mcl-1 protein levels after 24h of exposure to indicated concentrations of YM155. (B) Effect of YM155 on IL-6 signaling: p-Stat3, Stat3, p-Erk, p-Akt (Ser 473) and Mcl-1 protein levels assessed by immunoblot. Same lysates were used as in (Fig. 4); PARP and MCM7 blots are identical to (Fig. 4).
YM155 targets Mcl-1 in MM cells
As IL-6 stimulation sensitized OPM-2 cells to treatment with YM155, we examined the phosphorylation of STAT3 and the protein expression of survivin and Mcl-1 in IL-6 stimulated cells exposed to YM155. IL-6 strongly induced STAT3 phosphorylation in OPM-2 cells (Figure 7A). YM155 inhibited this effect at a concentration of 20 nM in accordance to the sensitization to YM155 seen in the previous cell growth assays. Mcl-1 expression has been reported to be regulated by IL-6/STAT3 signaling [33] Here, Mcl-1 was markedly induced upon IL-6 stimulation and this effect was completely abrogated by treatment with 20nM of YM155. The constitutive expression of survivin in OPM-2 cells was not significantly enhanced by IL-6 and was not altered by exposure to YM155 in this concentration range (Figure 7A). This data suggests that down regulation of Mcl-1 rather than that of survivin might be the mechanism of action of YM155 in OPM-2 cells.
Survivin and Mcl-1 are proteins with a high turnover rate and are rapidly eliminated by proteasomal degradation [34, 35]. Activation of the ER stress response leads to a general inhibition of protein translation. We asked whether ER stress alone can lead to changes in survivin and Mcl-1 levels. We observed a rapid decrease of Mcl-1 in U-266 and INA-6 cells upon treatment with thapsigargin (Figure 7B). In contrast, only a moderate decline in survivin protein expression was seen in U-266 cells and no significant change in INA-6. Thapsigargin did not change phosphorylation of STAT3. This indicates that the activation of the ER stress response triggered by YM155 also contributes to the down regulation of Mcl-1.
YM155 induces alterations in two pathways – triggering the ER stress/UPR and selectively abrogating STAT3-dependent transcription factor activity – that seem to ultimately result in a depletion of the anti-apoptotic protein Mcl-1 in MM cells. We next sought to determine whether ectopic Mcl-1 expression is able to attenuate the anti-MM cell activity of YM155. As shown in figure 7C, exogenous Mcl-1 remarkably attenuates the inhibitory effect of YM155 on U-266 cell growth. Exogenous Mcl-1 protein levels were maintained in transfected cells and displayed reduced apoptotic cell death as assessed by PARP cleavage (Figure 7D). In contrast, exogenous expression of survivin had no significant impact on YM155 efficacy in MM cell growth (Figure 7C). This confirms that Mcl-1 depletion has a crucial role in the mechanism of action of YM155 in MM cells.
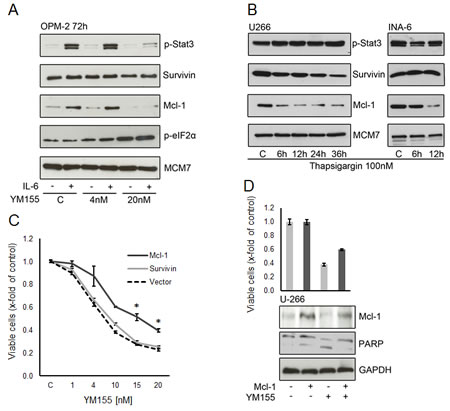
Figure 7: Mcl-1 is a critical target of YM155 in MM cells. (A) Effect of YM155 on OPM-2 cells stimulated with IL-6, treated with YM155 at indicated concentrations or vehicle only and lysates were subject to immunoblot analyses of indicate targets. (B) Mcl-1 protein expression under ER stress induced by thapsigargin. Cells were lysed after 6 and 12 hours (U-266 and INA-6) and after 24 and 36 hours (U-266 only, as INA-6 cells were very sensitive to thapsigargin 100 nM). (C) Effect of ectopic Mcl-1 expression on cell viability. U-266 cells were transfected with a plasmid containing full-length Mcl-1 or with a vector control, respectively. After 24 hours YM155 was added at the indicated concentrations. Viable cells were measured 48h after transfection. The statistical significance of differences observed in Mcl-1 transfected vs. control cells was determined using an unpaired Student t test (*, p > 0.01). (D) Cell viability and corresponding protein levels of Mcl-1. U-266 cells were transfected with control or full-length Mcl-1 plasmid, respectively. YM155 was added at 10nM after 24 hours, cells were harvested 48h after transfection and subject to immunoblot analyses as indicated. PARP cleavage was used to asses onset of apoptosis, GAPDH served as loading control.
MCL1 gene expression and survival
Though ubiquitous, there is a consistently lower level of expression in MM cells and myeloma cell lines when compared to normal bone marrow plasma cells (Suppl. Figure S4A). Contrary to survivin, MCL1 expression does not show an association with proliferation, but with adverse cytogenetic features (gain of 1q21 and presence of t(4;14)) and, at a statistically significant level despite a relatively small absolute difference, with gene expression based risk scores (Suppl. Figure S4B, S4C and S4D).
In 301 patients undergoing high-dose chemotherapy, MCL1 expression was associated with inferior EFS and OS (21.5 vs. 34.3 months, p = 0.005; 58.9 vs. n.r., p = 0.007) using an optimized cutoff defined by maximum logrank statistics. Similar results were obtained using data from 701 patients of the UAMS Arkansas group treated within the total therapy 2 and 3 protocols EFS of 65.1 vs. 79.2 months (p < 0.001) and OS of 86.4 vs. n.r. (p < 0.001) (Suppl. Figure S5A and S5B). This data confirm that elevated MCL1 expression is associated with inferior EFS and OS in MM patients.
YM155 in combination with other compounds
Bortezomib is known to trigger the ER stress response [36]. Low-dose YM155 (2nM and 10nM, respectively) was combined with low-doses of bortezomib (1nM and 2nM) in U-266 and OPM-2 cells. Isobologram analysis of cell growth demonstrated moderate antagonistic effects (CI 1.18 – 1.40) for this combination (Figure 8A). However, bortezomib requires the cleavage product of Mcl-1 to efficiently trigger apoptosis in MM cells [6] while YM155 abrogates Mcl-1 on a transcriptional level. In order to more specifically assess the potency of dual induction of ER stress, we combined YM155 (4nM and 20nM, respectively) with thapsigargin in both cell lines. As expected, moderately synergistic effects (CI 0.63 – 0.77) were seen with this combination (Figure 8A).
Recently, the co-dependency of MM cells on the anti-apoptotic Bcl-2 family members Mcl-1and Bcl-2/Bcl-xL has been reported [37]. Strikingly, co-treatment of OPM-2 cells with YM155 (20nM) and the antagonist of Bcl-2 proteins, ABT-263 (0.4 µM and 0.8 µM), led to a marked synergistic growth inhibition (CI 0.14 – 0.15) (Figure 8A) as well as to a highly synergistic activation of apoptotic signaling assessed by caspase 3/7 activity (Figure 8B and C). In contrast, the combination of these two compounds in U-266 cells was less effective but still synergistic (CI 0.60 – 0.61).
In summary, these results strongly support the early evaluation of YM155 in combination with inhibitors of other Bcl-2 family members in clinical trials.
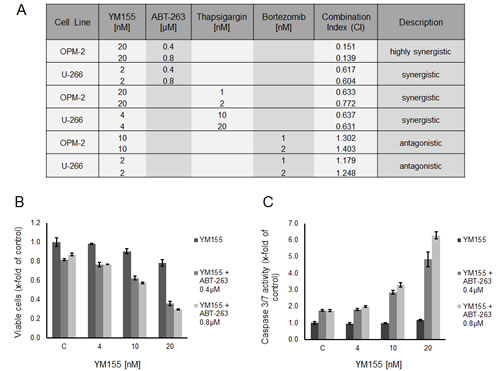
Figure 8: YM155 in combination treatments. (A) Treatment of U-266 and OPM-2 cells with combinations of YM155 and ABT-263, thapsigargin or bortezomib. Viable cells were measured after 48 hours of treatment. Combination indices were calculated as described in material and methods. (B) Synergism of YM155 and ABT-263 on OPM-2 cells. Cells were treated with YM155 alone or in combination with ABT-263 (0.4µM or 0.8 µM, respectively). Viable cells were assessed after 48 hours. (C) Caspase 3/7 activity after 48 hours in OPM-2 cells treated with YM155 alone or in combination with ABT-263.
Discussion
BIRC5/SURVIVIN is one of the most frequently over-expressed genes in all types of cancer [8]. The oncogenic activity of elevated survivin expression occurs through regulation of mitotic spindle assembly and cytokinesis, as well as through exerting anti-apoptotic effects by indirectly inhibiting caspase activation [38, 39]. In MM, BIRC5 expression, detectable in 43% of myeloma cell samples, is associated with proliferation, prognostically adverse chromosomal aberrations (del17p and gain of 1q21), and gene expression based high-risk scores. BIRC5 expression carries adverse prognostic significance regarding event-free and overall survival. It is thus a potential target for personalized and risk adapted treatment.
To explore survivin as a therapeutic target, we evaluated the cytotoxicity and mechanism of action of and some potential combination partners for the novel small molecule suppressant of survivin, YM155, now called sepantronium bromide. YM155 potently inhibits cell growth and induces apoptotic cell death in MM cells in a low nanomolar range, similar to what has been shown preclinically for prostate, colon, lung and a wide variety of other cancer entities [23]. These studies have already led to several phase II clinical trials with data now available on relapsed and refractory prostate cancer and diffuse large B-cell lymphoma [40, 41]. The excellent tolerability of the compound was demonstrated in these trials and single agent anti-tumor activity was reported. However, though there was one complete response in the lymphoma trial, overall relatively few patients responded. Ongoing clinical trials will therefore focus on drug combinations based on pre-clinically proven synergy.
We further aimed to dissect the molecular mechanism of action in MM cells. Although Cheng et al. describe YM155 as highly selective to suppress survivin promoter activity, the concomitant abrogation of Mcl-1 transcription was recently shown by Tang et al. in a panel of cell lines from solid tumors [42, 43]. While YM155 does suppress survivin levels in MM cells at higher concentrations, our data indicate that YM155 additionally triggers the ER stress response and abrogates both IL-6 induced as well as constitutive STAT3 signaling, ultimately converging on the inhibition of Mcl-1 expression, an anti-apoptotic factor that has been proven to be essential for MM cell survival [4, 44]. The potent anti-tumor activity of YM155, at least in MM, therefore seems to be mediated by the activation of the unfolded protein response and simultaneous abrogation of IL-6/STAT3 signaling. Mcl-1, rather than survivin, appears to play the major role in YM155-mediated growth inhibition and cell death in MM cells. This is further underlined by the reversal of YM-155 inhibition by forced expression of Mcl-1, whereas forced expression of survivin had no effect. In addition, knock-down of BIRC5/survivin by siRNA did not significantly inhibit growth and survival of MM cells (data not shown).
Based on these results, we re-evaluated the expression, association with molecular entities and prognostic significance of Mcl-1 in multiple myeloma. In contrast to survivin, MCL1 expression does not show an association with proliferation, but with adverse cytogenetic features. In line with these findings, high MCL1 expression is associated with adverse survival, confirming and extending data previously reported [45].
How does YM155 suppress expression of survivin and Mcl-1? Firstly, both Mcl-1 and survivin expression have been found to be regulated by STAT3 [33, 46, 47]. Specifically in MM, STAT3 dependent expression of Mcl-1 has been reported in INA-6 cells [48]. In addition, Mcl-1 and survivin, both proteins with short half-lives, are conceivably suppressed by the general inhibition of protein translation triggered by the unfolded protein response, as our data demonstrate that induction of ER stress alone leads to reduced protein levels of Mcl-1 and survivin. Furthermore, YM155 appears to disrupt the formation of a critical complex of transcription factors at the survivin core promoter by modulating subcellular localizations of Sp1 and ILF3/p54(nrb) [42, 49]. Interestingly, both Mcl-1 and survivin share transcriptional regulation by SP1, providing an additional molecular basis for the dual suppression of these critical pro-survival factors in cancer cells [50].
As the early first clinical trial data suggest that the single agent activity of YM155 is modest, appropriate combination partners based on preclinical synergy are likely to be required for the successful clinical application of this compound. Recently, a complex interaction pattern between anti-apoptotic Bcl-2 family members in MM cells has been reported [37]. This work describes a co-dependency of MM cells on Mcl-1 and Bcl-2/Bcl-xL. In line with these recent reports, our findings confirm a strikingly synergistic efficacy of the combination of YM155 with the antagonist of Bcl-2 proteins ABT-263, an orally active derivative of ABT-737 that selectively binds Bcl-2, Bcl-xL and Bcl-w [51, 52]. We therefore propose to further evaluate YM155 in combination with inhibitors of the Bcl-2 family in future clinical trials.
Taken together, these results provide the theoretical framework for clinical trials targeting survivin and Mcl-1 in patients with MM.
Materials and methods
Materials
YM155 and ABT-263 were purchased from Selleck Chemicals (Houston, TX). Thapsigargin, Hydroxychoroquin, IL-6, and IGF-I were obtained from Sigma Aldrich (St Louis, MO). Primary antibodies were obtained from Cell Signaling Technology (Danvers, MA): Survivin, p-STAT3, STAT3, p-ERK, p-AKT (Ser 473), PARP, p-eIF2α, CHOP and from Santa Cruz Biotechnology (Santa Cruz, CA): Actin, Mcl-1, GAPDH, MCM7, ATF-3.
Cell Culture
Human MM cell lines were purchased from the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany) and cultured as recommended. INA-6 cells, kindly provided by Dr. Renate Burger, University Hospital Schleswig Holstein Kiel (Kiel, Germany), were grown in RPMI-1640, 10% FCS (GIBCO/Invitrogen, Karlsruhe, Germany), with 2.5 ng/ml IL-6. The human MM cell line HG-1 was generated in the Multiple Myeloma Research Laboratory Heidelberg (unpublished data). All cell lines were periodically tested for contamination with mycoplasma by PCR and authenticated by DNA fingerprinting within our cell banking system according to the regulations of the German Cancer Research Center (DKFZ).
Primary cells, human myeloma cell lines, Gene Expression Profiling, iFISH
Patients with previously untreated MM (n=370), individuals with monoclonal gammopathy of unknown significance (MGUS, n=22), and 10 healthy donors presenting at the University Hospitals of Heidelberg and Montpellier were included in the study after written informed consent was obtained in accordance with the Declaration of Helsinki, which was approved by the institutional review boards of the University Hospital of Montpellier, France and of the Medical Faculty of the Ruprecht-Karls-University Heidelberg (#229/2003 and S-152/2010), Germany. A total of 301 patients underwent front line high-dose conditioning chemotherapy with 200 mg/m² melphalan and autologous stem cell transplantation. Purification of normal bone marrow plasma cells, MM cells, plasmablasts and bone marrow stromal cells as well as gene expression profiling were performed as previously described [2, 53-55]. Interphase fluorescence in situ hybridization analyses were done on CD138-purified plasma cells as published [56]. CD27+ memory B cells (MBCs, n=11) were purified from buffy coats purchased from the French Blood Center (Toulouse, France) and plasmablasts (PPC, n=10) generated using an in vitro model described previously [57]. Thirty two human myeloma cell lines were fully characterized and microarray profiled as described [58]. Microarray data on cell lines are available in the ArrayExpress public database under accession numbers E-TABM-937 and E-TABM-1088.
Statistical analysis
To assess for the presence or absence of gene expression, the “Presence-Absence calls with Negative Probesets” algorithm was used [59]. Differences in clinical parameters and cytogenetic analyses and between defined groups were investigated by exact Wilcoxon rank-sum test. Correlation was assessed using Pearson’s correlation or Kendall’s tau (for categorical variables) tests. The relationship between categorical variables was assessed using Fisher’s exact test. Differential gene expression was assessed using empirical Bayes statistics in linear models for microarray data [60]. P-values were adjusted for multiple testing controlling the false discovery rate as defined by Benjamini and Hochberg [61]. All computations were performed using R 2.14.1 (http://www.r-project.org/), and Bioconductor 2.9 [62]. EFS and OS were investigated using Cox’s proportional hazard model as previously published [53]. For EFS and OS, cut-offs were calculated as the mean cut-off from EFS and OS, respectively. Gene expression-based assessment of risk and proliferation as well as classifications of myeloma was performed as previously published [2]. Findings were confirmed using the same strategy on the independent group of 701 patients treated within the total therapy 2 and 3 protocols, respectively [63, 64]. Microarray data are available in the public databases under accession numbers E-TABM-1138 and GSE24080. An effect was considered as statistically significant if the P-value of its corresponding statistical test was not higher than 5 %.
Transfections
U-266 cell were transfected with the pCMV survivin, the pCMV Mcl-1 plasmid [65] or a control plasmid (pCMV-Tag4A, Stratagene, La Jolla, CA) using the Neon Transfection System (Invitrogen, Carlsbad, CA).
Cell-based Assays
Cell growth and Caspase 3/7 assays were conducted in 96-well plates according to the manufacturer’s instructions using CellTiter-Glo™ and Caspase-Glo 3/7™ assays from Promega (Madison, WI). Cell proliferation was assessed by measuring [3H]-thymidine uptake, as described in prior studies [36].
Isobologram analysis
For combination studies, data from CellTiter Glo™ assays were converted into values representing the fraction of growth affected (FA) in drug-treated versus untreated cells and analyzed using the CalcuSyn software program (Biosoft, Ferguson, MO) based on the Chou-Talalay method. A combination index (CI) < 0.9 indicates synergism, whereas 0.9 to 1.1 indicates additive effects.
SDS-PAGE and Western Immunoblot
Cell lysis, immunoprecipitation, and Western blot analysis were performed as described previously [36]. Briefly, after washing with phosphate-buffered saline, cells were pelleted and resuspended in lysis buffer (Tris/HCl pH 8.0 50 nM; NaCl 150 mM; Nonidet P40 1% (v/v); EDTA 1mM; sodium deoxycholate 0.25% (w/v)) containing protease and phosphatase inhibitors (Complete Protease Inhibitor Cocktail Tablets and PhoSTOP Phosphatase Inhibitor Cocktail Tablets, Roche, Basel, Switzerland). Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and afterwards blotted onto nitrocellulose membranes (GE Health Care, Buckinghamshire, UK). All membranes were blocked with 5% milk in TBST (Tris/HCl pH 7.4; NaCl 137 mM; KCl 2.7 mM; Tween 0.1% (v/v)) for one hour before incubation with the respective primary antibodies. The horseradish peroxidase conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and the ECL-Kit from Thermo Fisher Scientific (Thermo Fisher Scientific, Bonn, Germany).
Gene expression analysis
U-266 cells were treated with YM155 at concentrations of 4 nM and 50 nM for 6 and 12 hours as well as 36 hours (only 4nM). Control cells were cultivated with solvent only (DMSO, Merck, Darmstadt, Germany). Total RNA isolation using the RNeasy Kit (Qiagen, Hilden, Germany) was repeated independently and both biological replicates were used for microarray analysis using the Illumina chip HumanHT-12 v4 (Illumina, San Diego, CA). Data were analyzed using Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA). Only genes with a significant fold change (Benjamini-Hochberg adjusted p-value < 0.05) were included in the analysis.
Indirect immunofluorescence staining
U-266 and INA-6 cells were seeded on cover slips precoated with poly-l-lysin. Indirect immunofluorescence staining was carried out as previously described [66]. Fluorescence images were captured and processed using a Zeiss Axiovert 200 M microscope and Axiovison Software (Zeiss, Göttingen, Germany).
Acknowledgements
This work was supported in part by the Max-Eder-Program of Deutsche Krebshilfe (M.S.R), the Dietmar-Hopp Foundation (D.H., A.S., M.S.R., H.G.), the German Ministry of Education and Science (CAMPSIMM) (D.H., A.S., H.G.), the Deutsche Forschungsgemeinschaft (SFB/Transregio TRR79 (D.H., A.S., H.G.), the ARC (SL220110603450, Paris, France) and the ANR (2021-109/087437) (B.K.), and the European Community (FP7-OverMyR) (D.H., B.K.).
Contribution
V.W., D.H., and M.S.R. designed the research, performed the experiments, analyzed the data, and wrote the manuscript; A.S., L.W., I.B., A.K., and B.K. analyzed data and revised the manuscript; T.M. and T.R. performed the statistical analysis; K.P. contributed pivotal reagents; A.D.H. and H.G. reviewed the manuscript.
Conflict of interests
The others declare no conflict of interest.
References
1. Kumar SK, Rajkumar SV, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Zeldenrust SR, Dingli D, Russell SJ, Lust JA, Greipp PR, Kyle RA and Gertz MA. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008; 111(5):2516-2520.
2. Hose D, Reme T, Hielscher T, Moreaux J, Messner T, Seckinger A, Benner A, Shaughnessy JD, Jr., Barlogie B, Zhou Y, Hillengass J, Bertsch U, Neben K, et al. Proliferation is a central independent prognostic factor and target for personalized and risk-adapted treatment in multiple myeloma. Haematologica. 2011; 96(1):87-95.
3. Greipp PR, Katzmann JA, O’Fallon WM and Kyle RA. Value of beta 2-microglobulin level and plasma cell labeling indices as prognostic factors in patients with newly diagnosed myeloma. Blood. 1988; 72(1):219-223.
4. Zhang B, Gojo I and Fenton RG. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood. 2002; 99(6):1885-1893.
5. Le Gouill S, Podar K, Harousseau JL and Anderson KC. Mcl-1 regulation and its role in multiple myeloma. Cell cycle. 2004; 3(10):1259-1262.
6. Podar K, Gouill SL, Zhang J, Opferman JT, Zorn E, Tai YT, Hideshima T, Amiot M, Chauhan D, Harousseau JL and Anderson KC. A pivotal role for Mcl-1 in Bortezomib-induced apoptosis. Oncogene. 2008; 27(6):721-731.
7. Jourdan M, De Vos J, Mechti N and Klein B. Regulation of Bcl-2-family proteins in myeloma cells by three myeloma survival factors: interleukin-6, interferon-alpha and insulin-like growth factor 1. Cell death and differentiation. 2000; 7(12):1244-1252.
8. Velculescu VE. Essay: Amersham Pharmacia Biotech & Science prize. Tantalizing transcriptomes--SAGE and its use in global gene expression analysis. Science. 1999; 286(5444):1491-1492.
9. Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nature reviews Cancer. 2008; 8(1):61-70.
10. Sarela AI, Macadam RC, Farmery SM, Markham AF and Guillou PJ. Expression of the antiapoptosis gene, survivin, predicts death from recurrent colorectal carcinoma. Gut. 2000; 46(5):645-650.
11. Monzo M, Rosell R, Felip E, Astudillo J, Sanchez JJ, Maestre J, Martin C, Font A, Barnadas A and Abad A. A novel anti-apoptosis gene: Re-expression of survivin messenger RNA as a prognosis marker in non-small-cell lung cancers. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1999; 17(7):2100-2104.
12. Shariat SF, Lotan Y, Saboorian H, Khoddami SM, Roehrborn CG, Slawin KM and Ashfaq R. Survivin expression is associated with features of biologically aggressive prostate carcinoma. Cancer. 2004; 100(4):751-757.
13. Tanaka K, Iwamoto S, Gon G, Nohara T, Iwamoto M and Tanigawa N. Expression of survivin and its relationship to loss of apoptosis in breast carcinomas. Clinical cancer research : an official journal of the American Association for Cancer Research. 2000; 6(1):127-134.
14. Jourdan M, Reme T, Goldschmidt H, Fiol G, Pantesco V, De Vos J, Rossi JF, Hose D and Klein B. Gene expression of anti- and pro-apoptotic proteins in malignant and normal plasma cells. British journal of haematology. 2009; 145(1):45-58.
15. Dohi T, Okada K, Xia F, Wilford CE, Samuel T, Welsh K, Marusawa H, Zou H, Armstrong R, Matsuzawa S, Salvesen GS, Reed JC and Altieri DC. An IAP-IAP complex inhibits apoptosis. The Journal of biological chemistry. 2004; 279(33):34087-34090.
16. Tulu US, Fagerstrom C, Ferenz NP and Wadsworth P. Molecular requirements for kinetochore-associated microtubule formation in mammalian cells. Current biology : CB. 2006; 16(5):536-541.
17. Sampath SC, Ohi R, Leismann O, Salic A, Pozniakovski A and Funabiki H. The chromosomal passenger complex is required for chromatin-induced microtubule stabilization and spindle assembly. Cell. 2004; 118(2):187-202.
18. Hideshima T, Catley L, Raje N, Chauhan D, Podar K, Mitsiades C, Tai YT, Vallet S, Kiziltepe T, Ocio E, Ikeda H, Okawa Y, Hideshima H, et al. Inhibition of Akt induces significant downregulation of survivin and cytotoxicity in human multiple myeloma cells. British journal of haematology. 2007; 138(6):783-791.
19. Kardosh A, Soriano N, Liu YT, Uddin J, Petasis NA, Hofman FM, Chen TC and Schonthal AH. Multitarget inhibition of drug-resistant multiple myeloma cell lines by dimethyl-celecoxib (DMC), a non-COX-2 inhibitory analog of celecoxib. Blood. 2005; 106(13):4330-4338.
20. Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Richardson PG, Hideshima T, Munshi N, Treon SP and Anderson KC. Biologic sequelae of nuclear factor-kappaB blockade in multiple myeloma: therapeutic applications. Blood. 2002; 99(11):4079-4086.
21. Romagnoli M, Trichet V, David C, Clement M, Moreau P, Bataille R and Barille-Nion S. Significant impact of survivin on myeloma cell growth. Leukemia. 2007; 21(5):1070-1078.
22. Nakahara T, Kita A, Yamanaka K, Mori M, Amino N, Takeuchi M, Tominaga F, Hatakeyama S, Kinoyama I, Matsuhisa A, Kudoh M and Sasamata M. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer research. 2007; 67(17):8014-8021.
23. Nakahara T, Kita A, Yamanaka K, Mori M, Amino N, Takeuchi M, Tominaga F, Kinoyama I, Matsuhisa A, Kudou M and Sasamata M. Broad spectrum and potent antitumor activities of YM155, a novel small-molecule survivin suppressant, in a wide variety of human cancer cell lines and xenograft models. Cancer science. 2011; 102(3):614-621.
24. Shaughnessy JD, Jr., Zhan F, Burington BE, Huang Y, Colla S, Hanamura I, Stewart JP, Kordsmeier B, Randolph C, Williams DR, Xiao Y, Xu H, Epstein J, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007; 109(6):2276-2284.
25. Decaux O, Lode L, Magrangeas F, Charbonnel C, Gouraud W, Jezequel P, Attal M, Harousseau JL, Moreau P, Bataille R, Campion L, Avet-Loiseau H, Minvielle S, et al. Prediction of survival in multiple myeloma based on gene expression profiles reveals cell cycle and chromosomal instability signatures in high-risk patients and hyperdiploid signatures in low-risk patients: a study of the Intergroupe Francophone du Myelome. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008; 26(29):4798-4805.
26. Barlogie B, Hall R, Zander A, Dicke K and Alexanian R. High-dose melphalan with autologous bone marrow transplantation for multiple myeloma. Blood. 1986; 67(5):1298-1301.
27. Schwab G, Siegall CB, Aarden LA, Neckers LM and Nordan RP. Characterization of an interleukin-6-mediated autocrine growth loop in the human multiple myeloma cell line, U266. Blood. 1991; 77(3):587-593.
28. Alvarez JV and Frank DA. Genome-wide analysis of STAT target genes: elucidating the mechanism of STAT-mediated oncogenesis. Cancer biology & therapy. 2004; 3(11):1045-1050.
29. Meister S, Schubert U, Neubert K, Herrmann K, Burger R, Gramatzki M, Hahn S, Schreiber S, Wilhelm S, Herrmann M, Jack HM and Voll RE. Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer research. 2007; 67(4):1783-1792.
30. Raab MS, Breitkreutz I, Tonon G, Zhang J, Hayden PJ, Nguyen T, Fruehauf JH, Lin BK, Chauhan D, Hideshima T, Munshi NC, Anderson KC and Podar K. Targeting PKC: a novel role for beta-catenin in ER stress and apoptotic signaling. Blood. 2009; 113(7):1513-1521.
31. Klein B, Tarte K, Jourdan M, Mathouk K, Moreaux J, Jourdan E, Legouffe E, De Vos J and Rossi JF. Survival and proliferation factors of normal and malignant plasma cells. International journal of hematology. 2003; 78(2):106-113.
32. Podar K, Hideshima T, Chauhan D and Anderson KC. Targeting signalling pathways for the treatment of multiple myeloma. Expert opinion on therapeutic targets. 2005; 9(2):359-381.
33. Puthier D, Bataille R and Amiot M. IL-6 up-regulates mcl-1 in human myeloma cells through JAK / STAT rather than ras / MAP kinase pathway. European journal of immunology. 1999; 29(12):3945-3950.
34. Zhao J, Tenev T, Martins LM, Downward J and Lemoine NR. The ubiquitin-proteasome pathway regulates survivin degradation in a cell cycle-dependent manner. Journal of cell science. 2000; 113 Pt 23:4363-4371.
35. Yang-Yen HF. Mcl-1: a highly regulated cell death and survival controller. Journal of biomedical science. 2006; 13(2):201-204.
36. Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr., Lee KP and Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006; 107(12):4907-4916.
37. Morales AA, Kurtoglu M, Matulis SM, Liu J, Siefker D, Gutman DM, Kaufman JL, Lee KP, Lonial S and Boise LH. Distribution of Bim determines Mcl-1 dependence or codependence with Bcl-xL/Bcl-2 in Mcl-1-expressing myeloma cells. Blood. 2011; 118(5):1329-1339.
38. Lens SM, Vader G and Medema RH. The case for Survivin as mitotic regulator. Current opinion in cell biology. 2006; 18(6):616-622.
39. Altieri DC. Survivin and IAP proteins in cell-death mechanisms. The Biochemical journal. 2010; 430(2):199-205.
40. Tolcher AW, Quinn DI, Ferrari A, Ahmann F, Giaccone G, Drake T, Keating A and de Bono JS. A phase II study of YM155, a novel small-molecule suppressor of survivin, in castration-resistant taxane-pretreated prostate cancer. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2012; 23(4):968-973.
41. Cheson BD, Bartlett NL, Vose JM, Lopez-Hernandez A, Seiz AL, Keating AT, Shamsili S and Papadopoulos KP. A phase II study of the survivin suppressant YM155 in patients with refractory diffuse large B-cell lymphoma. Cancer. 2012; 118(12):3128-3134.
42. Cheng Q, Ling X, Haller A, Nakahara T, Yamanaka K, Kita A, Koutoku H, Takeuchi M, Brattain MG and Li F. Suppression of survivin promoter activity by YM155 involves disruption of Sp1-DNA interaction in the survivin core promoter. International journal of biochemistry and molecular biology. 2012; 3(2):179-197.
43. Tang H, Shao H, Yu C and Hou J. Mcl-1 downregulation by YM155 contributes to its synergistic anti-tumor activities with ABT-263. Biochemical pharmacology. 2011; 82(9):1066-1072.
44. Derenne S, Monia B, Dean NM, Taylor JK, Rapp MJ, Harousseau JL, Bataille R and Amiot M. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood. 2002; 100(1):194-199.
45. Wuilleme-Toumi S, Robillard N, Gomez P, Moreau P, Le Gouill S, Avet-Loiseau H, Harousseau JL, Amiot M and Bataille R. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia. 2005; 19(7):1248-1252.
46. Aoki Y, Feldman GM and Tosato G. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood. 2003; 101(4):1535-1542.
47. Epling-Burnette PK, Liu JH, Catlett-Falcone R, Turkson J, Oshiro M, Kothapalli R, Li Y, Wang JM, Yang-Yen HF, Karras J, Jove R and Loughran TP, Jr. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. The Journal of clinical investigation. 2001; 107(3):351-362.
48. Brocke-Heidrich K, Kretzschmar AK, Pfeifer G, Henze C, Loffler D, Koczan D, Thiesen HJ, Burger R, Gramatzki M and Horn F. Interleukin-6-dependent gene expression profiles in multiple myeloma INA-6 cells reveal a Bcl-2 family-independent survival pathway closely associated with Stat3 activation. Blood. 2004; 103(1):242-251.
49. Yamauchi T, Nakamura N, Hiramoto M, Yuri M, Yokota H, Naitou M, Takeuchi M, Yamanaka K, Kita A, Nakahara T, Kinoyama I, Matsuhisa A, Kaneko N, et al. Sepantronium bromide (YM155) induces disruption of the ILF3/p54(nrb) complex, which is required for survivin expression. Biochemical and biophysical research communications. 2012; 425(4):711-716.
50. Townsend KJ, Zhou P, Qian L, Bieszczad CK, Lowrey CH, Yen A and Craig RW. Regulation of MCL1 through a serum response factor/Elk-1-mediated mechanism links expression of a viability-promoting member of the BCL2 family to the induction of hematopoietic cell differentiation. The Journal of biological chemistry. 1999; 274(3):1801-1813.
51. Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005; 435(7042):677-681.
52. Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, Roberts L, Tahir SK, Xiao Y, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer research. 2008; 68(9):3421-3428.
53. Hose D, Reme T, Meissner T, Moreaux J, Seckinger A, Lewis J, Benes V, Benner A, Hundemer M, Hielscher T, Shaughnessy JD, Jr., Barlogie B, Neben K, et al. Inhibition of aurora kinases for tailored risk-adapted treatment of multiple myeloma. Blood. 2009; 113(18):4331-4340.
54. Seckinger A, Meissner T, Moreaux J, Goldschmidt H, Fuhler GM, Benner A, Hundemer M, Reme T, Shaughnessy JD, Jr., Barlogie B, Bertsch U, Hillengass J, Ho AD, et al. Bone morphogenic protein 6: a member of a novel class of prognostic factors expressed by normal and malignant plasma cells inhibiting proliferation and angiogenesis. Oncogene. 2009; 28(44):3866-3879.
55. Seckinger A, Meissner T, Moreaux J, Depeweg D, Hillengass J, Hose K, Reme T, Rosen-Wolff A, Jauch A, Schnettler R, Ewerbeck V, Goldschmidt H, Klein B, et al. Clinical and prognostic role of annexin A2 in multiple myeloma. Blood. 2012; 120(5):1087-1094.
56. Neben K, Jauch A, Hielscher T, Hillengass J, Lehners N, Seckinger A, Granzow M, Raab MS, Ho AD, Goldschmidt H and Hose D. Progression in smoldering myeloma is independently determined by the chromosomal abnormalities del(17p), t(4;14), gain 1q, hyperdiploidy, and tumor load. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013; 31(34):4325-4332.
57. Jourdan M, Caraux A, De Vos J, Fiol G, Larroque M, Cognot C, Bret C, Duperray C, Hose D and Klein B. An in vitro model of differentiation of memory B cells into plasmablasts and plasma cells including detailed phenotypic and molecular characterization. Blood. 2009; 114(25):5173-5181.
58. Moreaux J, Klein B, Bataille R, Descamps G, Maiga S, Hose D, Goldschmidt H, Jauch A, Reme T, Jourdan M, Amiot M and Pellat-Deceunynck C. A high-risk signature for patients with multiple myeloma established from the molecular classification of human myeloma cell lines. Haematologica. 2011; 96(4):574-582.
59. Warren P, Taylor D, Martini PGV, Jackson J and Bienkowska J. (2007). PANP - a New Method of Gene Detection on Oligonucleotide Expression Arrays. Bioinformatics and Bioengineering, 2007 BIBE 2007 Proceedings of the 7th IEEE International Conference on, pp. 108-115.
60. Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical applications in genetics and molecular biology. 2004; 3:Article3.
61. Hochberg Y and Benjamini Y. More powerful procedures for multiple significance testing. Statistics in medicine. 1990; 9(7):811-818.
62. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome biology. 2004; 5(10):R80.
63. Barlogie B, Tricot G, Rasmussen E, Anaissie E, van Rhee F, Zangari M, Fassas A, Hollmig K, Pineda-Roman M, Shaughnessy J, Epstein J and Crowley J. Total therapy 2 without thalidomide in comparison with total therapy 1: role of intensified induction and posttransplantation consolidation therapies. Blood. 2006; 107(7):2633-2638.
64. van Rhee F, Szymonifka J, Anaissie E, Nair B, Waheed S, Alsayed Y, Petty N, Shaughnessy JD, Jr., Hoering A, Crowley J and Barlogie B. Total Therapy 3 for multiple myeloma: prognostic implications of cumulative dosing and premature discontinuation of VTD maintenance components, bortezomib, thalidomide, and dexamethasone, relevant to all phases of therapy. Blood. 2010; 116(8):1220-1227.
65. Reynolds JE, Yang T, Qian L, Jenkinson JD, Zhou P, Eastman A and Craig RW. Mcl-1, a member of the Bcl-2 family, delays apoptosis induced by c-Myc overexpression in Chinese hamster ovary cells. Cancer research. 1994; 54(24):6348-6352.
66. Raab MS, Breitkreutz I, Anderhub S, Ronnest MH, Leber B, Larsen TO, Weiz L, Konotop G, Hayden PJ, Podar K, Fruehauf J, Nissen F, Mier W, et al. GF-15, a novel inhibitor of centrosomal clustering, suppresses tumor cell growth in vitro and in vivo. Cancer research. 2012; 72(20):5374-5385.