
CANCER METABOLISM
Despite diagnostic and therapeutic advances in the last decades, cancer is still one of the deadliest diseases around the world. The lack of a cure for cancer reflects the complexity of its molecular bases, as illustrated by the hallmarks of cancer since 2000 [1], and updated in 2011 [2]. Warburg effect, also called aerobic glycolysis, known since the 1920s [3], is by definition the shift presented by tumor cells from the complete oxidation of glucose to an incomplete oxidation to lactate, even in the presence of oxygen. This observation may be considered one of the hallmarks of cancer and shows one of the alterations related to metabolism presented by cancer cells [1, 4].
The Warburg effect and its consequences
Healthy cells in the presence of oxygen usually convert glucose to pyruvate, then acetyl-CoA, which in turn is completely oxidized in the TCA cycle inside the mitochondria, generating during this process 32 molecules of ATP per molecule of glucose. The consumption of oxygen is taken in a process called oxidative phosphorylation (OxPhos) inside the mitochondria. Under hypoxic conditions, those cells are able to convert glucose to lactate, generating 2 molecules of ATP per molecule of glucose.
Cancer cells present an altered behavior. Even in the presence of oxygen, these cells oxidize glucose to lactate, in a process that is several times less efficient to generate ATP and therefore consumes more glucose. This apparent paradox is explained in part by several other alterations in metabolism that occur besides the Warburg effect.
The main consequence of the Warburg effect is the increased consumption of glucose in cancer cells. This property was initially observed by Warburg’s experiments and it is now the theoretical basis for diagnostic purposes. The administration of [18F] fluoro-2-deoxy-glucose (FDG) in patients and the analysis by PET (Positron Emission Tomography) scan may reveal sites in the body that greatly incorporate glucose and thus may contain tumor cells. This technique, called FDG-PET, has been widely used to search for different types of cancers and is one of the most elegant proofs of the Warburg effect [5].
The increased consumption of glucose and the preference to oxidize it to lactate is actually observed in any proliferating cell [4, 6, 7]. The main reason for that is a detour from an energy production state to a biosynthetic state, where it is more important for the cell to acquire the building blocks of its structure, such as nucleotides, amino acids, lipids and NADPH - an important tool for biosynthesis - than ATP production. Accordingly, part of the glucose consumed through the Warburg effect enters the Pentose Phosphate Pathway (PPP), generating ribose-5-phosphate and NADPH (Figure 1), which are greatly demanded in proliferating cells that need to duplicate their DNA mass and actively synthesize RNA [4, 6, 7].
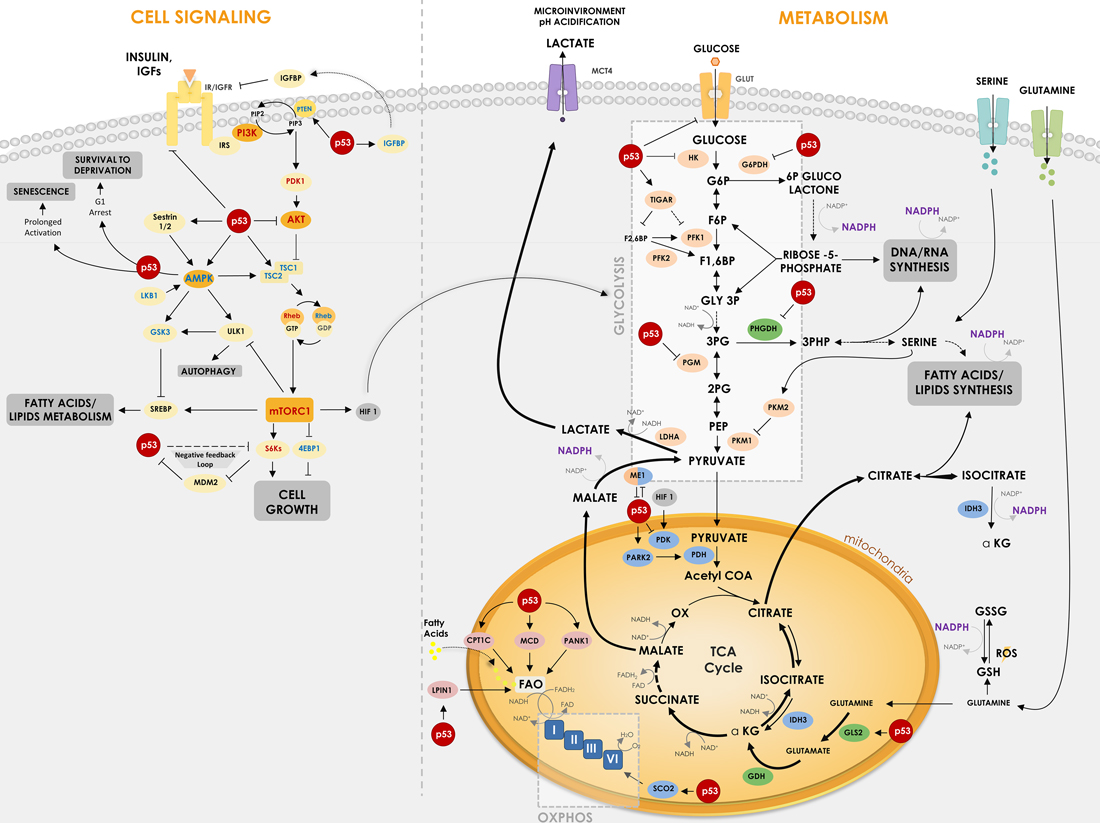
Figure 1: P53 regulates critical points in cell signaling and metabolism. p53 is involved in different critical points of metabolism control and cell signaling, being able to regulate key proteins and enzymes in cellular response to cancer. The Figure shows how p53 suppresses glycolysis and increases OxPhos (oxidative phosphorylation) while it regulates IGF signaling pathway. p53 regulates glucose uptake by some glucose transporters (GLUT1, GLUT3 and GLUT4) and regulates direct and indirectly enzymes in its downstream oxidation to pyruvate – HK, TIGAR, PFK1/2, G6PDH, PGM and ME1. p53 is also important for mitochondrial respiration through regulation of MCD, LPIN1, PANK1, CPT1C and SCO2. Furthermore, p53 regulates enzymes involved in amino acids metabolism such as PHGDH, which participates in serine metabolism derived from the 3PG, and GLS2, which catalyzes the hydrolysis of glutamine to glutamate, increasing TCA cycle flux. In cell signaling transduction, p53 reveals a crucial activity in suppressing downstream proteins of the IGF pathway, such as AKT, AMPK and S6Ks, modulating processes such as cell growth, autophagy and lipid metabolism. In cell signaling, proteins with positive relation with cancer are shown in red and negative relation in blue. Regular arrows represent activation and blunt arrows inhibition. Thicker arrows indicate increased reactions in cancer when compared to thin arrows.
The second most evident consequence of Warburg effect is the increased levels of lactate secretion. The secretion of lactate is important to maintain pH homeostasis inside the cell, but it also plays roles for cancer progression outside the cell. It has been demonstrated that extracellular environment acidosis precedes angiogenesis, and thus lactate may stimulate angiogenesis in a hypoxia-inducible factor 1 (HIF-1) independent manner [8]. Besides, acidosis of extracellular matrix may increase local invasion of the cancer cells [9] and inhibit infiltration of T lymphocytes [10], thus presenting additional advantages for tumor progression.
A third consequence of the Warburg effect is the reduced usage of the respiratory chain in the mitochondria, due to reduced levels of OxPhos and oxygen consumption. Accordingly, less reactive oxygen species (ROS) are produced by aerobic glycolysis compared to oxidative phosphorylation, which may lead the cancer cells to escape apoptosis and to enhance proliferation [11, 12].
Beyond Warburg effect
The alterations in metabolism in a cancer cell go beyond the Warburg effect. As it might be expected, the aerobic glycolysis shunt increases the energy demand by the cells, which is supplied by alternative metabolic pathways, like glutamine and fatty acid oxidation through the TCA cycle (Figure 1).
It has long been known that cancer cells and actually any proliferating cell have increased consumption of glutamine [13, 14]. Glutamine can enter the TCA cycle and be oxidized to lactate, which contributes for media acidification and also NAPDH production, through the cytoplasmic malic enzyme 1 (ME1), as seen in Figure 1 [15]. Besides, glutamine metabolism is an important source of glutathione (GSH), which is used by the cell to control oxidative stress [16]. NADPH is used in this case to regenerate (reduce) the oxidized form of GSH - named GSSG -back to GSH, after ROS clearance.
Dividing cells, including cancer cells, have also an increased demand for lipids, since increase in size and number demand phospholipids to form their plasma membrane and their inner membranes of organelles. The increased consumption of glutamine is an important source of carbon to generate acetyl-CoA, precursor for lipid synthesis, using alternative routes of the TCA cycle, as seen in Figure 1 [17]. Besides, lipid biosynthesis has a great demand for NADPH, which is supplied by PPP, ME1 and the cytoplasmic isoform 1 of isocitrate dehydrogenase (IDH1).
Glutamine metabolism through TCA cycle is also an important source of amino acids, which is a great demand in dividing cells since protein synthesis is increased. Alpha-ketoglutarate, oxaloacetate and pyruvate are precursors of several amino acids and thus their flux through TCA cycle is active in cancer cells. Nevertheless, TCA complete cycling is altered in cancer cells (see arrows patterns in Figure 1).
Another enzyme, that has been characterized as altered in tumors, is the pyruvate kinase (PK), the last step of the glycolytic pathway, responsible for phosphoenolpyruvate (PEP) conversion in pyruvate, generating ATP (Figure 1). The PKM2 isoform is a splicing variant of this enzyme that is found in proliferating cells, in embryonic tissues and in several types of cancer cells [18–20]. PKM2 has diminished activity compared to PKM1, thus slowing down the last reaction of the glycolytic pathway, which may look disadvantageous, but indeed is beneficial to cancer cells, since decreasing the speed of the last steps of glycolysis increases the flux of glycolysis intermediates to PPP, thus generating more NADPH and precursors for nucleotide biogenesis like ribose-5-phosphate [20–22]. Besides, accumulation of these intermediates, as 3-phosphoglycerate (3PG), favors serine/glycine de novo synthesis pathway, also important for nucleotide synthesis.
Serine/glycine metabolism is indeed another altered pathway in cancer (Figure 1). Serine and glycine are known precursors of phospholipids, nucleotides and GSH, which are important for cell growth and proliferation and redox control, as already discussed. It is well described that cancer cells have increased consumption of serine [23, 24]. Besides, the first enzyme of the serine pathway, called phosphoglycerate dehydrogenase (PHGDH), that converts 3-phosphoglycerate (3PG) to 3-phosphohydroxypyruvate (3PHP), is overexpressed in several types of tumors [25, 26]. Interestingly, serine, together with fructose-1,6-bisphosphate (F16BP), are allosteric activators of PKM2, meaning that serine starvation inhibits the flux through the glycolytic pathway, accumulating intermediates for de novo serine synthesis [22, 27] (Figure 1).
Proline is another amino acid that seems to have a role in cancer. Proline is produced either by glutamate or by arginine-derived ornithine, where pyrroline-5-carboxylate reductase (PYCR1) is one of the main enzymes of proline biosynthesis. Evidences suggest that PYCR1 is upregulated in several types of tumors [28]. Conversely, proline oxidase or dehydrogenase or p53-induced gene 6 (POX, PRODH, PIG6) expression, which participates in proline degradation, inhibits tumor growth inducing cell cycle arrest [29].
Overall, the alterations related to metabolism in cancer implicate complex relationships between metabolic pathways and regulatory networks, involving several metabolic intermediates and expanding Warburg’s first observations. As it will be discussed, the tumor suppressor p53 plays a pivotal role in regulating the expression and function of several of metabolic genes.
REGULATION OF METABOLISM BY P53
P53, encoded by the gene TP53, was first described as a cellular protein that interacts with an oncogenic viral protein [30–33]. The initial observation indicated that overexpression of p53 induced cell transformation and p53 was therefore considered an oncogene [34–36]. Nonetheless in the next few years other reports indicated an opposite effect of p53 and the cloned wild-type p53 was devoid of oncogenic activity [37]. It soon became clear that mutant forms of p53 were responsible for the early oncogenic observations. Indeed p53 is the most frequently mutated gene in cancer, reaching a prevalence of about 95% in serous ovarian cancer and with a mean frequency of 42% [38]. Gene deletion or truncation are frequently observed in tumor suppressors, like BRCA1 and Rb that show weak or no expression of the proteins, however, the most frequent alteration in p53 gene are missense mutations [39]. Instead of loss of expression, there´s an altered p53 protein with a “gain of function” (GOF) mutation that is tumorigenic and favors cell transformation.
Even though induction of apoptosis and cell cycle arrest are the hallmarks of p53 activity, it’s role in metabolism have been recognized and p53 is acknowledged as a general stress sensor, not only important to inhibit cancer progression, but also to respond during viral infection, starvation or oxidative stress, reducing cell proliferation, altering cellular metabolism and inhibiting survival [40]. As a general stress sensor, the ability to circumvent p53 is essential for the progression of DNA mutation, metabolism alteration, oxygen deprivation and viral infection.
Wild-type p53 is upregulated after DNA damage and induces cell cycle arrest and apoptosis and is considered the “guardian of the genome” [41]. p53 can directly bind to proteins involved in apoptotic signaling, however, its main action mechanism is through transcriptional regulation of genes involved in cell cycle, induction of apoptosis, angiogenesis, autophagy, immunomodulation and several other processes that can hamper cancer progression. There are in the human genome more than 4,000 p53 binding sites that regulate the expression of more than 500 genes [42, 43]. From these at least 75 are involved with biosynthesis and metabolism [44]. Figure 1 shows how p53 induces/inhibits key metabolic genes involved in glycolytic pathway, OxPhos, lipid and amino acids metabolism and cell growth. Figure 2 summarizes the p53 key target proteins involved in TCA cycle, ROS control, amino acids, fatty acids and glucose metabolism.
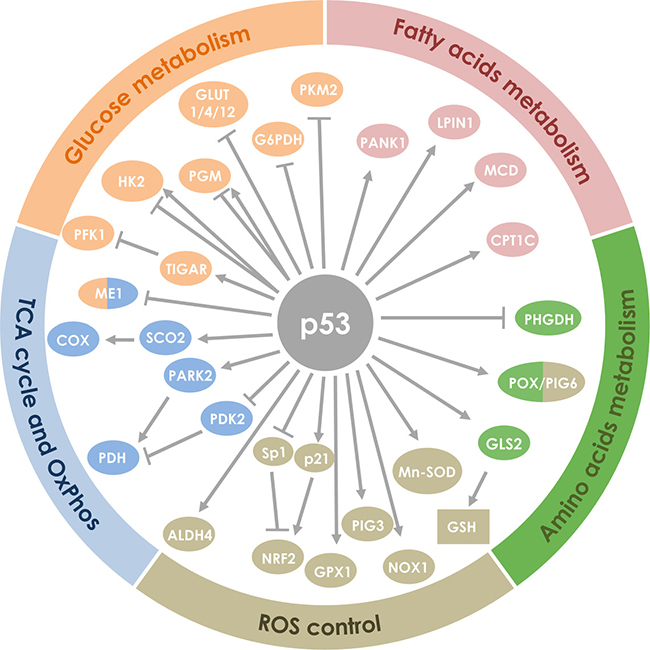
Figure 2: Regulation of proteins involved in cell metabolism by p53. Proteins (indicated by circles) and other molecules (indicated by squares) were grouped according to their involvement in the metabolism of glucose, amino acids and fatty acids, ROS regulation, TCA cycle and the oxidative phosphorylation. Regular arrows represent activation and blunt arrows inhibition.
p53 and glucose metabolism
TP53-induced glycolysis and apoptosis regulator (TIGAR) is one of the best-known proteins regulated by p53 that mediates its action in metabolism. Expression of TIGAR is stimulated early by low levels of p53 and plays a role in ROS inhibition and cell survival allowing time for DNA repair mechanisms. However, after prolonged stress, its expression is reduced and pro-apoptotic genes are induced instead. TIGAR induction leads to inhibition of the glycolysis enzyme Phosphofructokinase (PFK-1) and redirection of glucose to PPP, thus increasing NADPH and other intermediates, which are essential DNA precursors used during DNA repair. TIGAR also degrades fructose-2,6-bisphosphate (F26BP), a strong allosteric activator of PFK-1 [45] (Figure 1). However, this redirection to PPP may be abrogated by p53 direct inactivation of glucose 6-phosphate dehydrogenase (G6PDH); mutant p53 lacks this ability, thus increasing PPP flux and glucose consumption (Figure 1) [46].
A study has demonstrated that the loss of both p53 and PTEN in prostate cancer cells is responsible for an increase in expression of HK2, contributing for the aerobic glycolysis [47]. Phosphoglycerate mutase 1 (PGAM1 or PGM1), the enzyme that converts 3-phosphoglycerate (3PG) to 2-phosphoglycerate (2PG), is also downregulated by p53 (Figure 1) [48]. It is known that p53 loss increases PGAM1 expression and activity, hence increasing glycolysis and biosynthesis for tumor growth [49]. In the muscle, however, p53 seems to be a transcriptional activator of a tissue specific PGAM (PGMA2) [50]. HK2 and PGAM1 are examples of glycolytic enzymes downregulated by p53.
ME1, which converts malate to pyruvate and vice versa, an anaplerotic reaction linking glycolysis to the TCA cycle, has also a relationship with p53. A study has shown that ME1 and ME2 can regulate p53, where their downregulation induce p53 activation and p53 associated senescence but not apoptosis; alternatively they can be regulated by p53, where p53 represses their expression, regulating NADPH production and glutamine metabolism (Figure 1) [51].
Finally, it is known that glucose receptors like GLUT1 and GLUT4 may be downregulated by wild-type p53, while mutated forms of p53 induce cancer cells to increase expression of those receptors and consequently to increase the glucose consumption associated with Warburg effect [52]. GLUT3 has also been found to become indirectly down-regulated by p53 through inhibition of IKK [53] and, more recently, it has been shown that p53 is also able to bind and to repress the GLUT12 gene promoter [54]. Overall wild-type p53 acts to reduce glucose intake and aerobic glycolysis (Figure 1).
p53, TCA and the oxidative phosphorylation
p53 is also able to inhibit pyruvate dehydrogenase kinase 2 (PDK2), a negative regulator of pyruvate dehydrogenase (PDH), thus increasing activity of the later and the flux through TCA cycle (Figure 1) [55]. The increased conversion of pyruvate to acetyl-CoA favors the oxidative phosphorylation and diminishes conversion of pyruvate to lactate, therefore loss of p53 and consequent loss of PDK2 inhibition contributes to the Warburg effect [55]. Conversely, p53 is able to increase expression of Parkin (PARK2), a Parkinson disease associated gene that is able to activate PDH (Figure 1). It has been demonstrated that PARK2 deficiency due to p53 loss increases glycolysis and reduces oxidative phosphorylation, thus contributing to the Warburg effect [56].
Several studies have shown the regulation of oxidative phosphorylation by p53 through synthesis of cytochrome C oxidase 2 protein (SCO2), which induces the cytochrome C oxidase complex (COX) synthesis (Figure 1). COX catalyzes the transfer of electrons to oxygen molecules, in the complex IV of the electron transport chain, a crucial step for OxPhos completion. It has been demonstrated that, in normal cells, wild-type p53 induces SCO2 expression, ensuring increased levels of OxPhos [57]. However, in several types of cancer cells, loss of p53 decreases SCO2 expression and the synthesis of a complete electron transport chain, impairing OxPhos and contributing for the shift to the incomplete oxidation of glucose to lactate [57, 58]. Conversely, SCO2 re-expression in p53-deficient cancer cells is able to rescue oxidative phosphorylation and oxygen consumption levels [58].
p53 and amino acids metabolism
As already discussed, glutamine is a very important source of carbon, NADPH and glutathione for cancer and any proliferating cells, contributing to redox control. Glutaminase (GLS) is the first enzyme that participates in glutamine metabolism, converting it to glutamate. Glutamate is then converted to alpha-ketoglutarate by glutamate dehydrogenase (GDH). p53 is known to upregulate GLS2, a liver-specific isoform of glutaminase, increasing alpha-ketoglutarate flux through TCA cycle, mitochondria respiration and also redox control by increasing GSH levels (Figure 1) [59, 60]. Therefore, p53 loss in the liver may create an unbalance in the redox homeostasis and increase DNA damage, contributing to cancer progression [60]. Since GLS2 upregulation by p53 increases OxPhos due to increase of carbon flux trough TCA cycle and NADH and FADH2 production, the loss of p53 may also decrease OxPhos through GLS2, contributing to the Warburg effect [61].
Phosphoglycerate dehydrogenase (PHGDH), which participates in the serine metabolism derived from the glycolysis intermediate 3-phosphoglycerate (3PG), is also a target of p53, with p53 regulatory elements in its promoter (Figure 1) [62]. It has been recently demonstrated that p53 suppresses PHGDH expression, therefore inhibiting serine biosynthesis, while serine starvation enhances p53-mediated cell death in melanomas, an approach that may have therapeutic applications [63]. Cancer cells have the ability to inhibit glycolysis rapidly under serine starvation, activating the de novo serine synthesis. However, cancer cells lacking p53 have increased sensitivity to serine starvation, triggering oxidative stress and inhibiting proliferation [23].
It has also been demonstrated that p53 is able to upregulate, in response to genotoxic damage, the expression of POX, the first enzyme of proline catabolism, regulating the balance between proline and glutamate and their derivate alpha-ketoglutarate [64]. It has been proposed that proline becomes available to cells as a stress substrate of collagen degradation, and might be a signaling molecule for p53 pathway [65].
p53 and lipids metabolism
Several studies have demonstrated the role of p53 to promote fatty acid oxidation, contributing for cell survival under starvation of nutrients (Figure 1). As examples, p53 is able to activate: carnitine palmitoyltransferase 1C (CPT1C), responsible for fatty acids transportation for oxidation [66]; malonyl CoA decarboxylase (MCD), which converts malonyl-CoA to acetyl-CoA [67]; lipin 1 (LPIN1), which acts as a nuclear transcription coactivator regulating the expression of genes related to lipid metabolism [68]; and pantothenate kinase 1 (PANK1), which participates in CoA synthesis, important for β-oxidation [69, 70]. Accordingly, wild-type p53 increases fatty acid oxidation and oxidative phosphorylation, inhibiting Warburg effect.
p53 and ROS regulation
Tumor cells have higher levels of ROS compared to normal cells and this may be useful to predict overall survival [71–75]. However, ROS induction has to be tightly controlled, because intermediate levels of oxidants can induce DNA damage, leading to mutation and promoting tumorigenesis. On the other hand, elevated levels of oxidants contribute to extensive DNA damage, mitochondrial membrane permeabilization, activation of apoptotic signaling and induction of cell death [76–78]. Cell metabolism is essential to control oxidants levels. In cancer cells, ROS modulate key metabolic enzymes, like pyruvate kinase M2 (PKM2) mentioned above [79, 80], inhibiting it through oxidation of Cys358, causing an increase in the availability of G6P and redirecting it to pentose phosphate pathway. This leads to formation of macromolecules and NADPH, which is required to generate GSH for ROS detoxification, controlling ROS levels in a cyclic mechanism [81].
There is an interplay between ROS and p53. The DNA damage induced by ROS can activate p53, which can both inhibit and promote oxidant production [82]. In tumor cells it was shown that low levels of p53 up-regulate several anti-oxidant genes, while down-regulation of p53 increases intracellular ROS and genome instability [83]. In normal conditions p53 protects cells from oxidative stress directly by inducing anti-oxidant genes, like glutathione peroxidase (GPX1), aldehyde dehydrogenase 4 (ALDH4) and Mn superoxide dismutase (Mn-SOD) [84–86] or indirectly by inducing TIGAR and GLS2. By favoring PPP, TIGAR also increases NADPH and GSH production and ROS scavenging [45]. GLS2 regulates antioxidant defense function in cells by increasing reduced glutathione (GSH) levels and decreasing ROS levels [60].
At the same time, p53 has been shown to activate the expression of genes that induce ROS and cell death. Among these a group of distinct genes were named p53 Induced Genes (PIGs) [87]. One of these genes, PIG3, is closely related to an NADPH-quinone oxidoreductase [87], it induces ROS [88] and decreases mitochondrial membrane potential [89]. Even though its induction is linked to apoptosis [90, 91], it requires other pro-apoptotic genes to induce apoptosis and it has also been proposed that PIG 3 may play a role in cancer cell survival, inducing sub-lethal levels of ROS [92], actively participating in the PI3K/AKT/PTEN pathway in PTC (papillary thyroid carcinoma). Silencing of PIG3 increased expression of PTEN and reduced PI3K and phosphorylated AKT, it was suggested that PIG3 induces ROS at intermediate levels, which induces phosphorylation of AKT and activation of mTOR [93]. We have observed that PIG3 is induced by p53 in prostate cancer, but its expression is not a key factor for induction of apoptosis [94].
As already discussed, POX, which participates in proline catabolism, is another p53-regulated protein. POX is also considered another member of the PIGs genes and may be called PIG6. POX catalyzes the conversion of proline into Delta-1-pyrroline-5-carboxylate (P5C) after p53 activation in colon and bladder cancer cell lines [95, 96]. PIG 6 also induces ROS, which mobilizes calcium and Calcineurin, causing apoptosis [97], it induces mitochondrial superoxide radicals and induce intrinsic pathway apoptosis, but also induces TRAIL, DR5 and caspase 8 cleavage by the extrinsic pathway [98]. Apoptosis mediated by POX can be inhibited by Mn-SOD induced by p53 [98, 99]. On an opposite effect it has also been observed that under hypoxia POX induces protective autophagy dependent on ROS, allowing tumor cell survival [100].
Another transcription factor involved in ROS inhibition is NRF2 (NF-E2 related factor 2). After oxidative stress this protein is stabilized and translocated to the nucleus, and induces the expression of antioxidant genes [101–104]. p53 modulates NRF2 activity, it can inhibit the expression of NRF2 through inhibition of Sp1 binding to the promoter region of NRF2 gene [105], inhibiting the activation of NRF2 antioxidants target genes, like x-CT, NQ01 and GST-α1 genes [106]. However, p53 has also been described to increase NRF2 activity indirectly through p21 induction. p21 directly interacts with NRF2 and prevents its degradation by inhibiting its ubiquitination [107]. Another review suggested that there are two phases of modulation of NRF2 by p53, in a first moment low levels of p53 induces p21 and therefore NRF2, in a second moment after severe DNA damage and high p53 expression inhibits the survival response mediated by NRF2 and induces cell death [108].
Several drugs that induce apoptosis of cancer cells rely on ROS induction [109, 110]. Indeed, an essential component of p53’s mechanism of action is the induction of oxidants [82, 111–113]. ROS (peroxide and superoxide) induction mediated by p53 was shown to induce apoptosis independently of cytochrome-c release and can regulate mitochondrial membrane potential [114]. Induction of p53 by ROS has been shown to mediate necrotic cell death through PARP activation [115]. PIG3 and PIG6 are downstream targets of p53 that induce oxidants production; however, others and we have observed that expression of these genes is not essential to induction of apoptosis. We have observed that NADPH oxidase, especially NOX1, is induced by p53 and can induce apoptosis of prostate cancer cell lines [94]. Figure 3 shows how in normal conditions p53 activates genes involved in ROS detoxification and under stress p53 induces genes that increase ROS to toxic levels that induce cell death.
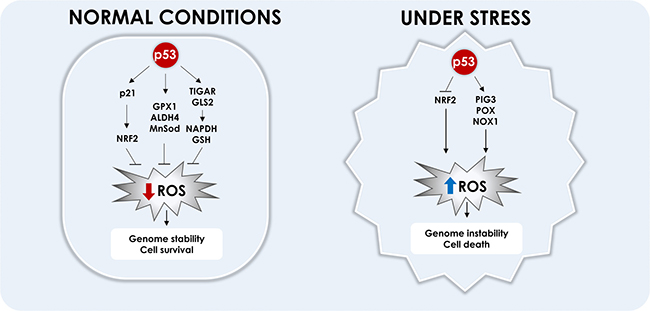
Figure 3: Modulation of ROS by p53. In normal cells, p53 was shown to reduce ROS levels, preventing DNA damage and promoting cell survival. p53 induces directly the anti-oxidant genes: GPX1, ALDH4 and Mn-SOD. p53 also induces TIGAR and GLS2, that redirects the cell metabolism to PPP, which increases NADPH and GSH production and ROS scavenging. p53 also induces p21 that prevents NRF2 degradation, which reduces ROS levels. In cancer cells or during stressful situations, p53 inhibits NRF2 expression and induces pro-oxidant genes: PIG3, POX and NOX1, which reduces mitochondrial membrane potential, induces DNA damage and apoptosis.
p53 and growth factors signaling
Growth factors are essential for cell homeostasis but their signaling is frequently deregulated during the tumorigenic process, and they are usually targets of tumor suppressors, such as p53 [1]. Activation of the signaling pathway of insulin-like growth factors (IGFs) is responsible for stimulating cell growth, proliferation and metabolism, sending pro-survival signals and contributing to increased glucose uptake [116, 117]. Insulin-like growth factors 1 and 2 are recognized by IGF1R, which in turn activates the MAPK and PI3K/AKT/mTOR signaling pathways (Figure 1). In addition, they have their activity regulated by co-stimulatory IGFBPs (1-6) proteins that have different effects on IGF signaling [117]. When activated, p53 is responsible for inhibiting the transcription and activation of a wide range of genes related to this pathway, including IGF1 and IGF1R [118–120]. p53 promotes as well the transcription of AMPK subunits, Tuberous Sclerosis 2 (TSC2), PTEN and IGFBP1 and 3, known negative regulators of IGF1/AKT/mTOR pathway and tumor suppressors [121–124]. mTOR down-regulation may lead to less activation of sterol regulatory element-binding proteins (SREBP), which can also be further degraded by GSK3, whose activation happens through inactivation of AKT by p53 [125–128]. Those two mechanisms simultaneously lead to reduced metabolism of fatty acids and cholesterol biosynthesis pathways [127].
Besides promoting expression of negative regulators of mTOR pathway, after genotoxic stress, DNA damage or in response to oxidative stress, p53 leads to AMPK phosphorylation and activation [129, 130]. Active AMPK phosphorylates TSC2, thus inhibiting mTOR pathway. Its activation is dependent of Sestrin 1 and 2, whose transcription is controlled by p53 [129, 131]. Sestrins 1 and 2 also participate on autophagy [132–135], by activating AMPK, which phosphorylates ULK1, an autophagy initiation kinase, and at the same time through mTOR inhibition, which is a negative regulator of ULK1 [136].
In a feedback mechanism, activated AMPK is also capable to phosphorylate p53 and induce G1 arrest when the cell is under glucose deprivation, allowing the cells to survive until growth conditions are restored [137]. Moreover, if conditions do not improve, AMPK persistent activation leads to p53 dependent senescence [137]. Another feedback mechanism linking energy status, cell growth and p53, involves p38αMAPK stimulation after DNA damage, and consequent mTOR/S6K1 activation [138]. In this case, activated S6K1 interacts with MDM2, blocking its inhibitory effect over p53. Working as a negative feedback loop, p53 can inhibit mTOR activation, S6K1 then releases MDM2 turning off p53 activity (Figure 1) [131, 138, 139].
Nevertheless, different levels of p53 activation can trigger different outcomes. High levels inhibit mTOR inducing reversible quiescence instead of senescence or cell death, while low levels are not able to inhibit mTOR, and with prolonged cell cycle arrest can lead to senescence, what is essential to understand the action of different chemotherapeutic agents as will be described below [140].
Not only PI3K/AKT/mTOR pathway affects classic chemotherapy, but also immunotherapy, having a significant impact on immune checkpoint blockade, that target CTLA-4 and PD-L1. PD-L1 expressed on the surface or tumor cells inhibits T-cells proliferation and effector function after its recognition by PD-1 [141]. PDL1 expression is correlated with tumor aggressiveness for different types of tumor [142, 143]. Presence of PD-1 in tumor infiltrating lymphocytes is associated with poor survival [144]. A recent review indicated that PI3K/AKT/mTOR pathway inhibitors reduce expression of checkpoint ligands, and is also important for T-regs immunosuppressive function, suggesting that combination with anti-PD-L1 and inhibitors of PI3K/AKT/mTOR could favor both therapies [145]. Another important aspect is that PD-L1 expression is correlated with p53 status [146], probably because p53 can inhibit PD-L1 through miR-34a [147]. Therefore, p53 and PI3K/AKT/mTOR pathways affect immune checkpoint blockade therapies.
Role of p53 in senescence and autophagy
Senescence is characterized by irreversible loss of the ability to proliferate in culture [148], and is more prone to happen in pre-malignant tumors. The senescent cells can be cleared by immune cells and have a more efficient tumor regression [149]. Interestingly senescent cells show reduced p53 transcriptional activity [150]. The first step for a cell to undergo senescence is related to p53 ability to promote cell cycle arrest through induction of p21, a cyclin-dependent kinase inhibitor that stops cell cycle at G1 phase [151]. However, cell cycle arrest is not enough to determine if a cell is senescent, as quiescent and differentiated cells also present the same characteristic [152]. Other senescence markers are senescence-associated beta-galactosidase and senescence-associated heterochromatic foci (SAHF); moreover p53 targets p21, promyelocytic leukemia protein (PML), plasminogen activator inhibitor (PAI-1) and deleted In Esophageal Cancer 1 (DEC1) [153].
Macroautophagy or simply autophagy is a process that controls degradation of proteins and organelles, necessary to recycle proteins and cell components and also represents an adaptive response to keep homeostasis after exposure to stressful conditions, it is essential for cell survival and at the same time its prolonged activation can also lead to cell death [154]. p53 can lead to autophagy as a protection against adverse growth conditions, keeping cells on a quiescent state until conditions improve. Inhibition of this protective autophagy redirects cells from quiescence to senescence on a mTOR dependent mechanism [155, 156].
Senescence and autophagy can be modulated by mTOR and p53. mTOR induces senescence and inhibits autophagy, while p53 was shown to induce autophagy, which inhibits senescence [156]. AKT was also shown to induce stabilization of p53 during nutrient starvation and silencing of p53 and AKT increased autophagy, indicating that the AKT-p53 axis may also play an opposite role in cell survival during nutrient starvation [157]. On Nutlin-3-resistant tumor cells, p53 activation leads to a protective autophagy and glycolysis stimulation, while on apoptosis sensitive cells, p53 activation decreases glycolysis, raises superoxide levels and inhibits autophagy. Using a combination of Nutlin-3 with glycolysis inhibitors, treatment of resistant tumor cells has the same effect presented on sensitive cells, with inhibition of autophagy and apoptosis triggering, indicating that metabolism plays an essential role in cell fate [158].
The switch from autophagy to apoptosis can be controlled by p53, regulating the expression pattern of autophagy related genes, ULK and ATG family [43], and apoptosis related ones, Bcl2, PUMA, Bax and others [159] depending on its activation signal. When it’s phosphorylated on Ser15, p53 dissociates from MDM2, inhibits Beclin1 and LC3 and activates apoptosis and inhibits autophagy [160]. In addition, when phosphorylation occurs on S392, p53 inhibits ULK1 directly, switching autophagy to apoptosis [161].
Autophagy usually has a protective effect against critical environmental conditions, allowing cells to survive until conditions improve. Nevertheless, cells can survive after losing the ability to activate autophagy and when it happens together with p53 inactivation this might trigger adenocarcinoma development, with changes in the metabolism focusing on anabolic pathways [162]. In fact, the relationship between autophagy and p53 determines cell fate, limiting tumor development when autophagy is blocked and p53 is functional, and promoting progression from benign lesions to invasive tumor when there is a lack of p53 combined with autophagy genes silencing [163].
In the end, if autophagy will prevent cells from dying or not depends on p53 status [164], and which will be the cell fate, autophagy, senescence, apoptosis, quiescence, depends of p53 combined with microenvironment conditions and other pathways alterations presented in the cell [165–169].
Modulation of p53 activity
p53 transcriptional activity can be modulated by its interaction with other transcription factors. One important interaction partner is the Peroxisome proliferator-activated receptor γ Coactivator 1-α (PGC-1α) protein. Interaction of PGC1α with p53 directs the expression of genes involved in cell cycle arrest and metabolism [170]. PGC1α positively regulates respiration, gluconeogenesis, mitochondrial biogenesis, metabolic processes and ROS inhibition [171–173]. Induction of PGC1α by starvation caused activation of the following p53 target genes: p21, GADD45α, TIGAR, SCO2 and Sestrin2 at early time points, later, if starvation continued, pro-apoptotic genes like BAX, PUMA and NOXA were activated. Silencing of PGC1α abrogated the induction of the pro-arrest and metabolic genes, while the pro-apoptotic genes were not altered [170]. There´s a feedback mechanism in these situations, p53 binds to and induces PPARGC1a promoter and induces expression of PGC1α. Depletion of the antioxidant GSH induced the p53-PGC1α-NFR2 axis [174]. It’s interesting, however, that p53 have also been shown to inhibit PGC1α, and induce oxidative stress of cardiomyocytes [175].
Mutant p53
Since the early studies of p53, It’s evident that its mutant forms may be just as important as the wild-type for cancer. Mutant forms of p53 have different expression profiles and may activate tumorigenic genes, showing an oncogenic “gain of function”, exhibiting a dominant-negative effect over the wild-type protein [176]. A recent review points out that there’s a “rainbow of mutants” of p53 and each one has a particular degree of gain of function and pathological consequence, implicating that knowledge of the specific mutations may have an impact on the therapeutic approach decision [177]. These mutant forms of p53 also regulate tumor cell metabolism and may modulate drug sensitivity.
Warburg effect and shift of cell metabolism to glycolysis are directly affected by p53. Expression of hexokinase 2 (HK2), the first enzyme of the glycolytic pathway, is induced by mutant p53, increasing the conversion of glucose to glucose-6-phosphate (G6P), which may diverts to glycolysis or PPP [178]. Cells expressing the mutant p53 increased expression and phosphorylation of PKM2 mediated by mTOR, while wild-type p53 had no positive influence in the expression levels of PKM2 and knockdown of wild-type p53 increased PKM2 phosphorylation [179]. Mutant p53 also increases PKM2 and promotes hepatocarcinogenesis [180]. The p53 target gene TIGAR that is upregulated by wild-type p53, is downregulated by different p53 mutants, including mutations in the DNA binding domain [181, 182] or mutations in the C-terminal, comprising the tetramerization and regulatory domains [44]. This differential expression of TIGAR in wild-type and mutant p53 has an impact in cancer treatment. The high expression levels of TIGAR mediated by wild-type p53 reduced the dependence on the glycolytic pathway and FDG efflux into the tumor cell. Cells expressing mutant p53 showed an increased dependence on the glycolytic pathway and sensitivity to the action of FX11, an inhibitor of the enzyme Lactate Dehydrogenase, which is responsible for the conversion of pyruvate to lactate and in the process regenerates NAD+ and divert pyruvate from conversion to acetyl-CoA that is important for oxidative phosphorylation [183]. Expression of mutant p53 increased glucose uptake, glycolytic rate and lactate production. These mutant forms promote GLUT1 and GLUT 4 translocation through RhoA/Rock activation. Inhibition of the RhoA/Rock/Glut1 axis abolished Warburg effect mediated by mutant p53 [184].
Garritano et al. compiled the results from the literature of wild-type and mutant p53 expression [185]. In their work, it was observed that POX, GLS2, SESN1, GAMT, NOTCH1, NOTCH3 and PLA2G2A have altered expression in cells expressing mutant p53. Pointing out the importance of GLS2 and POX to modulate metabolism to increase glycolysis and directing to Warburg effect. Tepper et al. [186] identified that PGM presents increased expression mediated by mutant p53. Several metabolic intermediates are tightly regulated by p53. Enzymes regulated by wild-type p53 including GAMT, GLS2 and POX are negatively regulated by mutant p53 [181, 182, 185]. In the high-grade serous ovarian cancer (HGS-OvCa) the p53 gene is mutated in almost all cases, reaching 96% [187]. It has been proposed that in HGS-OvCa the mutant p53 promotes sterol regulatory element binding proteins (SREBPs) and inhibits GAMT, promoting lipid anabolism and fatty acid oxidation [188]. As a net result there is a promotion of lipid anabolism that accelerates tumor growth and progression [188].
In normal cells, p53 exerts an important role in ROS detoxification, maintaining low levels of oxidants; while tumor cells exhibit increased levels of ROS. Cells expressing mutant p53 show repression of several important genes involved in ROS inhibition. Cells expressing mutant p53 showed a reduced expression of ALDH4A1 [182]. p53R273H reduced the NRF2 activity resulting in defect in ROS detoxification and cell survival [189], while mutants p53H179Y, p53L194R, p53S240R, p53R249S, p53A276D, and p53E286Q had no influence in the levels of NRF2 [105]. SESN1 and SESN2 expression was reduced by mutant p53 [44, 181, 185]. Some p53 mutants were not able to induce POX and activate the calcineurin pathway and apoptosis [97]. Downregulation of different genes involved in ROS detoxification may me important to maintain the intermediate levels of ROS that favors tumor cell growth.
Expression of the PGC1α transcription factor was reduced in cells expressing p53 missense mutations or with loss of p53 in a comparison of 28 lung adenocarcinoma cell lines with different p53 status [190]. However, one particular mutation p53P72R showed increased PGC1α function and increased metastatic capability [191]. Even though this mutation didn’t show evidence of increased glucose regulation compared to wild-type p53 [192].
The mTOR pathway is also modulated by p53. One of the main mechanisms involved in this regulation is through phosphorylation of AMPK. Inhibition of AMPK phosphorylation results in stimulation of the mTOR pathway. Expression of mutant p53 inhibited BECN1, DRAM1, ATG12, SESN1/2, repressed AMPK phosphorylation, increased phosphorylation of p70S6K and reduced autophagy [193, 194]. AMPK phosphorylation can also be induced by Sestrins [129, 195, 196], which have been observed to be increased by mutant p53 knockdown [194]. mTOR activity can also be modulated by EGFR, it has also been shown that p53R280K has an increased EGFR signaling [197]. Using an inhibitor of mTOR (Everolimus), the cells expressing mutant p53 were more sensitive to the drug, evidencing a stronger dependence in mTOR pathway [194]. Phosphorylation of AKT is induced by the mutant p53R273H, but not p53R175H [198]. In general, mTOR and S6Ks, known mediators of mTOR signaling, have been related to cancer progression in several cancer models [199–201].
Mutant p53 represses autophagy and is dependent on its cellular localization. Some mutant p53 have a preferential nuclear localization and fail to repress autophagy, while mutants that show a more cytoplasmic localization are able to repress autophagy. Mutants that have both cytoplasmic and nuclear localization are also able to inhibit autophagy, indicating that repression of autophagy occurs in the cytoplasm [202]. Interestingly while wild-type p53 is degraded through proteasome pathway, it was shown that mutant p53 (more than 20 different p53 forms have been tested) is deacetylated and degraded through lysosomal-dependent pathway in a chaperone-mediated autophagy (CMA) or through macroautophagy [203–205]. Collectively these data indicate that mutant p53 inhibits autophagy, which is responsible for the degradation of mutant p53, therefore prolonging its half-time.
Mutant p53 has also been shown to be important to increase drug resistance through different mechanisms. Mutant p53 induces activation of the Multi-drug resistance (MDR1) promoter. The MDR1 is a drug efflux pump that participates in the removal of drugs from the cell [206–208]. Patients with colorectal carcinoma showed a positive correlation among mutant p53, MDR1 (P-gp, ABCB1) and GST-pi expression [209]. Oxidative phosphorylation was shown to inhibit expression of the ABC transporters in cells expressing wild-type p53 and to increase expression of these transporters in cells expressing mutant p53 [210]. Increased drug resistance may also be modulated by mutant p53 through modulation of the expression of pro and anti-apoptotic genes like Fas and Bcl-XL [211]. Another important class of genes induced by mutant p53 is chromatin modulators. Post translational modification of histones are essential to regulate gene expression; methylation or acetylation of H3K4 are involved in gene activation, while methylation or acetylation of H3K9 and H3K27 are involved in gene repression [212]. The MDR1 is one of the genes activated by H3K4 methylation and acetylation [213]. Among the genes repressed by methylation of H3K9me3 and H3K27me3 are several genes involved in induction of apoptosis (FAS, Bcl2 and BAX). Silencing of KDM5D (JARID1D) (H3K4me3 demethylation agent) increases drug resistance [214]. As a net result, there is an increased chemoresistance [215–217]. The p53 GOF mutants induce expression of genes involved in histone methylation (H3K4me3 and H3K4me1) by MLL1 (kmt2a) and MLL2 (kmt2d) and histone acetylation (H3K9ac) by MOZ (kat6a) [218]. Mutant p53 induces PKM2, which interacted with H3 and caused H3K9me1 and H3K9Ac (Figure 4) [180]. Indicating that mutant p53 activates chromatin regulatory genes that repress important pro-apoptotic genes and induce drug efflux pumps. Figure 4 shows how mutant p53 induces or inhibits different genes that ultimately results in increased glycolysis, PPP, inhibition of autophagy, drug resistance and reduction of pro-apoptotic ROS and increase in pro-tumoral levels of ROS. Supplementary Table 1 lists the capacity of mutant p53 forms to induce/inhibit genes involved in metabolism, ROS and growth control, drug sensitivity, autophagy and cellular localization.
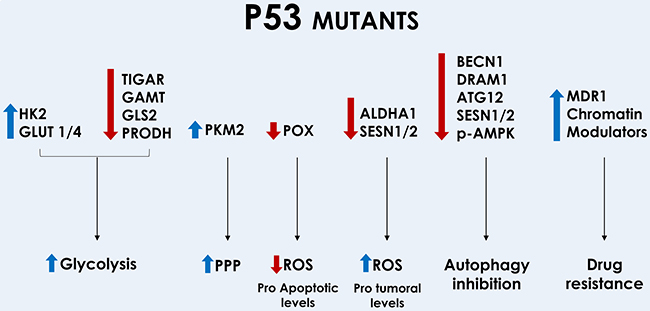
Figure 4: Modulation of metabolic genes by mutant forms of p53. Mutant p53 increases glycolysis through inhibition of TIGAR, GAMT, GLS2 and POX; and through induction of HK2 and GLUT1/4 translocation. Redirection of the PPP can be achieved through induction of PKM2. Some mutants of p53 reduce POX and ROS induction and ROS levels can be increased through inhibition of ALDHA1 and SESN 1/2. Autophagy is inhibited through downregulation of BECN1, DRAM1, ATG12, SESN1/2, and reduction of AMPK phosphorylation. Increased drug resistance is mediated by induction of MDR1 and chromatin modulators.
THERAPY TARGETING METABOLISM
Glucose metabolism
The molecular targets related to alterations in cancer metabolism that are currently under therapeutic investigation are summarized in Table 1 and Figure 5 correlates the drugs mentioned bellow with their targets in the metabolic pathways. As already mentioned, members of glucose transporters of the GLUT family are overexpressed in several types of cancers, suggesting that these transporters could be possible therapeutic targets [219]. Ritonavir, an HIV protease inhibitor that displays off-target inhibitory effects on GLUT4, can reduce the viability of myeloma cells and increase the sensitivity to the chemotherapeutic doxorubicin [220]. Ritonavir administration is associated with perturbation in the proteasomal activity and a small effect in p53 accumulation in several types of cancer [221]. It has also been found that Ritonavir inhibits AKT and induces senescence and oxidative stress of PBMCs [222, 223]. Phloretin is a naturally occurring dihydrochalcone that inhibits GLUT1 and, under hypoxia conditions, enhances daunorubicin anticancer effects [224]; it also induces cell cycle arrest in a p53 dependent manner [225]. WZB117 is another inhibitor of GLUT1 that leads to a lowered rate of glycolysis and cellular growth, showing synergistic anticancer effects when combined with cisplatin or paclitaxel, both drugs are known p53 inducers [226].
Table 1: Drugs acting over metabolism and the existence or not of a relation with p53
Glucose metabolism |
|||||
|---|---|---|---|---|---|
Compound |
Target |
Effects |
Stage of development |
Relation with p53 |
References |
Ritonavir |
GLUT4 |
Inhibits GLUT4 |
Clinical trial |
Yes |
[220] |
Phloretin |
GLUT1 |
Inhibits of GLUT1; enhances daunorubicin’s anticancer effects |
Preclinical |
Yes |
[224] |
WZB117 |
Inhibits GLUT1 |
Preclinical |
Yes |
[226] |
|
2DG |
Hexokinase |
Inhibits glucose metabolism |
Preclinical |
Yes |
|
3-bromopyruvate |
Inhibits glucose metabolism |
Preclinical |
Yes |
[250] |
|
Lonidamine |
Inhibits glucose metabolism; enhances doxorubicin anticancer effects |
Clinical trial |
Yes |
||
siRNA |
PFK2 |
Inhibits glucose metabolism |
Preclinical |
Yes |
[256] |
Phosphonomethyl analogue |
Phosphoglycerate mutase |
Inhibits glucose metabolism |
Preclinical |
Yes |
|
shRNA |
Pyruvate kinase |
Improves the therapeutic efficacy of cisplatin and docetaxel |
Preclinical |
No |
[261] |
TLN232 |
Inhibits glucose metabolism |
Clinical trial |
No |
[262] |
|
SAICAR |
Indirectly mediates antitumor effects |
Preclinical |
No |
||
Oxamate |
LDHA |
Inhibits LDHA |
Preclinical |
No |
[267] |
shRNA |
MCT4 |
Blocks the adaptive response to antiangiogenic drugs |
Preclinical |
No |
|
Phloretin |
Inhibits MCT4 |
Preclinical |
No |
[270] |
|
Lipid metabolism |
|||||
Cerulenin |
FASN |
Accumulates malonyl-CoA and induces ER stress |
Preclinical |
Yes |
|
C75 |
Accumulates malonyl-CoA and induces ER stress |
Preclinical |
Yes |
||
Orlistat |
Irreversibly inhibits the FASN activity, blocking FA synthesis |
Approved for obesity |
Yes |
||
EGCG |
Inhibits 50% of FASN activity, leading to apoptosis |
Clinical trial |
Yes |
||
SB-204990 |
ACLY |
Inhibits cholesterol and FA synthesis |
Preclinical |
Yes |
[291] |
ND-646 |
ACC |
Inhibits ACC enzymes (ACC1 and ACC2), suppressing FA synthesis |
Preclinical |
Yes |
[292] |
HC-3 |
CK |
Inhibits phosphatidylcholine synthesis |
Preclinical |
No |
|
CK37 |
Reduces the concentration of phosphocholine in transformed cells |
Preclinical |
No |
[293] |
|
Amino acid metabolism |
|||||
Phenylacetate |
Glutamine |
Reduces the biodisponibility of glutamine in the blood. |
Preclinical |
Yes |
|
BPTES |
GLS |
Inhibits GLS |
Preclinical |
No |
[301] |
968 |
Inhibitor of Rho GTPase-dependent cellular transformation, targets GLS |
Preclinical |
No |
[303] |
|
Arginine deiminase |
Arginine |
Converts arginine to citrulline and ammonia |
Clinical trial |
Yes |
[307] |
Asparaginase |
Asparagine |
Converts asparagines to aspartate and ammonia |
Approved |
Yes |
|
RNAi |
PHGDH |
Inhibits PHDGH |
Preclinical |
No |
|
Krebs cycle and oxidative phosphorylation |
|||||
DCA |
PDK1 |
Metabolic shift from cytoplasm-based glycolysis to mitochondria-based glucose oxidation |
Clinical Trial |
Yes |
|
Metformin |
Mitochondrial complex I, AMPK |
Activates AMPK since decreases mitochondrial respiration chain activity and ATP production |
Approved for diabetes and polycystic ovaries syndrome |
Yes |
|
HMS-101 |
IDH |
Specifically inhibits Isocitrate Dehydrogenase (IDH) |
Preclinical |
No |
[323] |
AG-120 |
Specifically inhibits Isocitrate Dehydrogenase (IDH) |
Clinical trial |
No |
||
AG-881 |
Specifically inhibits Isocitrate Dehydrogenase (IDH) |
Clinical trial |
No |
||
IDH305 |
Specifically inhibits Isocitrate Dehydrogenase (IDH) |
Clinical trial |
No |
||
Cell growth signaling |
|||||
Lapatinib |
EGFR |
Inhibits EGFR |
Preclinical |
Yes |
|
Trastuzumab |
Preclinical |
Yes |
|||
Rapamycin |
mTOR |
Inhibits mTORC1 and enhances the antitumor effects of cisplatin |
Approved as immunosuppressant |
Yes |
|
Rapalogues |
Inhibits mTOR, stimulating autophagy |
Approved as immunosuppressants. Temsirolimus and everolimus were approved for renal cell carcinoma, neuroendocrine tumors of pancreas and subependymal giant cell astrocytoma |
Yes |
[356] |
|
Acriflavine |
HIF-1 |
Inhibits HIF-1 |
Preclinical |
Yes |
|
PX478 |
Preclinical |
No |
|||
GDC-0941 |
PI3K |
Inhibits PI3K/AKT/mTOR axis |
Clinical trial |
No |
|
PX866 |
Clinical trial |
No |
|||
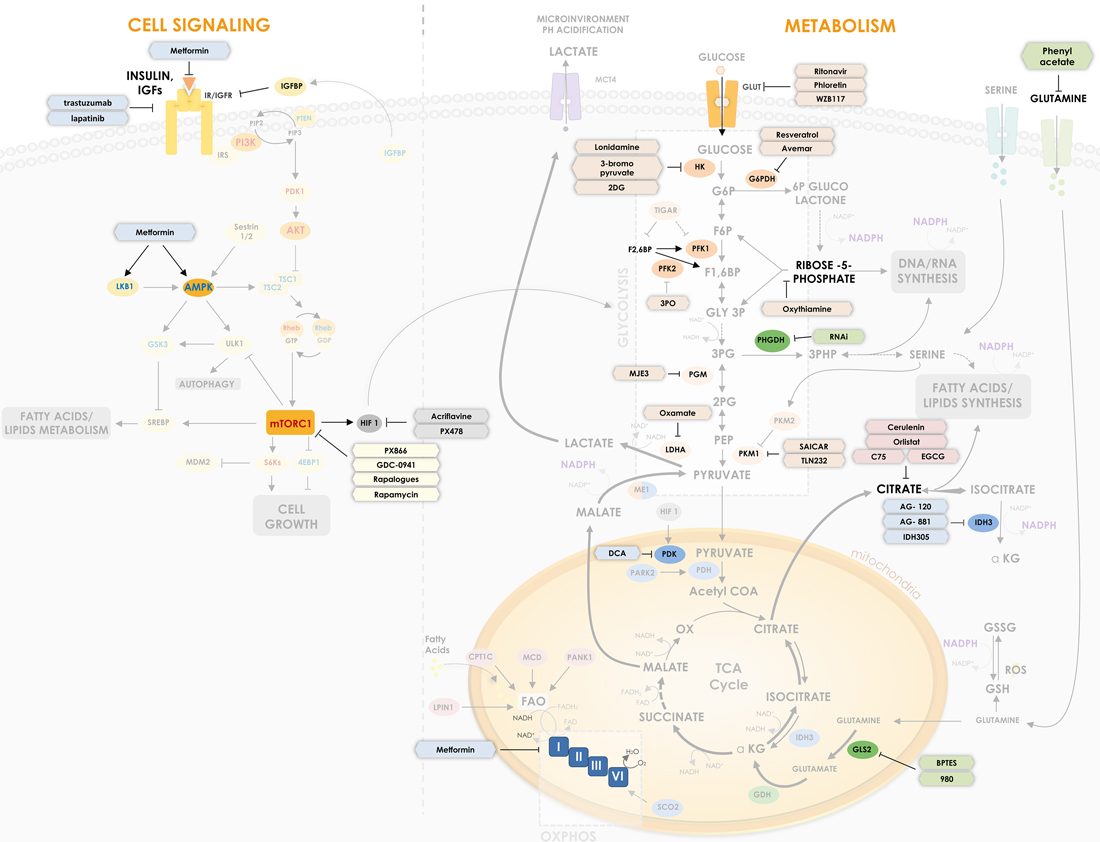
Figure 5: Metabolism based cancer therapies targets. Different therapies are being developed to regulate crucial metabolic targets. Some therapies are already validated for other diseases, such as metformin and DCA. The Figure shows molecular targets that are currently under therapeutic investigation in cancer. Drugs, their targets and relation with p53 are summarized in Table I.
G6PDH, the rate-limiting enzyme of PPP, allows the production of important molecules like NADPH and R5P, and is involved in the maintenance of GSH cellular levels. Breast cancer cells show a significant increase in oxidative PPP flux [227]. Over-expression of G6PDH leads to a significant resistance to apoptosis [228]. There are currently no inhibitors of PPP in clinical trials, even though it may be an attractive target. Some compounds like Resveratrol and Avemarare are known inhibitors of PPP enzymes like G6PDH [229, 230] and showed some positive results in colon cancer, leukemia and polycystic ovary syndrome cells by induction of apoptosis and cell cycle arrest [231, 232]. In 1999, an article described for the first time that Resveratrol increases endogenous levels of p53 in a cancer model [233]. The antiproliferative and pro-apoptotic role of Resveratrol has been demonstrated in several cancer types to be mediated by p53, including liver cancer, osteosarcoma and thyroid cancer [234–236]. In prostate cancer cells, Resveratrol induces cell cycle arrest and apoptosis when combined to docetaxel through p53 [237]. Finally, a study has demonstrated that wild-type and functional p53 is important to increase the anticancer effects of Resveratrol in cancer cells [238].
Transketolase-like protein 1 (TKTL1), an enzyme of the non-oxidative branch of the PPP, has been found to be upregulated in several tumor types [239], and is involved in tumor cell proliferation [240], sensitivity to ROS [241], and metabolic alteration mediated by TIGAR [242]. Oxythiamine, a thiamine antagonist, is able to bind TKTL1 and hinder tumor growth through inhibition of PPP and induction of cell cycle arrest [243, 244]. Thiamine antagonists are also able to trigger upregulation of p53 expression, phosphorylation and activation [245].
HK2 plays an important role both in glycolysis and apoptosis and its increased activity has been reported in several types of cancers, showing a direct relation between growth rates and HK2 activity [246]. 2-deoxy-d-glucose (2DG), an inhibitor of glucose metabolism, is a glucose analog that is phosphorylated by HK2, inhibiting it by competition and leading to growth arrest and/or apoptosis [247]. In addition, 2DG can potentiate the cytotoxic effects of chemotherapy, since combination of metformin and 2DG restored p53 function, inhibiting the overexpression of MDM2 and MDM4 and leading to cell growth arrest and apoptosis in MCF-7 cells resistant to doxorubicin [248, 249]. 3-bromopyruvate (3BP), another glycolysis and TCA inhibitor that targets HK2, leads to a depletion of the cellular ATP reserves, which was shown as a key determinant of chemoresistance in certain types of cancer [250]. In fact, multi-drug resistant cells have high expression of P-gp (P-glycoprotein 1), which requires ATP for its activity and therefore are more sensitive to ATP depletion [251, 252]. This inhibition of glycolysis not only enhances the cytotoxic effects of certain chemotherapeutics like daunorubicin and doxorubicin, but also markedly suppresses tumor growth when used with doxorubicin by limiting ABC transporters, like P-gp [253]. Lonidamine, a derivative of indole, is also an HK2 inhibitor. Results of clinical trials have shown that lonidamine combined with doxorubicin have good therapeutic efficiency for the treatment of certain types of cancer like breast, prostate, melanoma, brain and ovarian cancer [254]. 3BP and lonidamine presented diverse effects regarding p53 in glioblastoma cells. The first led to early cell death marked by p53 dephosphorylation, while the latter led to p53-dependent apoptosis favoring p53 translocation to mitochondria [255].
p53 promotes nucleotide biosynthesis through PPP in response to DNA damage by repressing the expression of FB3 isoform of PFK2 (PFKFB3), which is expressed in several cancers and is required for anchorage-independent growth of Ras-driven tumors [256]. Small-molecule inhibitors of PFKFB3, like 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO), have been reported to present a cytostatic effect on cancer cells [257].
As already discussed, PGM is upregulated by mutant forms of p53 [186] and regulates a unique step in glycolysis. Most of the glycolytic intermediates that are used as precursors for biosynthesis are upstream of this step. In several types of cancers, like hepatocellular carcinoma and colorectal cancer, its activity is increased [258]. The compound called MJE3 is a potent competitive inhibitor of PGMs that studies suggest to be a good candidate for chemotherapy [259, 260].
The pyruvate kinase isoform M2 (PKM2) is predominantly expressed in cancer cells as already mentioned. Several studies have shown a negative correlation between PKM2 expression and drug resistance, like cisplatin [261], suggesting that PKM2 is a potential target for adjuvant cancer therapy. TLN232 is an inhibitor of pyruvate kinase and has been evaluated as a stand-alone therapeutic intervention in patients with melanoma or metastatic renal cell carcinoma (ClinicalTrials.gov identifiers: NCT00735332; NCT00422786) [262]. Succinyl aminoimidazole carboxamide ribose-5’phosphate (SAICAR), an intermediate of the de novo purine nucleotide synthesis pathway, and the amino acid serine are known activators of PKM2, limiting the diversion of glycolytic intermediates towards the PPP and suggesting anticancer effects of serine starvation [263, 264].
Lactate dehydrogenase A (LDHA), the enzyme that converts pyruvate to lactate, is overexpressed in cancer, since the shift to oxidation of glucose to lactate is a well-known characteristic of the Warburg effect. The transcription factors, hypoxia-induced factor 1 (HIF-1) and Myc, known oncogenes, induce expression of LDHA [265]. The knockdown of human papillomavirus oncoprotein E6 in HeLa cells increased expression of p53 and decreased levels of miR-34a, which targets LDHA, thus reducing the Warburg effect [266]. Oxamate is a pyruvate analog that inhibits the conversion of pyruvate to lactate and has been reported to reverse the resistance associated to paclitaxel treatment associated with LDHA (Figure 5) [267].
Monocarboxylate transporter 4 (MCT4) is a membrane protein that exports lactate, allowing the preservation of cell pH homeostasis, and creating an acidic tumor extracellular environment [9]. Cells under hypoxia produce large amounts of lactate through LDHA, as mentioned before, and export them using MCT4. When oxygenated, cells may then absorb lactate from the extracellular medium using the MCT1 isoform, and convert it back to pyruvate for further oxidation [268]. Studies have shown that MCT4 knockdown could block the adaptive response to antiangiogenic drugs by disrupting the glycolytic flux [268, 269]. Phloretin, mentioned before, also has the capacity to inhibit MCT4, although mechanism insights and clinical trials are needed [270].
Lipid metabolism
As already discussed, deregulation of the lipid metabolism is one of the most common alterations in cancer, since fatty acids are required by cancer cells to form membranes and signaling molecules. As shown in Figure 5, the citrate generated in the TCA cycle is exported from the mitochondria to the cytosol, where the enzyme ATP citrate lyase (ACLY) converts it to acetyl-CoA, which is subsequently converted to cholesterol or catalyzed by acetyl-CoA carboxylase (ACC) to form malonyl-CoA. Fatty acid synthase (FASN) is a key enzyme that acts in the conversion of malonyl-CoA to fatty acids. In normal adult tissues, FASN expression is very low, while in several types of cancer, such as prostate and breast cancer, it is significantly upregulated [271, 272]. In bone tumors, FASN expression is significantly increased in a p53 and MAPK-dependent manner [273]. High expression of FASN is associated with the development and survival of tumor cells, as well as a poor cancer prognosis [274]. These data suggest that FASN may be a therapeutic target for cancer.
FASN inhibitors, such as cerulenin, C75, orlistat and epigallocatechin gallate (EGCG), demonstrated anti-tumor activity, leading to toxic accumulation of the malonyl-CoA intermediate, reduction of membranes synthesis and phospholipid function. Cerulenin, isolated from Cephalosporium caerulens, contains an epoxy group that irreversibly binds to FASN leading to its inhibition [275]. C75, derived from cerulenin, also interacts with FASN promoting its inhibition [276]. Both cerulenin and C75 lead to apoptosis by promoting endoplasmic reticulum stress and malonyl-CoA accumulation [277]. However, the anticancer effects of cerulenin is limited by its chemical instability and lack of systemic activity [276]. In combination with cerulenin, the p53-p21 pathway activation by oxaliplatin occurred in a smaller concentration and induced caspase-3 cleavage [278]. Growth arrest induced by C75 is modulated by p38 MAPK but not by p53 in human hepatocellular carcinoma [279]. Finally, cancer cells exposed to cerulenin or C75 were sensitized by the loss of p53, indicating that these compounds may be clinically useful against malignancies carrying p53 alterations [280].
Orlistat is classified as an anti-obesity drug, although recent studies have shown its anticancer effects, since orlistat is able to irreversibly inhibit FASN [281]. Orlistat promotes apoptosis, reduces cell growth and proliferation, angiogenesis and metastasis and modulates the expression of several genes including p53, Bcl2, PUMA and Caspase-3 [282–284]. Despite these effects, orlistat has a poor bioavailability and non-specific side effects, which may be a limiting factor for its use as an anticancer drug. Combinations of orlistat with other drugs have been studied in order to optimize its anticancer effects [285]. EGCG derived from green tea, is also an inhibitor of FASN activity and has anti-tumor effects, inducing apoptosis in several cancer cell lines and reducing tumors size [286, 287]. EGCG is able to induce apoptosis in the presence of wild-type and mutant p53, indicating that a p53-independent pathway may contribute to EGCG-induced apoptosis in colon cancer cells [288]. A study confirmed that the combination of EGCG and an siRNA targeting p53 in triple-negative breast cancer cells leads to activation of pro-apoptotic genes and the inhibition of pro-survival genes [289].
High expression of ACLY in cancer is observed, suggesting its oncogenic role, and ACLY silencing in cancer cells induced p53 activation and facilitated DNA damage-induced cell death, suggesting it may be a therapeutic target [290]. SB-204990, an ACLY inhibitor, reduces proliferation and survival of cancer cells, in vitro, and tumor growth and cells differentiation, in vivo [291]. An allosteric inhibitor of ACC, ND-646, suppresses fatty acid synthesis and tumor growth of non-small cell lung cancer, even in p53 null cells, in preclinical models [292].
Choline kinase (CK) participates in the phosphatidylcholine synthesis, which is an important constituent of the cell membrane. CK is overexpressed in tumors and is considered a potential target for cancer treatment [293]. Hemicholinium-3 (HC-3) is an inhibitor of CK activity, that has an oxazinium ring that occupies the choline binding site and is described to have anti-proliferative effects [294]. Another CK inhibitor, CK37, is able to disrupt the actin cytoskeleton, promoting ultrastructural changes in the plasma membrane and inhibiting cell proliferation and tumor growth [293].
Amino acids metabolism
Glutamine, as mentioned before, is highly consumed in cancer cells and may be oxidized to lactate. Its metabolism is an important source of GSH for redox control, citrate for fatty acids biosynthesis and TCA cycle intermediates for amino acids biosynthesis. Phenyl acetate is a drug that reduces the bioavailability of glutamine in the plasma, by condensing with the γ-amino group of glutamine and leading to its excretion in urine [295]. Phenylacetate inhibits the proliferation of tumor cells and promotes their differentiation in a phenotype that is usually associated with less aggressiveness [296]. A point to consider, however, is that glutamine depletion from the plasma may also increase cachexia conditions [297]. A Phenylacetate derivative called SCK6 inhibits proliferation of human lung cancer cells via G1 cell cycle arrest and apoptosis mediated by p53 and p21 [298]. In breast cancer and prostate cancer cells, however, phenylacetate increased p21 and p27Kip1 expression, respectively, without affecting the expression levels of p53 [299, 300].
GLS2, a key enzyme of the glutaminolysis process in rapidly growing cancer cells, is also an important therapeutic target. Bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl) ethyl sulfide (BPTES) is an allosteric inhibitor of GLS [301]. This compound preferentially inhibits the growth of cells with mutant isocitrate dehydrogenase IDH1 [302], but BPTES also decreases aerobic cell proliferation through induction of hypoxic cell death [267]. 968, a GLS inhibitor that is dependent on Rho GTPases, hinders the growth of human breast cancer and B lymphoma cells without affecting normal cells (Figure 5) [303].
Arginine is a non-essential amino acid in normal tissues, however, at least two types of cancer, hepatocellular carcinoma (HCC) and melanoma, require exogenous sources of arginine, leading to asparagine auxotrophy [304]. Those cancer cells do not express argininosuccinate synthetase 1 (ASS1), which is essential for endogenous arginine synthesis [305]. Therefore, depletion of arginine in the plasma of patients adversely affects the growth of HCC or melanoma tumors [306]. Clinical tests are exploring the anticancer effects of arginine deiminase, which converts arginine to citrulline and ammonia, resulting in the depletion of arginine and demonstrating anticancer effects [307]. Arginine deiminase upregulates p53 and p27Kip1 and downregulates cyclin D1, c-Myc and Bcl-xL in stomach adenocarcinoma cells [308]. In colon cancer cells, administration of arginine deiminase causes a p53-dependent cell cycle arrest by induction of microRNA-16 [309]. Finally, expression of arginine deiminase by a human telomerase reverse transcriptase promoter presented higher hepatoma targeting and oncolytic efficiency than expression of p53 by the same promoter in vivo [310].
Asparagine, like arginine, is a non-essential amino acid in humans, due to the presence of the asparagine synthetase (ASNS). In certain types of cancer, including leukemia, the activity of ASNS is decreased and cancer cells require asparagine uptake from the plasma to survive [304]. Asparaginase, a recombinant bacterial enzyme that converts asparagine to aspartate and ammonia, reducing the plasma levels of the first, has been approved by FDA for the treatment of childhood acute lymphoblastic leukemia [311]. The combination of 6-thioguanine, arabinoside and PEG-asparaginase is able to downregulate Bcl2 oncoprotein levels in both p53-null or p53-expressing leukemia cell lines [312]. In acute lymphoblastic leukemia cells, inhibition of autophagy enhances L-asparaginase-induced cytotoxicity and overcomes the acquired resistance to L-asparaginase, whereas a ROS-p53-positive feedback loop is an essential mechanism of this synergistic cytotoxicity [313]. Finally, the combination of asparaginase and the Nutlin RG7112, a known inhibitor of p53-MDM2 interaction that will be more discussed later, presented therapeutic enhancement against mixed-lineage leukemia-acute lymphoblastic leukemia (MLL-ALL) xenografts, demonstrating that p53 and reduction of asparagine cooperate in tumor inhibition [314].
PHGDH, the enzyme that catalyzes the first reaction in the de novo serine synthesis pathway from 3PG, is overexpressed in breast carcinomas and melanomas [25, 26]. Although the mechanisms whereby PHGDH exerts oncogenic effects remain unclear, PHGDH is a potential target for the development of novel anticancer drugs. RNAi based strategies against this enzyme have been tested, however, its inhibition fails to affect serine availability (Figure 5) [25, 26].
Krebs cycle and oxidative phosphorylation
PDK1 is a gate-keeping mitochondrial enzyme that regulates the flux of pyruvate into the mitochondria. PDK isoforms are remarkably overexpressed in multiple human tumor samples and they show low expression in normal tissues, which may minimize side effects of this enzyme inhibition [315]. Dichloroacetate (DCA) is a structural analog of pyruvate and inhibits PDK1, changing the cancer cells metabolism of cancer cells from cytoplasm-based glycolysis to mitochondria-based glucose oxidation (Figure 5) [316]. DCA indirectly stimulates the activity of pyruvate dehydrogenase (PDH), which is hyperactivated by Myc, RTK or HIF1 signaling [317]. DCA has been used in combination with other drugs in breast, ovarian, pancreatic and colorectal cancer [318]. DCA can alter the mitochondrial membrane potential in many cancer models, showing increased production of ROS and decreased efflux of proapoptotic mediators from the mitochondria [319]. DCA is able to increase extracellular pH by reducing lactate secretion [320], thus limiting local invasion [9]. Due to its low price and toxicity and long history of clinical application, this drug serves as a potential metabolic-targeting molecule for sensitizing cancer cells, especially in glioblastoma [321]. A study has shown that DCA is able to increase the transcriptional activity of p53 in cancer stem cells (CSCs), inducing Bax-depended apoptosis [322].
Isocitrate dehydrogenase (IDH) of the TCA cycle is an essential enzyme for mitochondria respiration. Different IDH inhibitors have been developed with encouraging in vitro efficacy. An IDH1 inhibitor, called HMS-101, was able to block colony formation of leukemia cells [323], while others like AG-120, AG-881 and IDH305 went to phase I of clinical development (Figure 5) [324]. A cancer clinical trial involving patients with cancer presented promising preliminary results [325].
Cytosolic malic enzyme (ME1) catalyzes the reversible oxidative decarboxylation of malate to pyruvate, generating carbon dioxide and NADPH and contributing for macromolecules biogenesis [326]. Pre-clinical studies targeting ME1 have shown positive effects on cancer cells, but as far as we know, no clinical therapy targeting inhibit ME1 have yet been approved [327, 328].
Metformin (1,1-dimethylbiguanide) was discovered in 1920’s and is derived from the alkaloid galegine or isoamylene guanidine, an active substance of Galega officinalis [329]. Belonging to the biguanide class of antidiabetic drugs, metformin is the most commonly prescribed therapy for type 2 diabetes patients [330]. Indeed, it has a broad use, including polycystic ovarian syndrome, metabolic syndrome and diabetes prevention. Increased glucose consumption is a hallmark of most cancer cells, and increased blood glucose and insulin levels observed in type 2 diabetes are associated with poor cancer prognosis [331]. Studies have shown that cancer mortality was substantially reduced in diabetic patients treated with metformin compared to other treatments [332, 333], bringing metformin to the spotlights of cancer therapy research.
Metformin acts directly in the reduction of insulin resistance and blood glucose concentration without causing hypoglycemia. Its mechanism of action is only now becoming clear, although it was first introduced in 1957 in Europe and in 1995 in USA [334]. This natural compound may suppress tumor progression by modulating whole body metabolic physiology or by acting directly in cancer cells, inducing a condition similar to caloric restriction [335]. Metformin impairs malignant growth indirectly by reduction of systemic glucose and insulin levels and directly by suppressing mTOR signaling, mitochondrial glucose oxidation and/or reducing stability of HIF under hypoxic conditions [336].
The first identified metformin cellular target in cancer came from the discovery of LKB1, a major upstream activator of AMPK [337]. The study has shown that metformin acts as a growth inhibitor in a dose-dependent manner by suppressing the mTOR/S6K pathway via LKB1–AMPK interaction in breast cancer cells, being the first to demonstrate its anticancer activity. LKB1-dependent and AMPK-dependent suppression of the mTOR pathway are possibly the most potent antineoplastic effects of metformin. Some types of cancers may have inactive LKB1 and defects in the LKB1–AMPK pathway, which may potentiate the risk of metabolic transformation of pre-neoplastic cells [338].
Another described mechanism of action of metformin besides LKB1 is through inhibition of the mitochondrial electron transport chain complex I [339]. This inhibition interrupts mitochondrial respiration and decreases proton-driven synthesis of ATP, causing a cellular energetic stress state and elevation of the AMP:ATP ratio. These changes result in allosteric activation of AMPK, a major sensor of cellular energy status. Therefore, metformin decreases growth signaling due to increased AMP:ATP ratio [340]. Metformin also diminishes glucose transport by inhibiting HK2 [339].
Through inhibition of mitochondrial complex I, metformin also reduces production of reactive oxygen species, oxidative stress and DNA damage. A recent study suggested that inhibition of mitochondrial glycerophosphate dehydrogenase (mGPD) could be the primary mechanism of metformin-mediated inhibition of gluconeogenesis [341]. Furthermore, metformin is able to inhibit both EMT and OxPhos markers, inducing a striking inhibition of proliferation and colony formation of acidic melanoma cells [342] and decreasing glucose oxidation through glutamine metabolism in prostate cancer cells [343].
Besides its action in mitochondrial complex I and LKB1, metformin has presented other possible targets. Many investigations suggest direct targets like ATM and Ragulator and several indirect targets like PKA, c-Myc, DICER, p53/REDD1 and NF-κB, revealing different mechanisms of action in cancer prevention and therapy [344]. Metformin is also able to decrease IGF1–insulin receptors signaling by lowering insulin levels and AMPK-dependent phosphorylation of IRS1 and to decrease mTORC1 signaling by inactivating Ragulator, mimicking the effects of amino acid starvation [344, 345]. A recent study described how p53/REDD1 axis causes mTORC1 downregulation through AMPK-independent mechanisms. In prostate cancer cell lines with wild-type p53, metformin may upregulate REDD1-mediated mTORC1 inhibition and cell-cycle arrest [346].
Several studies point the important relationship between metformin and p53 and highlight how p53 participates in metformin mechanisms of action, especially for cancer treatment. One study pointed a negative relation between metformin and p53, showing that AMPK activation by metformin diminishes p53 protein levels and oxidative stress [347]. The same study also has shown that metformin increases the deacetylation of p53 at a SIRT1-target site, which may turn it more susceptible to MDM2-mediated degradation. However, other studies have shown a positive correlation. In breast cancer, p53 is required for metformin-induced cell growth inhibition, further consolidating metformin use as an antitumor strategy [348]. Another study has shown that exposure of HepG2, a hepatoma cell line, to low doses of metformin results in induction of cell senescence, through AMPK pathway, in a p53-dependent manner [349]. As already described, the combination of metformin and 2DG selectively enhanced cytotoxicity of doxorubicin against MCF-7 cells through p53 [249]. The knowledge coming from research regarding metformin mechanism of action pathways currently allows the development of multiple therapeutic strategies for cancer treatment.
Cell growth signaling
Epidermal Growth Factor (EGF) is a well-known growth factor that is overexpressed in many types of cancer. Thus, EGF receptor (EGFR) family has been effectively targeted using drugs such as lapatinib (tyrosine kinase inhibitor active against EGFR and HER2) and trastuzumab (a humanized antibody targeting the HER2 receptor). Evidences suggest that these agents may be useful for clinical purposes, although studies suggest that they may increase resistance to other chemotherapeutics [350]. A study has shown that lapatinib downregulates and destabilizes mutant p53 via modulation of HSF1 activity in HER2-positive breast cancer cells, suggesting therapeutic benefits of the inhibitor [351]. Another one has shown that p53 expression could predict the complete response to chemotherapy in patients with HER2-positive breast cancer treated with sequential cycles of doxorubicin and cyclophosphamide followed by trastuzumab and paclitaxel [352].
mTOR is another well-known protein that is overexpressed in several types of tumors. It is regulated by nutrient availability and its activation stimulates a metabolic program to promote cell growth [201]. Rapamycin is a drug that inhibits the mTOR complex 1 (mTORC1) and is able to enhance the antitumor effects of cisplatin in gastric cancer (Figure 5) [353]. Data also suggest that mTOR inhibition with a dual PI3K/mTOR inhibitor, NVP-BEZ235, may revert chemoresistance in other types of cancer [354]. Furthermore, rapamycin increases PUMA expression and PARP cleavage, suggesting that MDM2 suppression by rapamycin stimulates p53-mediated apoptosis [355]. Rapalogues are any chemical agents that resemble rapamycin in its capacity to inhibit mTOR enzymatic activity. The therapeutic benefit provided by mTOR inhibitors may be limited by the intrinsic ability of these compounds to stimulate autophagy and hence render established tumors resistance to chemotherapeutics [356]. Inhibitors of mTOR downstream targets, like S6K, also presented positive effects against cancer. PF-4708671, a specific inhibitor of S6K1, is able to reduce proliferation of prostate, breast and lung cancer cells [199, 357, 358].
Hypoxia-inducible factor 1 (HIF1) is a transcription factor that controls the synthesis of several glycolytic enzymes and angiogenic factors in response to hypoxia and other stress conditions, increasing glucose uptake and its oxidation to lactate in order to generate ATP through an oxygen-independent mechanism [265]. T-lymphoblastic leukemia cell lines treatment with a HIF1 inhibitor called echinomycin inhibits cancer cells growth [359]. HIF1 inhibitors, such as acriflavine (ACF) and PX-478, have generated promising results in pre-clinical studies, yet have not entered clinical development [360, 361]. ACF is able to increase the expression of p21, and reverse the phosphorylation of CDK2 in a p53 dependent mechanism [362]. Inhibition of HIF-1α by PX-478 occurs in both normoxia and hypoxia and does not require p53 [363].
The phosphatidylinositol 3-kinase (PI3K) is a lipid kinase and an upstream activator of AKT and mTOR, as previously mentioned (Figure 5). This axis is one of the most commonly deregulated signaling networks in human cancers [364], since it promotes survival, glycolytic metabolism, fatty acid synthesis and cell growth mechanisms [4]. The therapeutic perspectives related to the PI3K pathway has been recently reviewed [365]. p53 induction is blocked by the PI3K inhibitor LY294002, implicating that the PI3K pathway is a critical mediator of p53 activation. Besides, LY294002 is able to inhibit p53 stabilization and functional activation in a variety of cell types and in response to several different DNA-damaging agents [366]. PI3K pathway inhibition can increase the sensitivity to adriamycin in HER-2/neu expressing breast tumor cells in a p53 dependent way [367]. GDC-0941 and PX866 are two drugs that inhibit PI3K currently in clinical trials (ClinicalTrials.gov identifiers: NCT00876109, NCT00726583). Data suggest that advanced solid tumors, like metastatic breast and non-Hodgkin’s lymphoma, are the most promising targets of those compounds [270].
THERAPY TARGETING P53
The ability of p53 to induce cell death has been thoroughly reviewed [368, 369]; however, its impact in metabolism is less discussed, even though it has an impact in cancer therapy. Here, we make a comprehensive review of how different therapies that induce p53 expression impact tumor cell metabolism. More specifically, how these therapies modulate key metabolic genes and how metabolism may dictate whether the cell undergo senescence, autophagy or apoptosis and more importantly resistance to chemotherapy.
Inducers of p53
As a general stress sensor protein, p53 is induced by DNA damage and several conventional anticancer therapies cause DNA damage, the most relevant in cancer therapy are ionizing radiation (IR) and genotoxic drugs.
Ionizing radiation
More than 50% of the cancer patients are treated with radiotherapy [370]. Ionizing radiation generates ROS causing DNA damage, cell death and recruitment of immune cells to the tumor site [371]. Ionizing radiation induces apoptosis and senescence of fibroblasts, which show phosphorylation of p53 and increased p21 expression, DNA damage was evident and senescence markers are evident on the first day of IR and progressively increases [372]. However, senescent tumor associated fibroblasts induced by low doses of IR express high levels of metalloproteinases (MMPs), which can favor malignant lung cancer cells growth in vitro and in vivo, indicating that senescent cells may enhance the growth of adjacent malignant cells [373].
Tumors derived from cells expressing wild-type p53 are more sensitive to gamma radiation, compared to cells with mutations in the p53 gene [374, 375]. Treatment with ionizing radiation induced senescence of Glioblastoma (GBM) cells expressing wild-type p53, but not cells with mutant p53 (T98) or cells expressing HPV E6 protein [376]. PTEN transcription is regulated by p53 as mentioned before [122, 377], and its status is important to determine IR effect. IR triggered apoptosis in PTEN positive cells, while in PTEN negative cells treatment led to AKT activation and high levels of ROS, which were essential to induce senescence. Interestingly, depletion of p53 prevented IR induced senescence in these cells [378]. p53 status is important to determine induction of senescence or cell death mediated by IR, as cells expressing mutant p53 had delayed and persistent induction of p21 after IR [379]. Senescence mediated by IR depends on p53 induction of CXCR2, which is important to induce phosphorylation of P38MAPK and senescence [380]. Apoptosis induced by IR is increased by knockdown of NRF2, showing the importance of ROS induction also in the apoptotic process. Autophagy mediated by IR showed an anti-apoptotic activity through induction of MAPK 1/2 phosphorylation and NRF2 upregulation. NRF2 mediated p53 inhibition and induced p65 and Bcl2 that exerted the anti-apoptotic activity (Figure 6) [381].
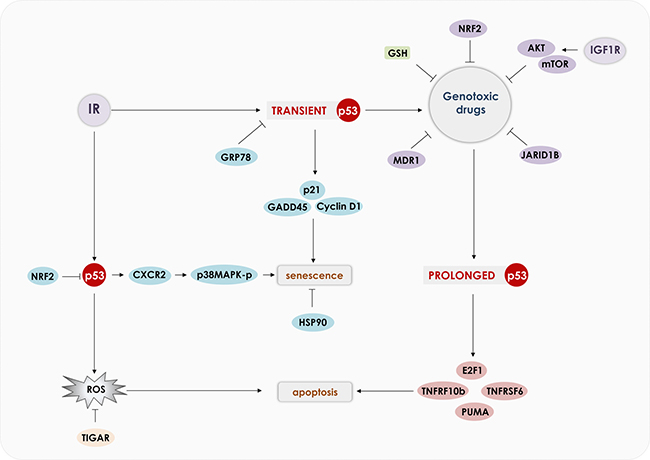
Figure 6: Mechanism of action of p53 inducers. Ionizing radiation (IR) induces p53, which increases ROS levels and induces apoptosis. TIGAR and NRF2 may prevent this activity. p53 induction by IR may also induce CXCR2, which induces phosphorylation of p38MAPK and senescence. Genotoxic drugs may have their activities modulated by NRF2, GSH, MDR1, JARID1B and the AKT/mTOR pathway, which is induced by IGF1R. Low drug concentration induces transient p53 expression and the target genes: p21, cyclin D1 and GADD45 that directs the cell to senescence, which can be inhibited by HSP90. Prolonged p53 expression induced by genotoxic drugs is involved in upregulation of E2F1, and the pro-apoptotic genes: TNFRSF10b (Killer-Dr5), TNFRSF6 (Fas-Apo) and PUMA.
Genotoxic drugs
p53 rapidly responds to DNA damage, promoting activation of cell cycle mediators, regulating metabolism and giving time to the DNA repair machinery to fix the damage, however, if the DNA damage is too extensive it will promote cell death. Several drugs cause DNA damage and p53 activation is one of the key events to trigger their actions.
Anthracyclines intercalate with DNA, inhibiting replication and transcription, they also interact with topoisomerase II leading to DNA breakage [382]. Epirubicin is an anthracycline, which induces TIGAR in a p53 dependent manner. Even though TIGAR is important to inhibit glycolysis, it also can reduce epirubicin toxicity, by reducing ROS levels, autophagy and apoptosis [383],showing that TIGAR induction by p53 can have pro-survival activity [384, 385]. The DNA damaging agents: etoposide and the anthracycline doxorubicin target OxPhos, promoting mitochondrial biogenesis, increasing ROS and inducing apoptosis, which are reduced by p53 deficiency [386]. Drug concentration is important to determine senescence or apoptosis. Low concentrations of doxorubicin induced senescence with increased genomic instability and increased expression of p53, p21 and Cyclin D1 [387]. Also, p21 was shown to be important to induce senescence and at the same time to reduce apoptosis mediaed by IR and doxorubicin [388]. Events underlying senescence mediated by doxorubicin are increased ROS levels, transient p53 activation, sustained p21 and ATM/ATR activation; while low and prolonged p53 and E2F1 upregulation with absence of p21 occurred in apoptotic cells [389]. Therefore p53 and p21 act as positive regulators of senescence, but are also not sufficient or absolutely required for doxorubicin mediated senescence [390]. Doxorubicin induced senescence dependent on p53 and p21 activation can be accelerated by inhibition of Heat shock protein 90 (HSP90), a chaperone that participates in DNA damage response [391]. Therapies that inhibit HSP90 may increase doxorubicin activity [391]. During senescence or apoptosis there is a different expression pattern mediated by p53. In normal senescent fibroblasts p53 can be found in the promoter of p21 and GADD45; after DNA damage mediated by doxorubicin, p53 binds to promoters of apoptotic genes like TNFRSF10b (DR5), TNFRSF6 (Fas-Apo) and PUMA [392].
Cisplatin (CPPD) is used in cancer therapy since 1971, it forms adducts with DNA and inhibits replication, transcription and cause DNA damage. It is used for treatment of several types of cancer; however, development of drug resistance poses as a major limitation for its wider use [393]. Cisplatin induced senescence in hepatoma cells is dependent on p53 and p21 activation and ROS induction [394, 395]. Different players modulate CPPD-induced senescence.IGF1R can promote proliferation and chemotherapy resistance and its inhibition increases p53 mediated apoptosis induced by CPPD and reduces senescence [396]. Glucose-regulated protein 78kda (GRP78), a chaperone localized in the ER, is an important inhibitor of senescence mediated by CPPD, responsible for reduced p53 expression and Ca2+ release inhibition from endoplasmic reticulum [397].
Drug resistance to CPPD is an important issue in the clinic, and is correlated to absence of apoptosis and is not due to autophagy, in fact pro-autophagic stimuli, such as inhibition of mTOR or AKT, induce cell death [398]. Rapamycin-insensitive companion of mTOR (Rictor) is a component of mTORC2, it is downregulated by CPPD in ovarian cancer cells and its knockdown in chemo-resistant cell lines sensitizes the cells to apoptosis dependent on p53 status, showing that mTOR pathway may be important in resistance to cisplatin [399]. Cells resistant to cisplatin were shown to be more sensitive to Nutlin, a drug responsible for disruption of MDM2 and p53 interaction, leading to p53 stabilization [158]. CPPD resistant cell lines also show increased activity of IGF1R/AKT signaling. Inhibitors of IGF1R or AKT induced apoptosis in CPPD-resistant cell lines in combination with Nutlin-3 by increasing p53 levels and inhibiting pro-survival autophagy [400]. UT-SSC26A is a cell line with enhanced chemoresistance to CPPD, it expresses a truncated p53 [401]. These cell lines have an upregulated expression of ABC transporters (ABCC2 and ABCG2), increased metabolic activity and GSH levels. Several chemotherapeutic agents, including CDDP, doxorubicin, methotrexate and vincristine can form conjugates with glutathione, which can be exported out of the cell by ABCC2, therefore inhibition of the ABC transporters recovers sensitivity to CPPD [402]. GSH may act not only as a cofactor in MRP2-mediated CPPD efflux, it may as well have a redox-regulating cytoprotective activity and function as a copper chelator [403].
JARID1B is a histone H3K4 demethylase [404]. It is interesting that multi-resistant cells show slow cycling properties, increased OxPhos and increased expression of JARID1B; direct knockdown of JARID1B or inhibition of mitochondrial respiration blocked the JARID1B subpopulation and sensitized the cells to chemotherapeutic drugs [405]. JARID1B has also been shown to inhibit p53 expression and JARID1B inhibition suppressed proliferation and invasion [406]. Expression levels of JARID1B were positively correlated with chemotherapy resistance in ovarian cancer patients [407].
As mentioned before mutant p53 has been shown to induce MDR1. Mutant p53 induces expression of MDR1 also through NF-κB upregulation, increased P-gp activity and showed increased resistance to doxorubicin [408]. Another gene involved in chemoresistance to cisplatin is NRF2. It was shown that mutant p53 increases NRF2 levels, which increases Bcl2 and Bcl-x, showing an unfavorable outcome after treatment with CPPD [105]. It is interesting that it had been observed that overexpression of either wild-type p53 or mutant p53 was shown to increase resistance to cisplatin, in this case cytoplasmic p53 inhibited caspase 9 activity and reduced chemo sensitivity [409]. Figure 6 shows how different agents may inhibit IR and genotoxic drugs activity and how different levels of p53 induces senescence or apoptosis.
Inhibitors of MDM2
MDM2 was firstly shown to interact with p53 and inhibit its transcriptional activity [410]. Their interaction happens through their N-terminal regions [411], on p53 at the residues F19, W23 and L26 [412]. The protein levels of MDM2 and p53 are tightly linked, as p53 activates the expression of MDM2 [413], which in turn acts as an E3 ubiquitin ligase, responsible to drive p53 degradation through proteasome, creating an auto regulated feedback loop [414, 415]. MDM2 induces ubiquitination of lysine residues in the p53 C-terminal region and alteration of these lysine residues to arginine generated a protein with high transcriptional activity and resistant to MDM2-mediated degradation [416]. Nutlins are cis-Imidazole molecules that mimic the p53 residues F19, W23 and L26 and have been shown to interact and inhibit MDM2 [417]. Some Nutlin derived compounds have reached clinical trials, RG7112 was developed by Roche and was tested in phase I clinical trials for different types of cancers, including, liposarcoma, soft tissue sarcoma, myelogenous leukemia and hematologic neoplasms, resulting in activation of p53, p21 and apoptosis induction [418, 419]. As mentioned before, its combination with asparaginase resulted in stronger antitumor activity [314].
Combination of Nutlin-3 and PI-103 (inhibitor of the PI3K/AKT/mTOR pathway) increased Bax activation, caspase-3 cleavage and apoptosis of AML cells, even though induction of several p53 target genes, like p21, NOXA, Bcl2, MDM2 and Survivin were reduced due to mTOR inactivation and attenuation of p53 protein synthesis. Nutlin-3 also cooperates with PI-103 in the blockage of p70S6K and 4E-BP1 phosphorylation, which also contributed to the reduction in protein synthesis [420]. Mantle cell lymphoma (MCL) cells with wild-type p53 status treated with only Nutlin-3 showed increased levels of p53 and activated p53 (p-ser15-p53), which induced phosphorylation of AMPK and decreased phosphorylation of rpS6, 4E-BP1, p70S6K and AKT. Cells with p53 mutated status and treated with Nutlin-3 did not show the same trend [421]. The intensity of p53 induction had different impacts in the modulation of the mTOR pathway [422]. This differential activity may modulate senescence dependent and independent of mTOR.
Treatment with Nutlin-3 induced cellular quiescence of human fibrosarcoma cells and non-tumorigenic human fibroblasts [423], inhibiting the phosphorylation of rpS6 and 4E-BP1, and combination of rapamycin and Nutlin-3a also suppressed senescence [424]. This suppression is mediated by TSC2, a p53 target gene that negatively regulates mTOR, indicating how p53 can inhibit mTOR and prevent mTOR mediated senescence [425]. In melanoma cell lines Nutlin-3a failed to inhibit mTOR and the cells underwent senescence, which could be converted to quiescence by combining Nutlin-3a and Rapamycin, which inhibited mTOR [425].
Glioblastoma multiforme (GBM), renal carcinoma and breast cancer cells treated with Nutlin-3 entered senescence in a mechanism that may be dependent on active mTOR [426–428]. Adult T-cell leukemia (ATL) cells treated with Nutlin-3 also undergo senescence, even in the absence of p16INK4a and p14ARF, important regulators of senescence. The p53 target genes, p21, PIG3 and TIGAR were induced by Nutlin-3, knockdown of TIGAR reduced apoptosis and senescence mediated by Nutlin-3, indicating that TIGAR may also be a mediator of senescence [429]. In leukemic cells Nutlin-3 was shown to down-regulate E2F-1 [430], which was shown to inhibit cellular senescence [431], indicating another pathway for senescence induction mediated by Nutlin-3. Nutlin-3 combined with metformin showed increased induction of senescence. Metformin up-regulated p53 and increased phosphorylation of AMPK and decreased in phosphorylation of mTOR [348]. Combination of Nutlin-3 and IR induced senescence of lung cancer cells [432].
Overall, the data from the literature indicate that mTOR plays an important role in senescence induction, and p53 modulation is an essential component in different therapies for the outcome of cell quiescence or senescence. High levels of p53 inhibit mTOR and prevent senescence, while medium levels of p53 do not inhibit mTOR and induces senescence in some cell lines [140, 422], while in other cell lines p53 activation induced senescence through different mechanisms [429, 431].
Nutlins can also induce autophagy. It has been suggested that autophagy inhibits apoptosis and promotes survival in cells treated with Nutlin [158]. However, other reports indicate that autophagy induced by Nutlin-3a can be pro-apoptotic and dependent of AMPK activation, which promoted autophagy through phosphorylation of ULK1 [433]. The pro-apoptotic autophagy was shown to be dependent on p53 positive status [434]. Metabolism plays an essential role in protective autophagy. Nutlin sensitive cells show increased activation of AMPK, induction of ROS, inhibition of glycolysis, mTOR and autophagy. Glycolysis plays a role to limit ROS induction, regulating expression of ATG and favoring autophagic flux. Therefore in cell lines that p53 do not limit glycolysis there is an increased autophagic flux and resistance to apoptosis [158]. ROS has also been shown to inhibit autophagy by decreasing ULK1 expression, which is induced by p53 after phosphorylation (S392) mediated by p70S6K [161]. Oxidants seem to play an essential role in the inhibition of autophagy and induction of senescence and apoptosis.
Modulators of mutant p53
Prima1 and Prima1-met (APR-246) are small molecules that rescue mutant p53 activity [435, 436], there are already early phase 1 clinical trials using Prima1-met for Esophageal carcinoma, acute myeloid leukemia, myelodysplastic syndrome and myeloproliferative neoplasm (ClinicalTrials.gov identifiers: NCT02999893, NCT03072043).
Prima1 and CP-31398 have shown chemopreventive activity by modulating p53 in tobacco carcinogen-induced lung adenocarcinoma model [437]. Proteomic analysis of breast carcinoma cell lines expressing mutants p53A278P, p53R280K or p53M385T treated with Prima1-met, showed differential expression of Annexin A1, Annexin A2, bifunctional methylenetetrahydrofolate dehydrogenase (MTHFD2), L-lactate dehydrogenase (LDH), GAPDH, malate dehydrogenase (MDH2) and voltage-dependent anion-selective channel protein 2 (VDAC2). GAPDH, LDH, MTHFD2, and MFH2 are responsible for regulating glycolysis progression, but are also linked to apoptosis, as apoptosis is ATP dependent, demanding high energy levels. Annexins were associated with activation of caspases, p38, and JNK signaling, while VDAC2 is associated with mitochondrial permeabilization, indicating that Prima1-met is able to activate genes involved in cell death and metabolism [438]. Prima1-met was also shown to be associated with down-regulation of O6-methylguanine-DNA methyltransferase (MGMT), which removes methyl adducts from guanine and is a major determinant of resistance to alkylating agents. In a glioblastoma cell line, low levels of Prima1-met induced senescence, and high concentrations induced massive cell death [439]. Prima 1 induced degradation of mutant p53 and autophagy in cell lines expressing wild type or mutant p53 [440, 441].
Hypoxia increases Prima-1 activity in breast cancer cells expressing mutant p53, possibly through oxidative stress, indicating that Prima-1 may prevent hypoxia-mediated chemoresistance [442]. The acute myeloid cell line KBM3 with mutant p53 treated with Prima1-met presented increased ROS levels, causing activation of heme oxygenase-1 (HMOX1) mediated by NRF2. Inhibitors of PI3K (wortmannin) and mTOR (rapamycin) prevented upregulation of HMOX1, increasing antitumor effects mediated by Prima1-met [443]. Prima1-met depleted GSH and induced ROS, independently of p53 [444]. ROS induction can also be achieved by modulation of TrxR1, an important enzyme that catalyzes thioredoxin that can be modulated by Prima1-met to exert a NADPH oxidase activity, an inductor of ROS, indicating one possible mechanism of how Prima1-met can induce ROS in a p53 independent manner [445]. Besides TrxR1 inhibition, Prima1-met may also inhibit other antioxidant enzymes, like Prx3 and GPx-1 [446]. Prima1 can have antitumor effects in cells with different p53 status, indicating that it has other mechanisms of action independent of mutant p53, but p53 status may modulate different outcomes, inducing apoptosis or necrosis [447, 448].
Mutant p53 have been shown to induce chemoresistance as discussed before. Prima1-met is also able to restore sensitivity to cisplatin and doxorubicin and 5-FU resistant cell lines expressing mutant p53. Its active form methylene quinuclidinone is able to directly interact and inhibit glutathione [449, 450] and prevents GSH mediated drug inhibition [402, 403].
p53 gene therapy and metabolism
In the early 1990s direct transfer of the p53 gene showed strong anti-tumor effects [451–456]. Soon viral vectors were employed to transfer p53 gene in clinical trials for patients with different types of cancer, using retroviral [457] or adenoviral vectors expressing p53 [458, 459]. Different adenoviral vectors expressing p53 (Advexin, Gendicine and SCH58500) have been tested in clinical trials, but only Gendicine is regularly used in the clinics [458, 460–461]. Differently from the approaches described before, this technique allows direct expression of the wild-type p53, without off target effects. At the same time, it can have strong antitumor effects even in cells with mutant or negative p53 status.
Transfer of the p53 gene conferred strong anti-tumor activity to a conditionally replicating adenovirus, this oncolytic adenovirus suppressed expression of p21 and showed strong induction of autophagy through DRAM activation [462]. ROS plays an essential role in p53 mediated cell death. First generation Adenovirus-p53 (Ad-p53) activates cell death through ROS induction and GSH depletion, which could be enhanced by combination with 2-deoxyglucose. This apoptotic activity can be partially inhibited by adenoviral vectors expressing catalase or glutathione peroxidase [463, 464]. Bcl-XL expression also prevented generation of ROS and inhibited induction of senescence mediated by an Ad-p53 [465]. Ad-catalase was shown to inhibit apoptosis mediated by DNA damaging agents; catalase increased p53 degradation and decreased its phosphorylation at serine 20 [466]. Higher levels of p53 mediated by an auto-regulated promoter induced ROS, that participated in prostate cancer cell lines apoptosis, even in p53 negative cell lines [94].
Ad-p53 has also been shown to increase drug sensitivity to 5-FU, doxorubicin, cisplatin, methotrexate, paclitaxel, carboplatin and induce cell death in chemoresistant cell lines. This effect is the result of the reduced expression of MDR1 [467–470]. It´s interesting to highlight that an Ad-p53 had a stronger effect in cisplatin and doxorubicin drugs resistant cell lines compared to sensitive cell lines. Drug resistant cell lines displayed higher transduction efficiency though [471].
Other p53 target genes have also been employed, like Ad-Puma, which induced ovarian cancer cells death, generating ROS, but as a protective mechanism also activated the NRF2/HMOX1 pathway [472]. Ad-p21 is capable to induce senescence, nevertheless, cells expressing p53 show increased levels of ROS and apoptosis, and the magnitude of ROS is important to determine cell fate between senescence and apoptosis [473].
CONCLUDING REMARKS
Like Janus, p53 has two faces, may be an oncogene or a tumor suppressor. In the beginning its mutant form rendered it an oncogenic function. However, wild-type p53 was revealed and aroused the notion of the tumor suppressors, turning p53 into the guardian of the genome. This transcription factor regulates genes involved in almost all hallmarks of cancer. Despite the fact that modulation of metabolism is one of the most underestimated activities of p53, it is a key factor to promote tumor survival, playing an important role in senescence, autophagy, apoptosis and resistance to chemotherapy.
Abbreviations
2PG: 2-phosphoglycerate; 3PG: 3-phosphoglycerate; 3PO: 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one; 3PHP: 3-phosphohydroxypyruvate; ALDH4: aldehyde dehydrogenase 4; AKT: AKT serine/threonine kinase; ASS1: argininosuccinate synthetase 1; ASNS: asparagine synthetase; ATG12: Autophagy Related 12; ACLY: ATP citrate lyase; BPTES: Bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl) ethyl sulfide; BCL2: BCL2, apoptosis regulator; PUMA: BCL2 binding component 3; BAX: BCL2 associated X, apoptosis regulator; BECN1: beclin 1; MTHFD2: bifunctional methylene tetrahydrofolate dehydrogenase; BRCA1: BRCA1, DNA repair associated; CPT1C: carnitine palmitoyltransferase 1C; CPPD: Cisplatin; p21: cyclin dependent kinase inhibitor 1A; p16INK4a/p14ARF: Cyclin Dependent Kinase Inhibitor 2A; COX: cytochrome C oxidase complex; SCO2: cytochrome C oxidase 2; CXCR2: C-X-C Motif Chemokine Receptor 2; P5C: Delta-1-pyrroline-5-carboxylate; DRAM1: DNA Damage Regulated Autophagy Modulator 1; E2F-1: E2F Transcription Factor 1; EGF: Epidermal Growth Factor; TNFRSF6: Fas cell surface death receptor; EGCG: epigallocatechin gallate; FASN: Fatty acid synthase; FDG: fluoro-2-deoxy-glucose; F16BP: fructose-1,6-bisphosphate; F26BP: fructose-2,6-bisphosphate; GOF: gain of function; G6PDH: glucose 6-phosphate dehydrogenase; GDH: glutamate dehydrogenase; GLS: glutaminase; GSH: glutathione; GPX1: glutathione peroxidase; HSP90: Heat shock protein 90; HMOX1: Heme Oxygenase 1; HIF-1: hypoxia-induced factor 1; IGFs: insulin-like growth factors; IDH1: isocitrate dehydrogenase; LPIN1: lipin 1; LDH: L-lactate dehydrogenase; JARID1B: Lysine Demethylase 5B; MLL1: Lysine Methyltransferase 2A; MLL2: Lysine Methyltransferase 2D; MOZ: Lysine Acetyltransferase 6A; ME1: malic enzyme 1; MCD: malonyl CoA decarboxylase; MDH2: malate dehydrogenase; mTOR: mechanistic Target Of Rapamycin kinase; MDM2: MDM2 proto-oncogene; MAPK: mitogen-activated protein kinase 1; Mn-SOD: Mn superoxide dismutase; MCT4: Monocarboxylate transporter 4; NOX1: NADPH oxidase; MDR1: Multi-drug resistance; NRF2: NF-E2 related factor 2; OxPhos: oxidative phosphorylation; PANK1: panthothene kinase 1; PGC-1α: Peroxisome proliferator-activated receptor γ Coactivator 1-α; PET: Positron Emission Tomography; NOXA: phorbol-12-myristate-13-acetate-induced protein 1; PTEN: phosphatase and tensin homolog; PI3K: phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit beta; PEP: phosphoenolpyruvate; PHGDH: phosphoglycerate dehydrogenase; PGAM1 or PGM1: phosphoglycerate mutase 1; POX: proline oxidase; PFK-1: proline dehydrogenase Phosphofructokinase; PRODH: proline dehydrogenase; PIG6: p53-induced gene 6; PIG3: p53-induced gene 3; PYCR1: pyrroline-5-carboxylate reductase; PDK2: pyruvate dehydrogenase kinase 2; PK: pyruvate kinase; PKM2: pyruvate kinase M2; Rb: Retinoblastoma transcriptional corepressor 1; ROS: reactive oxygen species; S6K1: ribosomal protein S6 kinase B1; p65: relA Proto-Oncogene, NF-KB Subunit; SESN: Sestrin; GLUT1: Solute Carrier Family 2 Member 1; Glut 4: Solute Carrier Family 2 Member 4; TIGAR: TP53-induced glycolysis and apoptosis regulator; TRAIL: TNF Superfamily Member 10; DR5: TNF receptor superfamily member 10b; TrxR1: Thioredoxin Reductase 1; TKTL1: Transketolase-like protein 1; TCA: tricarboxylic acid; TSC2: Tuberous Sclerosis 2; ULK1: unc-51 like autophagy activating kinase 1; VDAC2: voltage-dependent anion-selective channel protein 2.
ACKNOWLEDGMENTS
Financial support was received from the São Paulo Research Foundation, FAPESP (FMS 2012/13558-7; ICBP, 2015/00311-1; APM, 2017/04269-5, FRS 2015/16601-9; MGM, 2011/14416-9, 2016/25139-0; RET, 2011/21256-8). Financial support from CNPq (FMS, 447553/2014-3; RET, 442738/2014-5).
CONFLICTS OF INTEREST
None.
REFERENCES
1. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000; 100:57–70. https://doi.org/10.1016/S0092-8674(00)81683-9.
2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74. https://doi.org/10.1016/j.cell.2011.02.013.
3. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927; 8:519–30. https://doi.org/10.1085/jgp.8.6.519. PMID:19872213.
4. Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2012; 2:881–98. https://doi.org/10.1158/2159-8290.CD-12-0345.
5. Jadvar H, Alavi A, Gambhir SS. 18F-FDG uptake in lung, breast, and colon cancers: molecular biology correlates and disease characterization. J Nucl Med. 2009; 50:1820–27. https://doi.org/10.2967/jnumed.108.054098.
6. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008; 7:11–20. https://doi.org/10.1016/j.cmet.2007.10.002.
7. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324:1029–33. https://doi.org/10.1126/science.1160809.
8. Lu H, Forbes RA, Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem. 2002; 277:23111–15. https://doi.org/10.1074/jbc.M202487200.
9. Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, Bailey K, Balagurunathan Y, Rothberg JM, Sloane BF, Johnson J, Gatenby RA, Gillies RJ. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013; 73:1524–35. https://doi.org/10.1158/0008-5472.CAN-12-2796.
10. Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, Cova A, Canese R, Jachetti E, Rossetti M, Huber V, Parmiani G, Generoso L, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012; 72:2746–56. https://doi.org/10.1158/0008-5472.CAN-11-1272.
11. Brand KA, Hermfisse U. Aerobic glycolysis by proliferating cells: a protective strategy against reactive oxygen species. FASEB J. 1997; 11:388–95.
12. Ruckenstuhl C, Büttner S, Carmona-Gutierrez D, Eisenberg T, Kroemer G, Sigrist SJ, Fröhlich KU, Madeo F. The Warburg effect suppresses oxidative stress induced apoptosis in a yeast model for cancer. PLoS One. 2009; 4:e4592. https://doi.org/10.1371/journal.pone.0004592.
13. Newsholme EA, Crabtree B, Ardawi MS. The role of high rates of glycolysis and glutamine utilization in rapidly dividing cells. Biosci Rep. 1985; 5:393–400. https://doi.org/10.1007/BF01116556.
14. Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem. 1979; 254:2669–76.
15. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. 2007; 104:19345–50. https://doi.org/10.1073/pnas.0709747104.
16. Vaughn AE, Deshmukh M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat Cell Biol. 2008; 10:1477–83. https://doi.org/10.1038/ncb1807.
17. Anastasiou D, Cantley LC. Breathless cancer cells get fat on glutamine. Cell Res. 2012; 22:443–46. https://doi.org/10.1038/cr.2012.5.
18. Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005; 15:300–08. https://doi.org/10.1016/j.semcancer.2005.04.009.
19. Zwerschke W, Mazurek S, Massimi P, Banks L, Eigenbrodt E, Jansen-Dürr P. Modulation of type M2 pyruvate kinase activity by the human papillomavirus type 16 E7 oncoprotein. Proc Natl Acad Sci USA. 1999; 96:1291–96. https://doi.org/10.1073/pnas.96.4.1291.
20. Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008; 452:181–86. https://doi.org/10.1038/nature06667.
21. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011; 11:85–95. https://doi.org/10.1038/nrc2981.
22. Ye J, Mancuso A, Tong X, Ward PS, Fan J, Rabinowitz JD, Thompson CB. Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc Natl Acad Sci USA. 2012; 109:6904–09. https://doi.org/10.1073/pnas.1204176109.
23. Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, Vousden KH. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature. 2013; 493:542–46. https://doi.org/10.1038/nature11743.
24. Labuschagne CF, van den Broek NJ, Mackay GM, Vousden KH, Maddocks OD. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Reports. 2014; 7:1248–58. https://doi.org/10.1016/j.celrep.2014.04.045.
25. Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, Sasaki AT, Anastasiou D, Mullarky E, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011; 43:869–74. https://doi.org/10.1038/ng.890.
26. Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, Chen WW, Barrett FG, Stransky N, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011; 476:346–50. https://doi.org/10.1038/nature10350.
27. Amelio I, Cutruzzolá F, Antonov A, Agostini M, Melino G. Serine and glycine metabolism in cancer. Trends Biochem Sci. 2014; 39:191–98. https://doi.org/10.1016/j.tibs.2014.02.004.
28. Nilsson R, Jain M, Madhusudhan N, Sheppard NG, Strittmatter L, Kampf C, Huang J, Asplund A, Mootha VK. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat Commun. 2014; 5:3128. https://doi.org/10.1038/ncomms4128.
29. Liu Y, Borchert GL, Donald SP, Diwan BA, Anver M, Phang JM. Proline oxidase functions as a mitochondrial tumor suppressor in human cancers. Cancer Res. 2009; 69:6414–22. https://doi.org/10.1158/0008-5472.CAN-09-1223.
30. Kress M, May E, Cassingena R, May P. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J Virol. 1979; 31:472–83.
31. Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979; 278:261–63. https://doi.org/10.1038/278261a0.
32. Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 1979; 17:43–52. https://doi.org/10.1016/0092-8674(79)90293-9.
33. Melero JA, Stitt DT, Mangel WF, Carroll RB. Identification of new polypeptide species (48-55K) immunoprecipitable by antiserum to purified large T antigen and present in SV40-infected and -transformed cells. Virology. 1979; 93:466–80. https://doi.org/10.1016/0042-6822(79)90250-2.
34. Eliyahu D, Raz A, Gruss P, Givol D, Oren M. Participation of p53 cellular tumour antigen in transformation of normal embryonic cells. Nature. 1984; 312:646–49. https://doi.org/10.1038/312646a0.
35. Jenkins JR, Rudge K, Currie GA. Cellular immortalization by a cDNA clone encoding the transformation-associated phosphoprotein p53. Nature. 1984; 312:651–54. https://doi.org/10.1038/312651a0.
36. Parada LF, Land H, Weinberg RA, Wolf D, Rotter V. Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature. 1984; 312:649–51. https://doi.org/10.1038/312649a0.
37. Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989; 57:1083–93. https://doi.org/10.1016/0092-8674(89)90045-7.
38. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MD, Miller CA, Welch JS, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013; 502:333–39. https://doi.org/10.1038/nature12634.
39. Rivlin N, Brosh R, Oren M, Rotter V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer. 2011; 2:466–74. https://doi.org/10.1177/1947601911408889.
40. Humpton TJ, Vousden KH. Regulation of Cellular Metabolism and Hypoxia by p53. Cold Spring Harb Perspect Med. 2016; 6:a026146. https://doi.org/10.1101/cshperspect.a026146.
41. Lane DP. Cancer. p53, guardian of the genome. Nature. 1992; 358:15–16. https://doi.org/10.1038/358015a0.
42. Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, Liu J, Zhao XD, Chew JL, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006; 124:207–19. https://doi.org/10.1016/j.cell.2005.10.043.
43. Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, Sidow A, Attardi LD. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. Cold Spring Harbor Laboratory Press. 2013; 27:1016–31. https://doi.org/10.1101/gad.212282.112.
44. Wang B, Niu D, Lam TH, Xiao Z, Ren EC. Mapping the p53 transcriptome universe using p53 natural polymorphs. Cell Death Differ. 2014; 21:521–32. https://doi.org/10.1038/cdd.2013.132.
45. Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006; 126:107–20. https://doi.org/10.1016/j.cell.2006.05.036.
46. Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, Yang X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. 2011; 13:310–16. https://doi.org/10.1038/ncb2172.
47. Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L, Guo X, Chang LJ, Zhang Y, You MJ, Koochekpour S, Saleem M, Huang H, et al. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Reports. 2014; 8:1461–74. https://doi.org/10.1016/j.celrep.2014.07.053.
48. Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, Martinez D, Carnero A, Beach D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005; 65:177–85.
49. Hitosugi T, Zhou L, Elf S, Fan J, Kang HB, Seo JH, Shan C, Dai Q, Zhang L, Xie J, Gu TL, Jin P, Alečković M, et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell. 2012; 22:585–600. https://doi.org/10.1016/j.ccr.2012.09.020.
50. Ruiz-Lozano P, Hixon ML, Wagner MW, Flores AI, Ikawa S, Baldwin AS Jr, Chien KR, Gualberto A. p53 is a transcriptional activator of the muscle-specific phosphoglycerate mutase gene and contributes in vivo to the control of its cardiac expression. Cell Growth Differ. 1999; 10:295–306.
51. Jiang P, Du W, Mancuso A, Wellen KE, Yang X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature. 2013; 493:689–93. https://doi.org/10.1038/nature11776.
52. Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004; 64:2627–33. https://doi.org/10.1158/0008-5472.CAN-03-0846.
53. Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. 2008; 10:611–18. https://doi.org/10.1038/ncb1724.
54. Zawacka-Pankau J, Grinkevich VV, Hünten S, Nikulenkov F, Gluch A, Li H, Enge M, Kel A, Selivanova G. Inhibition of glycolytic enzymes mediated by pharmacologically activated p53: targeting Warburg effect to fight cancer. J Biol Chem. 2011; 286:41600–15. https://doi.org/10.1074/jbc.M111.240812.
55. Contractor T, Harris CR. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012; 72:560–67. https://doi.org/10.1158/0008-5472.CAN-11-1215.
56. Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, Hu W, Feng Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc Natl Acad Sci USA. 2011; 108:16259–64. https://doi.org/10.1073/pnas.1113884108.
57. Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science. 2006; 312:1650–53. https://doi.org/10.1126/science.1126863.
58. Ma W, Sung HJ, Park JY, Matoba S, Hwang PM. A pivotal role for p53: balancing aerobic respiration and glycolysis. J Bioenerg Biomembr. 2007; 39:243–46. https://doi.org/10.1007/s10863-007-9083-0.
59. Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y, Sugano S, Sato E, Nagao T, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA. 2010; 107:7461–66. https://doi.org/10.1073/pnas.1002459107.
60. Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA. 2010; 107:7455–60. https://doi.org/10.1073/pnas.1001006107.
61. Wang SJ, Gu W. To be, or not to be: functional dilemma of p53 metabolic regulation. Curr Opin Oncol. 2014; 26:78–85. https://doi.org/10.1097/CCO.0000000000000024.
62. Jun DY, Park HS, Lee JY, Baek JY, Park HK, Fukui K, Kim YH. Positive regulation of promoter activity of human 3-phosphoglycerate dehydrogenase (PHGDH) gene is mediated by transcription factors Sp1 and NF-Y. Gene. 2008; 414:106–14. https://doi.org/10.1016/j.gene.2008.02.018.
63. Ou Y, Wang SJ, Jiang L, Zheng B, Gu W. p53 Protein-mediated regulation of phosphoglycerate dehydrogenase (PHGDH) is crucial for the apoptotic response upon serine starvation. J Biol Chem. 2015; 290:457–66. https://doi.org/10.1074/jbc.M114.616359.
64. Raimondi I, Ciribilli Y, Monti P, Bisio A, Pollegioni L, Fronza G, Inga A, Campomenosi P. P53 family members modulate the expression of PRODH, but not PRODH2, via intronic p53 response elements. PLoS One. 2013; 8:e69152. https://doi.org/10.1371/journal.pone.0069152.
65. Phang JM, Donald SP, Pandhare J, Liu Y. The metabolism of proline, a stress substrate, modulates carcinogenic pathways. Amino Acids. 2008; 35:681–90. https://doi.org/10.1007/s00726-008-0063-4.
66. Sanchez-Macedo N, Feng J, Faubert B, Chang N, Elia A, Rushing EJ, Tsuchihara K, Bungard D, Berger SL, Jones RG, Mak TW, Zaugg K. Depletion of the novel p53-target gene carnitine palmitoyltransferase 1C delays tumor growth in the neurofibromatosis type I tumor model. Cell Death Differ. 2013; 20:659–68. https://doi.org/10.1038/cdd.2012.168.
67. Liu Y, He Y, Jin A, Tikunov AP, Zhou L, Tollini LA, Leslie P, Kim TH, Li LO, Coleman RA, Gu Z, Chen YQ, Macdonald JM, et al. Ribosomal protein-Mdm2-p53 pathway coordinates nutrient stress with lipid metabolism by regulating MCD and promoting fatty acid oxidation. Proc Natl Acad Sci USA. 2014; 111:E2414–22. https://doi.org/10.1073/pnas.1315605111.
68. Assaily W, Rubinger DA, Wheaton K, Lin Y, Ma W, Xuan W, Brown-Endres L, Tsuchihara K, Mak TW, Benchimol S. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol Cell. 2011; 44:491–501. https://doi.org/10.1016/j.molcel.2011.08.038.
69. Wang SJ, Yu G, Jiang L, Li T, Lin Q, Tang Y, Gu W. p53-Dependent regulation of metabolic function through transcriptional activation of pantothenate kinase-1 gene. Cell Cycle. 2013; 12:753–61. https://doi.org/10.4161/cc.23597.
70. Böhlig L, Friedrich M, Engeland K. p53 activates the PANK1/miRNA-107 gene leading to downregulation of CDK6 and p130 cell cycle proteins. Nucleic Acids Res. 2011; 39:440–53. https://doi.org/10.1093/nar/gkq796.
71. Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991; 51:794–98.
72. Zhou Y, Hileman EO, Plunkett W, Keating MJ, Huang P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood. 2003; 101:4098–104. https://doi.org/10.1182/blood-2002-08-2512.
73. Kamiguti AS, Serrander L, Lin K, Harris RJ, Cawley JC, Allsup DJ, Slupsky JR, Krause KH, Zuzel M. Expression and activity of NOX5 in the circulating malignant B cells of hairy cell leukemia. J Immunol. 2005; 175:8424–30. https://doi.org/10.4049/jimmunol.175.12.8424.
74. Patel BP, Rawal UM, Dave TK, Rawal RM, Shukla SN, Shah PM, Patel PS. Lipid peroxidation, total antioxidant status, and total thiol levels predict overall survival in patients with oral squamous cell carcinoma. Integr Cancer Ther. 2007; 6:365–72. https://doi.org/10.1177/1534735407309760.
75. Kumar B, Koul S, Khandrika L, Meacham RB, Koul HK. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 2008; 68:1777–85. https://doi.org/10.1158/0008-5472.CAN-07-5259.
76. Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J. Role of oxygen radicals in DNA damage and cancer incidence. Mol Cell Biochem. 2004; 266:37–56. https://doi.org/10.1023/B:MCBI.0000049134.69131.89.
77. Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007; 39:44–84. https://doi.org/10.1016/j.biocel.2006.07.001.
78. Roy K, Wu Y, Meitzler JL, Juhasz A, Liu H, Jiang G, Lu J, Antony S, Doroshow JH. NADPH oxidases and cancer. Clin Sci (Lond). 2015; 128:863–75. https://doi.org/10.1042/CS20140542.
79. Luo W, Semenza GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab. 2012; 23:560–66. https://doi.org/10.1016/j.tem.2012.06.010.
80. Wong N, De Melo J, Tang D. PKM2, a Central Point of Regulation in Cancer Metabolism. Int J Cell Biol. 2013; 2013:242513. https://doi.org/10.1155/2013/242513.
81. Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS, Thomas CJ, Vander Heiden MG, Cantley LC. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011; 334:1278–83. https://doi.org/10.1126/science.1211485.
82. Liu B, Chen Y, St Clair DK. ROS and p53: a versatile partnership. Free Radic Biol Med. 2008; 44:1529–35. https://doi.org/10.1016/j.freeradbiomed.2008.01.011.
83. Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005; 11:1306–13. https://doi.org/10.1038/nm1320.
84. Tan M, Li S, Swaroop M, Guan K, Oberley LW, Sun Y. Transcriptional activation of the human glutathione peroxidase promoter by p53. J Biol Chem. 1999; 274:12061–66. https://doi.org/10.1074/jbc.274.17.12061.
85. Hussain SP, Amstad P, He P, Robles A, Lupold S, Kaneko I, Ichimiya M, Sengupta S, Mechanic L, Okamura S, Hofseth LJ, Moake M, Nagashima M, et al. p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res. 2004; 64:2350–56. https://doi.org/10.1158/0008-5472.CAN-2287-2.
86. Yoon KA, Nakamura Y, Arakawa H. Identification of ALDH4 as a p53-inducible gene and its protective role in cellular stresses. J Hum Genet. 2004; 49:134–40. https://doi.org/10.1007/s10038-003-0122-3.
87. Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997; 389:300–05. https://doi.org/10.1038/38525.
88. Porté S, Valencia E, Yakovtseva EA, Borràs E, Shafqat N, Debreczeny JE, Pike AC, Oppermann U, Farrés J, Fita I, Parés X. Three-dimensional structure and enzymatic function of proapoptotic human p53-inducible quinone oxidoreductase PIG3. J Biol Chem. 2009; 284:17194–205. https://doi.org/10.1074/jbc.M109.001800.
89. Zhang Q, Cheng G, Qiu H, Zhu L, Ren Z, Zhao W, Zhang T, Liu L. The p53-inducible gene 3 involved in flavonoid-induced cytotoxicity through the reactive oxygen species-mediated mitochondrial apoptotic pathway in human hepatoma cells. Food Funct. 2015; 6:1518–25. https://doi.org/10.1039/C5FO00142K.
90. Venot C, Maratrat M, Dureuil C, Conseiller E, Bracco L, Debussche L. The requirement for the p53 proline-rich functional domain for mediation of apoptosis is correlated with specific PIG3 gene transactivation and with transcriptional repression. EMBO J. 1998; 17:4668–79. https://doi.org/10.1093/emboj/17.16.4668.
91. Ostrakhovitch EA, Cherian MG. Role of p53 and reactive oxygen species in apoptotic response to copper and zinc in epithelial breast cancer cells. Apoptosis. 2005; 10:111–21. https://doi.org/10.1007/s10495-005-6066-7.
92. Kotsinas A, Aggarwal V, Tan EJ, Levy B, Gorgoulis VG. PIG3: a novel link between oxidative stress and DNA damage response in cancer. Cancer Lett. 2012; 327:97–102. https://doi.org/10.1016/j.canlet.2011.12.009.
93. Xu J, Cai J, Jin X, Yang J, Shen Q, Ding X, Liang Y. PIG3 plays an oncogenic role in papillary thyroid cancer by activating the PI3K/AKT/PTEN pathway. Oncol Rep. 2015; 34:1424–30. https://doi.org/10.3892/or.2015.4096.
94. Tamura RE, Hunger A, Fernandes DC, Laurindo FR, Costanzi-Strauss E, Strauss BE. Induction of Oxidants Distinguishes Susceptibility of Prostate Carcinoma Cell Lines to p53 Gene Transfer Mediated by an Improved Adenoviral Vector. Hum Gene Ther. 2017; 28:639–53. https://doi.org/10.1089/hum.2016.139.
95. Donald SP, Sun XY, Hu CA, Yu J, Mei JM, Valle D, Phang JM. Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Res. 2001; 61:1810–15.
96. Macip S, Igarashi M, Berggren P, Yu J, Lee SW, Aaronson SA. Influence of induced reactive oxygen species in p53-mediated cell fate decisions. Mol Cell Biol. 2003; 23:8576–85. https://doi.org/10.1128/MCB.23.23.8576-8585.2003.
97. Rivera A, Maxwell SA. The p53-induced gene-6 (proline oxidase) mediates apoptosis through a calcineurin-dependent pathway. J Biol Chem. 2005; 280:29346–54. https://doi.org/10.1074/jbc.M504852200.
98. Liu Y, Borchert GL, Surazynski A, Hu CA, Phang JM. Proline oxidase activates both intrinsic and extrinsic pathways for apoptosis: the role of ROS/superoxides, NFAT and MEK/ERK signaling. Oncogene. 2006; 25:5640–47. https://doi.org/10.1038/sj.onc.1209564.
99. Liu Y, Borchert GL, Donald SP, Surazynski A, Hu CA, Weydert CJ, Oberley LW, Phang JM. MnSOD inhibits proline oxidase-induced apoptosis in colorectal cancer cells. Carcinogenesis. 2005; 26:1335–42. https://doi.org/10.1093/carcin/bgi083.
100. Liu W, Glunde K, Bhujwalla ZM, Raman V, Sharma A, Phang JM. Proline oxidase promotes tumor cell survival in hypoxic tumor microenvironments. 2012; 72:3677-86. https://doi.org/10.1158/0008-5472.CAN-12-0080.
101. Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005; 7:385–94. https://doi.org/10.1089/ars.2005.7.385.
102. Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004; 24:7130–39. https://doi.org/10.1128/MCB.24.16.7130-7139.2004.
103. Katoh Y, Iida K, Kang MI, Kobayashi A, Mizukami M, Tong KI, McMahon M, Hayes JD, Itoh K, Yamamoto M. Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Arch Biochem Biophys. 2005; 433:342–50. https://doi.org/10.1016/j.abb.2004.10.012.
104. Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev. 2006; 38:769–89. https://doi.org/10.1080/03602530600971974.
105. Tung MC, Lin PL, Wang YC, He TY, Lee MC, Yeh SD, Chen CY, Lee H. Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget. 2015; 6:41692–705. https://doi.org/10.18632/oncotarget.6150.
106. Faraonio R, Vergara P, Di Marzo D, Pierantoni MG, Napolitano M, Russo T, Cimino F. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J Biol Chem. 2006; 281:39776–84. https://doi.org/10.1074/jbc.M605707200.
107. Chen W, Sun Z, Wang XJ, Jiang T, Huang Z, Fang D, Zhang DD. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell. 2009; 34:663–73. https://doi.org/10.1016/j.molcel.2009.04.029.
108. Chen W, Jiang T, Wang H, Tao S, Lau A, Fang D, Zhang DD. Does Nrf2 contribute to p53-mediated control of cell survival and death? Antioxid Redox Signal. 2012; 17:1670–75. https://doi.org/10.1089/ars.2012.4674.
109. Jeong CH, Joo SH. Downregulation of Reactive Oxygen Species in Apoptosis. J Cancer Prev. 2016; 21:13–20. https://doi.org/10.15430/JCP.2016.21.1.13.
110. Zhang L, Li J, Zong L, Chen X, Chen K, Jiang Z, Nan L, Li X, Li W, Shan T, Ma Q, Ma Z. Reactive Oxygen Species and Targeted Therapy for Pancreatic Cancer. Oxid Med Cell Longev. 2016; 2016:1616781. https://doi.org/10.1155/2016/1616781.
111. Ladelfa MF, Toledo MF, Laiseca JE, Monte M. Interaction of p53 with tumor suppressive and oncogenic signaling pathways to control cellular reactive oxygen species production. Antioxid Redox Signal. 2011; 15:1749–61. https://doi.org/10.1089/ars.2010.3652.
112. Maillet A, Pervaiz S. Redox regulation of p53, redox effectors regulated by p53: a subtle balance. Antioxid Redox Signal. 2012; 16:1285–94. https://doi.org/10.1089/ars.2011.4434.
113. Vurusaner B, Poli G, Basaga H. Tumor suppressor genes and ROS: complex networks of interactions. Free Radic Biol Med. 2012; 52:7–18. https://doi.org/10.1016/j.freeradbiomed.2011.09.035.
114. Li PF, Dietz R, von Harsdorf R. p53 regulates mitochondrial membrane potential through reactive oxygen species and induces cytochrome c-independent apoptosis blocked by Bcl-2. EMBO J. 1999; 18:6027–36. https://doi.org/10.1093/emboj/18.21.6027.
115. Montero J, Dutta C, van Bodegom D, Weinstock D, Letai A. p53 regulates a non-apoptotic death induced by ROS. Cell Death Differ. 2013; 20:1465–74. https://doi.org/10.1038/cdd.2013.52.
116. Aaronson SA. Growth factors and cancer. Science. 1991; 254:1146–53. https://doi.org/10.1126/science.1659742.
117. LeRoith D, Yakar S. Mechanisms of disease: metabolic effects of growth hormone and insulin-like growth factor 1. Nat Clin Pract Endocrinol Metab. 2007; 3:302–10. https://doi.org/10.1038/ncpendmet0427.
118. Bruchim I, Sarfstein R, Werner H. The IGF Hormonal Network in Endometrial Cancer: Functions, Regulation, and Targeting Approaches. Front Endocrinol (Lausanne). 2014; 5:76. https://doi.org/10.3389/fendo.2014.00076.
119. Ohlsson C, Kley N, Werner H, LeRoith D. p53 regulates insulin-like growth factor-I (IGF-I) receptor expression and IGF-I-induced tyrosine phosphorylation in an osteosarcoma cell line: interaction between p53 and Sp1. Endocrinology. 1998; 139:1101–07. https://doi.org/10.1210/endo.139.3.5832.
120. Xiong L, Kou F, Yang Y, Wu J. A novel role for IGF-1R in p53-mediated apoptosis through translational modulation of the p53-Mdm2 feedback loop. J Cell Biol. 2007; 178:995–1007. https://doi.org/10.1083/jcb.200703044.
121. Baxter RC. Nuclear actions of insulin-like growth factor binding protein-3. Gene. 2015; 569:7–13. https://doi.org/10.1016/j.gene.2015.06.028.
122. Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ. The regulation of AMPK β1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007; 67:3043–53. https://doi.org/10.1158/0008-5472.CAN-06-4149.
123. Leu JI, George DL. Hepatic IGFBP1 is a prosurvival factor that binds to BAK, protects the liver from apoptosis, and antagonizes the proapoptotic actions of p53 at mitochondria. Genes Dev. 2007; 21:3095–109. https://doi.org/10.1101/gad.1567107.
124. Tavares MR, Pavan IC, Amaral CL, Meneguello L, Luchessi AD, Simabuco FM. The S6K protein family in health and disease. Life Sci. 2015; 131:1–10. https://doi.org/10.1016/j.lfs.2015.03.001.
125. Dong Q, Giorgianni F, Beranova-Giorgianni S, Deng X, O’Meally RN, Bridges D, Park EA, Cole RN, Elam MB, Raghow R. Glycogen synthase kinase-3-mediated phosphorylation of serine 73 targets sterol response element binding protein-1c (SREBP-1c) for proteasomal degradation. Biosci Rep. 2015; 36:e00284. https://doi.org/10.1042/BSR20150234.
126. Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J Biol Chem. 1998; 273:19929–32. https://doi.org/10.1074/jbc.273.32.19929.
127. Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, Downward J, Schulze A. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene. 2005; 24:6465–81. https://doi.org/10.1038/sj.onc.1208802.
128. Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, Schulze A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008; 8:224–36. https://doi.org/10.1016/j.cmet.2008.07.007.
129. Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008; 134:451–60. https://doi.org/10.1016/j.cell.2008.06.028.
130. Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA. National Academy of Sciences; 2005; 102:8204–9. https://doi.org/10.1073/pnas.0502857102.
131. Loayza-Puch F, Drost J, Rooijers K, Lopes R, Elkon R, Agami R, Zoncu R, Efeyan A, Sabatini D, Dazert E, Hall M, Menon S, Manning B, et al. p53 induces transcriptional and translational programs to suppress cell proliferation and growth. Genome Biol. 2013; 14:R32. https://doi.org/10.1186/gb-2013-14-4-r32.
132. Lee SO, Andey T, Jin UH, Kim K, Singh M, Safe S. The nuclear receptor TR3 regulates mTORC1 signaling in lung cancer cells expressing wild-type p53. Oncogene. 2012; 31:3265–76. https://doi.org/10.1038/onc.2011.504.
133. Li H, Liu S, Yuan H, Niu Y, Fu L. Sestrin 2 induces autophagy and attenuates insulin resistance by regulating AMPK signaling in C2C12 myotubes. Exp Cell Res. 2017; 354:18–24. https://doi.org/10.1016/j.yexcr.2017.03.023.
134. Xue R, Zeng J, Chen Y, Chen C, Tan W, Zhao J, Dong B, Sun Y, Dong Y, Liu C. Sestrin 1 ameliorates cardiac hypertrophy via autophagy activation. J Cell Mol Med. 2017; 21:1193–205. https://doi.org/10.1111/jcmm.13052.
135. Zhang XY, Wu XQ, Deng R, Sun T, Feng GK, Zhu XF. Upregulation of sestrin 2 expression via JNK pathway activation contributes to autophagy induction in cancer cells. Cell Signal. 2013; 25:150–58. https://doi.org/10.1016/j.cellsig.2012.09.004.
136. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011; 13:132–41. https://doi.org/10.1038/ncb2152.
137. Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005; 18:283–93. https://doi.org/10.1016/j.molcel.2005.03.027.
138. Lai KP, Leong WF, Chau JF, Jia D, Zeng L, Liu H, He L, Hao A, Zhang H, Meek D, Velagapudi C, Habib SL, Li B. S6K1 is a multifaceted regulator of Mdm2 that connects nutrient status and DNA damage response. EMBO J. 2010; 29:2994–3006. https://doi.org/10.1038/emboj.2010.166.
139. Akeno N, Miller AL, Ma X, Wikenheiser-Brokamp KA. p53 suppresses carcinoma progression by inhibiting mTOR pathway activation. Oncogene. 2015; 34:589–99. https://doi.org/10.1038/onc.2013.589.
140. Leontieva OV, Gudkov AV, Blagosklonny MV. Weak p53 permits senescence during cell cycle arrest. Cell Cycle. 2010; 9:4323–27. https://doi.org/10.4161/cc.9.21.13584.
141. Chen J, Jiang CC, Jin L, Zhang XD. Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol. 2016; 27:409–16. https://doi.org/10.1093/annonc/mdv615.
142. Kraft S, Fernandez-Figueras MT, Richarz NA, Flaherty KT, Hoang MP. PDL1 expression in desmoplastic melanoma is associated with tumor aggressiveness and progression. J Am Acad Dermatol. 2017; 77:534–42. https://doi.org/10.1016/j.jaad.2017.05.007.
143. Karpathiou G, Casteillo F, Giroult JB, Forest F, Fournel P, Monaya A, Froudarakis M, Dumollard JM, Prades JM, Peoc’h M. Prognostic impact of immune microenvironment in laryngeal and pharyngeal squamous cell carcinoma: immune cell subtypes, immuno-suppressive pathways and clinicopathologic characteristics. Oncotarget. 2017; 8:19310–22. https://doi.org/10.18632/oncotarget.14242.
144. Four M, Cacheux V, Tempier A, Platero D, Fabbro M, Marin G, Leventoux N, Rigau V, Costes-Martineau V, Szablewski V. PD1 and PDL1 expression in primary central nervous system diffuse large B-cell lymphoma are frequent and expression of PD1 predicts poor survival. Hematol Oncol. 2017; 35:487–96. https://doi.org/10.1002/hon.2375.
145. O’Donnell JS, Massi D, Teng MW, Mandala M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin Cancer Biol. 2018; 48:91–103. https://doi.org/10.1016/j.semcancer.2017.04.015.
146. Cha YJ, Kim HR, Lee CY, Cho BC, Shim HS. Clinicopathological and prognostic significance of programmed cell death ligand-1 expression in lung adenocarcinoma and its relationship with p53 status. Lung Cancer. 2016; 97:73–80. https://doi.org/10.1016/j.lungcan.2016.05.001.
147. Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y, Araujo L, Carbone DP, Shilo K, Giri DK, Kelnar K, Martin D, Komaki R, et al. PDL1 Regulation by p53 via miR-34. J Natl Cancer Inst. 2015; 108:djv303. https://doi.org/10.1093/jnci/djv303.
148. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965; 37:614–36. https://doi.org/10.1016/0014-4827(65)90211-9.
149. Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010; 10:51–57. https://doi.org/10.1038/nrc2772.
150. Huang B, Vassilev LT. Reduced transcriptional activity in the p53 pathway of senescent cells revealed by the MDM2 antagonist nutlin-3. Aging (Albany NY). 2009; 1:845–54. https://doi.org/10.18632/aging.100091.
151. Waga S, Hannon GJ, Beach D, Stillman B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature. 1994; 369:574–8. https://doi.org/10.1038/369574a0.
152. Qian Y, Chen X. Senescence regulation by the p53 protein family. Methods Mol Biol. 2013; 965:37–61. https://doi.org/10.1007/978-1-62703-239-1_3.
153. Qian Y, Chen X. Tumor suppression by p53: making cells senescent. Histol Histopathol. 2010; 25:515–26. https://doi.org/10.14670/HH-25.515.
154. Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010; 12:814–22. https://doi.org/10.1038/ncb0910-814.
155. Sui X, Han W, Pan H. p53-induced autophagy and senescence. Oncotarget. 2015; 6:11723–24. https://doi.org/10.18632/oncotarget.4170.
156. Sui X, Fang Y, Lou H, Wang K, Zheng Y, Lou F, Jin W, Xu Y, Chen W, Pan H, Wang X, Han W. p53 suppresses stress-induced cellular senescence via regulation of autophagy under the deprivation of serum. Mol Med Rep. 2015; 11:1214–20. https://doi.org/10.3892/mmr.2014.2853.
157. Sudhagar S, Sathya S, Gokulapriya G, Lakshmi BS. AKT-p53 axis protect cancer cells from autophagic cell death during nutrition deprivation. Biochem Biophys Res Commun. 2016; 471:396–401. https://doi.org/10.1016/j.bbrc.2016.02.064.
158. Duan L, Perez RE, Davaadelger B, Dedkova EN, Blatter LA, Maki CG. p53-regulated autophagy is controlled by glycolysis and determines cell fate. Oncotarget. 2015; 6:23135–56. https://doi.org/10.18632/oncotarget.5218.
159. Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003; 9030–40. https://doi.org/10.1038/sj.onc.1207116.
160. Li Z, Shi K, Guan L, Jiang Q, Yang Y, Xu C. Activation of p53 by sodium selenite switched human leukemia NB4 cells from autophagy to apoptosis. Oncol Res. 2013; 21:325–31. https://doi.org/10.3727/096504014X14024160459087.
161. Ci Y, Shi K, An J, Yang Y, Hui K, Wu P, Shi L, Xu C. ROS inhibit autophagy by downregulating ULK1 mediated by the phosphorylation of p53 in selenite-treated NB4 cells. Cell Death Dis. 2014; 5:e1542. https://doi.org/10.1038/cddis.2014.506.
162. Jonckheere N, Vincent A, Van Seuningen I. Of autophagy and in vivo pancreatic carcinogenesis: the p53 status matters! Clin Res Hepatol Gastroenterol. 2014; 38:423–25. https://doi.org/10.1016/j.clinre.2014.04.009.
163. Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, Adams PD, Anderson KI, Gottlieb E, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013; 504:296–300. https://doi.org/10.1038/nature12865.
164. Chakradeo S, Sharma K, Alhaddad A, Bakhshwin D, Le N, Harada H, Nakajima W, Yeudall WA, Torti SV, Torti FM, Gewirtz DA. Yet another function of p53—the switch that determines whether radiation-induced autophagy will be cytoprotective or nonprotective: implications for autophagy inhibition as a therapeutic strategy. Mol Pharmacol. 2015; 87:803–14. https://doi.org/10.1124/mol.114.095273.
165. Lane DP, Verma C, Fang CC. The p53 inducing drug dosage may determine quiescence or senescence. Aging (Albany NY). 2010; 2:748. https://doi.org/10.18632/aging.100229.
166. Clarke R, Cook KL, Hu R, Facey CO, Tavassoly I, Schwartz JL, Baumann WT, Tyson JJ, Xuan J, Wang Y, Wärri A, Shajahan AN. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Res. 2012; 72:1321–31. https://doi.org/10.1158/0008-5472.CAN-11-3213.
167. Kahlem P, Dörken B, Schmitt CA. Cellular senescence in cancer treatment: friend or foe? J Clin Invest. 2004; 113:169–74. https://doi.org/10.1172/JCI20784.
168. White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009; 15:5308–16. https://doi.org/10.1158/1078-0432.CCR-07-5023.
169. Purvis JE, Karhohs KW, Mock C, Batchelor E, Loewer A, Lahav G. p53 Dynamics Control Cell Fate. Science. 2012; 336:1440–4. https://doi.org/10.1126/science.1218351.
170. Sen N, Satija YK, Das S. PGC-1α, a key modulator of p53, promotes cell survival upon metabolic stress. Mol Cell. 2011; 44:621–34. https://doi.org/10.1016/j.molcel.2011.08.044.
171. Valle I, Alvarez-Barrientos A, Arza E, Lamas S, Monsalve M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc Res. 2005; 66:562–73. https://doi.org/10.1016/j.cardiores.2005.01.026.
172. Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 2006; 27:728–35. https://doi.org/10.1210/er.2006-0037.
173. St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006; 127:397–408. https://doi.org/10.1016/j.cell.2006.09.024.
174. Aquilano K, Baldelli S, Pagliei B, Cannata SM, Rotilio G, Ciriolo MR. p53 orchestrates the PGC-1α-mediated antioxidant response upon mild redox and metabolic imbalance. Antioxid Redox Signal. 2013; 18:386–99. https://doi.org/10.1089/ars.2012.4615.
175. Villeneuve C, Guilbeau-Frugier C, Sicard P, Lairez O, Ordener C, Duparc T, De Paulis D, Couderc B, Spreux-Varoquaux O, Tortosa F, Garnier A, Knauf C, Valet P, et al. p53-PGC-1α pathway mediates oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-A upregulation: role in chronic left ventricular dysfunction in mice. Antioxid Redox Signal. 2013; 18:5–18. https://doi.org/10.1089/ars.2011.4373.
176. Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012; 26:1268–86. https://doi.org/10.1101/gad.190678.112.
177. Sabapathy K, Lane DP. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat Rev Clin Oncol. 2018; 15:13–30. https://doi.org/10.1038/nrclinonc.2017.151.
178. Mathupala SP, Heese C, Pedersen PL. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J Biol Chem. 1997; 272:22776–80. https://doi.org/10.1074/jbc.272.36.22776.
179. Dando I, Cordani M, Donadelli M. Mutant p53 and mTOR/PKM2 regulation in cancer cells. IUBMB Life. 2016; 68:722–26. https://doi.org/10.1002/iub.1534.
180. Wu M, An J, Zheng Q, Xin X, Lin Z, Li X, Li H, Lu D. Double mutant P53 (N340Q/L344R) promotes hepatocarcinogenesis through upregulation of Pim1 mediated by PKM2 and LncRNA CUDR. Oncotarget. 2016; 7:66525–39. https://doi.org/10.18632/oncotarget.9089.
181. Schlereth K, Heyl C, Krampitz AM, Mernberger M, Finkernagel F, Scharfe M, Jarek M, Leich E, Rosenwald A, Stiewe T. Characterization of the p53 cistrome—DNA binding cooperativity dissects p53's tumor suppressor functions. PLoS Genet. 2013; 9:e1003726. https://doi.org/10.1371/journal.pgen.1003726.
182. Junk DJ, Vrba L, Watts GS, Oshiro MM, Martinez JD, Futscher BW. Different mutant/wild-type p53 combinations cause a spectrum of increased invasive potential in nonmalignant immortalized human mammary epithelial cells. Neoplasia. 2008; 10:450–61. https://doi.org/10.1593/neo.08120.
183. Rajeshkumar NV, Dutta P, Yabuuchi S, de Wilde RF, Martinez GV, Le A, Kamphorst JJ, Rabinowitz JD, Jain SK, Hidalgo M, Dang CV, Gillies RJ, Maitra A. Therapeutic Targeting of the Warburg Effect in Pancreatic Cancer Relies on an Absence of p53 Function. Cancer Res. 2015; 75:3355–64. https://doi.org/10.1158/0008-5472.CAN-15-0108.
184. Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, Lin M, Yu H, Liu L, Levine AJ, Hu W, Feng Z. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013; 4:2935. https://doi.org/10.1038/ncomms3935.
185. Garritano S, Inga A, Gemignani F, Landi S. More targets, more pathways and more clues for mutant p53. Oncogenesis. 2013; 2:e54. https://doi.org/10.1038/oncsis.2013.15.
186. Tepper CG, Gregg JP, Shi XB, Vinall RL, Baron CA, Ryan PE, Desprez PY, Kung HJ, deVere White RW. Profiling of gene expression changes caused by p53 gain-of-function mutant alleles in prostate cancer cells. Prostate. 2005; 65:375–89. https://doi.org/10.1002/pros.20308.
187. Bell D, Berchuck A, Birrer M, Chien J, Cramer DW, Dao F, Dhir R, DiSaia P, Gabra H, Glenn P, Godwin AK, Gross J, Hartmann L, et al, and Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474:609–15. https://doi.org/10.1038/nature10166.
188. Hu J, Liu Z, Wang X. Does TP53 mutation promote ovarian cancer metastasis to omentum by regulating lipid metabolism? Med Hypotheses. 2013; 81:515–20. https://doi.org/10.1016/j.mehy.2013.06.009.
189. Kalo E, Kogan-Sakin I, Solomon H, Bar-Nathan E, Shay M, Shetzer Y, Dekel E, Goldfinger N, Buganim Y, Stambolsky P, Goldstein I, Madar S, Rotter V. Mutant p53R273H attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high levels of reactive oxygen species. J Cell Sci. 2012; 125:5578–86. https://doi.org/10.1242/jcs.106815.
190. Taguchi A, Delgado O, Celiktaş M, Katayama H, Wang H, Gazdar AF, Hanash SM. Proteomic signatures associated with p53 mutational status in lung adenocarcinoma. Proteomics. 2014; 14:2750–59. https://doi.org/10.1002/pmic.201400378.
191. Basu S, Gnanapradeepan K, Barnoud T, Kung CP, Tavecchio M, Scott J, Watters A, Chen Q, Kossenkov AV, Murphy ME. Mutant p53 controls tumor metabolism and metastasis by regulating PGC-1α. Genes Dev. 2018; 32:230–43. https://doi.org/10.1101/gad.309062.117.
192. Antoun S, Atallah D, Tahtouh R, Alaaeddine N, Moubarak M, Khaddage A, Ayoub EN, Chahine G, Hilal G. Different TP53 mutants in p53 overexpressed epithelial ovarian carcinoma can be associated both with altered and unaltered glycolytic and apoptotic profiles. Cancer Cell Int. 2018; 18:14. https://doi.org/10.1186/s12935-018-0514-2.
193. Zhou G, Wang J, Zhao M, Xie TX, Tanaka N, Sano D, Patel AA, Ward AM, Sandulache VC, Jasser SA, Skinner HD, Fitzgerald AL, Osman AA, et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol Cell. 2014; 54:960–74. https://doi.org/10.1016/j.molcel.2014.04.024.
194. Cordani M, Oppici E, Dando I, Butturini E, Dalla Pozza E, Nadal-Serrano M, Oliver J, Roca P, Mariotto S, Cellini B, Blandino G, Palmieri M, Di Agostino S, Donadelli M. Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition. Mol Oncol. 2016; 10:1008–29. https://doi.org/10.1016/j.molonc.2016.04.001.
195. Sanli T, Linher-Melville K, Tsakiridis T, Singh G. Sestrin2 modulates AMPK subunit expression and its response to ionizing radiation in breast cancer cells. PLoS One. 2012; 7:e32035. https://doi.org/10.1371/journal.pone.0032035.
196. Morrison A, Chen L, Wang J, Zhang M, Yang H, Ma Y, Budanov A, Lee JH, Karin M, Li J. Sestrin2 promotes LKB1-mediated AMPK activation in the ischemic heart. FASEB J. 2015; 29:408–17. https://doi.org/10.1096/fj.14-258814.
197. Yallowitz AR, Li D, Lobko A, Mott D, Nemajerova A, Marchenko N. Mutant p53 Amplifies Epidermal Growth Factor Receptor Family Signaling to Promote Mammary Tumorigenesis. Mol Cancer Res. 2015; 13:743–54. https://doi.org/10.1158/1541-7786.MCR-14-0360.
198. Tan BS, Tiong KH, Choo HL, Chung FF, Hii LW, Tan SH, Yap IK, Pani S, Khor NT, Wong SF, Rosli R, Cheong SK, Leong CO. Mutant p53-R273H mediates cancer cell survival and anoikis resistance through AKT-dependent suppression of BCL2-modifying factor (BMF). Cell Death Dis. 2015; 6:e1826. https://doi.org/10.1038/cddis.2015.191.
199. Amaral CL, Freitas LB, Tamura RE, Tavares MR, Pavan IC, Bajgelman MC, Simabuco FM. S6Ks isoforms contribute to viability, migration, docetaxel resistance and tumor formation of prostate cancer cells. BMC Cancer. 2016; 16:602. https://doi.org/10.1186/s12885-016-2629-y.
200. Sridharan S, Basu A. S6 kinase 2 promotes breast cancer cell survival via Akt. Cancer Res. 2011; 71:2590–99. https://doi.org/10.1158/0008-5472.CAN-10-3253.
201. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011; 12:21–35. https://doi.org/10.1038/nrm3025.
202. Morselli E, Tasdemir E, Maiuri MC, Galluzzi L, Kepp O, Criollo A, Vicencio JM, Soussi T, Kroemer G. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle. 2008; 7:3056–61. https://doi.org/10.4161/cc.7.19.6751.
203. Rodriguez OC, Choudhury S, Kolukula V, Vietsch EE, Catania J, Preet A, Reynoso K, Bargonetti J, Wellstein A, Albanese C, Avantaggiati ML. Dietary downregulation of mutant p53 levels via glucose restriction: mechanisms and implications for tumor therapy. Cell Cycle. 2012; 11:4436–46. https://doi.org/10.4161/cc.22778.
204. Choudhury S, Kolukula VK, Preet A, Albanese C, Avantaggiati ML. Dissecting the pathways that destabilize mutant p53: the proteasome or autophagy? Cell Cycle. 2013; 12:1022–29. https://doi.org/10.4161/cc.24128.
205. Vakifahmetoglu-Norberg H, Kim M, Xia HG, Iwanicki MP, Ofengeim D, Coloff JL, Pan L, Ince TA, Kroemer G, Brugge JS, Yuan J. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013; 27:1718–30. https://doi.org/10.1101/gad.220897.113.
206. Wang LH, Okaichi K, Ihara M, Okumura Y. Sensitivity of anticancer drugs in Saos-2 cells transfected with mutant p53 varied with mutation point. Anticancer Res. 1998; 18:321–25.
207. Sampath J, Sun D, Kidd VJ, Grenet J, Gandhi A, Shapiro LH, Wang Q, Zambetti GP, Schuetz JD. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J Biol Chem. 2001; 276:39359–67. https://doi.org/10.1074/jbc.M103429200.
208. Chan KT, Lung ML. Mutant p53 expression enhances drug resistance in a hepatocellular carcinoma cell line. Cancer Chemother Pharmacol. 2004; 53:519–26. https://doi.org/10.1007/s00280-004-0767-4.
209. Zhang Z, Deng X, Ren X, Zhang B, Chen X, Yang J, Ding H, Sui J, Song X. Expression of mutant p53 and of the multidrug resistant proteins P-glycoprotein and glutathione S-transferase-pi correlated in colorectal adenocarcinoma. Scand J Gastroenterol. 2010; 45:925–34. https://doi.org/10.3109/00365521003734117.
210. Belkahla S, Haq Khan AU, Gitenay D, Alexia C, Gondeau C, Vo DN, Orecchioni S, Talarico G, Bertolini F, Cartron G, Hernandez J, Daujat-Chavanieu M, Allende-Vega N, Gonzalez MV. Changes in metabolism affect expression of ABC transporters through ERK5 and depending on p53 status. Oncotarget. 2018; 9:1114–29. https://doi.org/10.18632/oncotarget.23305.
211. Shetzer Y, Solomon H, Koifman G, Molchadsky A, Horesh S, Rotter V. The paradigm of mutant p53-expressing cancer stem cells and drug resistance. Carcinogenesis. 2014; 35:1196–208. https://doi.org/10.1093/carcin/bgu073.
212. Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007; 447:407–12. https://doi.org/10.1038/nature05915.
213. Henrique R, Oliveira AI, Costa VL, Baptista T, Martins AT, Morais A, Oliveira J, Jerónimo C. Epigenetic regulation of MDR1 gene through post-translational histone modifications in prostate cancer. BMC Genomics. 2013; 14:898. https://doi.org/10.1186/1471-2164-14-898.
214. Komura K, Jeong SH, Hinohara K, Qu F, Wang X, Hiraki M, Azuma H, Lee GS, Kantoff PW, Sweeney CJ. Resistance to docetaxel in prostate cancer is associated with androgen receptor activation and loss of KDM5D expression. Proc Natl Acad Sci USA. 2016; 113:6259–64. https://doi.org/10.1073/pnas.1600420113.
215. Huang SK, Scruggs AM, Donaghy J, Horowitz JC, Zaslona Z, Przybranowski S, White ES, Peters-Golden M. Histone modifications are responsible for decreased Fas expression and apoptosis resistance in fibrotic lung fibroblasts. Cell Death Dis. 2013; 4:e621. https://doi.org/10.1038/cddis.2013.146.
216. Benard A, Janssen CM, van den Elsen PJ, van Eggermond MC, Hoon DS, van de Velde CJ, Kuppen PJ. Chromatin status of apoptosis genes correlates with sensitivity to chemo-, immune- and radiation therapy in colorectal cancer cell lines. Apoptosis. 2014; 19:1769–78. https://doi.org/10.1007/s10495-014-1042-8.
217. Paschall AV, Yang D, Lu C, Choi JH, Li X, Liu F, Figueroa M, Oberlies NH, Pearce C, Bollag WB, Nayak-Kapoor A, Liu K. H3K9 Trimethylation Silences Fas Expression To Confer Colon Carcinoma Immune Escape and 5-Fluorouracil Chemoresistance. J Immunol. 2015; 195:1868–82. https://doi.org/10.4049/jimmunol.1402243.
218. Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, Barsyte-Lovejoy D, Al-awar R, Katona BW, Shilatifard A, Huang J, Hua X, Arrowsmith CH, Berger SL. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature. 2015; 525:206–11. https://doi.org/10.1038/nature15251.
219. Yamamoto T, Seino Y, Fukumoto H, Koh G, Yano H, Inagaki N, Yamada Y, Inoue K, Manabe T, Imura H. Over-expression of facilitative glucose transporter genes in human cancer. Biochem Biophys Res Commun. 1990; 170:223–30. https://doi.org/10.1016/0006-291X(90)91263-R.
220. McBrayer SK, Cheng JC, Singhal S, Krett NL, Rosen ST, Shanmugam M. Multiple myeloma exhibits novel dependence on GLUT4, GLUT8, and GLUT11: implications for glucose transporter-directed therapy. Blood. 2012; 119:4686–97. https://doi.org/10.1182/blood-2011-09-377846.
221. Gaedicke S, Firat-Geier E, Constantiniu O, Lucchiari-Hartz M, Freudenberg M, Galanos C, Niedermann G. Antitumor effect of the human immunodeficiency virus protease inhibitor ritonavir: induction of tumor-cell apoptosis associated with perturbation of proteasomal proteolysis. Cancer Res. 2002; 62:6901–08.
222. Srirangam A, Mitra R, Wang M, Gorski JC, Badve S, Baldridge L, Hamilton J, Kishimoto H, Hawes J, Li L, Orschell CM, Srour EF, Blum JS, et al. Effects of HIV protease inhibitor ritonavir on Akt-regulated cell proliferation in breast cancer. Clin Cancer Res. 2006; 12:1883–96. https://doi.org/10.1158/1078-0432.CCR-05-1167.
223. Lefèvre C, Auclair M, Boccara F, Bastard JP, Capeau J, Vigouroux C, Caron-Debarle M. Premature senescence of vascular cells is induced by HIV protease inhibitors: implication of prelamin A and reversion by statin. Arterioscler Thromb Vasc Biol. 2010; 30:2611–20. https://doi.org/10.1161/ATVBAHA.110.213603.
224. Kobori M, Shinmoto H, Tsushida T, Shinohara K. Phloretin-induced apoptosis in B16 melanoma 4A5 cells by inhibition of glucose transmembrane transport. Cancer Lett. 1997; 119:207–12. https://doi.org/10.1016/S0304-3835(97)00271-1.
225. Lin ST, Tu SH, Yang PS, Hsu SP, Lee WH, Ho CT, Wu CH, Lai YH, Chen MY, Chen LC. Apple Polyphenol Phloretin Inhibits Colorectal Cancer Cell Growth via Inhibition of the Type 2 Glucose Transporter and Activation of p53-Mediated Signaling. J Agric Food Chem. 2016; 64:6826–37. https://doi.org/10.1021/acs.jafc.6b02861.
226. Liu Y, Cao Y, Zhang W, Bergmeier S, Qian Y, Akbar H, Colvin R, Ding J, Tong L, Wu S, Hines J, Chen X. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther. 2012; 11:1672–82. https://doi.org/10.1158/1535-7163.MCT-12-0131.
227. Drabovich AP, Pavlou MP, Dimitromanolakis A, Diamandis EP. Quantitative analysis of energy metabolic pathways in MCF-7 breast cancer cells by selected reaction monitoring assay. Mol Cell Proteomics. 2012; 11:422–34. https://doi.org/10.1074/mcp.M111.015214.
228. Tian WN, Braunstein LD, Apse K, Pang J, Rose M, Tian X, Stanton RC. Importance of glucose-6-phosphate dehydrogenase activity in cell death. Am J Physiol. 1999; 276:C1121–31. https://doi.org/10.1152/ajpcell.1999.276.5.C1121.
229. Vanamala J, Radhakrishnan S, Reddivari L, Bhat VB, Ptitsyn A. Resveratrol suppresses human colon cancer cell proliferation and induces apoptosis via targeting the pentose phosphate and the talin-FAK signaling pathways-A proteomic approach. Proteome Sci. 2011; 9:49. https://doi.org/10.1186/1477-5956-9-49.
230. Shibuya N, Inoue K, Tanaka G, Akimoto K, Kubota K. Augmented pentose phosphate pathway plays critical roles in colorectal carcinomas. Oncology. 2015; 88:309–19. https://doi.org/10.1159/000369905.
231. Del Follo-Martinez A, Banerjee N, Li X, Safe S, Mertens-Talcott S. Resveratrol and quercetin in combination have anticancer activity in colon cancer cells and repress oncogenic microRNA-27a. Nutr Cancer. 2013; 65:494–504. https://doi.org/10.1080/01635581.2012.725194.
232. Saiko P, Ozsvar-Kozma M, Madlener S, Bernhaus A, Lackner A, Grusch M, Horvath Z, Krupitza G, Jaeger W, Ammer K, Fritzer-Szekeres M, Szekeres T. Avemar, a nontoxic fermented wheat germ extract, induces apoptosis and inhibits ribonucleotide reductase in human HL-60 promyelocytic leukemia cells. Cancer Lett. 2007; 250:323–28. https://doi.org/10.1016/j.canlet.2006.10.018.
233. Huang C, Ma WY, Goranson A, Dong Z. Resveratrol suppresses cell transformation and induces apoptosis through a p53-dependent pathway. Carcinogenesis. 1999; 20:237–42. https://doi.org/10.1093/carcin/20.2.237.
234. Kuo PL, Chiang LC, Lin CC. Resveratrol- induced apoptosis is mediated by p53-dependent pathway in Hep G2 cells. Life Sci. 2002; 72:23–34. https://doi.org/10.1016/S0024-3205(02)02177-X.
235. Shih A, Davis FB, Lin HY, Davis PJ. Resveratrol induces apoptosis in thyroid cancer cell lines via a MAPK- and p53-dependent mechanism. J Clin Endocrinol Metab. 2002; 87:1223–32. https://doi.org/10.1210/jcem.87.3.8345.
236. Alkhalaf M, Jaffal S. Potent antiproliferative effects of resveratrol on human osteosarcoma SJSA1 cells: novel cellular mechanisms involving the ERKs/p53 cascade. Free Radic Biol Med. 2006; 41:318–25. https://doi.org/10.1016/j.freeradbiomed.2006.04.019.
237. Singh SK, Banerjee S, Acosta EP, Lillard JW, Singh R. Resveratrol induces cell cycle arrest and apoptosis with docetaxel in prostate cancer cells via a p53/ p21WAF1/CIP1 and p27KIP1 pathway. Oncotarget. 2017; 8:17216–28. https://doi.org/10.18632/oncotarget.15303.
238. Ferraz da Costa DC, Casanova FA, Quarti J, Malheiros MS, Sanches D, Dos Santos PS, Fialho E, Silva JL. Transient transfection of a wild-type p53 gene triggers resveratrol-induced apoptosis in cancer cells. PLoS One. 2012; 7:e48746. https://doi.org/10.1371/journal.pone.0048746.
239. Coy JF, Dressler D, Wilde J, Schubert P. Mutations in the transketolase-like gene TKTL1: clinical implications for neurodegenerative diseases, diabetes and cancer. Clin Lab. 2005; 51:257–73.
240. Zhang S, Yang JH, Guo CK, Cai PC. Gene silencing of TKTL1 by RNAi inhibits cell proliferation in human hepatoma cells. Cancer Lett. 2007; 253:108–14. https://doi.org/10.1016/j.canlet.2007.01.010.
241. Xu X, Zur Hausen A, Coy JF, Löchelt M. Transketolase-like protein 1 (TKTL1) is required for rapid cell growth and full viability of human tumor cells. Int J Cancer. 2009; 124:1330–37. https://doi.org/10.1002/ijc.24078.
242. Wanka C, Steinbach JP, Rieger J. Tp53-induced glycolysis and apoptosis regulator (TIGAR) protects glioma cells from starvation-induced cell death by up-regulating respiration and improving cellular redox homeostasis. J Biol Chem. 2012; 287:33436–46. https://doi.org/10.1074/jbc.M112.384578.
243. Mariadasse R, Biswal J, Jayaprakash P, Rao GR, Choubey SK, Rajendran S, Jeyakanthan J. Mechanical insights of oxythiamine compound as potent inhibitor for human transketolase-like protein 1 (TKTL1 protein). J Recept Signal Transduct Res. 2016; 36:233–42. https://doi.org/10.3109/10799893.2015.1080272.
244. Raïs B, Comin B, Puigjaner J, Brandes JL, Creppy E, Saboureau D, Ennamany R, Lee WN, Boros LG, Cascante M. Oxythiamine and dehydroepiandrosterone induce a G1 phase cycle arrest in Ehrlich’s tumor cells through inhibition of the pentose cycle. FEBS Lett. 1999; 456:113–18. https://doi.org/10.1016/S0014-5793(99)00924-2.
245. Chornyy S, Parkhomenko Y, Chorna N. Thiamine antagonists trigger p53-dependent apoptosis in differentiated SH-SY5Y cells. Sci Rep. 2017; 7:10632. https://doi.org/10.1038/s41598-017-10878-x.
246. Bustamante E, Morris HP, Pedersen PL. Energy metabolism of tumor cells. Requirement for a form of hexokinase with a propensity for mitochondrial binding. J Biol Chem. 1981; 256:8699–704.
247. Aft RL, Zhang FW, Gius D. Evaluation of 2-deoxy-D-glucose as a chemotherapeutic agent: mechanism of cell death. Br J Cancer. 2002; 87:805–12. https://doi.org/10.1038/sj.bjc.6600547.
248. Mohanti BK, Rath GK, Anantha N, Kannan V, Das BS, Chandramouli BA, Banerjee AK, Das S, Jena A, Ravichandran R, Sahi UP, Kumar R, Kapoor N, et al. Improving cancer radiotherapy with 2-deoxy-D-glucose: phase I/II clinical trials on human cerebral gliomas. Int J Radiat Oncol Biol Phys. 1996; 35:103–11. https://doi.org/10.1016/S0360-3016(96)85017-6.
249. Xue C, Wang C, Sun Y, Meng Q, Liu Z, Huo X, Sun P, Sun H, Ma X, Ma X, Peng J, Liu K. Targeting P-glycoprotein function, p53 and energy metabolism: combination of metformin and 2-deoxyglucose reverses the multidrug resistance of MCF-7/Dox cells to doxorubicin. Oncotarget. 2017; 8:8622–32. https://doi.org/10.18632/oncotarget.14373.
250. Geschwind JF, Georgiades CS, Ko YH, Pedersen PL. Recently elucidated energy catabolism pathways provide opportunities for novel treatments in hepatocellular carcinoma. Expert Rev Anticancer Ther. 2004; 4:449–57. https://doi.org/10.1586/14737140.4.3.449.
251. Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002; 2:48–58. https://doi.org/10.1038/nrc706.
252. Kam Y, Das T, Tian H, Foroutan P, Ruiz E, Martinez G, Minton S, Gillies RJ, Gatenby RA. Sweat but no gain: inhibiting proliferation of multidrug resistant cancer cells with “ersatzdroges”. Int J Cancer. 2015; 136:E188–96. https://doi.org/10.1002/ijc.29158.
253. Nakano A, Tsuji D, Miki H, Cui Q, El Sayed SM, Ikegame A, Oda A, Amou H, Nakamura S, Harada T, Fujii S, Kagawa K, Takeuchi K, et al. Glycolysis inhibition inactivates ABC transporters to restore drug sensitivity in malignant cells. PLoS One. 2011; 6:e27222. https://doi.org/10.1371/journal.pone.0027222.
254. Nath K, Nelson DS, Heitjan DF, Leeper DB, Zhou R, Glickson JD. Lonidamine induces intracellular tumor acidification and ATP depletion in breast, prostate and ovarian cancer xenografts and potentiates response to doxorubicin. NMR Biomed. 2015; 28:281–90. https://doi.org/10.1002/nbm.3240.
255. Davidescu M, Macchioni L, Scaramozzino G, Cristina Marchetti M, Migliorati G, Vitale R, Corcelli A, Roberti R, Castigli E, Corazzi L. The energy blockers bromopyruvate and lonidamine lead GL15 glioblastoma cells to death by different p53-dependent routes. Sci Rep. 2015; 5:14343. https://doi.org/10.1038/srep14343.
256. Franklin DA, He Y, Leslie PL, Tikunov AP, Fenger N, Macdonald JM, Zhang Y. p53 coordinates DNA repair with nucleotide synthesis by suppressing PFKFB3 expression and promoting the pentose phosphate pathway. Sci Rep. 2016; 6:38067. https://doi.org/10.1038/srep38067.
257. Clem BF, O’Neal J, Tapolsky G, Clem AL, Imbert-Fernandez Y, Kerr DA 2nd, Klarer AC, Redman R, Miller DM, Trent JO, Telang S, Chesney J. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol Cancer Ther. 2013; 12:1461–70. https://doi.org/10.1158/1535-7163.MCT-13-0097.
258. Liu L, Wang S, Zhang Q, Ding Y. Identification of potential genes/proteins regulated by Tiam1 in colorectal cancer by microarray analysis and proteome analysis. Cell Biol Int. 2008; 32:1215–22. https://doi.org/10.1016/j.cellbi.2008.07.004.
259. Evans MJ, Saghatelian A, Sorensen EJ, Cravatt BF. Target discovery in small-molecule cell-based screens by in situ proteome reactivity profiling. Nat Biotechnol. 2005; 23:1303–07. https://doi.org/10.1038/nbt1149.
260. Evans MJ, Morris GM, Wu J, Olson AJ, Sorensen EJ, Cravatt BF. Mechanistic and structural requirements for active site labeling of phosphoglycerate mutase by spiroepoxides. Mol Biosyst. 2007; 3:495–506. https://doi.org/10.1039/b705113a.
261. Yoo BC, Ku JL, Hong SH, Shin YK, Park SY, Kim HK, Park JG. Decreased pyruvate kinase M2 activity linked to cisplatin resistance in human gastric carcinoma cell lines. Int J Cancer. 2004; 108:532–39. https://doi.org/10.1002/ijc.11604.
262. Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov. 2013; 12:829–46. https://doi.org/10.1038/nrd4145.
263. Keller KE, Tan IS, Lee YS. SAICAR stimulates pyruvate kinase isoform M2 and promotes cancer cell survival in glucose-limited conditions. Science. 2012; 338:1069–72. https://doi.org/10.1126/science.1224409.
264. Chaneton B, Hillmann P, Zheng L, Martin AC, Maddocks OD, Chokkathukalam A, Coyle JE, Jankevics A, Holding FP, Vousden KH, Frezza C, O’Reilly M, Gottlieb E. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature. 2012; 491:458–62. https://doi.org/10.1038/nature11540.
265. Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. 2007; 27:7381–93. https://doi.org/10.1128/MCB.00440-07.
266. Zhang R, Su J, Xue SL, Yang H, Ju LL, Ji Y, Wu KH, Zhang YW, Zhang YX, Hu JF, Yu MM. HPV E6/p53 mediated down-regulation of miR-34a inhibits Warburg effect through targeting LDHA in cervical cancer. Am J Cancer Res. 2016; 6:312–20.
267. Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013; 4:e532. https://doi.org/10.1038/cddis.2013.60.
268. Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, Wahl ML, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008; 118:3930–42. https://doi.org/10.1172/JCI36843.
269. Allen E, Miéville P, Warren CM, Saghafinia S, Li L, Peng MW, Hanahan D. Metabolic Symbiosis Enables Adaptive Resistance to Anti-angiogenic Therapy that Is Dependent on mTOR Signaling. Cell Reports. 2016; 15:1144–60. https://doi.org/10.1016/j.celrep.2016.04.029.
270. Tennant DA, Durán RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010; 10:267–77. https://doi.org/10.1038/nrc2817.
271. Bull JH, Ellison G, Patel A, Muir G, Walker M, Underwood M, Khan F, Paskins L. Identification of potential diagnostic markers of prostate cancer and prostatic intraepithelial neoplasia using cDNA microarray. Br J Cancer. 2001; 84:1512–19. https://doi.org/10.1054/bjoc.2001.1816.
272. Alo’ PL, Visca P, Marci A, Mangoni A, Botti C, Di Tondo U. Expression of fatty acid synthase (FAS) as a predictor of recurrence in stage I breast carcinoma patients. Cancer. 1996; 77:474–82. https://doi.org/10.1002/(SICI)1097-0142(19960201)77:33.0.CO;2-K.
273. Jiang Y, Yin X, Wu L, Qin Q, Xu J. MAPK/P53-mediated FASN expression in bone tumors. Oncol Lett. 2017; 13:4035–38. https://doi.org/10.3892/ol.2017.6015.
274. Kuhajda FP. Fatty acid synthase and cancer: new application of an old pathway. Cancer Res. 2006; 66:5977–80. https://doi.org/10.1158/0008-5472.CAN-05-4673.
275. Funabashi H, Kawaguchi A, Tomoda H, Omura S, Okuda S, Iwasaki S. Binding site of cerulenin in fatty acid synthetase. J Biochem. 1989; 105:751–55. https://doi.org/10.1093/oxfordjournals.jbchem.a122739.
276. Kuhajda FP, Pizer ES, Li JN, Mani NS, Frehywot GL, Townsend CA. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc Natl Acad Sci USA. 2000; 97:3450–54. https://doi.org/10.1073/pnas.97.7.3450.
277. Little JL, Wheeler FB, Fels DR, Koumenis C, Kridel SJ. Inhibition of fatty acid synthase induces endoplasmic reticulum stress in tumor cells. Cancer Res. 2007; 67:1262–69. https://doi.org/10.1158/0008-5472.CAN-06-1794.
278. Shiragami R, Murata S, Kosugi C, Tezuka T, Yamazaki M, Hirano A, Yoshimura Y, Suzuki M, Shuto K, Koda K. Enhanced antitumor activity of cerulenin combined with oxaliplatin in human colon cancer cells. Int J Oncol. 2013; 43:431–38. https://doi.org/10.3892/ijo.2013.1978.
279. Gao Y, Lin LP, Zhu CH, Chen Y, Hou YT, Ding J. Growth arrest induced by C75, A fatty acid synthase inhibitor, was partially modulated by p38 MAPK but not by p53 in human hepatocellular carcinoma. Cancer Biol Ther. 2006; 5:978–85. https://doi.org/10.4161/cbt.5.8.2883.
280. Li JN, Gorospe M, Chrest FJ, Kumaravel TS, Evans MK, Han WF, Pizer ES. Pharmacological inhibition of fatty acid synthase activity produces both cytostatic and cytotoxic effects modulated by p53. Cancer Res. 2001; 61:1493–99.
281. Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 2004; 64:2070–75. https://doi.org/10.1158/0008-5472.CAN-03-3645.
282. Carvalho MA, Zecchin KG, Seguin F, Bastos DC, Agostini M, Rangel AL, Veiga SS, Raposo HF, Oliveira HC, Loda M, Coletta RD, Graner E. Fatty acid synthase inhibition with Orlistat promotes apoptosis and reduces cell growth and lymph node metastasis in a mouse melanoma model. Int J Cancer. 2008; 123:2557–65. https://doi.org/10.1002/ijc.23835.
283. Seguin F, Carvalho MA, Bastos DC, Agostini M, Zecchin KG, Alvarez-Flores MP, Chudzinski-Tavassi AM, Coletta RD, Graner E. The fatty acid synthase inhibitor orlistat reduces experimental metastases and angiogenesis in B16-F10 melanomas. Br J Cancer. 2012; 107:977–87. https://doi.org/10.1038/bjc.2012.355.
284. Kant S, Kumar A, Singh SM. Fatty acid synthase inhibitor orlistat induces apoptosis in T cell lymphoma: role of cell survival regulatory molecules. Biochim Biophys Acta. 2012; 1820:1764–73. https://doi.org/10.1016/j.bbagen.2012.07.010.
285. Wright C, Iyer AK, Kaushik V, Azad N. Anti-Tumorigenic Potential of a Novel Orlistat-AICAR Combination in Prostate Cancer Cells. J Cell Biochem. 2017; 118:3834–45. https://doi.org/10.1002/jcb.26033.
286. Wang X, Tian W. Green tea epigallocatechin gallate: a natural inhibitor of fatty-acid synthase. Biochem Biophys Res Commun. 2001; 288:1200–06. https://doi.org/10.1006/bbrc.2001.5923.
287. Puig T, Turrado C, Benhamú B, Aguilar H, Relat J, Ortega-Gutiérrez S, Casals G, Marrero PF, Urruticoechea A, Haro D, López-Rodríguez ML, Colomer R. Novel Inhibitors of Fatty Acid Synthase with Anticancer Activity. Clin Cancer Res. 2009; 15:7608–15. https://doi.org/10.1158/1078-0432.CCR-09-0856.
288. Park SY, Jung CH, Song B, Park OJ, Kim YM. Pro-apoptotic and migration-suppressing potential of EGCG, and the involvement of AMPK in the p53-mediated modulation of VEGF and MMP-9 expression. Oncol Lett. 2013; 6:1346–50. https://doi.org/10.3892/ol.2013.1533.
289. Braicu C, Pileczki V, Pop L, Petric RC, Chira S, Pointiere E, Achimas-Cadariu P, Berindan-Neagoe I. Dual targeted therapy with p53 siRNA and Epigallocatechingallate in a triple negative breast cancer cell model. PLoS One. 2015; 10:e0120936. https://doi.org/10.1371/journal.pone.0120936.
290. Lee JH, Jang H, Lee SM, Lee JE, Choi J, Kim TW, Cho EJ, Youn HD. ATP-citrate lyase regulates cellular senescence via an AMPK- and p53-dependent pathway. FEBS J. 2015; 282:361–71. https://doi.org/10.1111/febs.13139.
291. Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA, Thompson CB. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005; 8:311–21. https://doi.org/10.1016/j.ccr.2005.09.008.
292. Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, Lombardo PS, Van Nostrand JL, Hutchins A, Vera L, Gerken L, Greenwood J, Bhat S, et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med. 2016; 22:1108–19. https://doi.org/10.1038/nm.4181.
293. Clem BF, Clem AL, Yalcin A, Goswami U, Arumugam S, Telang S, Trent JO, Chesney J. A novel small molecule antagonist of choline kinase-α that simultaneously suppresses MAPK and PI3K/AKT signaling. Oncogene. 2011; 30:3370–80. https://doi.org/10.1038/onc.2011.51.
294. Hong BS, Allali-Hassani A, Tempel W, Finerty PJ Jr, Mackenzie F, Dimov S, Vedadi M, Park HW. Crystal structures of human choline kinase isoforms in complex with hemicholinium-3: single amino acid near the active site influences inhibitor sensitivity. J Biol Chem. 2010; 285:16330–40. https://doi.org/10.1074/jbc.M109.039024.
295. Simell O, Sipilä I, Rajantie J, Valle DL, Brusilow SW. Waste nitrogen excretion via amino acid acylation: benzoate and phenylacetate in lysinuric protein intolerance. Pediatr Res. 1986; 20:1117–21. https://doi.org/10.1203/00006450-198611000-00011.
296. Samid D, Shack S, Myers CE. Selective growth arrest and phenotypic reversion of prostate cancer cells in vitro by nontoxic pharmacological concentrations of phenylacetate. J Clin Invest. 1993; 91:2288–95. https://doi.org/10.1172/JCI116457.
297. DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010; 29:313–24. https://doi.org/10.1038/onc.2009.358.
298. Chan HC, Kuo SC, Huang LJ, Liu CH, Hsu SL. A phenylacetate derivative, SCK6, inhibits cell proliferation via G1 cell cycle arrest and apoptosis. Eur J Pharmacol. 2003; 467:31–39. https://doi.org/10.1016/S0014-2999(03)01596-6.
299. Onishi T, Yamakawa K, Franco OE, Suzuki R, Kawamura J. p27Kip1 is the key mediator of phenylacetate induced cell cycle arrest in human prostate cancer cells. Anticancer Res. 2000; 20:3075–81.
300. Gorospe M, Shack S, Guyton KZ, Samid D, Holbrook NJ. Up-regulation and functional role of p21Waf1/Cip1 during growth arrest of human breast carcinoma MCF-7 cells by phenylacetate. Cell Growth Differ. 1996; 7:1609–15.
301. Shukla K, Ferraris DV, Thomas AG, Stathis M, Duvall B, Delahanty G, Alt J, Rais R, Rojas C, Gao P, Xiang Y, Dang CV, Slusher BS, Tsukamoto T. Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J Med Chem. 2012; 55:10551–63. https://doi.org/10.1021/jm301191p.
302. Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS, Rabinowitz JD, Dang CV, Riggins GJ. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010; 70:8981–87. https://doi.org/10.1158/0008-5472.CAN-10-1666.
303. Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, Wilson KF, Ambrosio AL, Dias SM, Dang CV, Cerione RA. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010; 18:207–19. https://doi.org/10.1016/j.ccr.2010.08.009.
304. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016; 23:27–47. https://doi.org/10.1016/j.cmet.2015.12.006.
305. Feun L, Savaraj N. Pegylated arginine deiminase: a novel anticancer enzyme agent. Expert Opin Investig Drugs. 2006; 15:815–22. https://doi.org/10.1517/13543784.15.7.815.
306. Izzo F, Marra P, Beneduce G, Castello G, Vallone P, De Rosa V, Cremona F, Ensor CM, Holtsberg FW, Bomalaski JS, Clark MA, Ng C, Curley SA. Pegylated arginine deiminase treatment of patients with unresectable hepatocellular carcinoma: results from phase I/II studies. J Clin Oncol. 2004; 22:1815–22. https://doi.org/10.1200/JCO.2004.11.120.
307. Ni Y, Schwaneberg U, Sun ZH. Arginine deiminase, a potential anti-tumor drug. Cancer Lett. 2008; 261:1–11. https://doi.org/10.1016/j.canlet.2007.11.038.
308. Kim JE, Kim SY, Lee KW, Lee HJ. Arginine deiminase originating from Lactococcus lactis ssp. lactis American Type Culture Collection (ATCC) 7962 induces G1-phase cell-cycle arrest and apoptosis in SNU-1 stomach adenocarcinoma cells. Br J Nutr. 2009; 102:1469–76. https://doi.org/10.1017/S0007114509990432.
309. Cui X, Witalison EE, Chumanevich AP, Chumanevich AA, Poudyal D, Subramanian V, Schetter AJ, Harris CC, Thompson PR, Hofseth LJ. The induction of microRNA-16 in colon cancer cells by protein arginine deiminase inhibition causes a p53-dependent cell cycle arrest. PLoS One. 2013; 8:e53791. https://doi.org/10.1371/journal.pone.0053791.
310. Jiang H, Guo S, Xiao D, Bian X, Wang J, Wang Y, Zhou H, Cai J, Zheng Z. Arginine deiminase expressed in vivo, driven by human telomerase reverse transcriptase promoter, displays high hepatoma targeting and oncolytic efficiency. Oncotarget. 2017; 8:37694–704. https://doi.org/10.18632/oncotarget.17032.
311. Masetti R, Pession A. First-line treatment of acute lymphoblastic leukemia with pegasparaginase. Biologics. 2009; 3:359–68.
312. Fu CH, Martin-Aragon S, Weinberg KI, Ardi VC, Danenberg PV, Avramis VI. Reversal of cytosine arabinoside (ara-C) resistance by the synergistic combination of 6-thioguanine plus ara-C plus PEG-asparaginase (TGAP) in human leukemia lines lacking or expressing p53 protein. Cancer Chemother Pharmacol. 2001; 48:123–33. https://doi.org/10.1007/s002800100289.
313. Takahashi H, Inoue J, Sakaguchi K, Takagi M, Mizutani S, Inazawa J. Autophagy is required for cell survival under L-asparaginase-induced metabolic stress in acute lymphoblastic leukemia cells. Oncogene. 2017; 36:4267–76. https://doi.org/10.1038/onc.2017.59.
314. Richmond J, Carol H, Evans K, High L, Mendomo A, Robbins A, Meyer C, Venn NC, Marschalek R, Henderson M, Sutton R, Kurmasheva RT, Kees UR, et al. Effective targeting of the P53-MDM2 axis in preclinical models of infant MLL-rearranged acute lymphoblastic leukemia. Clin Cancer Res. 2015; 21:1395–405. https://doi.org/10.1158/1078-0432.CCR-14-2300.
315. Zhang W, Zhang SL, Hu X, Tam KY. Targeting Tumor Metabolism for Cancer Treatment: Is Pyruvate Dehydrogenase Kinases (PDKs) a Viable Anticancer Target? Int J Biol Sci. 2015; 11:1390–400. https://doi.org/10.7150/ijbs.13325.
316. Sutendra G, Michelakis ED. Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front Oncol. 2013; 3:38. https://doi.org/10.3389/fonc.2013.00038.
317. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997; 7:261–69. https://doi.org/10.1016/S0960-9822(06)00122-9.
318. Kankotia S, Stacpoole PW. Dichloroacetate and cancer: new home for an orphan drug? Biochim Biophys Acta. 2014; 1846:617–29. https://doi.org/10.1016/j.bbcan.2014.08.005.
319. Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007; 11:37–51. https://doi.org/10.1016/j.ccr.2006.10.020.
320. Kumar A, Kant S, Singh SM. Novel molecular mechanisms of antitumor action of dichloroacetate against T cell lymphoma: implication of altered glucose metabolism, pH homeostasis and cell survival regulation. Chem Biol Interact. 2012; 199:29–37. https://doi.org/10.1016/j.cbi.2012.06.005.
321. Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer. 2008; 99:989–94. https://doi.org/10.1038/sj.bjc.6604554.
322. Morfouace M, Lalier L, Bahut M, Bonnamain V, Naveilhan P, Guette C, Oliver L, Gueguen N, Reynier P, Vallette FM. Comparison of spheroids formed by rat glioma stem cells and neural stem cells reveals differences in glucose metabolism and promising therapeutic applications. J Biol Chem. 2012; 287:33664–74. https://doi.org/10.1074/jbc.M111.320028.
323. Chaturvedi A, Araujo Cruz MM, Jyotsana N, Sharma A, Yun H, Görlich K, Wichmann M, Schwarzer A, Preller M, Thol F, Meyer J, Haemmerle R, Struys EA, et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood. 2013; 122:2877–87. https://doi.org/10.1182/blood-2013-03-491571.
324. Fujii T, Khawaja MR, DiNardo CD, Atkins JT, Janku F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med. 2016; 21:373–80.
325. Burris H, Mellinghoff I, Maher E, Wen P, Beeram M, Touat M, Faris J, Azad N, Cloughesy T, Gore L, Trent J, Von Hoff D, Goldwasser M, et al. The first reported results of AG-120, a first-in-class, potent inhibitor of the IDH1 mutant protein, in a Phase I study of patients with advanced IDH1-mutant solid tumors, including gliomas. Mol Cancer Ther. 2015; 14:PL04-05-PL04-05. https://doi.org/10.1158/1535-7163.TARG-15-PL04-05.
326. Al-Dwairi A, Pabona JM, Simmen RC, Simmen FA. Cytosolic malic enzyme 1 (ME1) mediates high fat diet-induced adiposity, endocrine profile, and gastrointestinal tract proliferation-associated biomarkers in male mice. PLoS One. 2012; 7:e46716. https://doi.org/10.1371/journal.pone.0046716.
327. Murai S, Ando A, Ebara S, Hirayama M, Satomi Y, Hara T. Inhibition of malic enzyme 1 disrupts cellular metabolism and leads to vulnerability in cancer cells in glucose-restricted conditions. Oncogenesis. 2017; 6:e329. https://doi.org/10.1038/oncsis.2017.34.
328. Zheng FJ, Ye HB, Wu MS, Lian YF, Qian CN, Zeng YX. Repressing malic enzyme 1 redirects glucose metabolism, unbalances the redox state, and attenuates migratory and invasive abilities in nasopharyngeal carcinoma cell lines. Chin J Cancer. 2012; 31:519–31. https://doi.org/10.5732/cjc.012.10088.
329. Witters LA. The blooming of the French lilac. J Clin Invest. 2001; 108:1105–07. https://doi.org/10.1172/JCI14178.
330. Morales DR, Morris AD. Metformin in cancer treatment and prevention. Annu Rev Med. 2015; 66:17–29. https://doi.org/10.1146/annurev-med-062613-093128.
331. Pollak MN. Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov. 2012; 2:778–90. https://doi.org/10.1158/2159-8290.CD-12-0263.
332. Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005; 330:1304–05. https://doi.org/10.1136/bmj.38415.708634.F7.
333. Franciosi M, Lucisano G, Lapice E, Strippoli GF, Pellegrini F, Nicolucci A. Metformin therapy and risk of cancer in patients with type 2 diabetes: systematic review. PLoS One. 2013; 8:e71583. https://doi.org/10.1371/journal.pone.0071583.
334. Bailey CJ. Metformin: historical overview. Diabetologia. 2017; 60:1566–76. https://doi.org/10.1007/s00125-017-4318-z.
335. Omar HA, Berman-Booty L, Kulp SK, Chen CS. Energy restriction as an antitumor target. Future Oncol. 2010; 6:1675–79. https://doi.org/10.2217/fon.10.130.
336. Luengo A, Sullivan LB, Heiden MG. Understanding the complex-I-ty of metformin action: limiting mitochondrial respiration to improve cancer therapy. BMC Biol. 2014; 12:82. https://doi.org/10.1186/s12915-014-0082-4.
337. Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006; 66:10269–73. https://doi.org/10.1158/0008-5472.CAN-06-1500.
338. Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005; 310:1642–46. https://doi.org/10.1126/science.1120781.
339. Salani B, Del Rio A, Marini C, Sambuceti G, Cordera R, Maggi D. Metformin, cancer and glucose metabolism. Endocr Relat Cancer. 2014; 21:R461–71. https://doi.org/10.1530/ERC-14-0284.
340. Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond). 2012; 122:253–70. https://doi.org/10.1042/CS20110386.
341. Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald MJ, Jurczak MJ, Camporez JP, Lee HY, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014; 510:542–46. https://doi.org/10.1038/nature13270.
342. Peppicelli S, Toti A, Giannoni E, Bianchini F, Margheri F, Del Rosso M, Calorini L. Metformin is also effective on lactic acidosis-exposed melanoma cells switched to oxidative phosphorylation. Cell Cycle. 2016; 15:1908–18. https://doi.org/10.1080/15384101.2016.1191706.
343. Fendt SM, Bell EL, Keibler MA, Davidson SM, Wirth GJ, Fiske B, Mayers JR, Schwab M, Bellinger G, Csibi A, Patnaik A, Blouin MJ, Cantley LC, et al. Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res. 2013; 73:4429–38. https://doi.org/10.1158/0008-5472.CAN-13-0080.
344. Pernicova I, Korbonits M. Metformin—mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014; 10:143–56. https://doi.org/10.1038/nrendo.2013.256.
345. Pierotti MA, Berrino F, Gariboldi M, Melani C, Mogavero A, Negri T, Pasanisi P, Pilotti S. Targeting metabolism for cancer treatment and prevention: metformin, an old drug with multi-faceted effects. Oncogene. 2013; 32:1475–87. https://doi.org/10.1038/onc.2012.181.
346. Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, Tanti JF, Giorgetti-Peraldi S, Bost F, Ben Sahra I, Regazzetti C, Robert G, Laurent K, et al. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011; 71:4366–72. https://doi.org/10.1158/0008-5472.CAN-10-1769.
347. Nelson LE, Valentine RJ, Cacicedo JM, Gauthier MS, Ido Y, Ruderman NB. A novel inverse relationship between metformin-triggered AMPK-SIRT1 signaling and p53 protein abundance in high glucose-exposed HepG2 cells. Am J Physiol Cell Physiol. 2012; 303:C4–13. https://doi.org/10.1152/ajpcell.00296.2011.
348. Li P, Zhao M, Parris AB, Feng X, Yang X. p53 is required for metformin-induced growth inhibition, senescence and apoptosis in breast cancer cells. Biochem Biophys Res Commun. 2015; 464:1267–74. https://doi.org/10.1016/j.bbrc.2015.07.117.
349. Yi G, He Z, Zhou X, Xian L, Yuan T, Jia X, Hong J, He L, Liu J. Low concentration of metformin induces a p53-dependent senescence in hepatoma cells via activation of the AMPK pathway. Int J Oncol. 2013; 43:1503–10. https://doi.org/10.3892/ijo.2013.2077.
350. Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, Sausen M, Phallen J, Hruban CA, Tokheim C, Niknafs N, Nesselbush M, Lytle K, et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015; 526:263–67. https://doi.org/10.1038/nature14969.
351. Li D, Marchenko ND. ErbB2 inhibition by lapatinib promotes degradation of mutant p53 protein in cancer cells. Oncotarget. 2017; 8:5823–33. https://doi.org/10.18632/oncotarget.12878.
352. Duman BB, Sahin B, Acikalin A, Ergin M, Zorludemir S. PTEN, Akt, MAPK, p53 and p95 expression to predict trastuzumab resistance in HER2 positive breast cancer. J BUON. 2013; 18:44–50.
353. Kamata S, Kishimoto T, Kobayashi S, Miyazaki M, Ishikura H. Possible involvement of persistent activity of the mammalian target of rapamycin pathway in the cisplatin resistance of AFP-producing gastric cancer cells. Cancer Biol Ther. 2007; 6:1036–43. https://doi.org/10.4161/cbt.6.7.4253.
354. Chiarini F, Grimaldi C, Ricci F, Tazzari PL, Evangelisti C, Ognibene A, Battistelli M, Falcieri E, Melchionda F, Pession A, Pagliaro P, McCubrey JA, Martelli AM. Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against T-cell acute lymphoblastic leukemia. Cancer Res. 2010; 70:8097–107. https://doi.org/10.1158/0008-5472.CAN-10-1814.
355. Kao CL, Hsu HS, Chen HW, Cheng TH. Rapamycin increases the p53/MDM2 protein ratio and p53-dependent apoptosis by translational inhibition of mdm2 in cancer cells. Cancer Lett. 2009; 286:250–59. https://doi.org/10.1016/j.canlet.2009.05.031.
356. Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov. 2011; 10:868–80. https://doi.org/10.1038/nrd3531.
357. Qiu Z, Sun R, Mo X, Li W. The p70S6K Specific Inhibitor PF-4708671 Impedes Non-Small Cell Lung Cancer Growth. PLoS One. 2016; 11:e0147185. https://doi.org/10.1371/journal.pone.0147185.
358. Hong SE, Kim EK, Jin HO, Kim HA, Lee JK, Koh JS, Seol H, Kim JI, Park IC, Noh WC. S6K1 inhibition enhances tamoxifen-induced cell death in MCF-7 cells through translational inhibition of Mcl-1 and survivin. Cell Biol Toxicol. 2013; 29:273–82. https://doi.org/10.1007/s10565-013-9253-2.
359. Yonekura S, Itoh M, Okuhashi Y, Takahashi Y, Ono A, Nara N, Tohda S. Effects of the HIF1 inhibitor, echinomycin, on growth and NOTCH signalling in leukaemia cells. Anticancer Res. 2013; 33:3099–103.
360. Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011; 11:393–410. https://doi.org/10.1038/nrc3064.
361. Palayoor ST, Mitchell JB, Cerna D, Degraff W, John-Aryankalayil M, Coleman CN. PX-478, an inhibitor of hypoxia-inducible factor-1alpha, enhances radiosensitivity of prostate carcinoma cells. Int J Cancer. 2008; 123:2430–37. https://doi.org/10.1002/ijc.23807.
362. Cao J, Lin G, Gong Y, Pan P, Ma Y, Huang P, Ying M, Hou T, He Q, Yang B. DNA-PKcs, a novel functional target of acriflavine, mediates acriflavine’s p53-dependent synergistic anti-tumor efficiency with melphalan. Cancer Lett. 2016; 383:115–24. https://doi.org/10.1016/j.canlet.2016.09.029.
363. Koh MY, Spivak-Kroizman T, Venturini S, Welsh S, Williams RR, Kirkpatrick DL, Powis G. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1alpha. Mol Cancer Ther. 2008; 7:90–100. https://doi.org/10.1158/1535-7163.MCT-07-0463.
364. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008; 27:5497–510. https://doi.org/10.1038/onc.2008.245.
365. Janku F, Yap TA, Meric-Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol. 2018; 15:273–91. https://doi.org/10.1038/nrclinonc.2018.28.
366. Bar J, Lukaschuk N, Zalcenstein A, Wilder S, Seger R, Oren M. The PI3K inhibitor LY294002 prevents p53 induction by DNA damage and attenuates chemotherapy-induced apoptosis. Cell Death Differ. 2005; 12:1578–87. https://doi.org/10.1038/sj.cdd.4401677.
367. Zheng L, Ren JQ, Li H, Kong ZL, Zhu HG. Downregulation of wild-type p53 protein by HER-2/neu mediated PI3K pathway activation in human breast cancer cells: its effect on cell proliferation and implication for therapy. Cell Res. 2004; 14:497–506. https://doi.org/10.1038/sj.cr.7290253.
368. Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018; 25:104–13. https://doi.org/10.1038/cdd.2017.169.
369. Lane DP, Cheok CF, Lain S. p53-based cancer therapy. Cold Spring Harb Perspect Biol. 2010; 2:a001222. https://doi.org/10.1101/cshperspect.a001222.
370. Jaffray DA. Image-guided radiotherapy: from current concept to future perspectives. Nat Rev Clin Oncol. 2012; 9:688–99. https://doi.org/10.1038/nrclinonc.2012.194.
371. Nesseler JP, Peiffert D, Vogin G, Nickers P. [Cancer, radiotherapy and immune system]. Cancer Radiother. 2017; 21:307–15. https://doi.org/10.1016/j.canrad.2017.02.002.
372. Cmielová J, Havelek R, Jiroutová A, Kohlerová R, Seifrtová M, Muthná D, Vávrová J, Rezáčová M. DNA damage caused by ionizing radiation in embryonic diploid fibroblasts WI-38 induces both apoptosis and senescence. Physiol Res. 2011; 60:667–77.
373. Papadopoulou A, Kletsas D. Human lung fibroblasts prematurely senescent after exposure to ionizing radiation enhance the growth of malignant lung epithelial cells in vitro and in vivo. Int J Oncol. 2011; 39:989–99. https://doi.org/10.3892/ijo.2011.1132.
374. Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher DE, Housman DE, Jacks T. p53 status and the efficacy of cancer therapy in vivo. Science. 1994; 266:807–10. https://doi.org/10.1126/science.7973635.
375. Cheng G, Kong D, Hou X, Liang B, He M, Liang N, Ma S, Liu X. The tumor suppressor, p53, contributes to radiosensitivity of lung cancer cells by regulating autophagy and apoptosis. Cancer Biother Radiopharm. 2013; 28:153–59. https://doi.org/10.1089/cbr.2012.1297.
376. Quick QA, Gewirtz DA. An accelerated senescence response to radiation in wild-type p53 glioblastoma multiforme cells. J Neurosurg. 2006; 105:111–18. https://doi.org/10.3171/jns.2006.105.1.111.
377. Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, Benchimol S, Mak TW. Regulation of PTEN transcription by p53. Mol Cell. 2001; 8:317–25. https://doi.org/10.1016/S1097-2765(01)00323-9.
378. Lee JJ, Kim BC, Park MJ, Lee YS, Kim YN, Lee BL, Lee JS. PTEN status switches cell fate between premature senescence and apoptosis in glioma exposed to ionizing radiation. Cell Death Differ. 2011; 18:666–77. https://doi.org/10.1038/cdd.2010.139.
379. Lehmann BD, McCubrey JA, Jefferson HS, Paine MS, Chappell WH, Terrian DM. A dominant role for p53-dependent cellular senescence in radiosensitization of human prostate cancer cells. Cell Cycle. 2007; 6:595–605. https://doi.org/10.4161/cc.6.5.3901.
380. Guo H, Liu Z, Xu B, Hu H, Wei Z, Liu Q, Zhang X, Ding X, Wang Y, Zhao M, Gong Y, Shao C. Chemokine receptor CXCR2 is transactivated by p53 and induces p38-mediated cellular senescence in response to DNA damage. Aging Cell. 2013; 12:1110–21. https://doi.org/10.1111/acel.12138.
381. Chen N, Zhang R, Konishi T, Wang J. Upregulation of NRF2 through autophagy/ERK 1/2 ameliorates ionizing radiation induced cell death of human osteosarcoma U-2 OS. Mutat Res. 2017; 813:10–17. https://doi.org/10.1016/j.mrgentox.2016.11.006.
382. Koleini N, Kardami E. Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget. 2017; 8:46663–80. https://doi.org/10.18632/oncotarget.16944.
383. Xie JM, Li B, Yu HP, Gao QG, Li W, Wu HR, Qin ZH. TIGAR has a dual role in cancer cell survival through regulating apoptosis and autophagy. Cancer Res. 2014; 74:5127–38. https://doi.org/10.1158/0008-5472.CAN-13-3517.
384. Qian S, Li J, Hong M, Zhu Y, Zhao H, Xie Y, Huang J, Lian Y, Li Y, Wang S, Mao J, Chen Y. TIGAR cooperated with glycolysis to inhibit the apoptosis of leukemia cells and associated with poor prognosis in patients with cytogenetically normal acute myeloid leukemia. J Hematol Oncol. 2016; 9:128. https://doi.org/10.1186/s13045-016-0360-4.
385. Zhang Y, Chen F, Tai G, Wang J, Shang J, Zhang B, Wang P, Huang B, Du J, Yu J, Zhang H, Liu F. TIGAR knockdown radiosensitizes TrxR1-overexpressing glioma in vitro and in vivo via inhibiting Trx1 nuclear transport. Sci Rep. 2017; 7:42928. https://doi.org/10.1038/srep42928.
386. Yadav N, Kumar S, Marlowe T, Chaudhary AK, Kumar R, Wang J, O’Malley J, Boland PM, Jayanthi S, Kumar TK, Yadava N, Chandra D. Oxidative phosphorylation-dependent regulation of cancer cell apoptosis in response to anticancer agents. Cell Death Dis. 2015; 6:e1969. https://doi.org/10.1038/cddis.2015.305.
387. Sliwinska MA, Mosieniak G, Wolanin K, Babik A, Piwocka K, Magalska A, Szczepanowska J, Fronk J, Sikora E. Induction of senescence with doxorubicin leads to increased genomic instability of HCT116 cells. Mech Ageing Dev. 2009; 130:24–32. https://doi.org/10.1016/j.mad.2008.04.011.
388. Wang Y, Blandino G, Givol D. Induced p21waf expression in H1299 cell line promotes cell senescence and protects against cytotoxic effect of radiation and doxorubicin. Oncogene. 1999; 18:2643–49. https://doi.org/10.1038/sj.onc.1202632.
389. Song YS, Lee BY, Hwang ES. Dinstinct ROS and biochemical profiles in cells undergoing DNA damage-induced senescence and apoptosis. Mech Ageing Dev. 2005; 126:580–90. https://doi.org/10.1016/j.mad.2004.11.008.
390. Chang BD, Xuan Y, Broude EV, Zhu H, Schott B, Fang J, Roninson IB. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene. 1999; 18:4808–18. https://doi.org/10.1038/sj.onc.1203078.
391. Sarangi U, Paithankar KR, Kumar JU, Subramaniam V, Sreedhar AS. 17AAG Treatment Accelerates Doxorubicin Induced Cellular Senescence: Hsp90 Interferes with Enforced Senescence of Tumor Cells. Drug Target Insights. 2012; 6:19–39. https://doi.org/10.4137/DTI.S9943.
392. Jackson JG, Pereira-Smith OM. p53 is preferentially recruited to the promoters of growth arrest genes p21 and GADD45 during replicative senescence of normal human fibroblasts. Cancer Res. 2006; 66:8356–60. https://doi.org/10.1158/0008-5472.CAN-06-1752.
393. Yan D, An G, Kuo MT. C-Jun N-terminal kinase signalling pathway in response to cisplatin. J Cell Mol Med. 2016; 20:2013–19. https://doi.org/10.1111/jcmm.12908.
394. Qu K, Lin T, Wang Z, Liu S, Chang H, Xu X, Meng F, Zhou L, Wei J, Tai M, Dong Y, Liu C. Reactive oxygen species generation is essential for cisplatin-induced accelerated senescence in hepatocellular carcinoma. Front Med. 2014; 8:227–35. https://doi.org/10.1007/s11684-014-0327-1.
395. Qu K, Lin T, Wei J, Meng F, Wang Z, Huang Z, Wan Y, Song S, Liu S, Chang H, Dong Y, Liu C. Cisplatin induces cell cycle arrest and senescence via upregulating P53 and P21 expression in HepG2 cells. Nan Fang Yi Ke Da Xue Xue Bao. 2013; 33:1253–59.
396. Davaadelger B, Duan L, Perez RE, Gitelis S, Maki CG. Crosstalk between the IGF-1R/AKT/mTORC1 pathway and the tumor suppressors p53 and p27 determines cisplatin sensitivity and limits the effectiveness of an IGF-1R pathway inhibitor. Oncotarget. 2016; 7:27511–26. https://doi.org/10.18632/oncotarget.8484.
397. Li W, Wang W, Li Y, Wang W, Wang T, Li L, Han Z, Wang S, Ma D, Wang H. Proteomics analysis of normal and senescent NG108-15 cells: GRP78 plays a negative role in cisplatin-induced senescence in the NG108-15 cell line. PLoS One. 2014; 9:e90114. https://doi.org/10.1371/journal.pone.0090114.
398. García-Cano J, Ambroise G, Pascual-Serra R, Carrión MC, Serrano-Oviedo L, Ortega-Muelas M, Cimas FJ, Sabater S, Ruiz-Hidalgo MJ, Sanchez Perez I, Mas A, Jalón FA, Vazquez A, Sánchez-Prieto R. Exploiting the potential of autophagy in cisplatin therapy: A new strategy to overcome resistance. Oncotarget. 2015; 6:15551–65. https://doi.org/10.18632/oncotarget.3902.
399. Im-aram A, Farrand L, Bae SM, Song G, Song YS, Han JY, Tsang BK. The mTORC2 component rictor contributes to cisplatin resistance in human ovarian cancer cells. PLoS One. 2013; 8:e75455. https://doi.org/10.1371/journal.pone.0075455.
400. Davaadelger B, Perez RE, Zhou Y, Duan L, Gitelis S, Maki CG. The IGF-1R/AKT pathway has opposing effects on Nutlin-3a-induced apoptosis. Cancer Biol Ther. 2017; 18:895–903. https://doi.org/10.1080/15384047.2017.1345397.
401. Mandic R, Schamberger CJ, Müller JF, Geyer M, Zhu L, Carey TE, Grénman R, Dünne AA, Werner JA. Reduced cisplatin sensitivity of head and neck squamous cell carcinoma cell lines correlates with mutations affecting the COOH-terminal nuclear localization signal of p53. Clin Cancer Res. 2005; 11:6845–52. https://doi.org/10.1158/1078-0432.CCR-05-0378.
402. Tonigold M, Rossmann A, Meinold M, Bette M, Märken M, Henkenius K, Bretz AC, Giel G, Cai C, Rodepeter FR, Beneš V, Grénman R, Carey TE, et al. A cisplatin-resistant head and neck cancer cell line with cytoplasmic p53(mut) exhibits ATP-binding cassette transporter upregulation and high glutathione levels. J Cancer Res Clin Oncol. 2014; 140:1689–704. https://doi.org/10.1007/s00432-014-1727-y.
403. Traverso N, Ricciarelli R, Nitti M, Marengo B, Furfaro AL, Pronzato MA, Marinari UM, Domenicotti C. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev. 2013; 2013:972913. https://doi.org/10.1155/2013/972913.
404. Xiang Y, Zhu Z, Han G, Ye X, Xu B, Peng Z, Ma Y, Yu Y, Lin H, Chen AP, Chen CD. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc Natl Acad Sci USA. 2007; 104:19226–31. https://doi.org/10.1073/pnas.0700735104.
405. Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Körbel C, Laschke MW, Gimotty PA, Philipp SE, Krause E, Pätzold S, Villanueva J, et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell. 2013; 23:811–25. https://doi.org/10.1016/j.ccr.2013.05.003.
406. Shen X, Zhuang Z, Zhang Y, Chen Z, Shen L, Pu W, Chen L, Xu Z. JARID1B modulates lung cancer cell proliferation and invasion by regulating p53 expression. Tumour Biol. 2015; 36:7133–42. https://doi.org/10.1007/s13277-015-3418-y.
407. Wang L, Mao Y, Du G, He C, Han S. Overexpression of JARID1B is associated with poor prognosis and chemotherapy resistance in epithelial ovarian cancer. Tumour Biol. 2015; 36:2465–72. https://doi.org/10.1007/s13277-014-2859-z.
408. Tsou SH, Hou MH, Hsu LC, Chen TM, Chen YH. Gain-of-function p53 mutant with 21-bp deletion confers susceptibility to multidrug resistance in MCF-7 cells. Int J Mol Med. 2016; 37:233–42. https://doi.org/10.3892/ijmm.2015.2406.
409. Chee JL, Saidin S, Lane DP, Leong SM, Noll JE, Neilsen PM, Phua YT, Gabra H, Lim TM. Wild-type and mutant p53 mediate cisplatin resistance through interaction and inhibition of active caspase-9. Cell Cycle. 2013; 12:278–88. https://doi.org/10.4161/cc.23054.
410. Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992; 69:1237–45. https://doi.org/10.1016/0092-8674(92)90644-R.
411. Chen J, Marechal V, Levine AJ. Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol. 1993; 13:4107–14. https://doi.org/10.1128/MCB.13.7.4107.
412. Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996; 274:948–53. https://doi.org/10.1126/science.274.5289.948.
413. Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993; 7:1126–32. https://doi.org/10.1101/gad.7.7a.1126.
414. Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997; 387:299–303. https://doi.org/10.1038/387299a0.
415. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997; 387:296–99. https://doi.org/10.1038/387296a0.
416. Rodriguez MS, Desterro JM, Lain S, Lane DP, Hay RT. Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol Cell Biol. 2000; 20:8458–67. https://doi.org/10.1128/MCB.20.22.8458-8467.2000.
417. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004; 303:844–48. https://doi.org/10.1126/science.1092472.
418. Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E. Clinical Overview of MDM2/X-Targeted Therapies. Front Oncol. 2016; 6:7. https://doi.org/10.3389/fonc.2016.00007.
419. Zhao Y, Aguilar A, Bernard D, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J Med Chem. 2015; 58:1038–52. https://doi.org/10.1021/jm501092z.
420. Kojima K, Shimanuki M, Shikami M, Samudio IJ, Ruvolo V, Corn P, Hanaoka N, Konopleva M, Andreeff M, Nakakuma H. The dual PI3 kinase/mTOR inhibitor PI-103 prevents p53 induction by Mdm2 inhibition but enhances p53-mediated mitochondrial apoptosis in p53 wild-type AML. Leukemia. 2008; 22:1728–36. https://doi.org/10.1038/leu.2008.158.
421. Drakos E, Atsaves V, Li J, Leventaki V, Andreeff M, Medeiros LJ, Rassidakis GZ. Stabilization and activation of p53 downregulates mTOR signaling through AMPK in mantle cell lymphoma. Leukemia. 2009; 23:784–90. https://doi.org/10.1038/leu.2008.348.
422. Serrano M. Shifting senescence into quiescence by turning up p53. Cell Cycle. 2010; 9:4256–57. https://doi.org/10.4161/cc.9.21.13785.
423. Korotchkina LG, Demidenko ZN, Gudkov AV, Blagosklonny MV. Cellular quiescence caused by the Mdm2 inhibitor nutlin-3A. Cell Cycle. 2009; 8:3777–81. https://doi.org/10.4161/cc.8.22.10121.
424. Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci USA. 2010; 107:9660–64. https://doi.org/10.1073/pnas.1002298107.
425. Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY). 2010; 2:344–52. https://doi.org/10.18632/aging.100160.
426. Villalonga-Planells R, Coll-Mulet L, Martínez-Soler F, Castaño E, Acebes JJ, Giménez-Bonafé P, Gil J, Tortosa A. Activation of p53 by nutlin-3a induces apoptosis and cellular senescence in human glioblastoma multiforme. PLoS One. 2011; 6:e18588. https://doi.org/10.1371/journal.pone.0018588.
427. Leontieva OV, Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY). 2010; 2:924–35. https://doi.org/10.18632/aging.100265.
428. Polański R, Noon AP, Blaydes J, Phillips A, Rubbi CP, Parsons K, Vlatković N, Boyd MT. Senescence induction in renal carcinoma cells by Nutlin-3: a potential therapeutic strategy based on MDM2 antagonism. Cancer Lett. 2014; 353:211–19. https://doi.org/10.1016/j.canlet.2014.07.024.
429. Hasegawa H, Yamada Y, Iha H, Tsukasaki K, Nagai K, Atogami S, Sugahara K, Tsuruda K, Ishizaki A, Kamihira S. Activation of p53 by Nutlin-3a, an antagonist of MDM2, induces apoptosis and cellular senescence in adult T-cell leukemia cells. Leukemia. 2009; 23:2090–101. https://doi.org/10.1038/leu.2009.171.
430. di Iasio MG, Zauli G. The non-genotoxic activator of the p53 pathway Nutlin-3 shifts the balance between E2F7 and E2F1 transcription factors in leukemic cells. Invest New Drugs. 2013; 31:458–60. https://doi.org/10.1007/s10637-012-9882-y.
431. Park C, Lee I, Kang WK. E2F-1 is a critical modulator of cellular senescence in human cancer. Int J Mol Med. 2006; 17:715–20.
432. Luo H, Yount C, Lang H, Yang A, Riemer EC, Lyons K, Vanek KN, Silvestri GA, Schulte BA, Wang GY. Activation of p53 with Nutlin-3a radiosensitizes lung cancer cells via enhancing radiation-induced premature senescence. Lung Cancer. 2013; 81:167–73. https://doi.org/10.1016/j.lungcan.2013.04.017.
433. Borthakur G, Duvvuri S, Ruvolo V, Tripathi DN, Piya S, Burks J, Jacamo R, Kojima K, Ruvolo P, Fueyo-Margareto J, Konopleva M, Andreeff M. MDM2 Inhibitor, Nutlin 3a, Induces p53 Dependent Autophagy in Acute Leukemia by AMP Kinase Activation. PLoS One. 2015; 10:e0139254. https://doi.org/10.1371/journal.pone.0139254.
434. Ringer L, Sirajuddin P, Tricoli L, Waye S, Choudhry MU, Parasido E, Sivakumar A, Heckler M, Naeem A, Abdelgawad I, Liu X, Feldman AS, Lee RJ, et al. The induction of the p53 tumor suppressor protein bridges the apoptotic and autophagic signaling pathways to regulate cell death in prostate cancer cells. Oncotarget. 2014; 5:10678–91. https://doi.org/10.18632/oncotarget.2528.
435. Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, Bergman J, Wiman KG, Selivanova G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002; 8:282–88. https://doi.org/10.1038/nm0302-282.
436. Lambert JM, Moshfegh A, Hainaut P, Wiman KG, Bykov VJ. Mutant p53 reactivation by PRIMA-1MET induces multiple signaling pathways converging on apoptosis. Oncogene. 2010; 29:1329–38. https://doi.org/10.1038/onc.2009.425.
437. Rao CV, Patlolla JM, Qian L, Zhang Y, Brewer M, Mohammed A, Desai D, Amin S, Lightfoot S, Kopelovich L. Chemopreventive effects of the p53-modulating agents CP-31398 and Prima-1 in tobacco carcinogen-induced lung tumorigenesis in A/J mice. Neoplasia. 2013; 15:1018–27. https://doi.org/10.1593/neo.131256.
438. Lee K, Wang T, Paszczynski AJ, Daoud SS. Expression proteomics to p53 mutation reactivation with PRIMA-1 in breast cancer cells. Biochem Biophys Res Commun. 2006; 349:1117–24. https://doi.org/10.1016/j.bbrc.2006.08.152.
439. Patyka M, Sharifi Z, Petrecca K, Mansure J, Jean-Claude B, Sabri S. Sensitivity to PRIMA-1MET is associated with decreased MGMT in human glioblastoma cells and glioblastoma stem cells irrespective of p53 status. Oncotarget. 2016; 7:60245–69. https://doi.org/10.18632/oncotarget.11197.
440. Russo D, Ottaggio L, Foggetti G, Masini M, Masiello P, Fronza G, Menichini P. PRIMA-1 induces autophagy in cancer cells carrying mutant or wild type p53. Biochim Biophys Acta. 2013; 1833:1904–13. https://doi.org/10.1016/j.bbamcr.2013.03.020.
441. Russo D, Ottaggio L, Penna I, Foggetti G, Fronza G, Inga A, Menichini P. PRIMA-1 cytotoxicity correlates with nucleolar localization and degradation of mutant p53 in breast cancer cells. Biochem Biophys Res Commun. 2010; 402:345–50. https://doi.org/10.1016/j.bbrc.2010.10.031.
442. Rieber M, Strasberg-Rieber M. Hypoxia, Mn-SOD and H(2)O(2) regulate p53 reactivation and PRIMA-1 toxicity irrespective of p53 status in human breast cancer cells. Biochem Pharmacol. 2012; 84:1563–70. https://doi.org/10.1016/j.bcp.2012.09.003.
443. Ali D, Mohammad DK, Mujahed H, Jonson-Videsäter K, Nore B, Paul C, Lehmann S. Anti-leukaemic effects induced by APR-246 are dependent on induction of oxidative stress and the NFE2L2/HMOX1 axis that can be targeted by PI3K and mTOR inhibitors in acute myeloid leukaemia cells. Br J Haematol. 2016; 174:117–26. https://doi.org/10.1111/bjh.14036.
444. Tessoulin B, Descamps G, Moreau P, Maïga S, Lodé L, Godon C, Marionneau-Lambot S, Oullier T, Le Gouill S, Amiot M, Pellat-Deceunynck C. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood. 2014; 124:1626–36. https://doi.org/10.1182/blood-2014-01-548800.
445. Peng X, Zhang MQ, Conserva F, Hosny G, Selivanova G, Bykov VJ, Arnér ES, Wiman KG. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013; 4:e881. https://doi.org/10.1038/cddis.2013.417.
446. Yoshikawa N, Kajiyama H, Nakamura K, Utsumi F, Niimi K, Mitsui H, Sekiya R, Suzuki S, Shibata K, Callen D, Kikkawa F. PRIMA-1MET induces apoptosis through accumulation of intracellular reactive oxygen species irrespective of p53 status and chemo-sensitivity in epithelial ovarian cancer cells. Oncol Rep. 2016; 35:2543–52. https://doi.org/10.3892/or.2016.4653.
447. Supiot S, Zhao H, Wiman K, Hill RP, Bristow RG. PRIMA-1(met) radiosensitizes prostate cancer cells independent of their MTp53-status. Radiother Oncol. 2008; 86:407–11. https://doi.org/10.1016/j.radonc.2008.01.001.
448. Cory AH, Chen J, Cory JG. Effects of PRIMA-1 on wild-type L1210 cells expressing mutant p53 and drug-resistant L1210 cells lacking expression of p53: necrosis vs. apoptosis. Anticancer Res. 2006; 26:1289–95.
449. Liu DS, Read M, Cullinane C, Azar WJ, Fennell CM, Montgomery KG, Haupt S, Haupt Y, Wiman KG, Duong CP, Clemons NJ, Phillips WA. APR-246 potently inhibits tumour growth and overcomes chemoresistance in preclinical models of oesophageal adenocarcinoma. Gut. 2015; 64:1506–16. https://doi.org/10.1136/gutjnl-2015-309770.
450. Mohell N, Alfredsson J, Fransson Å, Uustalu M, Byström S, Gullbo J, Hallberg A, Bykov VJ, Björklund U, Wiman KG. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 2015; 6:e1794. https://doi.org/10.1038/cddis.2015.143.
451. Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature. 1991; 352:345–47. https://doi.org/10.1038/352345a0.
452. Shaw P, Bovey R, Tardy S, Sahli R, Sordat B, Costa J. Induction of apoptosis by wild-type p53 in a human colon tumor-derived cell line. Proc Natl Acad Sci USA. 1992; 89:4495–99. https://doi.org/10.1073/pnas.89.10.4495.
453. Yonish-Rouach E, Grunwald D, Wilder S, Kimchi A, May E, Lawrence JJ, May P, Oren M. p53-mediated cell death: relationship to cell cycle control. Mol Cell Biol. 1993; 13:1415–23. https://doi.org/10.1128/MCB.13.3.1415.
454. Ryan JJ, Danish R, Gottlieb CA, Clarke MF. Cell cycle analysis of p53-induced cell death in murine erythroleukemia cells. Mol Cell Biol. 1993; 13:711–19. https://doi.org/10.1128/MCB.13.1.711.
455. Wang Y, Ramqvist T, Szekely L, Axelson H, Klein G, Wiman KG. Reconstitution of wild-type p53 expression triggers apoptosis in a p53-negative v-myc retrovirus-induced T-cell lymphoma line. Cell Growth Differ. 1993; 4:467–73.
456. Ramqvist T, Magnusson KP, Wang Y, Szekely L, Klein G, Wiman KG. Wild-type p53 induces apoptosis in a Burkitt lymphoma (BL) line that carries mutant p53. Oncogene. 1993; 8:1495–500.
457. Roth JA, Nguyen D, Lawrence DD, Kemp BL, Carrasco CH, Ferson DZ, Hong WK, Komaki R, Lee JJ, Nesbitt JC, Pisters KM, Putnam JB, Schea R, et al. Retrovirus-mediated wild-type p53 gene transfer to tumors of patients with lung cancer. Nat Med. 1996; 2:985–91. https://doi.org/10.1038/nm0996-985.
458. Zhang WW, Fang X, Mazur W, French BA, Georges RN, Roth JA. High-efficiency gene transfer and high-level expression of wild-type p53 in human lung cancer cells mediated by recombinant adenovirus. Cancer Gene Ther. 1994; 1:5–13.
459. Wilson JM. Gendicine: the first commercial gene therapy product. Hum Gene Ther. 2005; 16:1014–15. https://doi.org/10.1089/hum.2005.16.1014.
460. Wills KN, Maneval DC, Menzel P, Harris MP, Sutjipto S, Vaillancourt MT, Huang WM, Johnson DE, Anderson SC, Wen SF, Bookstein R, Shepard HM, Gregory RJ. Development and characterization of recombinant adenoviruses encoding human p53 for gene therapy of cancer. Hum Gene Ther. 1994; 5:1079–88. https://doi.org/10.1089/hum.1994.5.9-1079.
461. Peng Z. Current status of gendicine in China: recombinant human Ad-p53 agent for treatment of cancers. Hum Gene Ther. 2005; 16:1016–27. https://doi.org/10.1089/hum.2005.16.1016.
462. Hasei J, Sasaki T, Tazawa H, Osaki S, Yamakawa Y, Kunisada T, Yoshida A, Hashimoto Y, Onishi T, Uno F, Kagawa S, Urata Y, Ozaki T, Fujiwara T. Dual programmed cell death pathways induced by p53 transactivation overcome resistance to oncolytic adenovirus in human osteosarcoma cells. Mol Cancer Ther. 2013; 12:314–25. https://doi.org/10.1158/1535-7163.MCT-12-0869.
463. Ahmad IM, Abdalla MY, Aykin-Burns N, Simons AL, Oberley LW, Domann FE, Spitz DR. 2-Deoxyglucose combined with wild-type p53 overexpression enhances cytotoxicity in human prostate cancer cells via oxidative stress. Free Radic Biol Med. 2008; 44:826–34. https://doi.org/10.1016/j.freeradbiomed.2007.11.007.
464. Shatrov VA, Ameyar M, Bouquet C, Cai Z, Stancou R, Haddada H, Chouaib S. Adenovirus-mediated wild-type-p53-gene expression sensitizes TNF-resistant tumor cells to TNF-induced cytotoxicity by altering the cellular redox state. Int J Cancer. 2000; 85:93–97. https://doi.org/10.1002/(SICI)1097-0215(20000101)85:13.0.CO;2-I.
465. Jung MS, Jin DH, Chae HD, Kang S, Kim SC, Bang YJ, Choi TS, Choi KS, Shin DY. Bcl-xL and E1B-19K proteins inhibit p53-induced irreversible growth arrest and senescence by preventing reactive oxygen species-dependent p38 activation. J Biol Chem. 2004; 279:17765–71. https://doi.org/10.1074/jbc.M305015200.
466. Bai J, Cederbaum AI. Catalase protects HepG2 cells from apoptosis induced by DNA-damaging agents by accelerating the degradation of p53. J Biol Chem. 2003; 278:4660–67. https://doi.org/10.1074/jbc.M206273200.
467. Quist SR, Wang-Gohrke S, Köhler T, Kreienberg R, Runnebaum IB. Cooperative effect of adenoviral p53 gene therapy and standard chemotherapy in ovarian cancer cells independent of the endogenous p53 status. Cancer Gene Ther. 2004; 11:547–54. https://doi.org/10.1038/sj.cgt.7700727.
468. Yu ZW, Zhao P, Liu M, Dong XS, Tao J, Yao XQ, Yin XH, Li Y, Fu SB. Reversal of 5-flouroucial resistance by adenovirus-mediated transfer of wild-type p53 gene in multidrug-resistant human colon carcinoma LoVo/5-FU cells. World J Gastroenterol. 2004; 10:1979–83. https://doi.org/10.3748/wjg.v10.i13.1979.
469. Kraljević Pavelić S, Marjanović M, Poznić M, Kralj M. Adenovirally mediated p53 overexpression diversely influence the cell cycle of HEp-2 and CAL 27 cell lines upon cisplatin and methotrexate treatment. J Cancer Res Clin Oncol. 2009; 135:1747–61. https://doi.org/10.1007/s00432-009-0621-5.
470. Qi X, Chang Z, Song J, Gao G, Shen Z. Adenovirus-mediated p53 gene therapy reverses resistance of breast cancer cells to adriamycin. Anticancer Drugs. 2011; 22:556–62. https://doi.org/10.1097/CAD.0b013e328345b4e7.
471. Shirakawa T, Sasaki R, Gardner TA, Kao C, Zhang ZJ, Sugimura K, Matsuo M, Kamidono S, Gotoh A. Drug-resistant human bladder-cancer cells are more sensitive to adenovirus-mediated wild-type p53 gene therapy compared to drug-sensitive cells. Int J Cancer. 2001; 94:282–89. https://doi.org/10.1002/ijc.1453.
472. Yang J, Zhao X, Tang M, Li L, Lei Y, Cheng P, Guo W, Zheng Y, Wang W, Luo N, Peng Y, Tong A, Wei Y, et al. The role of ROS and subsequent DNA-damage response in PUMA-induced apoptosis of ovarian cancer cells. Oncotarget. 2017; 8:23492–506. https://doi.org/10.18632/oncotarget.15626.
473. Inoue T, Kato K, Kato H, Asanoma K, Kuboyama A, Ueoka Y, Yamaguchi S, Ohgami T, Wake N. Level of reactive oxygen species induced by p21Waf1/CIP1 is critical for the determination of cell fate. Cancer Sci. 2009; 100:1275–83. https://doi.org/10.1111/j.1349-7006.2009.01166.x.