
INTRODUCTION
GBM
Gliomas are the most common primary tumors that arise within the brain and have histologic features similar to normal glial cells (i.e., astrocytes, oligodendrocytes, and ependymal cells). The incidence rate of primary malignant brain tumors is about 6.5 cases per 100,000/year (2005–2009) [1]. In the USA the incidence of glioblastoma multiforme (GBM), grade IV glioma, is about 3.19 per 100,000 per year. The outcome for GBM patients is grim, with a median survival of 16.7 months and only 30% of patients surviving ≥ 24 months [2].
Current solutions are inadequate
Initial therapy
A graphic indicating the therapy and temporal pattern of administration in typical primary and salvage therapies of GBM is to be found in Supplementary Figure 1.
The Stupp protocol is the standard of care for the treatment of GBM and has led to significant survival improvements since its publication in 2005 [4, 5]. The original Stupp protocol consists of an aggressive resection of the GBM. Patients then undergo chemoradiotherapy some 2–8 weeks later; a combination of focal radiation (tumor volume + 2–3 cm margins) in 2 Gy daily fractions (5 days per week, typically Monday to Friday) for 6 weeks and TMZ, 75 mg/m2/day 1–2 hours before the radiation, for six weeks, with TMZ also taken during weekends. TMZ is then given post-radiotherapy (adjuvant) in six cycles consisting of 150–200 mg/m2/day for 5 days during each 28-day cycle; Supplementary Figure 1A. The main modification to this standard of care is the use of chemotherapy with radiotherapy, rather than after radiotherapy. The Stupp regime, in those patients that could tolerate the high radiation and TMZ, resulted in a significant survival improvement with 2-year-survival increasing from 10.4 to 26.5%.
Use of alkylating agents in GBM salvage therapy: Gliomas eventually develop resistance toward TMZ and patients then undergo salvage therapy that typically involves either additional chemotherapy or a combination of chemotherapy and re-radiation therapy. The alkylating agent CCNU is still a popular choice for salvage therapy, given as a single agent or part of a drug cocktail, with or without radiation. Two recent trials by Batchelor [3]. and by Taal [4]. use high dosages of CCNU, 56-days apart, to treat TMZ-resistant GBM, Supplementary Figure 1B. Dosage staggering is required because all alkylating agents attack bone marrow and therefore life-threatening complications are common. CCNU extends survival by ≈ 9.5 months, in patients who can tolerate the regime. PCV therapy is an extensively used salvage therapy and consists of Procarbazine (an alkylating agent), CCNU and Vincristine (tubulin disruptor), with drugs given in a specific sequence, over a 56-day cycle, Supplementary Figure 1C. Outcomes are slightly worse than aggressive CCNU therapy, with survival extended by ≈ 8.5 months [5–7], but this therapy is better tolerated by patients. Van den Bent and co-workers demonstrated that staggered doses of BCNU extended survival by 9.3 months in GBM patients with acquired TMZ resistance [8], but using the same protocol with a larger patient cohort extended survival by only 5.5 months [9], Supplementary Figure 1D. A trial by Hayat [10] of patients with either low-grade glioma (LGG) or GBM tumors used concomitant CCNU and re-radiation, followed by adjuvant CCNU. The low doses of CCNU caused few off-target complications and coupled with focal radiation of the tumor extended survival by ≈ 8 months, Supplementary Figure 1E.
The basis of chemotherapy: how alkylating agents work as chemotherapeutic drugs; alkylation and oxidative stress
Chemotherapeutic alkylating agents work because they alkylate a wide range of cellular nucleophiles such as thiols, alcohols and amines, including the nucleotide bases which make up DNA. Resistance to alkylating agents is typically due to one or more DNA repair systems being upregulated (repair function), or a pro-apoptotic signaling pathway (report function) linked to DNA repair being down-regulated in cancer cells [11, 12]. This means that DNA damage induced by a chemotherapeutic alkylating agents is either being repaired or tolerated in cancer cells more than in some of the non-cancerous cells, typically bone marrow, which is normally the most sensitive off-target tissue.
Elevated rates of reactive oxygen species (ROS) production are a hallmark of most cancer cells where they are associated with tumor development and progression [13]. Elevated steady-state ROS levels cause cancer cells to upregulate many DNA repair pathway proteins (reviews [14–16] and ROS preconditioning in glioma may be a driving force in innate drug-resistant phenotypes [17].
Why mono-therapeutic agents fail against glioma: evolution
Cancer cells, especially those present in a high-grade tumor, are the end products of evolutionary selection pressure that has acted on the descendants which arose from a single deviant cell. One can envision cancers as asexual pathogens. Computation evolutionary simulations and studies of asexual eukaryotic pathogens indicate that high mutation rates and low fidelity in DNA repair are hallmarks of asexual pathogens that have ‘evolved to evolve’ [18, 19]. High-grade tumors are heterogeneous, an indicator of rapid evolution, and tumor cells are characterized by high mutation loads, amplifications, deletions and structural rearrangements of swathes of DNA [20–24]. We postulated that aberrations in DNA repair pathways would be selected for in GBM, allowing these cancer cells to undergo rapid evolution and niche exploitation, but could be its ‘Achilles heel’ for a highly targeted chemotherapeutic drug strategy.
As an aid to the discussion of DNA repair pathways we have prepared graphics illustrating the mechanistic aspect of the Base Excision Repair (BER), Mismatch Repair (MMR) and Nucleotide Excision Repair (NER), which are shown in Supplementary Figure 2. As the Non-Homologous End-Joining (NHEJ), Micro-homology-Mediated End Joining (MMEJ) and Homologous Recombination (HR) and nucleophosmin-linked translesion DNA synthesis pathway in GBM [25] do not merit highly in the context of PAM-OBG treatment and we will not cover them in detail. We recommend the website maintained by KEGG.org for further information [26].
TMZ resistance mechanisms in glioma
TMZ is a prodrug version of the highly reactive methylation agent diazomethane, (Figure 1A (i)) which gives rise to a large number of DNA base adducts, including O6-methylguanine (O6MeG), Figure 1A (ii).
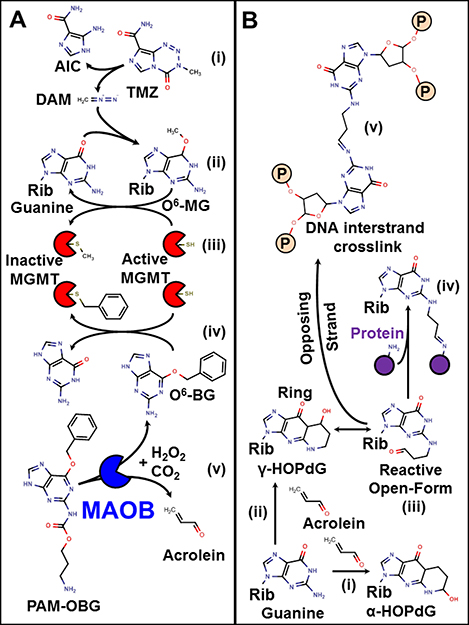
Figure 1: Mechanism of action of TMZ, O6-BG, PAM-OBG, and acrolein. (A) shows the action of TMZ, O6BG, and PAM-OBG. TMZ is a prodrug that spontaneously gives rise to the methylation agent diazomethane (i). DAM can react with DNA giving rise to O6MeG (ii). This toxic lesion is removed by MGMT which transfers the methyl adduct to a thiol in its active site (iii). MGMT can also react with O6BG, being deactivated, with a benzyl thio-ether formed (iv). PAM-OBG is oxidized by MAOB, producing CO2, O6BG, and acrolein (v). The reactions of acrolein and DNA are shown in (B). Guanine residues react with acrolein to generate HOPdG, the α- HOPdG bulky adduct is stable (i), but the γ-HOPdG has unreactive closed (ii) and open (iii) forms. Proteins, including DNA repair proteins, can become covalently linked to γ-HOPdG (iv), but the formation of DNA interstrand crosslinks (v) is the more toxic lesion.
In GBM the action of the enzyme O6-methylguanine DNA methyltransferase (MGMT) is the primary means of TMZ resistance. MGMT is a small suicide-enzyme that directly removes alkyl adducts from O6-guanine, via direct transfer of the alkyl to a cysteine thiol in the enzymes active site, Figure 1A (iii). Each MGMT can only repair a single lesion, as after adduct transfer and formation of alkyl thioether, MGMT is inactivated. MGMT also accepts an alkyl group from specific inhibitors such as O6-benzylguanine (O6BG), Figure 1A (iv).
MGMT repairs the O6MeG lesions generated by TMZ, in vitro and in vivo, limiting TMZ toxicity. MGMT gene expression levels are highly dependent on MGMT promoter methylation status, and in primary GBM biopsy samples, promoter methylation is found in some 35–45% of samples, with methylation of the promoter indicative of low MGMT mRNA levels [27–29]. However, mRNA levels do not directly correlate with the steady-state levels of the active MGMT, and this may in part be due to post-translational modification, including phosphorylation [30], altering MGMT biological half-life.
Levels of active MGMT in GBM
The steady-state levels of active MGMT in treatment naïve tumors depend on the relative rate of synthesis and rate of degradation, which is governed by ubiquitination rate. STAT3 activation causes phosphorylation of MGMT and this enhances MGMT lifetime, raising the steady-state levels of active MGMT some 5-fold [31]. MGMT/pMGMT levels correlate with TMZ treatment failure in GBM [31–41]. Thus, activated STAT3 may be a major mechanism of MGMT induced resistance to alkylating agents [31, 32].
Correlation of MGMT protein levels and MGMT promoter methylation is not statistically significant, and there is also no significant survival difference between patients whose GBM biopsy samples had high or low levels of MGMT if the MGMT is determined immunohistochemically [33, 34].
Mechanisms TMZ resistance
Evaluations of the changes in DNA-repair mechanisms in GBM tumors resected prior to chemoradiation, and then following failure of the standard Stupp protocol, indicates that upregulation of MGMT is not always the basis of TMZ drug resistance.
Generally, it appears that resistance to TMZ appears to arise from three mechanisms:
1) Upregulation of MGMT [28, 34–37].
2) Downregulation of canonical MMR (MutSa/MutLa). Agarwal [38] noted that after TMZ therapeutic failure there was generally an upregulation of MGMT or there was a downregulation of canonical MMR (or mutations in MutSa/MutLa) in second resection tumor samples.
3) Upregulation of BER, specifically the action of N-Methylpurine DNA Glycosylase (MPG), and its interaction with the Ataxia Telangiectasia Mutated Serine/Threonine Kinase (ATM) signaling system [39, 40]. Agnihotri and co-workers demonstrated, using samples drawn from pediatric GBM patients, that ATM/MPG form a complex and that MPG-BER may be able to remove O6MeG bases when MPG is phosphorylated via ATM [40].
Failure of MGMT inhibitors in the clinical setting
O6BG, a potent inhibitor of MGMT, is nontoxic when administered to animals or humans as a single agent [41]. A phase III trial utilizing O6BG to ablate MGMT activity throughout a patient’s tissues, prior to radiotherapy/BCNU + BCNU, was highly disappointing as O6BG did not provide an added benefit, but instead caused additional toxicity. Hematologic Grade 4 or higher toxicities quadrupled in patients receiving O6BG & BCNU vs. the BCNU arm. Several phase II trials utilizing an MGMT inhibitor combined with radiotherapy/TMZ have failed [42–44].
The combination of alkylating agents with an MGMT inhibitor has failed in clinical trials for a simple reason; alkylating agents are more toxic to non-target tissues, especially bone marrow, than toward tumor cells.
A MAOB activated bifunctional prodrug targeting MGMT and DNA
Design philosophy
Our aim was to design a therapeutic agent that would complement the existing best clinical treatment regime, potentiating TMZ chemoradiation toxicity within glioma cells but not systemically, making a chemotherapeutic to fit directly into the Stupp regime.
Strategy for a MAOB activated MGMT inhibitor
Expression of MAOB in LGG and GBM is highly elevated, and its levels correlate with the degree of tumor hypoxia [45–47]. Data from the TCGC database indicates that all major glioma sub-types have a similar MAOB mRNA distribution, with the minimum showing > 2.5-fold the level of normal brain tissue, the average ≈ 8-fold greater, and maximal > 30-fold higher (166 GBM, 283 LGG-WT, and 233 LGG-IDH mut) [48, 49]. Additionally, we find that MAOB and MGMT expression levels are highly correlated in GBM (p < 0.01) [48, 49]. We envisioned a prodrug that would become active after oxidation by MAOB and that after maturation would generate O6BG to inhibit MGMT would sensitize cells to TMZ chemoradiation. As the oxidative activation of a prodrug by MAOB also generates an aldehyde, we sought a design whereby the aldehyde produced from the prodrug maturation would be a DNA damaging agent in its own right.
Choice of MAOB as a maturation enzyme
Human bone marrow has almost no expression of MAOB, which makes a MAOB-based prodrug designed to potentiate alkylating agents highly attractive. Human MAOB and MGMT mRNA expression level [50], activity [51] and also protein half-life [52–54] are highly tissue dependent. Supplementary Figure 3A shows the levels of MGMT and MAOB mRNA (normalized to the liver) measured in human tissues [50, 55]. The MGMT:MAOB ratio of a tissue is a proxy for the off-target sensitivity expected from a MAOB-activated MGMT inhibitor, Supplementary Figure 3B. The very high levels of MGMT and low levels MAOB in bone marrow suggest that this would be the body’s least sensitive tissue.
MAOB maturation chemistry
We have identified two major MAO-specific prodrug chemical motifs that can be used in MAO specific prodrug design; tetrahydropyridines that become mitochondrial ‘smart bombs’ [56, 57] and immolative amine linkers that undergo ‘unmasking’. We have used the latter for the design of PAM-OBG, which allows an inactive ‘masked’ adduct of O6BG to be converted in O6BG only inside cells with MAOB activity. MAO’s oxidize propylamine ethers to give rise to acrolein and a primary alcohol, and this mechanism is the basis of the MAO-Glo™ (Luciferin) Assay (Promega Corporation, Madison, WI) [58, 59].
PAM-OBG is a MAOB specific prodrug version of O6BG that uses a similar immolative ‘unmasking’ route as used in MAO-Glo™. In PAM-OBG an amide-ether (carbamate) to a primary amine is used as the immolative propylamine linker, rather than using an ether linkage to a primary alcohol, as in the case of the aminopropylether luciferin MAO substrate used in MAO-Glo™.
Figure 1A (v) shows that PAM-OBG consists of an MAO-oxidizable propylamine linked to the 2-amino position of O6BG, via a carbamate. In silico modeling using the O6MeG hMGMT (C145S) crystal structure [60]. indicated that the addition to the 2-amino of O6-alkylguanine position would result in a sterically hindered molecule that could no longer fit the MGMT substrate pocket.
Toxicity of acrolein: ICLs and HOPdG
Acrolein is a highly reactive chemotherapeutic agent, generated by the cyclophosphamide class of nitrogen mustards (review [61]). Acrolein forms toxic DNA base adducts, notably α-HOPdG & γ-HOPdG. It also causes the formation of highly toxic DNA-protein crosslinks and interstrand cross-links (ICLs [61–66]), Figure 1B. Acrolein is extremely toxic in cells with DNA ICL repair defects, including cells derived from patients with Xeroderma pigmentosum (NER defects) [67] and Fanconi anemia (FA) [68–72].
Acrolein generated HOPdG lesions can be repaired directly via oxidation by three AlkBH enzymes, and indirectly by BER and MMR [61]. It has been shown that overexpression of ALKBH2 in glioma cell lines enhances resistance to TMZ, and conversely, siRNA-knockdown of ALKBH2 increases TMZ sensitivity [73]. Acrolein derived HOPdG is oxidized by ALKBH ~200x slower than are DNA methyl adducts [74]. Thus co-administration of acrolein/TMZ (in a Stupp protocol chemotherapeutic regime) will impose an opportunity cost, whereby the time taken by AlkBH to oxidize a single HOPdG adduct means that 200 methyl adducts will not be removed by this direct mechanism [75, 76].
The GBM157 model
All the in vitro and in vivo data presented herein derives from a de-identified human primary GBM. A GBM tumor was harvested from a patient and transplanted into mouse flank, and the resulting tumors were propagated and passaged in mouse flank and archived, frozen. Frozen tumor samples were then used to initiate flank tumors, which were then harvested and used to generate intracranial tumors. Cells from the original tumor were also used to generate tissue cultures, and these were archived, frozen, at 4–5 passages. The in vitro data that is presented using GBM157 are representative of experiments performed using other primary GBM cell lines.
GBM157 is one of the samples on our tumor microarray and the levels of MAOB are in the lowest quartile [45]. With regards to MGMT, we find total protein levels are in the highest quartile, as is HiF1-α, which is linked with MGMT expression levels [77]. We are currently further characterizing this line.
RESULTS
PAM-OBG: risk repair protein mRNA levels and patient survival in GBM
We examined the relationship between the mRNA levels of 181 DNA-repair genes and GBM patient survival using the data deposited in the TCGA database [2, 3]. This was to test the postulate that acrolein derived ICLs would be more toxic to GBM, than to non-cancerous cells, due to evolutionary pressures on glioma to down-regulate certain DNA repair functions.
Using the TCGA database (as of Dec 1st, 2017) we identified 110 (of 166) patients for which there was uncensored survival data (< 89 months) and whose treatment-naive tumors had been analyzed using RNA Seq V2. We examined the transcript levels of 181 genes, panned from the DNA repair pathway genes maintained by KEGG.org [26], involved in DNA repair. Survival curves were generated for each gene examined using ranked transcript data, comparing low and high mRNA levels and then assayed for statistical significance using the Cox-Mantel log-rank Chi-square test, (Mantel-Haenszel test). Raw and analyzed data is to be found in Supplementary Materials.
Of the 181 genes tested we found that 49 transcript levels impacted patient survival in a statistically significant manner (p < 0.05); 27 of these genes products (or pooled paralogues) were directly correlated with patient survival, (ALKBH1, ALKBH2, APEX1, APEX2, ASCC1, ASCC3, ATR, BLM, DCLRE1C, ERCC1, ERCC6, ERCC8, EXOG, GTF2H2C, MLH1, MPG, MSH4, MUTYH, NBN, PARP1&2, PMS1, POLDIP3, RAD23A, (RFC1-4), SLX1B, UBE2E1, UNG & XRCC5). Additionally, we identified 21 gene products (or pooled paralogues) where high levels correlated with poor patient survival, (ALKBH3, CHTF18, DMC1, HUS1, MGMT, MLH3, NEIL1, NEIL2, NEIL3, PAPD7, PARP3, PMS2, RAD17, RMI2, RPA3, RTEL1, SSBP1, TELO2, TERT, TOP3A, XPA & XRCC2).
In addition, we find that the PMS1/PMS2 and RAD23A/ERCC6 mRNA ratios, indicative of MutLβ/MutLα and of global/translation NER ratios respectively, to be independent risk factors in this GBM patient population.
Agnihotri and co-workers [39] found MPG to be a glioma risk factor, mainly examining pediatric tumors, and suggested that it could directly repair TMZ generated O6MeG lesions. We found that MPG expression also correlated with patient outcome, but that MPG effects were context dependent.
In Figure 2 we present a heat map of gene transcript levels, striated horizontally by patient survival outcome in quartiles and vertically by gene/function/pathway. The heat map was generated by taking the median mRNA level for each of the four patient groups. In Supplementary Figure 4 we show an additional heat map, also divided into survival quartiles, where we include the levels of transcripts for the 9-1-1 DNA repair pathway and telomere extension. Also included are the ratios of different core enzyme mRNA levels, which allow the ratios of repair pathways to be estimated in the different quartiles.
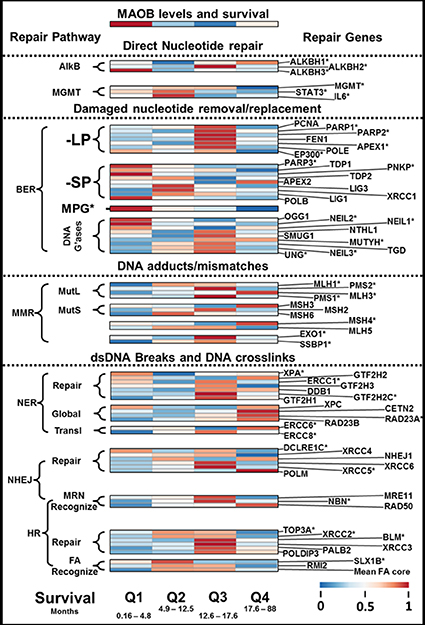
Figure 2: DNA repair enzyme transcript levels and GBM patient survival. Figure 2 presents the levels of different DNA repair enzymes or pathways, in the form of a heat map, striated by GBM patient outcome. At the top of the heat map is shown the relative levels of MAOB, the PAM-OBG activating enzyme. It can be noted that the poorest responding patients, Q1, have the least sophisticated expression of DNA repair pathways that can repair dsDNA breaks or ILCs.
We will describe the influence of DNA repair enzymes on patient outcome, stratified by survival in quartiles, except for the case of MGT, which will be described separately.
First quartile
The first quartile represents the patients with the worst outcome, and not reaching four and a half months survival after diagnosis. In these patients, we find the highest levels of MAOB and we also expected them to have high MGMT levels. However, these cells generally do not have high MGMT or STAT3/IL6. These tumors instead have high levels of another direct repair enzyme, ALKBH3. It has been shown by others that the pairing of ALKBH3 with a helicase (ASCC3) allows efficient removal of many dsDNA lesions, which are generated by TMZ, especially N1-MeA and N3-MeC [78]. In Q1 we find ALKBH3 paired with the helicase ASCC2, rather than the previously described ASCC3, and that the levels of ALKBH1, ALKBH2, ASCC1 and ASCC3 are low. In comparison with ALKBH1 and ALKBH2, ALKBH3 has far less non-repair functionality, as both ALKBH1 and 2 are involved in epigenetic modulation of tRNA, via demethylation of N1-methyladenosine RNA [79]. ALKBH1, but not ALKBH3, also demethylates histones, altering epigenetic gene expression [80]. ALKBH3 also lacks the potent AP-lyase activity found in ALKBH1 that can be pro-apoptotic [81].
High levels of the four DNA glycosylases, NEIL1, NEIL2, OGG1, and NTHL1 are found in the Q1 tumors, and these remove oxidized DNA bases and also TMZ generated N7-MeG and N3-MeA [82].
In Q1 tumors the main route of TMZ resistance appears to come from suppression of the canonical MMR pathway, halting futile cycling of O6MeG-DNA repair in DNA synthesis (see [83]), a well-reported mechanism of TMZ resistance [82, 84–87]. We find uniformly low levels of all MMR variants; MutLα, MutLβ and MutLγ, and also both MutSα and MutSβ, in this quartile.
Of the three major pathways that are capable of repairing dsDNA breaks, the NER pathway appears to predominate in these tumors. Moreover, global, rather than translational NER is suggested by the RAD23A/ERCC6 ratio. Low levels of the 9-1-1 complex proteins, including MUTYH1 [88], indicate that MUTYH1/CHK1/ATR dsDNA break sensor/signal pathway may not be operational in this cohort.
We find elevated levels of the canonical telomerase pathway genes; RTEL1, TELO2, and TERT expressed in this cohort, and it has been previously been shown that elevated levels of TERC, TERT and RTEL1, correlated with poor outcome in GBM [89].
The expected effect of PAM-OBG in the first quartile
The pattern of DNA repair pathways indicates poorly functional BER and MMR pathways and that PAM-OBG derived HOPdG lesions will slow ALKBH3 removal of TMZ generated DNA-methyl lesions. The ability of these cells in this cohort to remove ICLs is quite limited, given the very low expression levels of the components of the Fanconi anemia core complex, components of HR, and components of NHEJ [70]. ICLs, induced by acrolein, can be removed at modest rates via NER, as we know that cells derived from patients with xeroderma pigmentosum, with dysfunctional NER, show sensitivity toward acrolein [67].
The second and third quartiles
MGMT appears to be the main mechanism of TMZ resistance in Q2 and Q3, and the tumors in Q2 have high expression of STAT3/IL6, indicative of long MGMT biological half-life.
Short Patch BER (BER-SP) is DNA polymerase beta (POLB) dependent whereas PCNA-dependent Long Patch BER (BER-LP) is more promiscuous, using POLE, POLD or POLB [90]. The activity of POLB, also controlled by post-transcriptional modification and acylation by EP300, is high in Q3 and low in Q2, which essentially suppresses BER-SP but not BER-LP [91]. The difference in the usage of short/long patch BER in the two groups may lead to a tendency for evolutionary selection of XRCC1 based DNA repair, as POLB forms a complex with XRCC1 [92] or for PCNA based repair as POLE forms a tight complex with PCNA [93].
There is also a clear difference in the usage of MMR complexes, with the more aggressive Q2 GBM using MutLβ and MutLγ more than MutLα, and also MutSβ rather than MutSα. The canonical human MMR complex consists of MutSα (MSH2/MSH6) and MutLα (MLH1/PMS2), which is part of the ATR/CHK1 and ATM/CHK2 phosphorylation signaling system [94]. TMZ causes activation of the ATM and ATR kinases, phosphorylation of CHK1, CHK2, and p53, and triggering of the G(2)/M arrest via ATR/ATR MutSα/MutLα phosphorylation. Activation of ATM and of ATR has been implicated in the pro-apoptotic response to ionizing radiation. The lower MutSα/MutSβ ratio in Q3 compared to Q2, and loss of MSH6 signaling to ATR/CHK1 may explain these tumors sensitivity to chemoradiotherapy [95, 96]. In the Q1 cohort, NER appeared to be the major dsDNA break/ICL repair pathway, whereas in Q3 the GBM were biased toward NHEJ/MMEJ [97], and to HR, with FA core complex functionality [71]. Many of the tumors in Q2 lack major components of NER, HR and NHEJ/MMEJ pathways and may use hybrid repair process for eliminating ICLs and DNA double-strand breaks (DSBs).
The expected effect of PAM-OBG in the second and third quartiles
The expression of MGMT, with STAT3 and IL6, indicates that direct repair is the main means of overcoming TMZ toxicity. The generation of O6BG, from PAM-OBG, in these tumors will cause sensitization via MGMT inhibition. The tumors reliant on BER-LP will be highly sensitive to MMR repair futile cycling of O6MeG [71].
Acrolein lesions can be recognized by FA and repaired by HR in competent cells, but these repairs are slow and cause replication stalls, making cells highly vulnerable to daily cycles of chemoradiation.
The final quartile
These GBM have low MGMT and STAT3/IL6 levels explaining the better response to therapy. These tumors have poor BER and MMR pathway activity, and DSB/ICL repair appears to be a function of global, not translational, NER. They express some canonical NHEJ proteins but there is little evidence for FA core complex functionality.
The expected effect of PAM-OBG in the final quartile
Denuding the low levels of MGMT expressed by these cells will further sensitize them to TMZ. Their ability to deal with acrolein lesions, especially ICLs, appears to be poor.
MPG and patient outcome
In Supplementary Figure 5 we show three patient survival curves, with respect to MPG levels with Supplementary Figure 5A showing the whole patient population (n = 110). In the 71% of patients with low levels of MPG median survival is 11.8 months, the 29% of the population with high MPG have a median survival of 13.6 months, p = 0.023. This would appear to be in contradiction of the basic finding of Agnihotri and co-workers [39].
We stratified the patient population with respect to expression of XRCC1 (BER-SP), with one sub-population (n = 33) with high XRCC1 and the second sub-population (n = 77) with low XRCC1. We examined the effects of MPG in these two sub-populations.
In GBM with low BER-SP activity (low XRCC1 levels), MPG correlates with poor outcomes, with low MPG correlated to median survival of 15.2 months and high-level MPG to only 7.9 months, p = 0.02, Supplementary Figure 5B. In the subpopulation with high XRCC1 levels, MPG aided survival with patients with high MPG levels having a median survival of 14.1 months, but those with low MPG levels having a median survival of only 7.7 months, p = 0.015, Supplementary Figure 5C.
It, therefore, appears that MPG can remove toxic TMZ-adducts, including perhaps O6MeG, in cells utilizing BER-LP, whereas in GBM with efficient BER-SP, MPG sensitizes GBM to treatment. MPG toxicity in BER-SP, but not BER-LP, is most likely due to mitochondrial DNA repair directed apoptosis described by Fishel and co-workers [98]. It was shown that MPG repair of methyl lesions in mtDNA initiates mitochondrial driven apoptosis, in cases when the MPG glycosylase activity is higher than the downstream repair processes in BER-SP, causing an overload in AP-sites and DNA breaks.
The expected effect of PAM-OBG on MPG-based DNA repair
MPG can excise some oxidative adducts, including some generated by acrolein [99]. MPG levels correlate with POLB, MGMT, and ALKBH3 Supplementary Materials, the former being complexed with XRCC1 in BER-SP and the latter pair directly removing adducts from DNA. In these cells, the generation of O6BG- and acrolein adducts, from PAM-OBG, will sensitize the tumor cells to chemoradiation.
PAM-OBG sensitizes GBM cells toward alkylating agents in vitro via a MAOB mechanism
We titrated primary GBM cells, GBM157, with TMZ or BCNU in the presence and absence of PAM-OBG or O6BG, Figure 3A and 3B. GBM157 cells are resistant to both TMZ and BCNU, as they have high levels of MGMT and the IC50 values are ≈ 720 μM and > 150 μM, respectively.
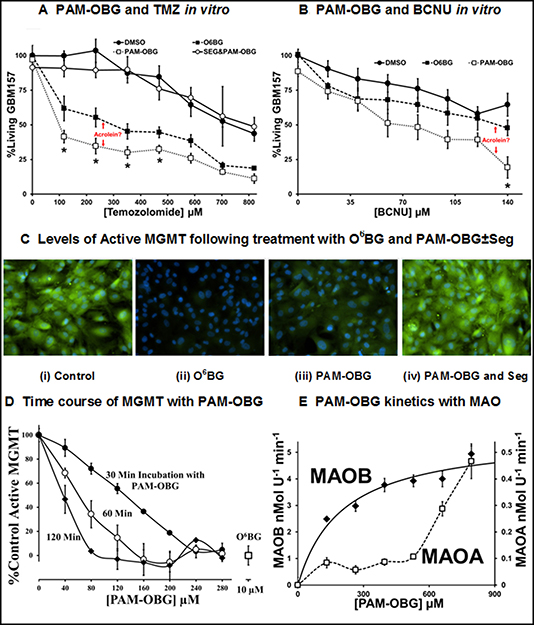
Figure 3: PAM-OBG is a MAOB sensitive prodrug. (A and B) show the differential effects of O6BG and PAM-OBG, on the toxicity of TMZ (A) and BCNU (B), measured at 24 hours, n = 3 and 4 respectively, in primary GBM cell cultures. In both cases, PAM-OBG is a better potentiation agent than is O6BG. (A) also shows that the synergistic effect of co-administration of PAM-OBG and TMZ is MAOB dependent, as the addition of selegiline abolishes potentiation. The t-test was used to compare cell numbers of the O6BG and PAM-OBG treated cultures at each TMZ or BCNU concentration and those pairs that are statistically significantly different at the p < 0.05 level are identified with a ‘*’. (C and D) demonstrate that PAM-OBG depletes active MGMT levels, with active MGMT labeled by O6PGG and visualized by ‘clicking’ with azido-FITC. The green FITC signal seen in (C) is due to the presence of active MGMT in GBM cell cultures. Treatment with 10 μM O6BG (ii) or 100 μM PAM-OBG (iii) drops this fluorescence with respect to the control (i). The loss in signal MGMT caused to PAM-OBG can be abolished by inhibiting MAOB with a pre-incubation of 10 μM Selegiline (iv). The effect of incubation time and PAM-OBG concentration on the loss of active MGMT in cultured glioma, at n = 4, has the line-shape of an enzymatic transformation of PAM-OBG, (D). Using recombinant human MAOA or MAOB, n = 4, the kinetics of PAM-OBG oxidation show that PAM-OBG is a good MAOB substrate, and the fit indicates that the Km is 200 μM for MAOB, but MAOA is a very poor substrate with a Km of > 700 μM (E).
In cells incubated with 10 μM O6BG, there was sensitization toward TMZ and BCNU, with IC50 values dropping to ≈ 280 μM and ≈ 130 μM, respectively. These data are consistent with the known MGMT drug resistance to TMZ and BCNU because TMZ is more efficient at generating O6G adducts than is BCNU [82, 100, 101].
We demonstrate that the sensitization observed in the presence of PAM-OBG is due to MAOB directed catalysis by pre-incubating cells with selegiline, a MAOB specific inhibitor, followed by PAM-OBG/TMZ. When MAOB is inhibited we observe no synergistic effect of PAM-OBG and either DNA alkylating agent.
PAM-OBG was a better sensitizer than was O6BG with both chemotherapeutics, with the IC50 falling to ≈ 30 μM for TMZ and to only ≈ 60 μM in the case of BCNU. We believe that this enhancement of toxicity observed with PAM-OBG vs. O6BG is due to the generation of acrolein from the MAOB catalytic oxidation of PAM-OBG.
PAM-OBG only denudes glioma cells MGMT when MAOB is active
It has been demonstrated that ubiquitination, and subsequent proteolysis, of alkylated MGMT is more rapid than that of the native, active enzyme [102, 103]. In initial experiments, we incubated primary GBM157 cells in BCNU, O6BG, and PAM-OBG ± Seg and observed that PAM-OBG resulted in higher than canonical MGMT molecular weight bands, which we interpret as evidence for ubiquitination [103, 104]. Western Blots interrogating MGMT, as shown in Supplementary Figure 6, appear to show ubiquitinated high molecular weight bands as observed by Kuo and coworkers [105].
To better observe the alkylation and inactivation of MGMT by PAM-OBG we developed a method to determine the levels of active MGMT using a substrate, O6-propargyl-guanine (O6PGG) that is readily amiable to visualization by linking an azido-functionalized fluorophore via click-chemistry [106, 107]. O6-propargyl-guanine reacts with MGMT to generate a propargyl-thioether that can be covalently linked to azido-PEG-FITC by incubation with Cu(II), and therefore be visualized using fluorescence microscopy.
Primary glioma cells were incubated with an MGMT substrate, either O6BG or PAM-OBG, for a defined time period and then 100 μM O6PGG was added, and 10 minutes later the cells were fixed with paraformaldehyde (PFA). Following permeabilization and washing, thioether-propargyl MGMT was covalently linked to FITC-PEG-azide using Cu(II). Figure 3C (i) shows a representative cell field and the green fluorescence labels active MGMT, and blue DAPI signal, DNA.
Pre-incubation of cells with 10 μM O6BG before application of O6PGG causes (almost) complete loss of active MGMT signal, Figure 3C (ii). A 75% loss of active MGMT is observed with cells incubated with 100 μM PAM-OBG show, Figure 3C (iii). This MAOB dependent loss of active MGMT with PAM-OBG is demonstrated in Figure 3C (iv) where we show that co-incubation of MAOB inhibitor selegiline and PAM-OBG does not alter MGMT levels.
The relationship between PAM-OBG concentration, incubation time, and levels of active MGMT was explored by titrating glioma with PAM-OBG for 0.5, 1 and 2 hours, prior to addition of O6PGG. In Figure 3D we show the drop in (active) propargyl-MGMT levels as a result of PAM-OBG conversion to O6BG. The line-shapes resemble truncated and inverted Michaelis–Menten kinetic plots and suggest that the Km for MAOB with PAM-OBG is between 160 and 200 μM (see 30-minute plot).
We confirmed this kinetic parameter using classical enzymology, titrating hrMAOA and hrMAOB with PAM-OBG and measuring the H2O2 generated from the reaction, via the HRP/Amplex Red method [56], Figure 3D. PAM-OBG is a good MAOB substrate but is a poor MAOA substrate. The data indicate that the Km of MAOB for PAM-OBG is ≈ 200 μM and its Vmax ≈ 13% of that observed with control MAOB substrate, benzylamine.
Acrolein derived from PAM-OBG causes DNA damage
The role of NER in ICL repair is the subject of two reviews, the first by Wood (NER) [108] and the second by Williams, Gottesman, and Gautier (Global/Translation NER and overlap with NHEJ/HR/FA) [109]. CDK7 functionality is integral to translational NER repair, being a core component of the TFIIH complex, and the drug compound SNS-032 is a good inhibitor of this kinase [110–112].
In addition to inhibiting CDK7, SNS-032 also inhibits the cyclin-dependent kinases CDK2, CDK5 and CDK9 [113], however, even though SNS-032 can alter glioma response to hypoxia, it is not toxic toward U87 cells even at very high drug concentrations (500 nM) [114].
In primary acute myeloid leukemia (ALM) SNS-032 acts in a synergistic fashion toward the deoxycytidine memetic drug Cytarabine [115], and it is also known that AML sensitivity, and patient outcome with cytarabine therapy, is linked to polymorphisms in nucleotide excision repair genes [116]. When CDK7 siRNA is used in cells it is found that translational NER is essentially halted, and the cells are sensitized to UV-induced interstrand crosslinks [117]. Translational NER is the main pathway used in the repair of cisplatin-induced ICLs [118], and treatment of cells with both CDK7 siRNA and cisplatin results in a 14-fold increase in toxicity [119].
The potential synergistic effects of PAM-OBG with SNS-032 was examined to determine if acrolein dependent ICLs are generated by the action of MAOB on PAM-OBG. The growth of GBM157 treated with 100 nM SNS-032 slowed by 17% over 24 hours (p < 0.024), but there was no noticeable effect on cell death; see DAPI nuclei in Figure 4A (i) and (ii). GBM157 cells incubated for 24 hours in 280 μM PAM-OBG showed a 10% drop in cell numbers (p < 0.01) and again there was no increase in cell death rates, Figure 4A (iii). Co-incubation of SNS-032 with PAM-OBG is devastating; we observed a 75% drop in viable cell numbers and the nuclei in surviving cells show abnormal structures and evidence of mitotic catastrophe [120, 121], Figure 4A (iv).
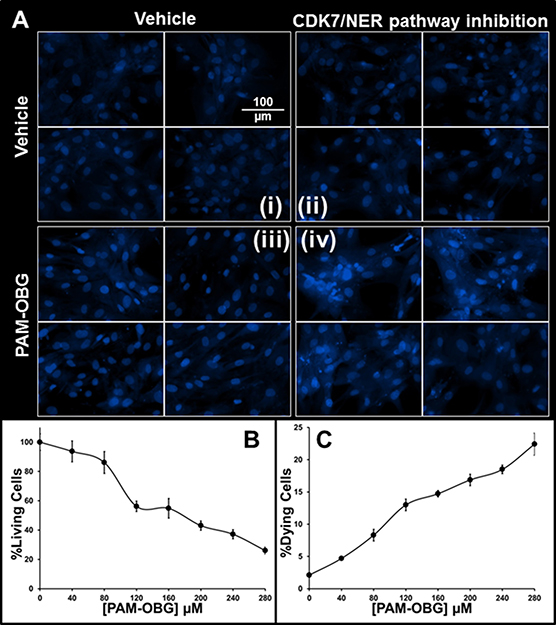
Figure 4: PAM-OBG derived acrolein is toxic in SNS-032 treated cells. (A) shows that acrolein derived from PAM-OBG is DNA damaging and these lesions are normally removed by a CDK7-sensitive NER repair process. Cells were incubated with/without 100 nM SNS-032, an inhibitor of CDK7, to an n = 6, and titrated with PAM-OBG, and labeled with Hoechst 33342 viability dye. In (A) we show four panels of high-resolution images of glioma cell nuclei in control cells (i), cells incubated with SNS-032 (ii), with 280 μM PAM-OBG (iii) and SNS-032 with PAM-OBG (iv). Only in glioma incubated for 24 hours with both PAM-OBG and the CDK7 inhibitor do we observe apoptotic cell nuclei and fragmented nuclei that are diagnostic of mitotic catastrophe. The fall in viable cells numbers and increase in dying cells 24 hours after treating GBM157 cells with SNS-032 and PAM-OBG is shown in (B and C), respectively.
In Figure 4B and 4C, we show the viable cells numbers and levels of dead/dying observed in cells treated with SNS-032 and titrated with PAM-OBG. PAM-OBG is highly toxic in cells with NER pathway inhibition. As we know that PAM-OBG generates acrolein, and we also know that acrolein generates ICLs and that the NER DNA repair pathway is sensitive toward SNS-032, it is reasonable to conclude that in glioma cells PAM-OBG gains part of its toxicity from acrolein-induced DNA-adducts.
Sensitizing intracranial primary GBM tumors to BCNU and CCNU with PAM-OBG
In Figure 5A we show animal survival curves of mice with intracranial GBM157 xenografts treated with BCNU or CCNU with/without PAM-OBG.
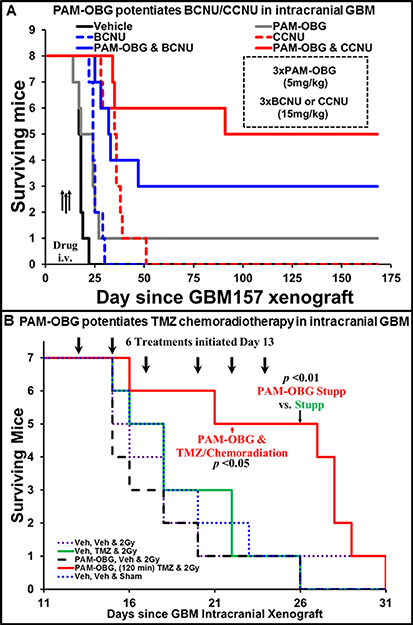
Figure 5: PAM-OBG potentiates the treatment of intracranial GBM tumors in mice. Figure 5 reports the survival curves are two studies where mice with intracranial GBM157 tumors were treated with PAM-OBG. (A) shows that in addition to potentiating the anti-cancer activity of BCNU and CCNU, it can increase survival of tumor-bearing animals as a mono-agent. Analysis of the survival curves shows that PAM-OBG increases survival of the animals by 6-fold, compared with alkylating agent monotherapy. (B) shows the effectiveness of PAM-OBG in a TMZ chemoradiotherapy intracranial mouse model. Beginning on day 13, post-xenograft, mice were given six rounds of intravenous PAM-OBG, TMZ by gavage two hours later, and then an hour later, 2Gy whole head radiation. This combined therapy gave a half-survival time of 27.5 days, more than 8 days more than animals who received only TMZ/2Gy irradiation. This highly aggressive GBM was unresponsive to treatment with radiation, PAM-OBG as monotherapy or to TMZ and 2Gy, with all control groups being statistically identical.
Control animals received two tail vein injections of vehicle, two hours apart, on days 10, 12 and 14 post-xenograft. This cohort reached an ethical end-point at a median time of 16.8 ± 1.6 days. Mice that were given PAM-OBG (5 mg/Kg) performed better, reaching a median survival time of 22.17 ± 4.85 days, and one animal was symptom-free, and healthy, for the full length of the 170-day study. Three intravenous injections of 15 mg/Kg of BCNU or CCNU were able to extend life some 7 and 18 days beyond the vehicle controls, with the median ethical end-points being reached at 24 ± 2.5 and 35 ± 4.8 days respectively. If the survival enhancement of combining PAM-OBG with the alkylating agent was additive we would expect that mice treated with BCNU/PAM-OBG to have a survival midpoint of 29.2 ± 6.7 days, yet the cohort that received this treatment had a 50% survival value of 92 ± 44 days. CCNU in combination with PAM-OBG proved to perform much better than the additive 39.9 ± 9.2 day midpoint, as this combination gives a value of 152.6 ± 64 days for 50% animal survival.
In addition to improving survival the combination of BCNU/CCNU with PAM-OBG in these mice we noted general health indicators, pink eyes, highly active behavior during handling, and they also put on weight, gaining ≈ 4.5 and 6 grams respectively.
Sensitizing intracranial primary GBM tumors to TMZ/radiation with PAM-OBG
Patients are of course typically treated with TMZ and radiation, and not BCNU or CCNU, and we wished to use PAM-OBG with chemoradiation in a mouse intracranial GBM xenograft model.
Without knowing the pharmacokinetics of PAM-OBG in this model we carried out a provisional study where we varied the time between intravenous PAM-OBG injection and receipt of TMZ by gavage, followed by whole head radiation an hour later. As we did not know the effectiveness of individual therapies (radiation, TMZ and PAM-OBG), we used a low GBM inoculum in the xenograft and treated the animals early after this procedure, on days 8, 10 and 12. This was done to stagger mouse survival between the groups by allowing treatments to have high efficacy on the model.
The survival curves of this initial study are shown in Supplementary Figure 7 and clearly demonstrated that the optimum time between injecting PAM-OBG and TMZ gavage was between 90 and 150 minutes. The study was halted after 170 days due to two animals reaching an ethical end-point unrelated to the xenograft (two of the mice had ulcerated bite wounds).
In Figure 5B we show the results of PAM-OBG on chemoradiotherapy, in a ‘mini-Stupp’ protocol.
We deliberately used a late treatment intracranial model, where the animals have a high tumor burden before treatment, in an attempt to recapture the conditions found in GBM patients who have very poor outcomes. In this model, we initiated therapy four days before the typical median survival of untreated mice. For the study, we required 35 animals, in 5 groups of 7 animals (the number of slots in our mouse-body radiation shield). However, we gave 50 mice primary GBM157 xenografts, a 43% excess because we knew a large fraction would reach an ethical end-point prior to the start of treatment. On day 13, when we randomized the heaviest 35 mice into five treatment groups of n = 7 animals, 11 mice (22%) had arrived at an ethical end-point.
Treatment was given three days a week (M/W/F), for two weeks, but mice were examined daily throughout the study. In this ‘mini-Stupp’ model the treatment group of mice received 5 mg/Kg intravenous PAM-OBG, two hours later they were given 10 mg/Kg of TMZ by gavage, and then one hour later they received 2Gy of whole head radiation. There were four groups of control animals, the vehicle group received Veh/Veh/sham irradiation and reached a median lifespan of 18.7 ± 3.8 days. The radiation group, Veh/Veh/2Gy had a survival mid-point of only 17.2 ± 3 days. In mice given six treatments of TMZ chemoradiation, Veh/TMZ/2Gy, survival was 18.9 ± 3.9 days. PAM-OBG monotherapy indicated that PAM-OBG is not a radiosensitizer as giving PAM-OBG three hours before radiation, resulting in a median lifespan of only 16.7 ± 3.2 days. There was no statistically significant difference in the survival curves of any of the four control cohorts. In the treated group, PAM-OBG/TMZ/2Gy, the mid-point survival was very much extended, reaching 27.5 ± 7.79 days, and this group achieved statistical significance, with respect to the chemoradiation group on day 22 at p < 0.05 and day 26 the p-value fell below 0.01, as on day 26 there were no survivors in the Veh/TMZ/2Gy group, but 70% of the treatment group were alive.
DISCUSSION
An extensive analysis of mRNA expression of different components of DNA repair pathways and GBM patient outcome indicates defects in the ability of aggressive GBM to repair ICLs. Our analysis supports the postulate that GBMs have evolved the ability to rapidly evolve by downregulating some DNA repair pathways so as to increase genomic instability and mutation rates. A loss in DNA replication fidelity, due to downregulation of DNA repair pathways as a general feature in cancer has been postulated by others [122–124]. The most aggressive and lethal GBM’s are characterized by having high levels of ALKBH3, low levels of canonical MMR and appear to rely on NER for the repair of dsDNA breaks and the removal of ICLs. In patients that succumb to GBM between 5–12 months MGMT, perhaps potentiated by STAT3 phosphorylation increases in pre-treatment MGMT steady-state levels, and short patch BER appear to the main means of TMZ chemotherapeutic resistance. Patients who survive between 12.5 and 18 months tend to have relatively high levels of MGMT, but no STAT3 potentiation, and are characterized by having high MMR levels. Those patients who survive longer than 18 months are characterized as having low MGMT levels, fully functional MMR and a wide range of repair systems for dsDNA breaks and ICLs.
PAM-OBG is a MAOB specific prodrug that generates O6BG and acrolein after enzymatic oxidation. MAOB levels are higher in low-grade glioma and in GBM than in the body’s other tissues. Thus PAM-OBG can ablate MGMT activity and create acrolein-DNA adducts in glioma cells, but not in tissues with low levels of MAOB. We have demonstrated that PAM-OBG is able to generate O6BG inside glioma, via MAOB, and that this O6BG then reacts with MGMT inactivating it. PAM-OBG treatment of glioma sensitizes cells to alkylating agents in vitro and in vivo. The acrolein generated by MAOB catalyzed immolation of PAM-OBG causes DNA damage and these acrolein derived lesions are highly toxic. Acrolein generated by PAM-OBG potentiates the toxicities of alkylating agents BCNU and TMZ and we demonstrate that the acrolein generated ICLs are normally repaired by the glioma translational NER pathway.
PAM-OBG ablates glioma MGMT in mouse intracranial models, potentiating TMZ chemoradiation, however, PAM-OBG does not itself potentiate radiotherapy. The increased survival of animals treated with PAM-OBG and TMZ/radiation is due to the generation of O6BG, inhibiting MGMT, increasing the steady state level of TMZ-induced O6MeG in DNA and also to the formation of acrolein adducts.
We believe that subject to the results of toxicity studies, that PAM-OBG could aid the treatment of GBM patients as an addition to current best treatment, the Stupp protocol. In addition, PAM-OBG may be useful in salvage therapy when used in combination with β-chloro-nitrosoureas like BCNU and CCNU or cisplatin-based PVC therapy.
MATERIALS AND METHODS
Materials
Unless otherwise indicated reagents were sourced from Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO, USA). Mice were obtained from Charles River Laboratories, Inc. (Wilmington, MA, USA).
Human rights and informed consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964 and later versions. Informed consent or substitute for it was obtained from all patients for being included in the study.
Permission
The animal research in mice was conducted according to Institutional Animal Care and Use Committee (IACUC) Protocol AUP-0315-0016, approved by the IACUC of Methodist Hospital. All animal care procedures conformed to the Guide for the Care and Use of Laboratory Animals (National Research Council, National Academy Press, Washington DC 1996, USA). Glioma tumors and resulting cultures used were from de-identified patient tissues and have no identifiable private information under IRB Protocol 00014547.
Tumor samples
A GBM tumor was taken at the time of excision and given the laboratory ID of GBM157. It was washed in Phosphate Buffered Saline (PBS, Fisher Scientific, Waltham, MA), chopped with a scalpel and homogenized in a BeadBug™ homogenizer (Benchmark Scientific, Inc. Edison, NJ) using 1.5 mm Zirconium beads, in an equal volume of PBS. An aliquot was diluted 1:1 in Matrigel™ and injected into the flanks of two Balb/c mice. Tumors were passaged twice in Balb/c mice and then expanded in nude mice (NU-Foxn1Nu) for three passages, harvested, frozen and archived.
Intracranial models
For the generation of GBM intracranial xenografts flank tumors were resurrected from frozen stocks and passaged in nude mice (NU-Foxn1Nu) twice and a donor tumor was harvested when the volume was approximately 1.5 cm3. The tumor was homogenized using a BeadBug™ homogenizer, in an equal volume of PBS and then diluted 1:1 in Matrigel™ and mice were given intracranial xenografts with 5 μl percutaneous injections directly into the brain via the postglenoid foramen [57, 125, 126].
PAM-OBG and BCNU and CCNU were given as intravenous injections of 100 μl using 20% Kolliphor® EL emulsification agent. TMZ was given as 100 μl oral dose dissolved in commercial gavage solution.
A pair of brain tissue sections, labeled with H&E or with DAP/V9 antibody, taken from a control animal with an intercranial xenograft is shown in Supplementary Figure 8.
Irradiation
We used a modification of the mouse irradiation holding system demonstrated by Grasso and co-workers [127]. Mice were anesthetized using an Isoflurane chamber and were then given 1 mg/kg of the liquid anesthetic Dexmedetomidine. Mice were transferred into 50 ml Conical Centrifuge Tubes, which had had the ends cut off, allowing their noses to protrude from the conical ends. Domestic pipe-lagging expanded polyurethane insulation was custom cut to hold the mice in position. Seven mice were placed into a pre-warmed (37°C), custom-made radiation-shield, which has seven ports to hold the mice in centrifuge tubes, with their heads above the shield. The shield housing was made from hollow aluminum centrifuge tube holder and the inner void is filled with nickel-coated lead bird shot. This design of shield was chosen as it can be fabricated in workshops that lack the environmental protection measures against aerosol lead particles. The shield containing the mice was placed in the pre-calibrated, irradiation chamber ‘safe’ on its turntable and irradiated with 2Gy radiation from a cesium source rod.
After irradiation the mice were removed from the tube, given 1 mg/Kg Antisedan (Dexmedetomidine antidote), and placed in cages, lined with paper towels, sitting on heating pads (37°C). They were allowed to gradually come to, before being transferred into their cages.
Cultured cells
Cells from the second aliquot of GBM157 homogenate were grown in Dulbecco’s modified Eagle’s medium (DMEM) with fetal bovine serum (FBS, 20%), 1U GlutaMax™, sodium pyruvate (1 mM), penicillin (100 U/ml), and streptomycin (100 mg/ml).GBM157cells are spontaneously immortal and were frozen at the fourth passage and used between seventh and ninth passages.
Glioma cells were grown to achieve confluency 24 hours after drug/vehicle dosing in Costar 96-well plates (Corning, NYC, NY, USA).
Fluorescence microscopy
Images were captured using a Nikon Eclipse TE2000-E at 4×, 20× or 30× magnification using a CoolSnap ES digital camera system (Roper Scientific) containing a CCD-1300-Y/HS 1392-1040 imaging array that is cooled by Peltier. Images were recorded and analyzed using Nikon NIS-Elements software (Elements 3.22.11). All images were saved as JPEG2000 files using Nikon NIS-Elements. The emission of FITC-labeled MGMT was collected using ex 450–490 nm, em 500–550 nm and Hoechst 33342 using ex 325–375 nm, em 435–485 nm.
Cell viability
Cells were incubated for 30 minutes with Hoechst 33342 (10 μM) and then were fixed with ice-cold 4% paraformaldehyde. We conducted cell counts in center field at ×4 or ×20 magnification, depending on cell density. Dead/dying cells were identified as having condensed nuclei with signal intensities over threefold that of the median cell nuclei or being identified as fragmented.
Measurement of active MGMT levels
Active MGMT was labeled by incubating cells with 100 μM O6PGG for 10 minutes, prior to fixation. The copper-catalyzed azide-alkyne cycloaddition was performed in 100 μM CuSO4, 500 μM THPTA ligand, 5 mM ascorbate and 10 μM FITC-azido PEG, typically overnight at room temperature [128].
Measurements of MAO activity by H2O2 generation
Amplex Red (150 mM), and HRP (3 U/ml) were incubated with rhMAOs (1 U/ml, Sigma) in pH 7.4 buffer (50 mM KPi, 0.015% lauryl maltoside) at 37°C for 15 min in 96-well format (75 mL volume per well) [56]. The formation of fluorescent resorufin (ex 500–560 nm, em 565–625 nm) was measured in a BioTeck Synergy HT spectrophotometer. The maximal signal generated from Amplex Red (150 nmol/ml) was established by the addition of H2O2 (1 mM) to each well at the end of the assay period.
Synthesis
O6-propargylguanine
O6-Propargylguanine was synthesized as reported by Griffin and colleagues [129]. Propargyl alcohol (1.2 mmol) was dissolved in anhydrous THF and cooled to 0°C. A solution of sodium hydride (2 mol equiv) in dry THF was added and stirred for 30 min at 0oC. 2-Amino-6-chloropurine (1 mol equiv) was added and the mixture was heated to reflux under N2 until TLC analysis confirmed consumption of starting material. The reaction mixture was neutralized with glacial acetic acid, and the solvent was removed in vacuo. The product was purified by chromatography on silica (EtOAc: EtOH 9:1) and recrystallization from ethanol.
FITC-azido-PEG
For ease of purification we used amino-PEG5000-azide (Sigma Cat#JKA5239) as our starting material for an azido-clickable fluorophore, as the product can easily be separated from starting materials. Amino-PEG5000-azide trifluoroacetate salt was added to fluorescein isothiocyanate in N,N-diisopropyl ethylamine (200 μL) and 5 ml anhydrous DMF, then stirred overnight. The resulting dark orange reaction mixture was dialyzed against 500 ml deionized water (MWCO = 0.5–1 kDa), then further concentrated and washed by ultrafiltration (Millipore filter packs, MWCO = 3000), centrifuged at 4000 rpm for 6 h.
PAM-OBG
We will publish the synthetic route used and the purification methodology in a future manuscript. Briefly, commercial O6BG is purified by crystallization in ethanol and the N9-secondary amine is protected with chloromethyl pivalate. Aminopropanol was protected using trifluoroacetate. Triphosgene is used to couple the two, via a carbamate, and deprotection of both the pivalate and trifluoroacetate was performed by hydrolysis in 0.1M NaOH. The crude PAM-OBG was then purified by HPLC/recrystallization giving > 2-gram quantities of pharmaceutical grade.
Abbreviations
Alkylation repair homologs (ALKBH), aminopropyl O6-benzylguanine N - carbamate (PAM-OBG), Ataxia Telangiectasia Mutated Serine/Threonine Kinase (ATM), Base Excision Repair (BER), Cyclin-dependent kinase 7 (CDK7), DNA interstrand cross-link (ICL), DNA methyltransferase (MGMT), DNA double strand breaks (DSB), Fanconi anaemia pathway (FA), Fluorescein isothiocyanate (FITC), glioblastoma multiforme (GBM), Homologous Recombination (HR), α/β hydroxy-1, N2-cyclic propano-2′-deoxyguanosine (α-HOPdG & γ-HOPdG), Isocitrate Dehydrogenase (IDH), Long Patch BER (BER-LP), low-grade glioma (LGG), mismatch repair (MMR), Monoamine Oxidase (MAO), N-Methylpurine DNA Glycosylase (MPG), Non-Homologous End-Joining (NHEJ), Nucleotide Excision Repair (NER), O6-benzylguanine (O6BG), O6-methylguanine (O6MeG), O6-propargyl-guanine (O6PGG), Procarbazine, CCNU and Vincristine therapy (PCV), reactive oxygen species (ROS), Selegiline (Seg), Sham irradiation (Sham), Short Patch BER (BER-SP), Temozolomide (TMZ), Tris (3-hydroxypropyltriazolyl-methyl)amine (THPTA), Vehicle (Veh).
Author contributions
M.A.S: Analysis of tumor databases and target selection. in silico modeling and drug design. Also formulated experiments, supervised technicians for all cell and mouse experiments.
D.S.B: Design of prodrug, model selection, data analysis and animal study design.
S.R: Synthesis of GMP PAM-OBG synthetic route. Synthesis, purification, and validation of purity. Synthesis of O6PGG/azido-PEG-FITC purification and validation of purity. MGMT assay development.
ACKNOWLEDGMENTS
Work at the Houston Methodist Hospital: This work was supported by the Donna and Kenneth R. Peak Foundation, The Kenneth R. Peak Brain and Pituitary Center at Houston Methodist Hospital, The Taub Foundation, The Blanche Green Estate Fund of the Pauline Sterne Wolff Memorial Foundation, The John S. Dunn Foundation, The American Brain Tumor Association, The Marilee A. and Gary M. Schwarz Foundation, The Houston Methodist Hospital Foundation, and the Verelan Foundation. We are grateful to the many patients and families whose lives have been changed by the devastating effects of brain tumors who have helped us with our research.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007; 114:97–109.
2. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D, Brandes AA, Hilton M, Abrey L, et al. Bevacizumab plus Radiotherapy–Temozolomide for Newly Diagnosed Glioblastoma. The New England Journal of Medicine. 2014; 370:709–722.
3. Batchelor TT, Mulholland P, Neyns B, Nabors LB, Campone M, Wick A, Mason W, Mikkelsen T, Phuphanich S, Ashby LS, DeGroot J, Gattamaneni R, Cher L, et al. van den Bent. Phase III Randomized Trial Comparing the Efficacy of Cediranib As Monotherapy, and in Combination With Lomustine, Versus Lomustine Alone in Patients With Recurrent Glioblastoma. Journal of Clinical Oncology. 2013; 31:3212–3218.
4. Taal W, Oosterkamp HM, Walenkamp AM, Dubbink HJ, Beerepoot LV, Hanse MC, Buter J, Honkoop AH, Boerman D, de Vos FY, Dinjens WN, Enting RH, Taphoorn MJ, et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a randomised controlled phase 2 trial. Lancet Oncol. 2014; 15:943–953.
5. Kappelle AC, Postma TJ, Taphoorn MJ, Groeneveld GJ, van den Bent MJ, van Groeningen CJ, Zonnenberg BA, Sneeuw KC, Heimans JJ. PCV chemotherapy for recurrent glioblastoma multiforme. Neurology. 2001; 56:118–120.
6. Schmidt F, Fischer J, Herrlinger U, Dietz K, Dichgans J, Weller M. PCV chemotherapy for recurrent glioblastoma. Neurology. 2006; 66:587–589.
7. Brada M, Stenning S, Gabe R, Thompson LC, Levy D, Rampling R, Erridge S, Saran F, Gattamaneni R, Hopkins K, Beall S, Collins VP, Lee SM. Temozolomide versus procarbazine, lomustine, and vincristine in recurrent high-grade glioma. J Clin Oncol. 2010; 28:4601–4608.
8. van den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MC, Kros JM, Carpentier AF, Clement PM, Frenay M, Campone M, Baurain JF, Armand JP, Taphoorn MJ, Tosoni A, et al. Randomized Phase II Trial of Erlotinib Versus Temozolomide or Carmustine in Recurrent Glioblastoma: EORTC Brain Tumor Group Study 26034. Journal of Clinical Oncology. 2009; 27:1268–1274.
9. Reithmeier T, Graf E, Piroth T, Trippel M, Pinsker MO, Nikkhah G. BCNU for recurrent glioblastoma multiforme: efficacy, toxicity and prognostic factors. BMC Cancer. 2010; 10:30.
10. Hayat K, Jones B, Bisbrown G, Baria K, Pigott T. Retreatment of patients with intracranial gliomas by external beam radiotherapy and cytotoxic chemotherapy. Clinical Oncology. 1997; 9:158–163.
11. Mao H, LeBrun DG, Yang J, Zhu VF, Li M. Deregulated Signaling Pathways in Glioblastoma Multiforme: Molecular Mechanisms and Therapeutic Targets. Cancer Investigation. 2012; 30:48–56.
12. Zhang D, Tang B, Xie X, Xiao YF, Yang SM, Zhang JW. The interplay between DNA repair and autophagy in cancer therapy. Cancer Biology & Therapy. 2015; 16:1005–1013.
13. Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010; 44:479–496.
14. Sosa V, Moliné T, Somoza R, Paciucci R, Kondoh H, Lleonart ME. Oxidative stress and cancer: An overview. Ageing Research Reviews. 2013; 12:376–390.
15. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013; 12:931–947.
16. Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: How are they linked? Free Radical Biology & Medicine. 2010; 49:1603–1616.
17. Bobola MS, Kolstoe DD, Blank A, Chamberlain MC, Silber JR. Repair of 3-methyladenine and abasic sites by base excision repair mediates glioblastoma resistance to temozolomide. Front Oncol. 2012; 2:176.
18. André JB, Godelle B. The Evolution of Mutation Rate in Finite Asexual Populations. Genetics. 2006; 172:611–626.
19. Gervais C, Roze D. Mutation Rate Evolution in Partially Selfing and Partially Asexual Organisms. Genetics. 2017; 207:1561–1575.
20. Soeda A, Hara A, Kunisada T, Yoshimura SI, Iwama T, Park DM. The Evidence of Glioblastoma Heterogeneity. Scientific Reports. 2015; 5:7979.
21. Inda MDM, Bonavia R, Seoane J. Glioblastoma Multiforme: A Look Inside Its Heterogeneous Nature. Cancers. 2014; 6:226–239.
22. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suvà ML, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014; 344:1396.
23. Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, Asthana S, Jalbert LE, Nelson SJ, et al. Mutational Analysis Reveals the Origin and Therapy-driven Evolution of Recurrent Glioma. Science. 2014; 343:189–193.
24. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, et al. The Somatic Genomic Landscape of Glioblastoma. Cell. 2013; 155:462–477.
25. Ziv O, Zeisel A, Mirlas-Neisberg N, Swain U, Nevo R, Ben-Chetrit N, Martelli MP, Rossi R, Schiesser S, Canman CE, Carell T, Geacintov NE, Falini B, et al. Identification of novel DNA-damage tolerance genes reveals regulation of translesion DNA synthesis by nucleophosmin. Nature Communications. 2014; 5:5437.
26. Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Research. 2012; 40:D109–D114.
27. Uno M, Oba-Shinjo SM, Camargo AA, Moura RP, de Aguiar PH, Cabrera HN, Begnami M, Rosemberg S, Teixeira MJ, Marie SK. Correlation of MGMT promoter methylation status with gene and protein expression levels in glioblastoma. Clinics. 2011; 66:1747–1755.
28. Switzeny OJ, Christmann M, Renovanz M, Giese A, Sommer C, Kaina B. MGMT promoter methylation determined by HRM in comparison to MSP and pyrosequencing for predicting high-grade glioma response. Clinical Epigenetics. 2016; 8:49.
29. Bady P, Sciuscio D, Diserens AC, Bloch J, van den Bent MJ, Marosi C, Dietrich PY, Weller M, Mariani L, Heppner FL, McDonald DR, Lacombe D, Stupp R, et al. MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta Neuropathologica. 2012; 124:547–560.
30. Mullapudi SR, Ali-Osman F, Shou J, Srivenugopal KS. DNA repair protein O6-alkylguanine-DNA alkyltransferase is phosphorylated by two distinct and novel protein kinases in human brain tumour cells. Biochem J. 2000; 351:393–402.
31. Kohsaka S, Wang L, Yachi K, Mahabir R, Narita T, Itoh T, Tanino M, Kimura T, Nishihara H, Tanaka S. STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol Cancer Ther. 2012; 11:1289–1299.
32. Lee ES, Ko KK, Joe YA, Kang SG, Hong YK. Inhibition of STAT3 reverses drug resistance acquired in temozolomide-resistant human glioma cells. Oncology Letters. 2011; 2:115–121.
33. Rodriguez FJ, Thibodeau SN, Jenkins RB, Schowalter KV, Caron BL, O'Neill BP, James CD, Passe S, Slezak J, Giannini C. MGMT immunohistochemical expression and promoter methylation in human glioblastoma. Appl Immunohistochem Mol Morphol. 2008; 16:59–65.
34. Hemati S, Sayadi A, Mahzooni P, Sirous M. Evaluation of O6-methylguanine-DNA methyltransferase enzyme expression effect on survival of patients with Grade 4 brain astrocytoma. Journal of Research in Medical Sciences. 2014; 19:42–432.
35. Bobola MS, Alnoor M, Chen JY, Kolstoe DD, Silbergeld DL, Rostomily RC, Blank A, Chamberlain MC, Silber JR. O6-methylguanine-DNA methyltransferase activity is associated with response to alkylating agent therapy and with MGMT promoter methylation in glioblastoma and anaplastic glioma. BBA Clinical. 2015; 3:1–10.
36. Qiu ZK, Shen D, Chen YS, Yang QY, Guo CC, Feng BH, Chen ZP. Enhanced MGMT expression contributes to temozolomide resistance in glioma stem-like cells. Chin J Cancer. 2014; 33:115–122.
37. Lalezari S, Chou AP, Tran A, Solis OE, Khanlou N, Chen W, Li S, Carrillo JA, Chowdhury R, Selfridge J, Sanchez DE, Wilson RW, Zurayk M, et al. Combined analysis of O6-methylguanine-DNA methyltransferase protein expression and promoter methylation provides optimized prognostication of glioblastoma outcome. Neuro-Oncology. 2013; 15:370–381.
38. Agarwal S, Suri V, Sharma MC, Sarkar C. Therapy and progression--induced O6-methylguanine-DNA methyltransferase and mismatch repair alterations in recurrent glioblastoma multiforme. Indian J Cancer. 2015; 52:568–573.
39. Agnihotri S, Gajadhar AS, Ternamian C, Gorlia T, Diefes KL, Mischel PS, Kelly J, McGown G, Thorncroft M, Carlson BL, Sarkaria JN, Margison GP, Aldape K, et al. Alkylpurine–DNA–N-glycosylase confers resistance to temozolomide in xenograft models of glioblastoma multiforme and is associated with poor survival in patients. The Journal of Clinical Investigation. 2012; 122:253–266.
40. Agnihotri S, Burrell K, Buczkowicz P, Remke M, Golbourn B, Chornenkyy Y, Gajadhar A, Fernandez NA, Clarke ID, Barszczyk MS, Pajovic S, Ternamian C, Head R, et al. ATM regulates 3-Methylpurine-DNA glycosylase and promotes therapeutic resistance to alkylating agents. Cancer Discovery. 2014; 4:1198–1213.
41. Friedman HS, Kokkinakis DM, Pluda J, Friedman AH, Cokgor I, Haglund MM, Ashley DM, Rich J, Dolan ME, Pegg AE, Moschel RC, McLendon RE, Kerby T, et al. Phase I trial of O6-benzylguanine for patients undergoing surgery for malignant glioma. J Clin Oncol. 1998; 16:3570–3575.
42. Ranson M, Hersey P, Thompson D, Beith J, McArthur GA, Haydon A, Davis ID, Kefford RF, Mortimer P, Harris PA, Baka S, Seebaran A, Sabharwal A, et al. Randomized trial of the combination of lomeguatrib and temozolomide compared with temozolomide alone in chemotherapy naive patients with metastatic cutaneous melanoma. J Clin Oncol. 2007; 25:2540–2545.
43. Quinn JA, Pluda J, Dolan ME, Delaney S, Kaplan R, Rich JN, Friedman AH, Reardon DA, Sampson JH, Colvin OM, Haglund MM, Pegg AE, Moschel RC, et al. Phase II Trial of Carmustine Plus O6-Benzylguanine for Patients With Nitrosourea-Resistant Recurrent or Progressive Malignant Glioma. Journal of Clinical Oncology. 2002; 20:2277–2283.
44. Quinn JA, Jiang SX, Reardon DA, Desjardins A, Vredenburgh JJ, Rich JN, Gururangan S, Friedman AH, Bigner DD, Sampson JH, McLendon RE, Herndon JE 2nd, Walker A, et al. Phase II trial of temozolomide plus O6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J Clin Oncol. 2009; 27:1262–1267.
45. Sharpe MA, Baskin DS. Monoamine oxidase B levels are highly expressed in human gliomas and are correlated with the expression of HiF-1alpha and with transcription factors Sp1 and Sp3. Oncotarget. 2016; 7:3379–3393. https://doi.org/10.18632/oncotarget.6582.
46. Callado L, Garibi J, Meana JJ. Gliomas: Role of Monoamine Oxidase B in Diagnosis. In Tumors of the Central Nervous System, Volume 1. Hayat MA, editor. Springer Netherlands. 2011; 53–59.
47. Gabilondo AM, Hostalot C, Garibi JM, Meana JJ, Callado LF. Monoamine oxidase B activity is increased in human gliomas. Neurochem Int. 2008; 52:230–234.
48. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discovery. 2012; 2:401.
49. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:11.
50. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson I, Edlund K, Lundberg E, et al. Tissue-based map of the human proteome. Science. 2015; 347:1260419–9.
51. Christmann M, Verbeek B, Roos WP, Kaina B. O6-Methylguanine-DNA methyltransferase (MGMT) in normal tissues and tumors: Enzyme activity, promoter methylation and immunohistochemistry. Biochim Biophys Acta. 2011; 1816:179–190.
52. Freedman NM, Mishani E, Krausz Y, Weininger J, Lester H, Blaugrund E, Ehrlich D, Chisin R. In vivo measurement of brain monoamine oxidase B occupancy by rasagiline, using (11)C-l-deprenyl and PET. J Nucl Med. 2005; 46:1618–1624.
53. Kuhar MJ, Joyce AR. Slow onset of CNS drugs: can changes in protein concentration account for the delay? Trends in Pharmacological Sciences. 2001; 22:450–456.
54. Menzies RA, Gold PH. The Turnover of Mitochondria in a Variety of Tissues of Young Adult and Aged Rats. The Journal of Biological Chemistry. 1971; 246:2425–2429.
55. McFaline-Figueroa JL, Braun CJ, Stanciu M, Nagel ZD, Mazzucato P, Sangaraju D, Cerniauskas E, Barford K, Vargas A, Chen Y, Tretyakova N, Lees JA, Hemann MT, et al. Samson. Minor Changes in Expression of the Mismatch Repair Protein MSH2 Exert a Major Impact on Glioblastoma Response to Temozolomide. Cancer Research. 2015; 75:3127.
56. Sharpe MA, Han J, Baskin AM, Baskin DS. Design and Synthesis of a MAO-B-Selectively Activated Prodrug Based on MPTP: A Mitochondria-Targeting Chemotherapeutic Agent for Treatment of Human Malignant Gliomas. ChemMedChem. 2015; 10:621–628.
57. Sharpe MA, Livingston AD, Gist TL, Ghosh P, Han J, Baskin DS. Successful Treatment of Intracranial Glioblastoma Xenografts With a Monoamine Oxidase B-Activated Pro-Drug. EBioMedicine. 2015; 2:1122–1132.
58. Valley MP, Zhou W, Hawkins EM, Shultz J, Cali JJ, Worzella T, Bernad L, Good T, Good D, Riss TL, Klaubert DH, Wood KV. A bioluminescent assay for monoamine oxidase activity. Anal Biochem. 2006; 359:238–246.
59. Zhou W, Valley MP, Shultz J, Hawkins EM, Bernad L, Good T, Good D, Riss TL, Klaubert DH, Wood KV. New Bioluminogenic Substrates for Monoamine Oxidase Assays. Journal of the American Chemical Society. 2006; 128:3122–3123.
60. Daniels DS, Woo TT, Luu KX, Noll DM, Clarke ND, Pegg AE, Tainer JA. DNA binding and nucleotide flipping by the human DNA repair protein AGT. Nat Struct Mol Biol. 2004; 11:714–720.
61. Moghe A, Ghare S, Lamoreau B, Mohammad M, Barve S, McClain C, Joshi-Barve S. Molecular Mechanisms of Acrolein Toxicity: Relevance to Human Disease. Toxicological Sciences. 2015; 143:242–255.
62. Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer. 2011; 11:467–480.
63. Kehrer JP, Biswal SS. The Molecular Effects of Acrolein. Toxicological Sciences. 2000; 57:6–15.
64. Krokan H, Grafstrom RC, Sundqvist K, Esterbauer H, Harris CC. Cytotoxicity, thiol depletion and inhibition of O6-methylguanine-DNA methyltransferase by various aldehydes in cultured human bronchial fibroblasts. Carcinogenesis. 1985; 6:1755–1759.
65. Lee HW, Wang HT, Weng MW, Hu Y, Chen WS, Chou D, Liu Y, Donin N, Huang WC, Lepor H, Wu XR, Wang H, Beland FA, et al. Acrolein- and 4-Aminobiphenyl-DNA adducts in human bladder mucosa and tumor tissue and their mutagenicity in human urothelial cells. Oncotarget. 2014; 5:3526–3540. https://doi.org/10.18632/oncotarget.1954.
66. Minetti CA, Remeta DP, Johnson F, Iden CR, Breslauer KJ. Impact of α-Hydroxy-Propanodeoxyguanine Adducts on DNA Duplex Energetics: Opposite Base Modulation and Implications for Mutagenicity and Genotoxicity. Biopolymers. 2010; 93:370–382.
67. Curren RD, Yang LL, Conklin PM, Grafstrom RC, Harris CC. Mutagenesis of xeroderma pigmentosum fibroblasts by acrolein. Mutation Research Letters. 1988; 209:17–22.
68. Chen CC, Taniguchi T, D'Andrea A. The Fanconi anemia (FA) pathway confers glioma resistance to DNA alkylating agents. J Mol Med (Berl). 2007; 85:497–509.
69. Kondo N, Takahashi A, Ono K, Ohnishi T. DNA damage induced by alkylating agents and repair pathways. J Nucleic Acids. 2010; 2010:543531.
70. Kottemann MC, Smogorzewska A. Fanconi anemia and the repair of Watson and Crick crosslinks. Nature. 2013; 493:356–363.
71. Michl J, Zimmer J, Tarsounas M. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. The EMBO Journal. 2016; 35:909–923.
72. Patil AA, Sayal P, Depondt ML, Beveridge RD, Roylance A, Kriplani DH, Myers KN, Cox A, Jellinek D, Fernando M, Carroll TA, Collis SJ. FANCD2 re-expression is associated with glioma grade and chemical inhibition of the Fanconi Anaemia pathway sensitises gliomas to chemotherapeutic agents. Oncotarget. 2014; 5:6414–6424. https://doi.org/10.18632/oncotarget.2225.
73. Johannessen TC, Prestegarden L, Grudic A, Hegi ME, Tysnes BB, Bjerkvig R. The DNA repair protein ALKBH2 mediates temozolomide resistance in human glioblastoma cells. Neuro Oncol. 2013; 15:269–278.
74. Duncan T, Trewick SC, Koivisto P, Bates PA, Lindahl T, Sedgwick B. Reversal of DNA alkylation damage by two human dioxygenases. Proceedings of the National Academy of Sciences of the United States of America. 2002; 99:16660–16665.
75. Fedeles BI, Singh V, Delaney JC, Li D, Essigmann JM. The AlkB Family of Fe(II)/α-Ketoglutarate-dependent Dioxygenases: Repairing Nucleic Acid Alkylation Damage and Beyond. The Journal of Biological Chemistry. 2015; 290:20734–20742.
76. Singh V, Fedeles BI, Li D, Delaney JC, Kozekov ID, Kozekova A, Marnett LJ, Rizzo CJ, Essigmann JM. Mechanism of Repair of Acrolein- and Malondialdehyde-Derived Exocyclic Guanine Adducts by the α-Ketoglutarate/Fe(II) Dioxygenase AlkB. Chemical Research in Toxicology. 2014; 27:1619–1631.
77. Persano L, Pistollato F, Rampazzo E, Della Puppa A, Abbadi S, Frasson C, Volpin F, Indraccolo S, Scienza R, Basso G. BMP2 sensitizes glioblastoma stem-like cells to Temozolomide by affecting HIF-1α stability and MGMT expression. Cell Death & Disease. 2012; 3:e412.
78. Dango S, Mosammaparast N, Sowa ME, Xiong LJ, Wu F, Park K, Rubin M, Gygi S, Harper JW, Shi Y. DNA unwinding by ASCC3 helicase is coupled to ALKBH3 dependent DNA alkylation repair and cancer cell proliferation. Molecular Cell. 2011; 44:373–384.
79. Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, Peer E, Kol N, Ben-Haim MS, Dai Q, Di Segni A, Salmon-Divon M, Clark WC, Zheng G, Pan T, Solomon O, et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature. 2016; 530:441–446.
80. Ougland R, Lando D, Jonson I, Dahl JA, Moen MN, Nordstrand LM, Rognes T, Lee JT, Klungland A, Kouzarides T, Larsen E. ALKBH1 is a Histone H2A Dioxygenase Involved in Neural Differentiation. Stem Cells. 2012; 30:2672–2682.
81. Müller TA, Meek K, Hausinger RP. Human AlkB homologue 1 (ABH1) exhibits DNA lyase activity at abasic sites. DNA Repair. 2010; 9:58–65.
82. Zhang JM. Stevens FG, Bradshaw TD. Temozolomide: Mechanisms of Action, Repair and Resistance. Current Molecular Pharmacology. 2012; 5:102–114.
83. Nickoloff JA, Jones D, Lee SH, Williamson EA, Hromas R. Drugging the Cancers Addicted to DNA Repair. J Natl Cancer Inst. 2017; 109.
84. Liu L, Markowitz S, Gerson SL. Mismatch Repair Mutations Override Alkyltransferase in Conferring Resistance to Temozolomide but not to 1,3-Bis(2-chloroethyl)nitrosourea. Cancer Research. 1996; 56:5375.
85. Liu L, Taverna P, Whitacre CM, Chatterjee S, Gerson SL. Pharmacologic Disruption of Base Excision Repair Sensitizes Mismatch Repair-deficient and -proficient Colon Cancer Cells to Methylating Agents. Clinical Cancer Research. 1999; 5:2908–17.
86. Liu L, Gerson SL. Targeted modulation of MGMT: Clinical implications. Clin Cancer Res. 2006; 12:328–331.
87. Reese JS, Qin X, Ballas CB, Sekiguchi M, Gerson SL. MGMT expression in murine bone marrow is a major determinant of animal survival after alkylating agent exposure. J Hematother Stem Cell Res. 2001; 10:115–123.
88. Hwang BJ, Jin J, Gunther R, Madabushi A, Shi G, Wilson GM, Lu AL. Association of the Rad9-Rad1-Hus1 Checkpoint Clamp with MYH DNA Glycosylase and DNA. DNA Repair. 2015; 31:80–90.
89. Walsh KM, Wiencke JK, Lachance DH, Wiemels JL, Molinaro AM, Eckel-Passow JE, Jenkins RB, Wrensch MR. Telomere maintenance and the etiology of adult glioma. Neuro-Oncology. 2015; 17:1445–1452.
90. Fortini P, Pascucci B, Parlanti E, Sobol RW, Wilson SH, Dogliotti E. Different DNA Polymerases Are Involved in the Short- and Long-Patch Base Excision Repair in Mammalian Cells. Biochemistry. 1998; 37:3575–3580.
91. Carter RJ, Parsons JL. Base Excision Repair, a Pathway Regulated by Posttranslational Modifications. Molecular and Cellular Biology. 2016; 36:1426–1437.
92. Pascucci B, Russo MT, Crescenzi M, Bignami M, Dogliotti E. The accumulation of MMS-induced single strand breaks in G(1) phase is recombinogenic in DNA polymerase β defective mammalian cells. Nucleic Acids Research. 2005; 33:280–288.
93. Fujisawa R, Ohashi E, Hirota K, Tsurimoto T. Human CTF18-RFC clamp-loader complexed with non-synthesising DNA polymerase ε efficiently loads the PCNA sliding clamp. Nucleic Acids Research. 2017; 45:4550–4563.
94. Yan S, Sorrell M, Berman Z. Functional interplay between ATM/ATR-mediated DNA damage response and DNA repair pathways in oxidative stress. Cellular and Molecular Life Sciences. 2014; 71:3951–3967.
95. Smith J, Mun Tho L, Xu N, Gillespie DA. Chapter 3 - The ATM–Chk2 and ATR–Chk1 Pathways in DNA Damage Signaling and Cancer. In Advances in Cancer Research. G. F. Vande Woude, and Klein G, editors. Academic Press. 2010; 73–112.
96. Myers K, Gagou ME, Zuazua-Villar P, Rodriguez R, Meuth M. ATR and Chk1 Suppress a Caspase-3–Dependent Apoptotic Response Following DNA Replication Stress. PLoS Genetics. 2009; 5:e1000324.
97. Sharma S, Javadekar SM, Pandey M, Srivastava M, Kumari R, Raghavan SC. Homology and enzymatic requirements of microhomology-dependent alternative end joining. Cell Death & Disease. 2015; 6:e1697.
98. Fishel ML, Seo YR, Smith ML, Kelley MR. Imbalancing the DNA Base Excision Repair Pathway in the Mitochondria; Targeting and Overexpressing N-Methylpurine DNA Glycosylase in Mitochondria Leads to Enhanced Cell Killing. Cancer Research. 2003; 63:608.
99. Tudek B, Zdżalik-Bielecka D, Tudek A, Kosicki K, Fabisiewicz A, Speina E. Lipid peroxidation in face of DNA damage, DNA repair and other cellular processes. Free Radical Biology & Medicine. 2017; 107:77–89.
100. Wu M, Kelley MR, Hansen WK, Martin WJ. Reduction of BCNU toxicity to lung cells by high-level expression of O6-methylguanine-DNA methyltransferase. Am J Physiol Lung Cell Mol Physiol. 2001; 280:L755–L761.
101. Ishiguro K, Zhu YL, Shyam K, Penketh PG, Baumann RP, Sartorelli AC. Quantitative relationship between guanine O6-alkyl lesions produced by Onrigin™ and tumor resistance by O6-alkylguanine-DNA alkyltransferase. Biochemical Pharmacology. 2010; 80:1317–1325.
102. Cheng J, Ye F, Dan G, Zhao Y, Zhao J, Zou Z. Formation and degradation of nitrogen mustard-induced MGMT-DNA crosslinking in 16HBE cells. Toxicology. 2017; 389:67–73.
103. Srivenugopal KS, Yuan XH, Friedman HS, Ali-Osman F. Ubiquitination-dependent proteolysis of O6-methylguanine-DNA methyltransferase in human and murine tumor cells following inactivation with O6-benzylguanine or 1,3-bis(2-chloroethyl)-1-nitrosourea. Biochemistry. 1996; 35:1328–1334.
104. Velu CS, Niture SK, Bailey NI, Srivenugopal KS. MGMT protein turnover in human tumors is mediated by the ubiquitin-proteasome pathway: Phosphorylation-dependent Ub-conjugation by the Skp2-SCF complex. Cancer Research. 2005; 65:1283.
105. Kuo CC, Liu JF, Shiah HS, Ma LC, Chang JY. Tamoxifen accelerates proteasomal degradation of O6-methylguanine DNA methyltransferase in human cancer cells. International Journal of Cancer. 2007, 121:2293–2300.
106. Binder WH, Kluger C. Azide/Alkyne-"Click"; Reactions: Applications in Material Science and Organic Synthesis. Current Organic Chemistry. 2006; 10:1791–1815.
107. Thirumurugan P, Matosiuk D, Jozwiak K. Click Chemistry for Drug Development and Diverse Chemical Biology Applications. Chemical Reviews. 2013; 113:4905–4979.
108. Wood RD. Nucleotide excision repair proteins and interstrand crosslink repair. Environmental and Molecular Mutagenesis. 2010; 51:520–526.
109. Williams HL, Gottesman ME, Gautier J. The differences between ICL repair during and outside of S-Phase. Trends in Biochemical Sciences. 2013; 38:386–393.
110. Arab HH, Wani G, Ray A, Shah ZI, Zhu Q, Wani AA. Dissociation of CAK from Core TFIIH Reveals a Functional Link between XP-G/CS and the TFIIH Disassembly State. PLoS One. 2010; 5:e11007.
111. Compe E, Egly JM. TFIIH: when transcription met DNA repair. Nat Rev Mol Cell Biol. 2012; 13:343.
112. Kodym E, Kodym R, Reis AE, Habib AA, Story MD, Saha D. The small-molecule CDK inhibitor, SNS-032, enhances cellular radiosensitivity in quiescent and hypoxic non-small cell lung cancer cells. Lung Cancer. 2009; 66:37–47.
113. Conroy A, Stockett DE, Walker D, Arkin MR, Hoch U, Fox JA, Hawtin RE. SNS-032 is a potent and selective CDK 2, 7 and 9 inhibitor that drives target modulation in patient samples. Cancer Chemotherapy and Pharmacology. 2009; 64:723–732.
114. Ali MA, Reis A, Ding LH, Story MD, Habib AA, Chattopadhyay A, Saha D. SNS-032 prevents hypoxia-mediated glioblastoma cell invasion by inhibiting hypoxia inducible factor-1alpha expression. Int J Oncol. 2009; 34:1051–1060.
115. Walsby E, Lazenby M, Pepper C, Burnett AK. The cyclin-dependent kinase inhibitor SNS-032 has single agent activity in AML cells and is highly synergistic with cytarabine. Leukemia. 2011; 25:411–419.
116. Strom SS, Estey EH, Outschoorn UM, Guillermo GM. AML outcome: role of nucleotide excision repair polymorphisms in intermediate risk patients. Leukemia & Lymphoma. 2010; 51:598–605.
117. Mourgues S, Gautier V, Lagarou A, Bordier C, Mourcet A, Slingerland J, Kaddoum L, Coin F, Vermeulen W, Gonzales de Peredo A, Monsarrat B, Mari PO, Giglia-Mari G. ELL, a novel TFIIH partner, is involved in transcription restart after DNA repair. Proc Natl Acad Sci U S A. 2013; 110:17927–17932.
118. Enoiu M, Jiricny J, Schärer DO. Repair of cisplatin-induced DNA interstrand crosslinks by a replication-independent pathway involving transcription-coupled repair and translesion synthesis. Nucleic Acids Research. 2012; 40:8953–8964.
119. Liu WX, Liu XY, Yu HU, Chen Y, Hao Q. Effect of cyclin-dependent kinase 7 silencing on cisplatin sensitivity in endometrial carcinoma cells. Molecular Medicine Reports. 2015; 11:1745–1751.
120. Fragkos M, Beard P. Mitotic Catastrophe Occurs in the Absence of Apoptosis in p53-Null Cells with a Defective G1 Checkpoint. PLoS One. 2011; 6:e22946.
121. Milanović D, Firat E, Grosu AL, Niedermann G. Increased radiosensitivity and radiothermosensitivity of human pancreatic MIA PaCa-2 and U251 glioblastoma cell lines treated with the novel Hsp90 inhibitor NVP-HSP990. Radiation Oncology (London, England). 2013; 8:42–42.
122. Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M, Van Allen E, Kryukov GV, Sboner A, et al. Punctuated Evolution of Prostate Cancer Genomes. Cell. 2013; 153:666–677.
123. Broustas CG, Lieberman HB. DNA Damage Response Genes and the Development of Cancer Metastasis. Radiation Research. 2014; 181:111–130.
124. Tubbs A, Nussenzweig A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell. 2017; 168:644–656.
125. Iwami K, Momota H, Natsume A, Kinjo S, Nagatani T, Wakabayashi T. A novel method of intracranial injection via the postglenoid foramen for brain tumor mouse models. J Neurosurg. 2012; 116:630–635.
126. Nilewski LG, Singh M, Baskin DS, Tour JM, Sharpe MA. Transfer of Dyes and Drugs into Cells Using EGFR-Targeted Nanosyringes. ACS Chemical Neuroscience. 2018; 9:107–117.
127. Grasso C, Fabre MS, Collis SV, Castro ML, Field CS, Schleich N, McConnell MJ, Herst PM. Pharmacological Doses of Daily Ascorbate Protect Tumors from Radiation Damage after a Single Dose of Radiation in an Intracranial Mouse Glioma Model. Frontiers in Oncology. 2014; 4:356.
128. Presolski SI, Hong VP, Finn MG. Copper-Catalyzed Azide–Alkyne Click Chemistry for Bioconjugation. Current Protocols in Chemical Biology. 2011; 3:153–162.
129. Griffin RJ, Arris CE, Bleasdale C, Boyle FT, Calvert AH, Curtin NJ, Dalby C, Kanugula S, Lembicz NK, Newell DR, Pegg AE, Golding BT. Resistance-Modifying Agents. 8. Inhibition of O6-Alkylguanine-DNA Alkyltransferase by O6-Alkenyl-, O6-Cycloalkenyl-, and O6-(2-Oxoalkyl)guanines and Potentiation of Temozolomide Cytotoxicity in vitro by O6-(1-Cyclopentenylmethyl)guanine. Journal of Medicinal Chemistry. 2000; 43:4071–4083.